

Abstract
Craniosynostosis is a developmental craniofacial anomaly, resulting in impairment of brain development and abnormally shaped skull. The main cause of craniosynostosis is premature closure of one or more cranial sutures. It usually occurs as an isolated condition, but may also be associated with other malformations as part of complex syndromes. When left untreated, craniosynostosis can cause serious complications, such as developmental delay, facial abnormality, sensory, respiratory and neurological dysfunction, anomalies affecting the eye, and psychological disturbances. Thus, early diagnosis, expert surgical techniques, postoperative care, and adequate follow-up are of vital importance in treating craniosynostosis.
Keywords: Craniosynostosis, development, classification, diagnosis, treatment
INTRODUCTION
During the embryonic development, the cranial vault develops from the mesenchymal tissue. It is first arranged as a capsular membrane around developing brain. Gradually, the outer mesenchymal layer is formed through the process of intramembranous ossification [1-5]. This intramembranous bone growth depends mainly on the direction of the forces that are defined by the growth of the brain. In the developmental period, the brain is surrounded by dural fibers, which are closely related and strongly attached to the sutural system. Calvarial sutures are formed during the embryonic development at the sites of approximation of the membranous bones and later represent the major sites of bone expansion. This process is a combination of i) deposition of osteoid at the sutural margins, ii) surface apposition and resorption (remodeling) of the bone, and iii) centrifugal displacement by the expanding brain [4-10].
The fusion of the sutures is mainly regulated by the dura mater, which interacts with the overlying tissues of the cranial vault. The dura mater provides many important regulators of growth, such as intercellular signals (for example, signaling mediated by fibroblast growth factor [FGF] and transforming growth factor beta [TGF-β] have been suggested to be vital in this process), mechanical signals, and cells which undergo transformation and migrate to the sutures. This complex signaling cascade can be disrupted by a large number of genetic mutations, leading to an abnormal development of the cranial sutures [9-14]. Finally, this may result in a premature fusion of one or more sutures, which is called craniosynostosis [5,8].
CRANIOSYNOSTOSIS
A craniosynostosis is a developmental anomaly which occurs as a consequence of abnormal and non-physiological sutural fusion. In a newborn, the membranous bones of the cranial vault are separated by the intervening sutures. Such arrangement enables the infant’s skull to pass more easily through the birth canal and allows the compensatory growth of skull during the brain growth. When one or more sutures are prematurely closed, the compensatory growth starts perpendicular to the patent sutures since the brain still grows and expands in the direction of lower resistance. The result is an abnormally shaped skull and also, in more severe cases, increased intracranial pressure (ICP), as well as sensory, respiratory and neurological dysfunctions [1,8,9,15,16]. The prevalence of craniosynostosis is 1 in 2100-2500 births [17,18]. According to different studies, the cumulative prevalence of craniosynostoses has risen significantly with no obvious cause [19]. Both environmental factors (e.g., intrauterine fetal head constraint, abnormal position, oligohydramnios, prenatal exposures to teratogens, maternal smoking, and antiepileptic drugs such as valproic acid and phenytoin) and genes (single gene mutations, chromosome abnormalities, and polygenic background) may all be predisposing factors for the disease. Genetic causes account for approximately 20% of all craniosynostoses and are associated with many complications. Most of the genes linked to craniosynostosis are inherited in an autosomal dominant manner. However, in about 50% of the cases, new mutations may also occur [15,20-23]. Craniosynostoses are classified according to the sutures and the frequencies of these different types of craniosynostoses are as follows: Sagittal (≈60%), coronal (≈25%), metopic (≈15%), and lambdoid (≈2%) [1,24,25].
CLASSIFICATION OF CRANIOSYNOSTOSES
Different classifications of craniosynostosis are used depending on the underlying mechanism, presence of other disorders, or number of fused sutures. For instance, if a craniosynostosis develops due to a primary defect of the ossification process it is called primary craniosynostosis. On the other hand, secondary craniosynostosis is the result of known systemic diseases with hematologic or metabolic dysfunction, such as rickets and hypothyroidism. Secondary craniosynostosis can also develop in newborns with microcephaly due to a failure of brain growth or following shunt placement in children with hydrocephalus. Furthermore, craniosynostosis can be classified into syndromic, e.g., as part of Apert, Crouzon, or Pfeiffer syndrome, and more commonly encountered, non-syndromic craniosynostosis, where it develops as an isolated disorder. Simple craniosynostosis is a term used when only one suture fuses prematurely, while complex craniosynostosis is used to describe a premature fusion of multiple sutures [8,26-30].
DIAGNOSTICS
The diagnosis of a typical craniosynostosis is usually clinical and it is commonly diagnosed in the first year of life. The clinical assessment determines the following: i) whether a craniosynostosis is present, ii) whether there are additional features suggesting an associated syndrome, and iii) whether urgent or elective management is required [15,31]. At first, a careful medical history is obtained. To determine the etiology of craniosynostosis, the focus is on the family history of unusual head shapes, prenatal exposure to teratogens (e.g., valproic acid), and evidence of intrauterine constraint due to multiple pregnancy, primiparity, abnormal fetal position, or oligohydramnios. The history also includes possible complications of pregnancy, as well as the evidence of achieved milestones during early life. The clinical examination is an important part where typical signs are looked for, especially possible congenital anomalies (e.g., a broad, radially deviated thumb in Pfeiffer syndrome or syndactyly in Apert syndrome), dysmorphic features of the face (hyper or hypotelorism, hypoplasia of midface, asymmetry, the position, shape and size of the ears), the skull shape from all directions, and the measurement of the head circumference for calculating the cephalic index (the ratio of maximum breadth to maximum length of the skull). Any sutural ridging, prominent blood vessels on the scalp, and the size, shape and tension of the fontanels should also be assessed. For evaluating ICP, ophthalmological examination is of great importance. In cases with increased ICP, papilledema is present [32]. When assessing the functional consequences of the condition, the most important clinical information includes possible airway obstruction, feeding difficulties, eye protection, and signs of increased ICP. Although, the diagnosis of craniosynostoses in typical cases can be made after clinical evaluation, many surgeons also confirm the diagnosis radiologically, especially when a surgical treatment is planned [15,33].
The computed tomography (CT) with three-dimensional (3D) reconstruction is considered the most complete and accurate imaging to diagnose craniosynostosis [34]. With this method, all sutures can be assessed for patency. In addition, the CT scanning allows evaluating the brain for possible structural abnormalities (e.g. ventriculomegaly, agenesis of the corpus callosum, and craniocerebral asymmetry). However, because of the radiation risk, this method should be indicated carefully [15,35].
In comparison to CT, plain radiography is less accurate in visualizing the cranial sutures. It is nevertheless a cost-effective method in infants with a low risk of craniosynostosis. One of its benefits is that general anesthesia is not required [33].
The magnetic resonance imaging (MRI) is an excellent technique for the evaluation of brain, albeit less accurate in visualizing the cranial sutures compared to CT. Generally, MRI is reserved for patients in which CT reveals cerebral anomalies [15,33]. Furthermore, it is an important imaging modality in combination with ultrasound (US) examination, especially in infants with suspected intracranial anomalies and associated complications of craniosynostosis (i.e., syndromic craniosynostoses). Recently, a novel MRI technique has been described, namely gradient- and spin-echo (GRASE) imaging. GRASE imaging can identify the cranial sutures similarly to other 2D imaging modalities. This type of MRI technique minimizes the soft tissue contrast and enhances the bone-soft tissue boundaries. It can therefore reveal normal cranial sutures as hyperintense signal distinguished from the signal void of the cranial bones [36].
US examination is a fast, low-cost, radiation-free method that requires no sedation. However, it is applicable only in cases with open fontanelles. Standard ultrasonography of the calvarial sutures, in the absence of other craniofacial malformations, may be a feasible method for diagnosing simple, non-syndromic craniosynostosis in utero [37]. An important advantage of ultrasonography is the possibility to visualize and follow-up the cranial sutures on every examination, to obtain the best scan for measuring the skull and one or more prematurely fused sutures. In the experienced hands, US may be as reliable as CT [38]. The diagnosis is possible after the first trimester [39].
When a syndromic craniosynostosis is suspected, genetic testing is usually performed. Based on some results, genetic testing of FGF receptor genes (FGFR3 and FGFR2) is advised. It is recommended to all patients presenting with coronal or multisuture synostosis, since these two types are often genetically determined [15,21]; the FGFR2 and FGFR3 as well as the genes for transcription factors (i.e., TWIST and MSX2) are the most frequently mutated genes in these disorders [5,6,8,21]. During the last decades, the understanding of molecular and genetic pathways associated with common forms of craniosynostosis has been increased. To date, mutations in 57 genes have been identified as an underlying cause of craniosynostosis. These genes and transcription factors are fundamental in the skull morphogenesis and, among others, include the FGFR genes, TWIST, MSX2, bone morphogenetic protein (BMP) genes, TGFB2, ERF of ETS transcription factor family, RUNX2, EFNB1, FAM20C, and LMX1B gene [40-43].
TYPICAL FEATURES OF VARIOUS TYPES OF CRANIOSYNOSTOSES
The typical features of various types of craniosynostoses are briefly described below (Table 1 and Figure 1).
TABLE 1.
Clinical features of various types of craniosynostoses

FIGURE 1.
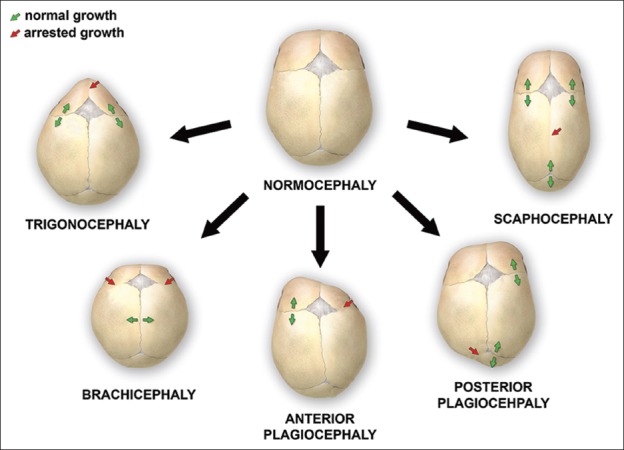
Various deformations of the skull, associated with single-suture synostoses
Scaphocephaly
In this type of synostosis, there is a premature fusion of the sagittal suture. It is commonly observed in premature infants. The head is typically elongated in the anterior-posterior direction and shortened in the bilateral direction. In some children, frontal bossing is present and the ridging of the sagittal suture is palpable. Boys are more frequently affected than girls, with a ratio of 3.5:1 [31,35,44,45].
Anterior plagiocephaly
Anterior plagiocephaly results from a premature fusion of the coronal suture. On the affected side, the forehead is flattened because of arrested growth, and higher supraorbital margins form a characteristic sign on radiographs, known as the Harlequin sign. On the opposite side, the forehead is pushed forward. Additional findings include flat cheeks on the side of synostosis and nasal septum deviation towards the normal side. It is more common in girls than in boys (ratio 2:1). In the case when bicoronal fusion closes prematurely, the condition is called brachycephaly. Here, the head is typically shorter and wider [18,31,35,44-46].
Posterior plagiocephaly
Posterior plagiocephaly is a unilateral lambdoid synostosis. Frontal and occipital bossing can develop contralateral to the affected side. The ipsilateral ear and mastoid can be displaced downward. In the majority of cases, the ear is also displaced in the anteroposterior direction. Clinically, the shape of the head from above can resemble a trapezoid [16,47,48].
Positional plagiocephaly
It is essential yet sometimes difficult to distinguish between synostotic and positional plagiocephaly, which is more common. The latter is caused by repeated pressure to the same area before or after birth. The ipsilateral ear and forehead are usually displaced anteriorly, giving the head a parallelogram shape. The ipsilateral occipital flattening can be accompanied by a contralateral occipital bossing. The male-to-female ratio is 3:1. The effects of the positional plagiocephaly are primarily cosmetic and do not require surgical intervention [16,48].
The number of infants with this type of plagiocephaly has risen over the past several years, which has been associated with the “back to sleep” campaign to reduce the incidence of sudden infant death syndrome (SIDS). The presence of torticollis, prematurity, and gross motor delay can also predispose an infant to positional plagiocephaly [16,44,48,49].
Trigonocephaly
Trigonocephaly results from a premature fusion of the metopic suture. The back of the head is broad and the forehead is narrow and pointed. When viewed from above, the forehead has a triangular shape. The orbits are abnormally close together (hypotelorism) [16,31,35].
Brachycephaly
Brachycephaly is a bilateral coronal synostosis. As a result of the fused coronal suture, the skull is short. The forehead and occipital part are flattened and the frontal bone is prominent and elongated in a vertical direction. The orbits are abnormally separated (hypertelorism) and the Harlequin malformation of the orbits is seen on radiographs [33].
INFLUENCE ON CHILD DEVELOPMENT
Craniosynostosis, especially the syndromic type, is often associated with a higher risk of impaired cognitive development. This is due to restricted growth of the brain and a secondary deformation of the brain tissue caused by overgrown sutures and increased ICP. Many studies have examined the neurological status of children with craniosynostoses. Recently, a longitudinal study was launched, testing children at the start of their schooling. The study followed the neuropsychological development of 182 children with a simple form of craniosynostosis and of a control group, from early childhood to school age [50]. The results showed that, on average, children with a simple form of craniosynostosis exhibited a mild cognitive and academic deficit [50,51].
The developmental gap varies depending on the subtype of craniosynostosis. Children with metopic, unicoronal, or lambdoid synostosis have a higher likelihood of learning disabilities in comparison to children with sagittal synostosis. The highest variations have been found in the intelligence quotient and in computer skills. On the other hand, speech and reading were not significantly affected. In 58% of children, learning difficulties were not detected. Understanding the cognitive development of a child helps in determining the potential risk for developmental gap. With prompt identification and support programs, the cognitive and academic deficit may be prevented or at least considerably reduced [35,50,51].
TREATMENT AND COMPLICATIONS
Similarly to the underlying causes and clinical characteristics, the treatment of craniosynostoses is also very heterogeneous. Most of the uncomplicated, non-syndromic forms may be treated electively. On the other hand, some cases of the syndromic forms require urgent interventions. In severe cases, the focus is on maintaining the airway and nutritional support, eye protection and normal ICP [15].
The most important factors in determining the extent of surgery and surgical modality are the patient age and presentation [52]. Although the surgical treatment of craniosynostoses is most commonly used, the conservative approach may be adopted first, especially in patients with positional plagiocephaly and in cases in which unilateral synostosis is not very pronounced. Normally, the positional plagiocephaly may not require a treatment, although remodeling helmets are beneficial in some severe deformities. In addition, a minimally invasive approach, especially endoscopic suturectomy, may be used in some cases to correct the positional deformities surgically. The remodeling helmets can be used for very young patients (i.e., younger than 6 months) alone or in combination with suturectomy [52-54].
The main objectives are to achieve a normal brain development by providing sufficient space within the skull and a cosmetically acceptable appearance [16,31,35]. The optimal period for the operation remains a subject of debate and is considered to be between 6 and 12 months of age in the case when no signs of increased ICP are present [55]. In addition, this time period represents the most active phase of the brain and head development and is thus optimal for a surgical correction of the head shape and re-ossification of bone defects [56].
According to the clinical criteria and surgical capabilities, an open craniotomy and reconstruction or an endoscopic procedure are considered. After the operation, additional corrections with the helmets may be required and this usually lasts from 4 to 6 months. Both open surgery and endoscopy have advantages and disadvantages. The endoscopic intervention is more appropriate until the age of 6 months. In that period, the cranial bones are still sufficiently flexible and can be manipulated by the endoscope. After 6 months, open surgery is preferred due to the stiffness of the bones. The advantage of endoscopic intervention is a shorter duration of surgery, less blood loss, and faster postoperative recovery. However, frequently, it should be combined with a postoperative use of a remodeling helmet and is suitable only for very young children. After 6 month of age, this technique is usually not effective. The upper limit is about 5-6 months of age and open surgery should be employed thereafter [52-54], because open surgery is more suitable for children older than 6 months as well as for those with complex or syndromic synostosis (Figure 2). Moreover, this approach still provides better possibilities of wide remodeling of the cranial vault and skull base [35,57].
FIGURE 2.
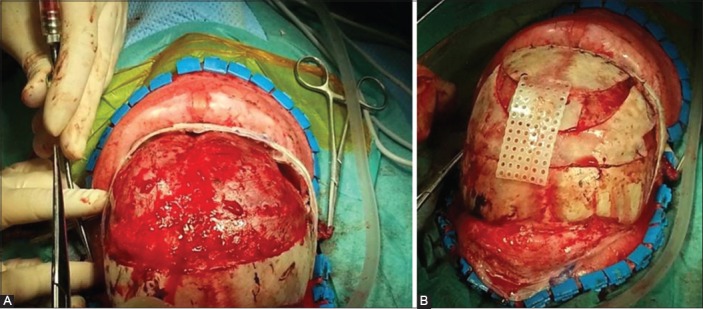
The surgical reconstruction of anterior plagiocephaly. During the operation, the child is placed in a supine position. A coronal skin incision is performed and after the periosteal dissection, the fused right coronal suture is recognized. The bone flap is removed and an extensive remodeling of the orbital bone and forehead follows (A). The final appearance of the skull after the completed surgical reconstruction, filling of bone defects, and their connection with resorbable plates (B).
The complications of surgical treatment include postoperative hyperthermia as the most common complication, next infections such as meningitis, subgaleal and subcutaneous hematoma, rupture of the dura mater, cerebrospinal fluid leakage, and blood loss. Several complications may occur in patients in which a reoperation is needed (Table 2). In general, the complications vary depending on the type of surgery. The endoscopically supported removal of cranial sutures is the intervention with minimal complications even though it does not allow a complete remodeling [58]. The mortality and morbidity rate is 0.1% and may reach up to 50% in the case of severe blood loss [59].
TABLE 2.
Possible complications of surgical treatment in craniosynostoses

After surgery, the treatment is not completed. It is necessary to follow up the patients regularly and to control the growth and circumference of the head, observe possible symptoms of increased ICP and other potential complications. Only through regular follow-ups it is possible to detect prematurely fusing cranial sutures in time and offer the child a reoperation; nevertheless, this is still performed only rarely [33].
CONCLUSION
Craniosynostosis is a rare craniofacial anomaly which may lead to various complications, deformations, and neurological impairment during the child’s development. Early identification and appropriate treatment are therefore vital. The aim of the surgical treatment is to enable the normal brain development and to achieve an acceptable cosmetic effect.
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
- 1.Kjaer I. Neuro-osteology. Crit Rev Oral Biol Med. 1998;9(2):224–44. doi: 10.1177/10454411980090020501. https://doi.org/10.1177/10454411980090020501. [DOI] [PubMed] [Google Scholar]
- 2.Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235(9):2353–75. doi: 10.1002/dvdy.20833. https://doi.org/10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- 3.Nah HD, Pacifici M, Gerstenfeld LC, Adams SL, Kirsch T. Transient chondrogenic phase in the intramembranous pathway during normal skeletal development. J Bone Miner Res. 2000;15(3):522–33. doi: 10.1359/jbmr.2000.15.3.522. https://doi.org/10.1359/jbmr.2000.15.3.522. [DOI] [PubMed] [Google Scholar]
- 4.Gong SG. Cranial neural crest: Migratory cell behaviour and regulatory networks. Exp Cell Res. 2014;325(2):90–5. doi: 10.1016/j.yexcr.2014.03.015. https://doi.org/10.1016/j.yexcr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 5.Kouskoura T, Fragou N, Alexiou M, John N, Sommer L, Graf D, et al. The genetic basis of craniofacial and dental abnormalities. Schweiz Monatsschr Zahnmed. 2011;121(7-8):636–46. [PubMed] [Google Scholar]
- 6.Twigg SR, Wilkie AO. A Genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet. 2015;97(3):359–77. doi: 10.1016/j.ajhg.2015.07.006. https://doi.org/10.1016/j.ajhg.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao X, Qu Z, Tickner J, Xu J, Dai K, Zhang X. The role of SATB2 in skeletogenesis and human disease. Cytokine Growth Factor Rev. 2014;25(1):35–44. doi: 10.1016/j.cytogfr.2013.12.010. https://doi.org/10.1016/j.cytogfr.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: A brief review. Plast Reconstr Surg. 2008;121(4):170e–8. doi: 10.1097/01.prs.0000304441.99483.97. https://doi.org/10.1097/01.prs.0000304441.99483.97. [DOI] [PubMed] [Google Scholar]
- 9.van Adrichem LN, Hoogeboom AJ, Wolvius EB. Genetics of craniofacial development. [Article in Dutch] Ned Tijdschr Tandheelkd. 2008;115(2):61–8. [PubMed] [Google Scholar]
- 10.Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: Insights from human genetics and experimental studies. J Anat. 2005;207(5):637–53. doi: 10.1111/j.1469-7580.2005.00475.x. https://doi.org/10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogle RC, Tholpady SS, McGlynn KA, Ogle RA. Regulation of cranial suture morphogenesis. Cells Tissues Organs. 2004;176(1-3):54–66. doi: 10.1159/000075027. https://doi.org/10.1159/000075027. [DOI] [PubMed] [Google Scholar]
- 12.Chim H, Manjila S, Cohen AR, Gosain AK. Molecular signaling in pathogenesis of craniosynostosis: The role of fibroblast growth factor and transforming growth factor-ß. Neurosurg Focus. 2011;31(2):E7. doi: 10.3171/2011.5.FOCUS1197. https://doi.org/10.3171/2011.5.FOCUS1197. [DOI] [PubMed] [Google Scholar]
- 13.Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219(4):472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. https: //doi.org/10.1002/ 1097-0177 (2000) 9999:9999 <:AID-DVDY1073 > 3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Delashaw JB, Persing JA, Jane JA. Cranial deformation in craniosynostosis. A new explanation. Neurosurg Clin N Am. 1991;2(3):611–20. [PubMed] [Google Scholar]
- 15.Johnson D, Wilkie AO. Craniosynostosis. Eur J Human Genet. 2011;19(4):369–76. doi: 10.1038/ejhg.2010.235. https://doi.org/10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma RK. Craniosynostosis. Indian J Plast Surg. 2013;46(1):18–27. doi: 10.4103/0970-0358.113702. https://doi.org/10.4103/0970-0358.113702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet A. 2008;146A(8):984–91. doi: 10.1002/ajmg.a.32208. https://doi.org/10.1002/ajmg.a.32208. [DOI] [PubMed] [Google Scholar]
- 18.Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of nonsyndromic corronal craniosynostosis. Am J Med Genet. 1995;55(4):500–4. doi: 10.1002/ajmg.1320550422. https://doi.org/10.1002/ajmg.1320550422. [DOI] [PubMed] [Google Scholar]
- 19.Cornelissen M, Ottelander BD, Rizopoulos D, van der Hulst R, Mink van der Molen A, van der Horst C, et al. Increase of prevalence of craniosynostosis. J Craniomaxillofac Surg. 2016;44(9):1273–9. doi: 10.1016/j.jcms.2016.07.007. https://doi.org/10.1016/j.jcms.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Lara PA, Carmichael SL, Graham JM, Jr, Lammer EJ, Shaw GM, Ma C, et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet A. 2010;152A(2):394–400. doi: 10.1002/ajmg.a.33246. https://doi.org/10.1002/ajmg.a.33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391–400. doi: 10.1542/peds.2009-3491. https://doi.org/10.1542/peds.2009-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael SL, Ma C, Rasmussen SA, Honein MA, Lammer EJ, Shaw GM. National Birth Defects Prevention Study. Craniosynostosis and maternal smoking. Birth Defects Res A Clin Mol Teratol. 2008;82(2):78–85. doi: 10.1002/bdra.20426. https://doi.org/10.1002/bdra.20426. [DOI] [PubMed] [Google Scholar]
- 23.Gedzelman E, Meador KJ. Antiepileptic drugs in women with epilepsy during pregnancy. Ther Adv Drug Saf. 2012;3(2):71–87. doi: 10.1177/2042098611433192. https://doi.org/10.1177/2042098611433192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Semin Pediatr Neurol. 2007;14(3):150–61. doi: 10.1016/j.spen.2007.08.008. https://doi.org/10.1016/j.spen.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Greenwood J, Flodman P, Osann K, Boyadjiev SA, Kimonis V. Familial incidence and associated symptoms in a population of individuals with nonsyndromic craniosynostosis. Genet Med. 2014;16(4):302–10. doi: 10.1038/gim.2013.134. https://doi.org/10.1038/gim.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang SG, Kang JK. Current and future perspectives in craniosynostosis. J Korean Neurosurg Soc. 2016;59(3):247–9. doi: 10.3340/jkns.2016.59.3.247. https://doi.org/10.3340/jkns.2016.59.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saal HM. Genetic evaluation for craniofacial conditions. Facial Plast Surg Clin North Am. 2016;24(4):405–25. doi: 10.1016/j.fsc.2016.06.001. https://doi.org/10.1016/j.fsc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Ryoo HG, Kim SK, Cheon JE, Lee JY, Wang KC, Phi JH. Slit ventricle syndrome and early-onset secondary craniosynostosis in an infant. Am J Case Rep. 2014;15:246–53. doi: 10.12659/AJCR.890590. https://doi.org/10.12659/AJCR.890590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen MM., Jr Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47(5):581–616. doi: 10.1002/ajmg.1320470507. https://doi.org/10.1002/ajmg.1320470507. [DOI] [PubMed] [Google Scholar]
- 30.Panchal J, Uttchin V. Management of craniosynostosis. Plast Reconstr Surg. 2003;111(6):2032–48. doi: 10.1097/01.PRS.0000056839.94034.47. https://doi.org/10.1097/01.PRS.0000056839.94034.47. [DOI] [PubMed] [Google Scholar]
- 31.Esparza J, Hinojosa J, García-Recuero I, Romance A, Pascual B, Martínez de Aragón A, et al. Surgical treatment of isolated and syndromic craniosynostosis. Results and complications in 283 consecutive cases. Neurocirugia (Astur) 2008;19(6):509–29. doi: 10.1016/s1130-1473(08)70201-x. https://doi.org/10.1016/S1130-1473(08)70201-X. [DOI] [PubMed] [Google Scholar]
- 32.Governale LS. Craniosynostosis. Pediatr Neurol. 2015;53(5):394–401. doi: 10.1016/j.pediatrneurol.2015.07.006. https://doi.org/10.1016/j.pediatrneurol.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Zaleckas L, Neverauskienė A, Daugelavicius V, Šidlovskaitė-Baltakė D, Raugalas R, Vištartaitė B, et al. Diagnosis and treatment of craniosynostosis: Vilnius team experience. Acta Med Litu. 2015;22(2):111–21. https://doi.org/10.6001/actamedica.v22i2.3126. [Google Scholar]
- 34.Tartaro A, Larici AR, Antonucci D, Merlino B, Colosimo C, Bonomo L. Optimization and diagnostic accuracy of computerized tomography with tridimensional spiral technique in the study of craniostenosis. [Article in Italian] Radiol Med. 1998;96(1-2):10–7. [PubMed] [Google Scholar]
- 35.Burokas L. Craniosynostosis: Caring for infants and their families. Crit Care Nurse. 2013;33(4):39–50. doi: 10.4037/ccn2013678. https://doi.org/10.4037/ccn2013678. [DOI] [PubMed] [Google Scholar]
- 36.Kim HJ, Roh HG, Lee IW. Craniosynostosis: Updates in radiologic diagnosis. J Korean Neurosurg Soc. 2016;59(3):219–26. doi: 10.3340/jkns.2016.59.3.219. https://doi.org/10.3340/jkns.2016.59.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stelnicki EJ, Mooney MP, Losken HW, Zoldos J, Burrows AM, Kapucu R, et al. Ultrasonic prenatal diagnosis of coronal suture synostosis. J Craniofac Surg. 1997;8(4):252–8. doi: 10.1097/00001665-199707000-00004. https://doi.org/10.1097/00001665-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Accardi MC, Lo Magno E, Ermito S, Dinatale A, Cacciatore A, Cavaliere A, et al. Echotomography of craniosynostosis: Review of literature. J Prenat Med. 2009;3(2):31–3. [PMC free article] [PubMed] [Google Scholar]
- 39.Miller C, Losken HW, Towbin R, Bowen A, Mooney MP, Towbin A, et al. Ultrasound diagnosis of craniosynostosis. Cleft Palate Craniofac J. 2002;39(1):73–80. doi: 10.1597/1545-1569_2002_039_0073_udoc_2.0.co_2. https://doi.org/10.1597/1545-1569(2002) 039<0073:UDOC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 40.Kosty J, Vogel TW. Insights into the development of molecular therapies for craniosynostosis. Neurosurg Focus. 2015;38(5):E2. doi: 10.3171/2015.2.FOCUS155. https://doi.org/10.3171/2015.2.FOCUS155. [DOI] [PubMed] [Google Scholar]
- 41.Marie PJ, Kaabeche K, Guenou H. Roles of FGFR2 and twist in human craniosynostosis: Insights from genetic mutations in cranial osteoblasts. Front Oral Biol. 2008;12:144–59. doi: 10.1159/000115036. https://doi.org/10.1159/000115036. [DOI] [PubMed] [Google Scholar]
- 42.Ciurea AV, Toader C. Genetics of craniosynostosis: Review of the literature. J Med Life. 2009;2(1):5–17. [PMC free article] [PubMed] [Google Scholar]
- 43.Katsianou MA, Adamopoulos C, Vastardis H, Basdra EK. Signaling mechanisms implicated in cranial sutures pathophysiology: Craniosynostosis. BBA Clin. 2016:165–76. doi: 10.1016/j.bbacli.2016.04.006. https://doi.org/10.1016/j.bbacli.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ursitti F, Fadda T, Papetti L, Pagnoni M, Nicita F, Iannetti G, et al. Evaluation and management of nonsyndromic craniosynostosis. Acta Peadiatr. 2011;100(9):1185–94. doi: 10.1111/j.1651-2227.2011.02299.x. https://doi.org/10.1111/j.1651-2227.2011.02299.x. [DOI] [PubMed] [Google Scholar]
- 45.Shillito J, Jr, Matson DD. Craniosynostosis: A review of 519 surgical patients. Pediatrics. 1968;41(4):829–53. [PubMed] [Google Scholar]
- 46.Spazzapan P, Bosnjak R, Velnar T. Craniofacial reconstruction of the skull in anterior plagiocephaly: A case report. Br J Med Med Res. 2016;18(2):1–7. https://doi.org/10.9734/BJMMR/2016/28075. [Google Scholar]
- 47.Haas-Lude K, Wolff M, Will B, Bender B, Krimmel M. Clinical and imaging findings in children with non-syndromic lambdoid synostosis. Eur J Pediatr. 2014;173(4):435–40. doi: 10.1007/s00431-013-2186-1. https://doi.org/10.1007/s00431-013-2186-1. [DOI] [PubMed] [Google Scholar]
- 48.Mulliken JB, Vander Woude DL, Hansen M, LaBrie RA, Scott RM. Analysis of posterior plagiocephaly: Deformational versus synostotic. Plast Reconstr Surg. 1999;103(2):371–80. doi: 10.1097/00006534-199902000-00003. https://doi.org/10.1097/00006534-199902000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Aarnivala H, Vuollo V, Harila V, Heikkinen T, Pirttiniemi P, Holmström L, et al. The course of positional cranial deformation from 3 to 12 months of age and associated risk factors: A follow-up with 3D imaging. Eur J Pediatr. 2016;175(12):1893–903. doi: 10.1007/s00431-016-2773-z. https://doi.org/10.1007/s00431-016-2773-z. [DOI] [PubMed] [Google Scholar]
- 50.Speltz ML, Collett BR, Wallace ER, Starr JR, Cradock MM, Buono L, et al. Intellectual and academic functioning of school-age children with single-suture craniosynostosis. Pediatrics. 2015;135(3):e615–23. doi: 10.1542/peds.2014-1634. https://doi.org/10.1542/peds.2014-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Starr JR, Collett BR, Gaither R, Kapp-Simon KA, Cradock MM, Cunningham ML, et al. Multicenter study of neurodevelopment in 3-year-old children with and without single-suture craniosynostosis. Arch Pediatr Adolesc Med. 2012;166(6):536–42. doi: 10.1001/archpediatrics.2011.1800. https://doi.org/10.1001/archpediatrics.2011.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chong S, Wang KC, Phi JH, Lee JY, Kim SK. Minimally invasive suturectomy and postoperative helmet therapy: Advantages and limitations. J Korean Neurosurg Soc. 2016;59(3):227–32. doi: 10.3340/jkns.2016.59.3.227. https://doi.org/10.3340/jkns.2016.59.3.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rottgers SA, Lohani S, Proctor MR. Outcomes of endoscopic suturectomy with postoperative helmet therapy in bilateral coronal craniosynostosis. J Neurosurg Pediatr. 2016;18(3):281–6. doi: 10.3171/2016.2.PEDS15693. https://doi.org/10.3171/2016.2.PEDS15693. [DOI] [PubMed] [Google Scholar]
- 54.Jimenez DF, Barone CM. Endoscopic technique for sagittal synostosis. Childs Nerv Syst. 2012;28(9):1333–9. doi: 10.1007/s00381-012-1768-y. https://doi.org/10.1007/s00381-012-1768-y. [DOI] [PubMed] [Google Scholar]
- 55.Utria AF, Mundinger GS, Bellamy JL, Zhou J, Ghasemzadeh A, Yang R, et al. The importance of timing in optimizing cranial vault remodelling in syndromic craniosynostosis. Plast Reconstr Surg. 2015;135(4):1077–84. doi: 10.1097/PRS.0000000000001058. https://doi.org/10.1097/PRS.0000000000001058. [DOI] [PubMed] [Google Scholar]
- 56.Garza RM, Khosla RK. Nonsyndromic craniosynostosis. Semin Plast Surg. 2012;26(2):53–63. doi: 10.1055/s-0032-1320063. https://doi.org/10.1055/s-0032-1320063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Honeycutt JH. Endoscopic-assisted craniosynostosis surgery. Semin Plast Surg. 2014;28(3):144–9. doi: 10.1055/s-0034-1384810. https://doi.org/10.1055/s-0034-1384810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esparza J, Hinojosa J. Complications in the surgical treatment of craniosynostosis and craniofacial syndromes: Apropos of 306 transcranial procedures. Childs Nerv Syst. 2008;24(12):1421–30. doi: 10.1007/s00381-008-0691-8. https://doi.org/10.1007/s00381-008-0691-8. [DOI] [PubMed] [Google Scholar]
- 59.Czerwinski M, Hopper RA, Gruss J, Fearon JA. Major morbidity and mortality rates in craniofacial surgery: An analysis of 8101 major procedures. Plast Reconstr Surg. 2010;126(1):181–6. doi: 10.1097/PRS.0b013e3181da87df. https://doi.org/10.1097/prs.0b013e3181da87df. [DOI] [PubMed] [Google Scholar]