

Graphical abstract

Keywords: Mitochondria, Protease, Shotgun, Top-down, Electroelution
Highlights
-
•
Proteases regulate mitochondrial function.
-
•
Protein fragments are identified after dopamine treatment.
-
•
Entire proteins may be electroeluted from gel slices
Abstract
Mitochondria possess a proteolytic system that contributes to the regulation of mitochondrial dynamics, mitochondrial biogenesis and mitophagy. We aimed at the identification by bottom-up proteomics of altered protein processing due to the activation of mitochondrial proteases in a cellular model of impaired dopamine homeostasis. Moreover, we optimized the conditions for top-down proteomics to identify the cleavage site sequences.
Proteomics and genomics investigations for the identification and characterization of proteases and their substrates are globally referred to as degradomics. Proteases are not only involved in the degradation of unfolded or damaged proteins, but serve to provide a system by which protein function is regulated. Indeed, proteases catalyze an irreversible hydrolysis reaction regulating activity and localisation of many proteins [1]. Mitochondria possess a highly conserved, intraorganelle proteolytic system that conducts the surveillance of protein quality control within mitochondria [2]. These proteases degrade damaged or unfolded proteins to peptides, which are subsequently either transferred out of the organelle or further degraded by mitochondrial oligopeptidases [3]. In addition to their conventional role in the degradation of misfolded or damaged mitochondrial proteins, many mitochondrial proteases take part in the regulation of proteins that orchestrate mitochondrial dynamics, mitochondrial biogenesis and mitophagy [4]. An alteration of mitochondrial proteases activity is detrimental to cell health and has been linked to aging and to various diseases, including neurological disorders like spinocerebellar ataxia and Parkinson's disease (PD) [5], [6]. Indeed, mutations in the mitochondrial serine protease HTRA2 have been associated with PD in sporadic patients [7]. We previously reported a selective degradation of voltage-dependent anion-selective channel proteins (VDACs) induced by dopamine toxicity, in a cellular model of PD [8]. However, by the combination of gel-based and gel-free techniques, we observed an accumulation of VDACs proteolytic fragments inside mitochondria, together with proteolytic fragments belonging to other mitochondrial proteins [9].These findings suggested that the impairment of dopamine homeostasis, one of the putative causes of sporadic PD, could induce an alteration of mitochondrial protease functions, deeply compromising the mitochondrial quality control systems. From this point of view, a more comprehensive characterization of mitochondrial proteases could provide new insights into PD pathogenesis.
This work was developed in the context of the biology and disease action of the mitochondrial human proteome project (mt-HPP) [10]. To understand protease functions it is of primary importance to determine their substrates and their cleavage site(s). In order to identify which substrates were differentially processed by mitochondrial proteases in a cellular model for the impairment of dopamine homeostasis [11], we combined an in-gel fractionation of mitochondrial proteins, followed by shotgun proteomics of selected molecular weight ranges.
SH-SY5Y human neuroblastoma cells were treated or not with 250 μM dopamine, in the presence of 700 U/mL of catalase for 24 h, in three independent replicates per condition. Mitochondrial enriched fractions were obtained by differential centrifugation according to standardized guidelines developed by the mt-HPP team [9]. Samples were lysed in RIPA buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% sodium deoxycholate, 1% NP-40 and 0.1% SDS) and 30 μg of mitochondrial proteins were separated by SDS-PAGE electrophoresis on acrylamide/bisacrylamide 16% gel. Three slices in different molecular weight ranges (i.e., 40–25, 25–15 and 15–10 kDa) were manually excised (Fig. 1A), washed twice with 100 mM NH4HCO3 in 50% v/v acetonitrile (ACN) for 10 min and subjected to trypsin digestion. Cysteines were reduced with 10 mM DTT in 100 mM NH4HCO3 for 45 min at 56 °C and alkylated with 55 mM iodoacetamide (IAA) in 100 mM NH4HCO3 at room temperature for 30 min in the dark. Gel slices were washed twice as described above and dried in a vacuum centrifuge. The dry gel pieces were incubated with digestion buffer (12 ng/μL porcine trypsin, 100 mM NH4HCO3, 10 mM CaCl2 and 5% ACN) overnight at 37 °C. After a brief centrifugation, supernatants containing hydrophylic peptides were saved in a new microcentrifuge tube. Hydrophobic peptides were extracted by adding 1% trifluoroacetic acid (TFA) and incubating at room temperature for 10 minutes in agitation. Supernatants were collected and pooled with the first ones. Eventually, 60% v/v ACN and 0.1% v/v TFA were added to the gel pieces and after 10 minutes vortexing the supernatants were collected, pooled together and vacuum dried. The digested samples were analyzed by nano LC-MS/MS using a Proxeon EASY-nLCII (Thermo Fisher Scientific, Milan, Italy) coupled to the maXis HD UHR-TOF mass spectrometer (Bruker Daltonics). Five μL of sample were injected and concentrated on a trapping C18-A1 EASY Column (2 cm, 100 μm I.D., 5 μm p.s., Thermo Fisher Scientific). Trapped peptides were subsequently separated on a C18-Acclaim PepMap (25 cm, 75 μm I.D., 5 μm p.s., Thermo Fisher Scientific) at 0.3 μL/min with a gradient from 2 to 45% B in 20 min (eluents: A, 0.1% FA in H2O; B, 0.1% FA in CH3CN). The raw data were processed with PEAKS 7.5 and searched against UniProt SwissProt database (release 2015_03 including common MS contaminants − 20,441 entries). The tolerances for the mass error were set to 15 ppm and 0.05 Da for precursors and fragments respectively, two missed cleavage allowed, carboamidomethylation of Cys as fixed modification and oxidation of Met, deamidation of Asn and Gln and acetylation of Lys were selected as variable modifications. −10logP value for peptides was adjusted to provide an FDR lower than 0.1% at peptide and protein level.
Fig. 1.

Bottom-up analysis of mitochondrial proteolytic fragments. (A) Schematic representation of gel slicing in three fractions with different molecular weight range (i.e., 40–25, 25–15 and 15–10 kDa). (B) HSPD1 proteolytic peptide levels in the 40–25 kDa fraction (bottom) and 25–15 kDa (top). (C) VDAC1 proteolytic peptide levels in the 40–25 kDa fraction (bottom) and 25–15 kDa (top). CTR: cells under control conditions, DA: cells treated with 250 μM dopamine. Mean and SEM refer to three independent replicates. For further details see text.
We employed a bottom-up approach to detect the proteins that were present at a lower molecular weight compared to their theoretical one, indicating a possible proteolytic processing. Among all identified proteolytic fragments, only the mitochondrial 60 kDa heat shock protein (HSPD1) and the mitochondrial porin VDAC1 showed a different behaviour between dopamine treated cells and controls. Although HSPD1 resulted processed by proteases in both conditions, with proteolytic fragments present in all gel-fractions, dopamine induced an accumulation of HSPD1 peptides in the 25–15 kDa region (Fig. 1B). In line with our previous reports [8], [9], dopamine induced a decrease of full length VDAC1 (30 kDa) along with an accumulation of VDAC1 small peptides (in the 25–15 kDa fraction) inside mitochondria (Fig. 1C).
Since all peptides identified by shotgun proteomics were trypsin digested, we were not able to reconstruct the aminoacidic sequence of processed substrates, so to detect the consensus site of proteases. To overcome these limitations and identify proteolytic fragments generated in the mitochondria after dopamine treatment, we moved towards a top-down approach, where proteins are separated by electrophoretic or chromatographic techniques without any prior digestion of the sample. However, the measurement of intact proteins presents many technical challenges, especially for complex multiprotein samples, thus we focused our efforts on establishing a reliable procedure for the preparation of protein samples for top-down proteomics. We decided to separate proteins through SDS–PAGE, cut thin slices at the level of lower MW and electroelute each protein (or fragments) out of the gel. To this purpose, a homemade electroeluter was designed and assembled using a polymethylmethacrilate support, milled to obtain the elution cell (1 × 2 × 0.2 cm). The cell was then drilled to allow the insertion of two platinum electrodes, plugged to a power supply. Electroelution condition setting was optimized using prestained protein MW markers (PageRuler Plus Prestained Protein Ladder, 10 to 250 kDa, Thermo-Scientific), separated in an acrylamide/bisacrylamide 16% gel. Protein bands were manually excised, placed in the eluter cell and covered with 200 μL elution buffer (500 mM NH4HCO3). By applying a constant current (10 mA), we observed that 20 min were needed to elute the 25 kDa protein-marker, 25 min for the 35 kDa protein and 30 min for the 70 kDa protein. The buffer was collected as soon as the gel bands became colourless indicating the complete elution of the proteins. In order to remove SDS residues that could interfere with subsequent MS analysis, samples were added with 20% v/v ethanol and loaded into an ultrafiltration cell (Vivaspin 500 MWCO 3000 Da PES, Sartorius) and centrifuged at 15000 × g to a final volume of 50 μL. Then, retained samples were collected in new tubes and vacuum dried. To verify the efficiency of the protein elution, dried samples were re-suspended in Laemmli buffer and loaded on an acrylamide/bisacrylamide 16% gel. After blue-silver staining [12], we observed single protein bands at the same molecular weight of the eluted proteins, confirming that proteins were correctly eluted and recovered (Fig. 2).
Fig. 2.
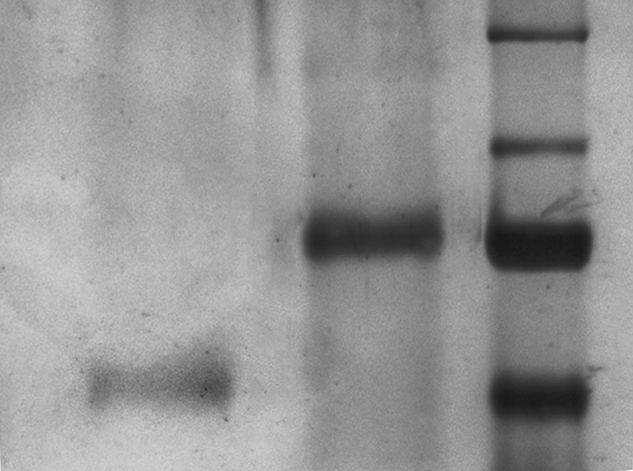
Validation of the electroelution procedure. Prestained protein MW markers (70 and 55 kDa) were excised from an acrylamide/bisacrylamide 16% gel. After elecroelution, samples were cleaned from SDS residues by ultrafiltration (MWCO 3 kDa) with 20% v/v ethanol. Samples were vacuum dried, resuspended with Laemmli buffer and separated by SDS-PAGE. First lane: 55 kDa marker; second lane: 70 kDa marker.
Future work will be devoted to the fragmentation of isolated proteins to obtain sequence information. The availability of terminal sequences of a proteolytic fragment by top-down analysis could be used to identify the protease potentially responsible for its formation using the Merops database (http://merops.sanger.ac.uk/). In this database, for a specific protease are reported all its substrates and vice versa. The information in the database is retrieved from the literature, therefore some reported data might not have physiological relevance.
As a whole, the present communication describes a strategy for the identification for cleaved protease substrates by combining a shotgun analysis of pre-fractionated low-molecular-weight protein components with electroelution of full-length proteins for top-down characterization. The proposed procedure should constitute a basis for both identification and quantification of proteins that migrate at a molecular weight lower than that expected, and the identification of their sequence termini, thus permitting the assessment of putative proteases responsible for their cleavage.
Contributor Information
Tiziana Alberio, Email: tiziana.alberio@uninsubria.it.
Mauro Fasano, Email: mauro.fasano@uninsubria.it.
References
- 1.López-Otín C., Overall C.M. Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002;3:509–519. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 2.Koppen M., Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit. Rev. Biochem. Mol. Biol. 2007;42:221–242. doi: 10.1080/10409230701380452. [DOI] [PubMed] [Google Scholar]
- 3.Tatsuta T., Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anand R., Langer T., Baker M.J. Proteolytic control of mitochondrial function and morphogenesis. Biochim. Biophys. Acta. 2013;1833:195–204. doi: 10.1016/j.bbamcr.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 5.Shanbhag R., Shi G., Rujiviphat J., McQuibban G.A. The emerging role of proteolysis in mitochondrial quality control and the etiology of Parkinson's disease. Parkinsons Dis. 2012;2012:382175. doi: 10.1155/2012/382175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinelli P., Rugarli E.I. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim. Biophys. Acta. 2010;1797:1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Strauss K.M., Martins L.M., Plun-Favreau H., Marx F.P., Kautzmann S., Berg D., Gasser T., Wszolek Z., Müller T., Bornemann A., Wolburg H., Downward J., Riess O., Schulz J.B., Krüger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 8.Alberio T., Mammucari C., D'Agostino G., Rizzuto R., Fasano M. Altered dopamine homeostasis differentially affects mitochondrial voltage-dependent anion channels turnover. Biochim. Biophys. Acta. 2014;1842:1816–1822. doi: 10.1016/j.bbadis.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 9.Alberio T., Bondi H., Colombo F., Alloggio I., Pieroni L., Urbani A., Fasano M. Mitochondrial proteomics investigation of a cellular model of impaired dopamine homeostasis, an early step in Parkinson's disease pathogenesis. Mol. Biosyst. 2014;10:1332–1344. doi: 10.1039/c3mb70611g. [DOI] [PubMed] [Google Scholar]
- 10.Urbani A., De Canio M., Palmieri F., Sechi S., Bini L., Castagnola M., Fasano M., Modesti A., Roncada P., Timperio A.M., Bonizzi L., Brunori M., Cutruzzolà F., Di Ilio V., Di Ilio C., Federici G., Folli F., Foti S., Gelfi C., Lauro D., Lucacchini A., Magni F., Messana I., Pandolfi P.P., Papa S., Pucci, P., Sacchetta P., Italian Mt-Hpp Study Group-Italian Proteomics Association (www. itpa. it) The mitochondrial Italian Human Proteome Project initiative (mt-HPP) Mol. Biosyst. 2013;9:1984–1992. doi: 10.1039/c3mb70065h. [DOI] [PubMed] [Google Scholar]
- 11.Alberio T., Lopiano L., Fasano M. Cellular models to investigate biochemical pathways in Parkinson's disease. FEBS J. 2012;279:1146–1155. doi: 10.1111/j.1742-4658.2012.08516.x. [DOI] [PubMed] [Google Scholar]
- 12.Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G.M., Carnemolla B., Orecchia P., Zardi L., Righetti P.G. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]