

Summary
Hematopoietic stem cells (HSCs) sustain hematopoiesis throughout life. HSCs exit dormancy to restore hemostasis in response to stressful events, such as acute blood loss, and must return to a quiescent state to prevent their exhaustion and resulting bone marrow failure. HSC activation is driven in part through the phosphatidylinositol 3-kinase (PI3K)/AKT/mTORC1 signaling pathway, but less is known about the cell-intrinsic pathways that control HSC dormancy. Here, we delineate an ERK-dependent, rate-limiting feedback mechanism that controls HSC fitness and their re-entry into quiescence. We show that the MEK/ERK and PI3K pathways are synchronously activated in HSCs during emergency hematopoiesis and that feedback phosphorylation of MEK1 by activated ERK counterbalances AKT/mTORC1 activation. Genetic or chemical ablation of this feedback loop tilts the balance between HSC dormancy and activation, increasing differentiated cell output and accelerating HSC exhaustion. These results suggest that MEK inhibitors developed for cancer therapy may find additional utility in controlling HSC activation.
Keywords: hematopoietic stem cells, emergency hematopoiesis, hematopoietic stem cell activation, hematopoietic stem cell exhaustion, oxidative stress, mitochondrial fitness, intracellular signaling, ERK pathway, AKT/mTORC1 pathway, feedback regulation of signaling
Graphical Abstract
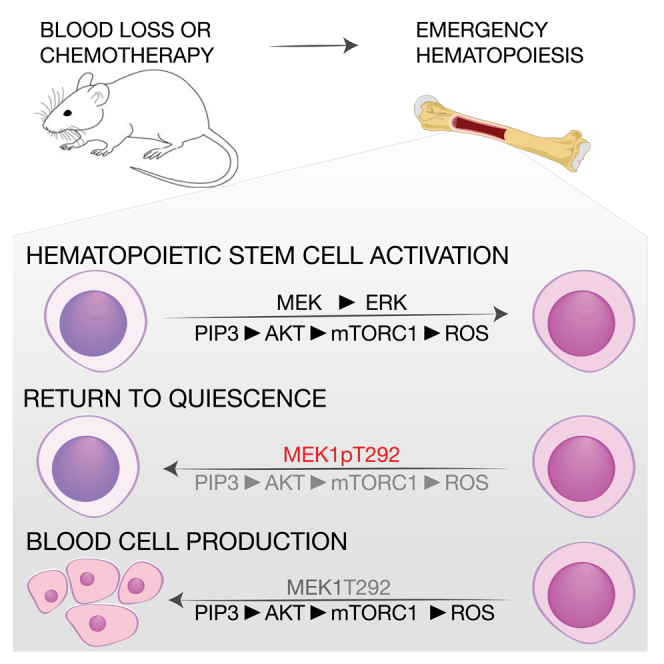
Highlights
-
•
MEK/ERK and AKT/mTORC1 are reversibly activated during hematopoiesis
-
•
MEK1 prevents HSC exhaustion during stress hematopoiesis
-
•
Feedback phosphorylation of MEK1 by ERK limits AKT/mTORC1 activation
-
•
ERK-mediated MEK1 phosphorylation returns activated HSCs to quiescence
Baccarini and colleagues identify a cell-intrinsic feedback mechanism limiting the strength of MEK/ERK and AKT/mTORC1 signals during the activation of hematopoietic stem cells. The mechanism hinges on the negative feedback phosphorylation of MEK1 by activated ERK and is required to prevent HSC exhaustion.
Introduction
The production of blood cells depends on the fitness of the hematopoietic stem cell (HSC) compartment, which contributes to multilineage hematopoiesis throughout life (Bernitz et al., 2016, Busch et al., 2015, Sawai et al., 2016, Sun et al., 2014). HSCs are mostly quiescent but can be reversibly activated to meet the increased demand imposed on the organism by stresses, such as infections, blood loss, or chemotherapy-induced myelotoxicity (Baldridge et al., 2010, Essers et al., 2009, Wilson et al., 2008). HSCs exist in at least three different states: a dormant state, characterized by quiescence and by the highest regenerative potential in the HSC cell compartment (Bernitz et al., 2016, Takizawa et al., 2011, Wilson et al., 2008); an active or homeostatic state, reached by gradual transition from the dormant state, in which the HSCs are still quiescent but “primed” for replication (Cabezas-Wallscheid et al., 2017, Wilson et al., 2008); and a fully activated state, characterized by active cycling and typically induced during emergency hematopoiesis. Metabolic changes play an important role in HSC activation. Quiescent HSCs rely primarily on anaerobic glycolysis. They contain low numbers of mitochondria, limited essentially by mitophagy (Ho et al., 2017, Ito et al., 2016, Warr et al., 2013), and low levels of reactive oxygen species (ROS). Conversely, HSC activation is characterized by a continuous increase in biosynthetic activity and correlates with mitochondrial expansion, increased reliance on oxidative phosphorylation, and increased ROS levels (Bigarella et al., 2014, Cabezas-Wallscheid et al., 2017, Chandel et al., 2016, Schultz and Sinclair, 2016). The mTOR pathway, which stimulates mitochondrial biogenesis and counteracts autophagy, is a major player in HSC activation and differentiation (Chen et al., 2008, Kharas et al., 2010, Rodgers et al., 2014, Yilmaz et al., 2006); in contrast, retinoic acid-vitamin A signaling antagonizes HSC proliferation and promotes the return to quiescence after activation (Cabezas-Wallscheid et al., 2017).
The functionality of the HSC compartment hinges on the balance between quiescence and activation. A reduction in the ability of HSCs to exit quiescence will result in insufficient blood cell production, whereas an increased propensity to exit quiescence or failure to re-enter it will cause HSC exhaustion and bone marrow failure. The identity of the cell-intrinsic mechanism(s) required to terminate activation and re-enter dormancy and the signaling pathways that mediate this transition remain key unresolved questions in the field.
The MEK/ERK and phosphatidylinositol 3-kinase (PI3K) pathways are common targets of growth factors regulating hematopoiesis (Chung and Kondo, 2011, Polak and Buitenhuis, 2012). In mouse models, constitutive activation of these pathways promotes HSC proliferation at the expense of quiescence. Both pathways are activated downstream of RAS, and at least one of the RAS paralogs, KRAS, is required in HSCs for adult hematopoiesis (Damnernsawad et al., 2016); constitutively activated KRASG12D sensitizes HSCs and progenitors to cytokines (Van Meter et al., 2007), whereas NRASG12D has a bimodal effect on HSCs, increasing regenerative potential in a slow-cycling pool and decreasing it in a fast-cycling pool (Li et al., 2013). In contrast, constitutive activation of the PI3K pathway by deletion of its antagonist PTEN decreases regenerative potential and causes HSC exhaustion, primarily via mTOR activation (Kalaitzidis et al., 2012, Kharas et al., 2010, Lee et al., 2010, Magee et al., 2012, Yilmaz et al., 2006, Zhang et al., 2006). However, mTORC1 activity is required for hematopoiesis (Guo et al., 2013, Hoshii et al., 2012, Kalaitzidis et al., 2012). The roles ascribed to the RAF/MEK/ERK in hematopoiesis so far are lineage specific. Oncogenic BRAFV600E promotes monocyte expansion (Kamata et al., 2013), and BRAF and ERK are required for myelopoiesis (Kamata et al., 2005, Staser et al., 2013); in contrast, epiblast-restricted ablation of MEK1 causes extramedullary hematopoiesis and myeloproliferation (Zmajkovicova et al., 2013). Finally, RAF1 is required for erythroid differentiation (Kolbus et al., 2002, Rubiolo et al., 2006).
From all of the above, it is clear that efficient hematopoiesis requires the fine-tuning of the MEK/ERK and PI3K pathways. However, the mechanisms involved in this regulation are currently unknown. We have previously shown that activated ERK can dim the activation of both pathways by feedback phosphorylation of a specific residue on MEK1 (Catalanotti et al., 2009, Zmajkovicova et al., 2013), suggesting that these feedbacks may play a role in hematopoiesis.
Results
MEK1 Prevents HSC Exhaustion during Emergency Hematopoiesis
To study the cell-autonomous effects of MEK1 in hematopoietic cells in detail, we used a conditional knockout (cKO) mouse model inducing hematopoietic system-restricted MEK1 deletion (Vav1iCRE;Map2klF/F mice, hereafter termed MEK1-cKO; de Boer et al., 2003). MEK1 was efficiently deleted in MEK1-cKO bone marrow (BM), whereas expression of the paralog MEK2 was unaffected (Figure S1A). One-year-old MEK1-cKO showed a moderate peripheral pancytopenia (Figure S1B), which correlated with reduced HSC numbers and loss of label-retaining cells (Figures S1C and S1D; for full HSC fluorescence-activated cell sorting [FACS] gating strategy, see Figure S1E). We next tested the regenerative capacity of MEK1-deficient HSCs by performing serial competitive reconstitution assays, in which CRE+, F/F, or cKO Ly5.2+ donor BM cells (each containing 100 HSCs) were mixed with F/F Ly5.1+ competitor BM and injected into lethally irradiated Ly5.1+ recipient mice (Figure 1A). MEK1-cKO cells could contribute to all lineages but yielded low levels of peripheral blood, BM, and HSC chimerism (Figure 1B) and exhausted after the second round of transplantation (Figure 1C). Consistent with this defect in HSC regenerative capacity, MEK1 ablation suppressed chemotherapy-induced emergency hematopoiesis. Repetitive exposure to the myelotoxic drug 5-FU (Figure 1D) caused HSC expansion in F/F animals, whereas in MEK1-cKO mice, the HSC compartment contracted dramatically, leading to BM failure and premature death (Figures 1E and 1F). In the initial phases of the treatment, however, the output of differentiated cells in both BM and peripheral blood of MEK1-cKO animals was higher than that of controls (Figure 1E).
Figure 1.

MEK1 Ablation Increases HSC Proliferation and Differentiation, Leading to HSC Exhaustion
(A) Serial transplantation protocol.
(B and C) Blood chimerism (left), lineage distribution (center) in peripheral blood, BM cellularity, and HSC chimerism in lethally irradiated recipients reconstituted with F/F, CRE+, or cKO BM analyzed during the first (B) or second (C) round of transplantation.
(D) Repetitive (rep) 5-FU treatment protocol.
(E) HSCs per femur, lineage+ cells per femur, and peripheral blood parameters (Hb, hemoglobin; PLT, platelets; WBC, white blood cells) during repetitive 5-FU treatment.
(F) Kaplan-Meier survival curve. Median survival time (MST): F/F = 84 days; MEK1-cKO = 39 days; p < 0.001 according to the log rank (Mantel-Cox) test.
(G) Colony-forming units (CFUs) and % lineage+ cells derived from HSCs in LTC.
(H) Cell cycle distribution of HSCs harvested 12 weeks after transplantation (Transpl), 12 days after the third 5-FU injections (rep 5-FU), or after 6 weeks in LTC.
Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 comparing CRE+ or F/F to cKO. See also Figure S1.
A more detailed analysis of the hematopoietic compartment showed that all other stem and precursor cell subsets analyzed behaved similarly to HSCs, with numbers indistinguishable from the F/F controls in young animals and significant contraction occurring in aging, chemotherapy, and transplantation (Mendeley Data, https://doi.org/10.17632/7rdg6mjk5h.1).
The defect caused by MEK1 ablation was cell intrinsic and could be recapitulated in long-term cultures (LTCs) of HSCs seeded on F/F feeder layers. In these experiments, MEK1-cKO HSCs produced a significantly higher number of lineage+ cells than F/F cultures, whereas the number of cells capable of generating colony-forming units (CFUs) steadily decreased (Figure 1G). Increased output of differentiated cells and HSC exhaustion correlated with reduced numbers of HSCs in G0 in all the systems investigated (Figure 1H).
MEK1 Guards against HSC Exhaustion by Regulating the Expression of Genes Promoting Cell Cycle and Oxidative Phosphorylation
To gain further insight on the role of MEK1, we next focused on mice recovering from a single 5-FU injection, which promotes the reversible activation of virtually all dormant HSCs (Wilson et al., 2008). In this setup, the temporal correlation among contraction of the HSC compartment, increased HSC proliferation, and increased output of differentiated cells in MEK1-cKO mice was readily apparent (Figures 2A–2C). To explore the mechanisms driving HSC exhaustion in an unbiased manner, we performed transcriptome-wide RNA sequencing (RNA-seq) experiments comparing F/F and MEK1-cKO HSCs from untreated animals or from animals recovering from one single 5-FU injection administered 9 days before harvesting the cells, at the time of maximum HSC expansion in F/F BM. 483 genes were upregulated and 450 were downregulated in 5-FU-treated F/F HSCs (complete gene list in Table S1); among these genes, two groups were clearly impacted by MEK1 ablation (Figures 2D, 2E, and S2). The first group included genes connected with increased cell cycle activity. The upregulation of cell-cycle-promoting genes and the downregulation of cell cycle inhibitors, validated by qRT-PCR, correlated with the reduction in G0 populations in all stress systems investigated (Figure 2F, top). Upregulation of cell cycle genes was also observed in HSCs isolated from untreated MEK1-cKO animals, where the percentage of HSCs in G0 was indistinguishable between F/F and cKO (compare Figure 2B with Figure 2F, top). Thus, MEK1-cKO HSCs are in a state reminiscent of the GAlert (Rodgers et al., 2014) or “active” as opposed to “dormant” state (Cabezas-Wallscheid et al., 2017), i.e., they are primed for cell cycle entry. This is consistent with the higher numbers of cycling MEK1-cKO HSCs and with the initial burst of lineage+ cells observed in all activation models.
Figure 2.

MEK1 Ablation Delays Return to Quiescence during Recovery from a Single 5-FU Injection and Promotes the Expression of Cell Cycle and OXPHOS Genes
(A–C) BM cellularity and HSCs per femur (A), HSC cell cycle distribution (B), and blood parameters (C) during the recovery from 5-FU injection.
(D and E) Heatmap (D) and scatterplots (E) of genes differentially expressed in F/F and cKO HSCs from untreated or 5-FU-treated mice (9 days prior isolation). Blue, increased expression in F/F HSCs; red, increased expression in cKO HSCs.
(F) qRT-PCR of selected cell cycle and OXPHOS genes in FAC-sorted HSCs. Data are normalized to expression levels in F/F HSCs from untreated mice (dotted line).
Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 comparing CRE+ or F/F to cKO. White asterisks, comparison of 5-FU-treated versus untreated animals of the same genotype. See also Figure S2 and Table S1 for a complete gene list.
The second group of upregulated genes encoded mitochondrial components, most notably proteins of the electron transport chain involved in oxidative phosphorylation (OXPHOS) (Murphy, 2009). Upregulation of these genes was only observed in HSCs isolated from animals undergoing stress-induced hematopoiesis or cultured on feeder layers in vitro (Figures 2F, bottom, and S2), consistent with the notion that full-fledged HSC activation relies on mitochondrial expansion, increased OXPHOS, and higher intracellular ROS levels (Chandel et al., 2016, Ito and Suda, 2014).
Increased ROS Levels and Mitochondrial Mass in MEK-cKO HSCs Undergoing Emergency Hematopoiesis
Indeed, ROS levels/cell increased in HSCs recovering from 5-FU myeloablation, peaking at day 9 in correspondence with the maximum output of differentiated cells and returning to normal by day 21. The increase in intracellular ROS levels was more pronounced in MEK1-cKO than in F/F HSCs (Figure 3A) and correlated with a transient increase in DNA damage, but not apoptosis or senescence (Figures S3A–S3E), in line with the concept that HSCs incur and repair DNA damage when induced to exit quiescence (Walter et al., 2015). Similar results were obtained in the other models tested (Figures 3B and S3). Treatment with the antioxidant N-acetylcysteine (NAC), which counteracts ROS-induced damage, significantly increased the survival of MEK1-cKO mice undergoing myelotoxic chemotherapy, showing that increased ROS production contributes to the phenotype in vivo (Figure 3C). The increase in ROS levels was observed in all MEK1-deficient, lineage-BM cells (Figure S3F).
Figure 3.

MEK1 Ablation HSCs Promote Oxidative Stress and Decrease Mitophagy in HSCs
(A) ROS levels in HSCs recovering from 5-FU injection.
(B) Representative FACS histograms of ROS levels in HSCs exposed to chronic stress as in Figure 1. Values represent mean fluorescence intensity (MFI) ± SD.
(C) Kaplan-Meier survival curve of mice. MST: F/F = 93 days; cKO = 39 days; F/F + NAC = 92 days; cKO + NAC = 65 days. ∗∗p < 0.01 according to the log rank (Mantle-Cox) test; n = 10.
(D and E) Mitochondrial mass (Mitotracker Green, D, and Δψ per mitochondrial mass, E) in HSCs isolated from mice exposed to stress or grown in LTC.
(F) FACS analysis of PINK1 protein levels in HSCs.
(G) Representative confocal images and quantification of the co-localization of mitochondria (TOM20+) and lysosomes (LAMP1+) in HSCs exposed to a single 5-FU injection (9 days before isolation) and chronic stresses as defined in the legend to Figure 1.
Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. White asterisks, comparison of 5-FU-treated versus untreated animals of the same genotype. See also Figure S3.
Mitochondria are the major producers of intracellular ROS; these radicals, in turn, can cause mitochondrial damage (Murphy, 2009). We therefore analyzed whether ROS production correlated with increased mitochondrial mass and/or mitochondrial damage (defined as a decrease in mitochondrial membrane potential, Δψ) during emergency hematopoiesis.
Mitochondrial mass and mitochondrial membrane potential/cell did not significantly differ in F/F and cKO HSCs recovering from a single 5-FU injection (Figure S3G). However, mitochondrial mass was increased and membrane potential (Δψ) was decreased in MEK1-cKO HSCs and multipotent progenitors upon repetitive 5-FU stress, as well as in cKO HSCs in LTCs (Figures 3D, 3E, and S3F), suggesting an accumulation of damaged mitochondria in these models. Damaged mitochondria are cleared by PINK1/PARK2-mediated mitophagy (Pickrell and Youle, 2015), which supports HSC maintenance downstream of the PML/PPARδ pathway (Ito et al., 2012, Ito et al., 2016). The PINK1 gene was downregulated in the cKO (Table S1), and PINK1 protein expression was reduced in MEK1-cKO HSCs from 5-FU-treated mice or from LTCs (Figure 3F). Confocal microscopy revealed reduced co-localization of mitochondria with lysosomes (labeled with Tom20 [translocase of outer membrane 20] and LAMP1 [lysosomal-associated membrane protein 1] antibodies, respectively) in cKO HSCs from 5-FU-treated mice or from LTCs (Figure 3G), suggesting decreased mitophagy.
MEK/ERK and AKT/mTORC1 Are Reversibly Activated during Emergency Hematopoiesis
The data above show that MEK1 dials down proliferation, OXPHOS, and the accumulation of ROS and damaged mitochondria in HSC stimulated to proliferate in vivo and in vitro. To elucidate the MEK1-dependent mechanisms governing these key parameters of reversible HSC activation, we combined surface marker and intracellular (phospho)protein FACS analysis and focused on MEK-related HSC signaling during the recovery from a single 5-FU injection (for antibody validation, see Mendeley Data, https://doi.org/10.17632/7rdg6mjk5h.1). We analyzed MEK phosphorylation on the RAF-dependent activation sites (S218/222) and MEK1 phosphorylation on T292, the ERK-dependent site required for MEK1/MEK2 inactivation and for efficient PTEN-mediated PIP3 turnover at the membrane (Catalanotti et al., 2009, Zmajkovicova et al., 2013). In addition, we analyzed the activation of AKT (defined as the phosphorylation of S473) and of its downstream target mTORC1, which is involved in HSC mobilization through ROS control and mitochondrial biogenesis (Chandel et al., 2016, Ito and Suda, 2014). Activation of mTORC1 was defined as phosphorylation of mTOR and of the mTORC1 target S6.
In HSCs of 5-FU-treated mice, phosphorylation of MEK1, ERK, AKT, and mTORC1 paralleled HSC proliferation, output of differentiated cells, and ROS increase (compare Figure 4A with Figures 2A–2C and 3A). Activation of ERK, AKT, and mTORC1 was also significantly increased in MEK1-cKO HSCs isolated from 5-FU-treated or transplanted mice or from LTCs (Figure S4A). Furthermore, the transcription factor FOXO3A, which promotes HSC maintenance (Miyamoto et al., 2007, Warr et al., 2013) and is negatively regulated by ERK (Yang et al., 2008) and AKT (Brunet et al., 1999), was phosphorylated in HSC from 5-FU-treated mice and more robustly so in MEK1-cKO HSC in this and all other models tested (Figures 4B and S4B). On day 9 after 5-FU injection, FOXO3A was excluded from the nuclei of both F/F and cKO HSCs (Figure 4C) and the expression of its target genes Sod2 and Catalase (involved in limiting ROS levels), as well as of Pink1 (Ito and Suda, 2014, Ito et al., 2016, Mei et al., 2009, Miyamoto et al., 2007, Rimmelé et al., 2015, Tothova and Gilliland, 2007, Warr et al., 2013), was downregulated, particularly in the cKO (Figure 4D). Thus, coordinated activation of the ERK and AKT pathways coincides with increased proliferation, ROS production, and the control of mitophagy during the recovery from myelotoxic 5-FU treatment. In line with this, phosphorylation of the RAF-dependent and ERK-dependent sites on MEK1 as well as ERK and AKT phosphorylation were lowest in HSCs and increased with differentiation in the BM of untreated F/F mice (Figures S4C and S4D).
Figure 4.

The MEK/ERK and AKT Pathways Are Transiently Activated in HSCs Recovering from Myeloablation
(A and B) Expression and phosphorylation of (A) MEK, ERK, AKT, and mTORC1 (mTOR and S6) and (B) FOXO3A (ERK and AKT target sites) in HSCs recovery from 5-FU (n = 25, 5 experiments, 5 animals each). Data represent fold change relative to untreated F/F HSCs. Phosphorylation of most molecules increased significantly in F/F HSCs from days 6 to 12 (p < 0.001; in the case of phospho-S6 from days 3 to 9 comparing treated versus untreated F/F).
(C) Representative confocal images and percentage of FAC-sorted HSCs with FOXO3A nuclear localization. Scale bars, 2μm.
(D) FOXO3A target gene expression in HSCs from untreated and 5-FU-treated animals.
In (C) and (D), n = 4. Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 comparing F/F versus cKO. White asterisks, treated versus untreated animals of the same genotype. See also Figure S4.
Control of mTORC1-Dependent ROS Production by MEK1 Regulates HSC Activation and Regenerative Potential
To explore the relationship among HSC activation, OXPHOS and ROS increase, and ERK/AKT signaling, we silenced MEK1 or inhibited MEK in mouse or human HSC LTCs. It is important to note here that the MEK inhibitor was titrated to achieve fine-tuning rather than complete inhibition of ERK activation (about 50%; Figures 5C and 7C), which is not tolerated by the cells. Both treatments mimicked the defects induced by CRE-mediated MEK1 ablation, reducing the number of CFUs, increasing ROS levels and mitochondrial mass, and decreasing mitochondrial fitness (Figures S5A–S5D). Inhibition of OXPHOS and mTOR by metformin (Foretz et al., 2014) or addition of NAC increased CFU numbers in mouse and human LTCs, normalized ROS levels, and rescued the mitochondrial phenotype caused by MEK1 deficiency or MEK inhibition without affecting ERK or AKT activation (Figures S6A–S6F). Thus, increased ROS production is responsible for mitochondrial damage in our system and is the consequence, not the cause, of the increased ERK/AKT signaling induced by MEK1 silencing or inhibition. MEK1 downregulation activated both the ERK and the AKT pathway, whereas chemical MEK inhibition prevented activation of ERK and phosphorylation of MEK1 on the ERK-dependent site T292 (Catalanotti et al., 2009, Zmajkovicova et al., 2013) and activated the AKT pathway exclusively (Figures S5E and S5F). These results strongly suggested that AKT pathway activation, rather than the sustained MEK2/ERK phosphorylation characteristic of MEK1-deficient cells, was responsible for the phenotypes observed. Indeed, inhibition of PI3K by LY294002 or of mTORC1 by rapamycin (Chiang and Abraham, 2005, Shimobayashi and Hall, 2014) fully rescued the biological phenotypes of MEK1-deficient mouse HSCs (Figures 5A and 5B, left) or MEK-inhibited human HSCs (Figures 5A and 5B, right). PI3K inhibition also selectively reduced the phosphorylation of mTOR and of FOXO3A on the AKT-dependent site and restored PINK1 protein expression (Figure 5C).
Figure 5.

Increased AKT and mTORC1 Activity Underlie the Phenotype of MEK1-Deficient or MEK-Inhibited HSCs
Mouse F/F and cKO HSCs and DMSO or iMEK-treated human HSCs were treated with LY294002 (iPI3K) or rapamycin (RAPA; mTORC1 inhibitor) in LTC.
(A) After 6 weeks, cells were harvested and their ability to form colonies was determined in a 10-day CFU assay in the absence of inhibitors.
(B and C) ROS levels and Δψ per mitochondrial mass (B) and intracellular signaling (C) were also determined and are shown for the HSC and multipotent progenitor (MPP) subset. Data show fold change relative to mouse F/F or human untreated cells (normalized as 1; dotted line; n = 3).
Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. White asterisks, untreated versus iP3K/RAPA-treated cultures of the same genotype. See also Figures S5 and S6.
Figure 7.

ERK-Dependent Phosphorylation of MEK1 T292 Feeds Back on AKT/mTORC1/FOXO3A Signaling to Balance HSC Quiescence and Differentiation
Effect of lentiviral expression of MEK1 and FOXO3A (wild-type or phosphosite mutants) and of PINK1 on mouse F/F and cKO HSCs or on iMEK-treated human HSCs.
(A–C) CFUs (A), ROS levels and Δψ per mitochondrial mass (B), and intracellular signaling (C) were determined after 6 weeks. Data show fold change relative to mouse cKO or human iMEK-treated HSCs (normalized as 1; dotted line; n = 3). Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, comparing vector-infected HSCs with HSCs infected with the indicated constructs.
(D) Model: differentiation-promoting events in red; see text for details.
To determine whether the control of mTORC1 signaling, OXPHOS, and ROS by MEK1 is causally related to the increased HSC attrition observed in the MEK1-cKO mice, we treated animals exposed to single or multiple 5-FU injections with NAC or rapamycin. Both NAC and rapamycin reduced the rebound of HSC numbers as well as the percentage of actively cycling HSCs in control F/F mice recovering from a single 5-FU injection, indicating that the transient increase in mTORC1 signaling and ROS is causally related to the production of HSCs (Figure 6A). Importantly, both inhibitors reduced ROS production, demonstrating that mTORC1 is responsible for the transient increase in ROS during HSC activation (Figures S7A and S7B). In MEK1-cKO mice, NAC and rapamycin had the opposite effect; they reduced the percentage of cycling HSC, increased the numbers of BM HSCs (Figure 6A), and decreased the output of differentiated cells (Figures S7C and S7D), essentially erasing the difference in the reaction of MEK1-cKO and F/F mice to myelotoxic 5-FU treatment and rescuing the phenotype. NAC and rapamycin treatment also protected MEK1 cells from attrition during repetitive 5-FU chemotherapy (Figures 6B and S7E). Most importantly, treatment of recipient mice with NAC and rapamycin fully rescued the reconstituting potential of MEK1-deficient HSCs in a competitive transplantation experiment (Figures 6C, 6D, S7G, and S7H).
Figure 6.

Increased mTORC1 Activation and ROS Production Are Responsible for the Attrition of cMEK1 KO HSCs in Emergency Hematopoiesis
(A and B) Impact of NAC and rapamycin (RAPA) treatment on HSC per femur (top) and HSC cell cycle distribution (bottom) in F/F and MEK1-cKO mice recovering from a single 5-FU injection (A) or subjected to chronic 5-FU treatment (B).
(C and D) Regenerative potential of F/F and MEK1-cKO BM in lethally irradiated recipient mice treated with NAC (C) or rapamycin (D). Top: blood chimerism is shown; bottom: HSC chimerism and cell cycle distribution are shown. Recipients were treated with vehicle/NAC or vehicle/RAPA once per day between week 2 and week 12 after transplantation.
Error bars represent the SD of the mean. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Vehicle versus NAC or RAPA-treated mice of the same genotype is shown. See also Figure S7.
ERK-Dependent Phosphorylation of MEK1 T292 Is the Linchpin of the Intracellular Negative Feedback Mechanism that Allows Reversible HSC Activation
To gain insight into the mechanism underlying the MEK1-dependent negative feedback, we attempted to rescue the phenotypes induced by MEK1 ablation in mouse HSCs (Figures 7A–7C, left) or by MEK1 inhibition in human HSCs (Figures 7A–7C, right) by expressing wild-type MEK1 and its phosphorylation site mutants T292D (mimicking ERK-dependent feedback phosphorylation) or T292A (preventing ERK-dependent feedback phosphorylation). T292D MEK1 rescued all biochemical and biological phenotypes induced by MEK1 ablation or MEK inhibition, whereas T292A MEK1 failed to do so. Wild-type MEK1, on the other hand, rescued the phenotypes induced by MEK1 deficiency, but not those due to MEK1 inhibition, which prevents T292 phosphorylation (Figures 7A–7C, compare the left with the right). FOXO3A mutants that cannot be phosphorylated by AKT, but not those that cannot be phosphorylated by ERK, significantly increased the number of CFUs in MEK1-deficient mouse HSC LTCs as well as in MEK-inhibited human HSC LTCs (Figure 7A). This correlated with a decrease in damaged mitochondria (Δψ per mitochondrial mass; Figure 7B) and with restored expression of the FOXO3A target PINK1 (Figure 7C). ROS levels remained high in FOXO3A (AKT)-transduced HSCs (Figure 7B). Ectopic PINK1 expression also reduced the number of damaged mitochondria without affecting ROS production and increased the number of CFUs recovered from mouse MEK1-deficient LTCs (Figures 7A and 7B, left). In MEK-inhibitor-treated human HSC LTCs, on the other hand, PINK1 expression only marginally reduced damaged mitochondria and failed to restore CFU numbers (Figures 7A and 7B, right). Whereas the reason for this discrepancy is currently unknown, it is worth noting in this context that ERK inhibition can decrease PINK1 stability and PARK2 recruitment (Park et al., 2017).
Discussion
Collectively, our data show that the ERK and the PI3K pathways are coordinately activated in HSCs and that a cell-intrinsic negative feedback signaling loop involving phosphorylation of MEK1 by activated ERK limits the strength and duration of PI3K/AKT/mTORC1 signaling to allow activated HSCs to return to quiescence. If this ERK-dependent feedback is disabled, HSCs shift from a dormant to an active or primed state (Cabezas-Wallscheid et al., 2017), which has been described as “GALERT,” interestingly induced by mTORC1, in other stem cell populations (Rodgers et al., 2014). The mechanism is at work during homeostasis, as shown by the contraction of the hematopoietic compartment in untreated, aged MEK1-cKO mice; however, its essential role in preventing HSC burnout becomes obvious when HSCs are induced to proliferate in vivo by chemotherapy or transplantation and in in vitro in LTCs. In all these models, exhaustion is driven by a combination of mTORC1-dependent ROS production, which damages the mitochondria, and decreased FOXO3A/PINK1-dependent clearance of these organelles (Figure 7D).
Our data exemplify the relevance of paralog-specific signaling, and more precisely of paralog-specific pathway cross-talk, in the ERK pathway. In the RAF/MEK/ERK cascade, multiple paralogs are able to carry out common function, consisting in MEK phosphorylation in the case of the three RAF kinases and in ERK phosphorylation in the case of the two MEK kinases. This raises the question of why so many paralogs have survived evolution. In evolutionary terms, the paralogs RAF1 and MEK1 can be considered “moonlighting proteins,” multifunctional proteins that have evolved to carry out at least one essential function that is separate from their role within the ERK pathway. Specifically, RAF1 and MEK1 are required to regulate the activity of other pathways (RHO-dependent pathways in the case of RAF1; PI3K in the case of MEK1) via protein-protein interaction (Desideri et al., 2015, Dorard et al., 2017). Intriguingly, it is the activation of the core pathway (RAF/MEK/ERK) that generates the phosphorylated forms of RAF1 and MEK1 required for the cross-talk with the RHO and with PI3K pathway, respectively (Varga et al., 2017, Zmajkovicova et al., 2013). In the case of MEK1, T292 phosphorylation promotes the formation of a ternary complex (MEK1/MAGI100/PTEN) required for the translocation of PTEN to the membrane, where the phosphatase turns over PIP3 and dims signaling (Zmajkovicova et al., 2013). T292 first appears in amniota and is then conserved in this clade, hinting at an important role for this residue in the control of signal strength and diversification in complex organisms. In the specific context of HSC development, it is worth noting that the earliest fossil evidence of a hematopoietic bone marrow, found in a sarcopterygian fish with a close evolutionary relationship to the tetrapoda (Sanchez et al., 2014), precedes the appearance of MEK1 T292, making it tempting to speculate that this residue might have evolved to represent an advantage in this context. Much progress has been made in understanding the signals mobilizing HSC in homeostasis and in emergency hematopoiesis in the context of the bone marrow niche (Crane et al., 2017). The emerging picture is exceedingly complex, including signals delivered through soluble factors and through cell-cell contact. Our results show that these stimuli, whatever their nature, converge on the MEK/ERK and PI3K modules. T292 phosphorylation by activated ERK might have evolved as a robust system of checks and balances, ensuring that these two crucial pathways will be coordinately reset irrespectively of the stimulus that activated them; the only requirement is the presence of the protein-protein interaction partners mediating the feedback (i.e., MAGI100), which determines the tissue specificity of the phenotypes. In favor of this hypothesis, it is worth noting that the defective repopulating potential of both MEK1 cKO HSCs (Figure 6) and PTEN KO HSCs (Lee et al., 2010, Yilmaz et al., 2006) is caused by excessive mTORC1 activation as exemplified by the rescue of this function through rapamycin treatment of the recipients.
On a more translational note, the complexity of the signals that regulate hematopoiesis limits the design of interventions aimed at modulating the entry or exit of HSCs from quiescence, whereas the feedback mechanism described here is druggable. At least in LTCs, chemical inhibition of MEK mimics the effect of ablation, preventing the inactivation of the AKT/mTORC1 pathway and increasing HSC output. This suggests that targeting the MEK/ERK pathway could be developed into a strategy to fine-tune the HSC compartment, for instance by remobilizing dysfunctional HSCs with reduced differentiation potential, as observed in aging (Akunuru and Geiger, 2016). In the context of leukemia, oncogenic NRASG12D, the common activator of MEK and PI3K, is considered an early, predisposing mutation potentially promoting the expansion of pre-leukemic clones. NRASG12D promotes the proliferation and reduces the repopulating potential of a subset of HSCs while reducing the proliferation and increasing the repopulating potential of another subset. It is tempting to speculate that, in the latter subset, the strict feedback control of PI3K signaling regulates proliferation, preserving a pool of self-renewing clones, whereas in fast-proliferating HSC pools, reduced feedback control would tilt the balance toward sustained PIP3 signaling, fast proliferation, and ultimately exhaustion. In line with this hypothesis, MEK1 ablation or inhibition restricts NRAS-driven disease (Nowacka et al., 2016, Wang et al., 2013) and mTOR activation suppresses leukaemogenesis in PTEN-deficient animals (Kalaitzidis et al., 2012, Lee et al., 2010); targeting MEK/ERK to modulate both pathways could promote the exit of malignant stem cells from quiescence to sensitize them to chemotherapy and/or stimulate their differentiation.
STAR★Methods
Key Resources Table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| α-mouse Gr-1-Biotin | eBioscience | clone RB6-8C5; RRID:AB_469757 |
| α-mouse CD5-Biotin | eBioscience | clone 53-7.3; RRID:AB_466338 |
| α-mouse CD2-Biotin | eBioscience | clone RPA-2.10; RRID:AB_466314 |
| α-mouse CD3ε-Biotin | eBioscience | clone 145-2C11; RRID:AB_466320 |
| α-mouse Ter119-Biotin | eBioscience | clone TER-119; RRID:AB_466796 |
| α-mouse CD8a-Biotin | eBioscience | clone 53-6.7; RRID:AB_466345 |
| α-mouse B220-Biotin | eBioscience | clone RA3-6B2; RRID:AB_466450 |
| PerCP Streptavidin | BD Biosciences | Cat#554064; RRID:AB_2336918 |
| α-mouse Sca1-PE | eBioscience | clone D7; RRID:AB_466085 |
| α-mouse cKIT-APC-eFluor780 | eBioscience | clone 2B8; RRID:AB_1272177 |
| α-mouse CD150-APC | BioLegend | clone TC15-12F12.2; RRID:AB_493460 |
| α-mouse CD48-PE-Cy7 | BD Biosciences | clone HM48-1; RRID:AB_1727501 |
| α-human CD34-APC | eBiosciences | clone 4H11; RRID:AB_2016604 |
| α-mouse CD34-FITC | eBioscience | clone RAM34; RRID:AB_465020 |
| α-human CD16/32-PE | eBioscience | clone 93; RRID:AB_465568 |
| α-human CD38- PE-Cy7 | BD Biosciences | clone HB7; RRID:AB_399969 |
| α-mouse Gr-1-PE | eBioscience | clone RB6-8C5; RRID:AB_466046 |
| α-mouse CD3ε-APC-eFluor 780 | eBioscience | clone 145-2C11; RRID:AB_11149132 |
| α-mouse B220-APC | eBioscience | clone RA3-6B2; RRID:AB_469394 |
| α-mouse Mac1-PerCP-Cy5.5 | eBioscience | clone M1/70; RRID:AB_953560 |
| α-mouse CD45.1-FITC | eBioscience | clone A20; RRID:AB_465059 |
| α-mouse CD45.2-AlexaFluor 700 | eBioscience | clone 104; RRID:AB_657752 |
| rabbit α-cleaved caspase 3 | Cell Signaling Technology | clone 5A1E; RRID:AB_2070042 |
| mouse α- α-MEK1 | Cell Signaling Technology | clone 61B12; RRID:AB_10693788 |
| rabbit α-pMEK1/2 S218/222 | Cell Signaling Technology | Cat #9121; RRID:AB_331648 |
| rabbit α-MEK1/2 | Cell Signaling Technology | Cat #9122; RRID:AB_823567 |
| rabbit α-pMEK1 T292 | Upstate | Cat #07-852; RRID:AB_568845 |
| rabbit α-MEK2 | Cell Signaling Technology | clone 13E3; RRID:AB_2140641 |
| rabbit α-ERK1/2 | Cell Signaling Technology | Cat #9102; RRID:AB_330744 |
| rabbit α-pERK1/2 | Cell Signaling Technology | Cat #9101; RRID:AB_331646 |
| rabbit α-AKT | Cell Signaling Technology | Cat #9272; RRID:AB_329827 |
| rabbit α-pAKT S473 | Cell Signaling Technology | Cat #9271; RRID:AB_329825 |
| rabbit α-FOXO3a | Cell Signaling Technology | Cat #2497; RRID:AB_836876 |
| rabbit α-pFOXO3a S294 | Cell Signaling Technology | Cat #5538; RRID:AB_10696878 |
| rabbit α-pFOXO3a S253 | Cell Signaling Technology | Cat #9466; RRID:AB_2106674 |
| rabbit α-mTOR | Cell Signaling Technology | Cat #2983; RRID:AB_2105622 |
| rabbit α-p-mTOR S2448 | Cell Signaling Technology | Cat #5536; RRID:AB_10691552 |
| rabbit α-S6 | Cell Signaling Technology | Cat #2217; RRID:AB_331355 |
| rabbit α-pS6 | Cell Signaling Technology | Cat #4858; RRID:AB_916156 |
| rabbit α-PINK1 | Abcam | Cat #ab23707; RRID:AB_447627 |
| rabbit mAb IgG isotype control | Cell Signaling Technology | Cat #3900; RRID:AB_1550038 |
| mouse mAb IgG isotype control | Cell Signaling Technology | Cat #5415; RRID:AB_10829607 |
| goat α-rabbit IgG Alexa Fluor 488 | ThermoFisher Scientific | Cat #R37116; RRID:AB_2556544 |
| goat α-mouse IgG Alexa Fluor 488 | ThermoFisher Scientific | Cat #A28175; RRID:AB_2536161 |
| goat α-rabbit IgG Alexa Fluor 647 | ThermoFisher Scientific | Cat #A27040; RRID:AB_2536101 |
| goat α-mouse IgG Alexa Fluor 647 | ThermoFisher Scientific | Cat #A28181; RRID:AB_2536165 |
| mouse α-TOMM20 | Abcam | Cat #ab56783; RRID:AB_945896 |
| rabbit α-LAMP1 | Abcam | Cat #ab24170; RRID:AB_775978 |
| mouse α-p-Histone H2A.X (S139) | Millipore | clone JBW301; RRID:AB_309864 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-Fluorouracil | Sigma | Cat #F6627 |
| N-acetyl-L-cysteine | Sigma | Cat #A7250 |
| 5-bromodeoxyuridine | Sigma | Cat #B5002 |
| DMSO | Sigma | Cat #276855 |
| U0126 | Cell Signaling Technology | Cat #9903S |
| LY294002 | Cell Signaling Technology | Cat #9901S |
| Metformin | Sigma | Cat #PHR1084 |
| Rapamycin | Enzo Life Science | Cat #BML-A275-0005/0025 |
| Polyethylene glycol (PEG) 400 | Sigma | Cat #91893 |
| TweenR 80 | Sigma | Cat #P1754 |
| CellROX Green Reagent | ThermoFisher Scientific | Cat #C10444 |
| CellROX Deep Red Reagent | ThermoFisher Scientific | Cat #C10422 |
| MitoTracker Green FM | ThermoFisher Scientific | Cat #M7514 |
| MitoTracker Deep Red FM | ThermoFisher Scientific | Cat #M22426 |
| TMRE | Sigma | Cat #87917 |
| recombinant murine SCF | PeproTech | Cat #250-03 |
| recombinant murine TPO | PeproTech | Cat #315-14 |
| qRT-PCR primers | Sigma | Table S2 |
| Critical Commercial Assays | ||
| FITC BrdU Flow Kit | BD Biosciences | Cat #559619 |
| MethoCult M3434 | StemCell Technologies | Cat #03434 |
| MethoCult H4435 Enriched | StemCell Technologies | Cat #04435 |
| Mouse LTC assay | StemCell Technologies | https://cdn.stemcell.com/media/files/manual/MA28417-Mouse_Long_Term_Culture_Initiating_Cell_Assays_Procedure.pdf |
| Human LTC assay | StemCell Technologies | https://cdn.stemcell.com/media/files/manual/MA28412-Human_Long_Term_Culture_Initiating_Cell_Assay.pdf |
| Fixable Viability Dye eFluor 450 | eBioscience | Cat #65-0863-14 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat #554714 |
| FITC Mouse α-Ki-67 Set | BD Biosciences | Cat #556026 |
| α-Biotin MicroBeads | Miltenyi Biotec | Cat #130-090-485 |
| Deposited Data | ||
| RNA-seq data | this paper | GEO: GSE112842 |
| Analysis of the F/F and MEK1 cKO hematopoietic compartments in different stress models | this paper; Mendeley data | https://doi.org/10.17632/7rdg6mjk5h.1 |
| Validation of the antibodies used in the phosflow experiments | this paper; Mendeley data | https://doi.org/10.17632/7rdg6mjk5h.1 |
| Experimental Models: Cell Lines | ||
| Human: CD34+ bone marrow progenitors | CellSystems | http://cellsystems.de/category/cd34-cells-1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: MAP2k1 (MEK1) F/F | Catalanotti et al., 2009 | N/A |
| Mouse: VAV1-iCRE | de Boer et al., 2003 | N/A |
| Mouse: C57/BL6 Ly5.1 | Charles River Laboratories | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Manuela Baccarini (manuela.baccarini@univie.ac.at).
Experimental Model and Subject Details
To obtain conditional knockout (cKO) mice lacking MEK1 in the hematopoietic system, MEK1 F/F mice (Catalanotti et al., 2009) were crossed with Vav1-iCre transgenic mice (de Boer et al., 2003). Genotyping of MEK1 was performed by using 3 different primers: primer 1, 5′-GACGTGGTGAACAGGAAAGGGATTGGG-3′; primer 2, 5′-TGGAGCTGGAGT CACGGGTGGTTGTAA-3′; and primer 3, 5-GCGAACTGGGAGCTGGCAACGGTG GAG-3′. Primer pair 1 and 3 amplifies a 470-bp 'fragment of the endogenous Map2k1 allele and a 520-bp fragment of the floxed allele, whereas primer pair 1 and 2 amplifies a 600-bp fragment of the null Mek1 allele (Catalanotti et al., 2009). The following primers were used for Vav1-iCre PCR: forward 5′-GCCTGCCCTCCCTGTGGATGCCACCT-3′and reverse 5′-GTGGCAGAAGGGGCAGC CACACCATT-3′. Both mouse strains (MEK1 F/F and MEK1 cKO) were on a pure C57/BL6 Ly5.2 (= CD45.2) background. Unless otherwise specified, all experiments were performed with 8-12 weeks old body weight-matched mice from both genders. For transplantation experiments, 12 weeks old Ly5.1 (= CD45.1) C57/BL6 recipient mice were used. All animals were maintained at the MFPL animal facility under specific pathogen-free conditions and all experiments were performed in accordance with a protocol authorized by the Austrian Ministry of Science and Communications. The health status was determined by using sentinel animals housed under the same conditions. Human bone marrow derived CD34+ progenitor cells from young healthy female and male donors were obtained from CellSystems.
Method Details
5-FU treatment
To induce emergency hematopoiesis, 5-Fluorouracil (5-FU; Sigma) was dissolved in 0.9% NaCl for 15 min at 55°C and 150 mg/kg were injected intraperitoneally either as a single dose or as multiple doses (up to 7, once every 12 days; repetitive 5-FU).
N-acetyl-L-cysteine treatment in vivo
N-acetyl-L-cysteine (NAC; Sigma) was dissolved in PBS and 100 mg/kg was administered daily by subcutaneous injection. To study the effect of ROS on the recovery from a single 5-FU injection, animals were pre-treated with NAC for 7 days.
Rapamycin treatment in vivo
Rapamycin (RAPA; Enzo Life Science) was dissolved in absolute ethanol (10 mg/ml), further diluted in 5% PEG-400/5% TweenR80/PBS and 0.4 mg/kg were administered daily by intraperitoneal injection. To study the effect of mTOR signaling on the recovery from a single 5-FU injection, animals were pre-treated with RAPA for 7 days.
Serial competitive stem cell transplantation assay
A 1:1 mixture of Ly5.2+ donor (F/F, cKO, or CRE) and Ly5.1+ competitor BM cells (2 × 106 total BM cells containing 100 donor and 100 competitor HSCs) were prepared in HBSS and transplanted by tail-vein injection into lethally irradiated, two-three months old C57BL/6 Ly5.1 ( = CD45.1) recipient mice. Starting three weeks after transplantation, peripheral blood was collected once per week in EDTA tubes and blood counts were performed on a V-Sight analyzer (Menarini Diagnostics, Italy). Donor chimerism in the peripheral blood was determined by FACS analysis. BM analysis was performed 12 weeks after transplantation. At this time point, 2 × 106 BM cells isolated from primary recipients were re-injected into lethally irradiated Ly5.1+ secondary recipient mice and monitored by the same protocol. In selected experiments, recipients were treated with NAC, RAPA, or the respective vehicle once per day starting from 2 weeks after transplantation.
5-bromodeoxyuridine label experiment
To determine the numbers of 5-bromodeoxyuridine (BrdU, Sigma) label-retaining cells, a single dose of BrdU (200 mg/kg i.p.) was administered to 5-month old animals, followed by 1 mg/ml BrdU in drinking water (14 days; BrdU water was exchanged every 3 days). After a chase period of 200 days, BM cells were isolated and stained with surface antigen markers for mouse HSPC characterization. BrdU incorporation was measured by flow cytometry using the FITC BrdU Flow Kit (BD Biosciences) according to the manufacturer’s instructions. BM cells from non-BrdU injected mice were used as a control.
In vitro long-term co-culture (LTC) assay
LTC assays were performed according to the protocol from StemCell Technologies (https://cdn.stemcell.com/media/files/manual/MA28412-Human_Long_Term_Culture_Initiating_Cell_Assay.pdf). For mouse HSCs (LSK CD150+ CD48-), Ly5.1 BM stromal feeder layer cells were generated in 96-well plates (flat bottom, tissue culture treated) according to the protocol. The M2-10B4 mouse fibroblast cell line (ATCC® CRL-1972; ATCC) was used as stromal feeder layer for human HSCs (CD34+CD38- cells). After irradiation of the feeder layers, 100 HSCs per well were FACS-sorted directly onto the feeder layer and incubated for up to 10 weeks at 33°C (5% CO2) in MyeloCult medium (M5300 for mouse cells or M5100 for human cells, StemCell Technologies, supplemented with 1μM hydrocortisone). Half of the medium was replaced with fresh medium once per week. In selected experiments, cells were treated with DMSO (1:10,000 dilution; Sigma) iMEK ( = U0126, 250 nM; Cell Signaling Technologies = CST), iPI3K ( = LY294002, 1 μM; CST), Rapamycin (500 nM; Enzo Life Science), Metformin (500 μM; Sigma), and NAC (100 μM; Sigma), either alone or in different combinations.
Cells were harvested at different time points (2, 4, 6, 8, 10 weeks) and transferred to cytokine-supplemented methylcellulose medium (MethoCult GF M3434 or MethoCult H4435 Enriched, StemCell Technologies). Colony forming units (CFUs) were scored after 10 days. For further functional characterization after 6 weeks in co-culture, ROS levels, mitochondrial mass, mitochondrial membrane potential, and intracellular signaling in HSCs identified as Ly5.2+ LSK CD48- (mouse) or CD34+ CD38- cells (human) were determined by FACS.
HSC senescence was assessed by single cell-sorting LSK CD150+ CD48- cells into cytokine-supplemented methylcellulose medium (MethoCult GF M3434) and counting the number of colonies derived from 100 sorted single-cells after 10 days of incubation.
Flow cytometry analysis and sorting of mouse and human HSCs
Mouse BM cells were recovered by crushing of femora and tibiae in 5 mL of 2% FCS/IMDM medium using a mortar and pestle. The cell suspension was filtered through a 70 μm cell strainer and washed twice in 2% FCS/IMDM. Viability staining was performed following red blood cell lysis (eBioscience) using the fixable viability dye eFluor 450 (eBioscience). Gating started with a FSC/SSC dot plot visualizing all acquired events. After doublet discrimination using FSC-A/FSC-W, singlets were gated for live cells. Live single cells were then plotted against surface/intracellular markers. The boundary between positive and negative cells was determined by fluorescence-minus one (FMO) and isotype controls. For FACS analysis of SLAM-marker-defined mouse HSPCs, cells were stained with a cocktail of biotinylated lineage-specific antibodies against Gr-1 (RRID:AB_469757), CD5 (RRID:AB_466338), CD2 (RRID:AB_466314), CD3ε (RRID:AB_466320), Ter119 (RRID:AB_466796), CD8a (RRID:AB_466345), and B220 (RRID:AB_466450) (all from eBioscience), washed thrice in 1% FCS/PBS, and subsequently stained with streptavidin-PercP (RRID:AB_2336918, BD Bioscience) together with fluorophore-labeled antibodies against Sca1 (RRID:AB_466085, eBioscience), c-KIT (RRID:AB_1272177, eBiosciences), CD150 (RRID:AB_493460, BioLegend) and CD48 (RRID:AB_1727501, BD Biosciences) for immature progenitors or fluorophore-labeled antibodies against Sca1, c-KIT, CD34 (RRID:AB_2016604 or RRID:AB_465020, eBioscience) and CD16/32 (RRID:AB_465568, eBioscience) for mature progenitors. For FAC-sorting, BM cells were first stained with the biotinylated lineage-cocktail followed by lineage-depletion using α-biotin beads and an AutoMACS cell separator (Miltenyi Biotec). Lineage-depleted BM cells were then labeled with a panel of antibodies to identify HSPCs (Sca1, c-KIT, CD150, CD48). Mouse HSPCs were defined as follows: lin+, lineage-positive (differentiated cells); lin-, lineage-negative cells; LK, lin- Sca1- c-KIT+ (mature progenitors) = MEP, lin- Sca1+ c-KIT- CD34- CD16/32-; CMP, lin- Sca1+ c-KIT- CD34dim CD16/32-dim; GMP, lin- Sca1+ c-KIT- CD34+ CD16/32+; LSK, lin- Sca1+ c-KIT+ (immature stem/progenitor cells) = HPC1, LSK CD150- CD48+ (hematopoietic progenitor cell 1); HPC2, LSK CD150+ CD48+ (hematopoietic progenitor cell 2); MPP, LSK CD150- CD48- (multipotent progenitor); HSC, LSK CD150+ CD48- (hematopoietic stem cell) (Oguro et al., 2013). The FACS gating strategy for mouse HSPCs is shown in Figure S1E. Human BM CD34+ HSPCs from young healthy donors were purchased from CellSystems. For FACS analysis and sorting of human HSCs, CD34-pre-enriched cells were stained with antibodies against CD34 (RRID:AB_2016604 or RRID:AB_465020, eBioscience) and CD38 (RRID:AB_399969, eBioscience). Human HSCs were defined as CD34+ CD38- cells. The purity of sorted HSCs populations, determined by FACS analysis of post-sort fractions, was 89%–98% for mouse and 81%–97% for human HSCs, respectively.
Differentiation was analyzed by staining with conjugated antibodies against specific surface markers as defined above plus α-Mac1 (RRID:AB_953560, eBiosciences). B cells were identified as B220+, T cells as CD3ε+, and myeloid cells as Mac1/Gr1+. In competitive transplantation experiments, peripheral blood chimerism was assessed by co-staining with antibodies against Ly5.1 (RRID:AB_465059) and Ly5.2 (RRID:AB_657752; both from eBiosciences).
Cell cycle analysis
Surface marker stained BM cells were washed in phosphate-buffered saline (PBS; pH 7.4), fixed in Cytofix/Cytoperm buffer (BD Bioscience) for 30 min at room temperature (RT), washed twice with PermWash buffer (BD Bioscience), and incubated in PermWash buffer with Ki-67-FITC antibody (1:20, BD Bioscience) for 1h at RT. Cells were then washed twice with PermWash buffer, resuspended in 1% BSA/PBS containing 1μg/ml DAPI, and incubated for further 15 min before analysis.
ROS, MitoTracker and mitochondrial membrane potential measurement
Freshly isolated BM or cells from LTCs washed in PBS and incubated for 30 min at 37°C with CellROX Green or CellROX Deep Red (1 μM, Thermo Fisher Scientific), MitoTracker Green or MitoTracker Deep Red (20 nM, Thermo Fisher Scientific), or tetramethylrhodamine ethyl ester perchlorate (TMRE; 5 nM, Sigma) in 2% FCS/IMDM. After labeling, cells were washed three times with PBS and stained with cell surface markers for mouse or human HSCs. A minimum of 100 HSCs per condition were analyzed by FACS.
FACS intracellular staining for (phospho)-proteins
For intracellular staining, BM cells were stained with HSC surface markers, fixed for 20 min at RT in Cytofix/Cytoperm buffer (BD Bioscience), washed twice with PermWash buffer (BD Bioscience), and incubated for 1h at RT with the following primary antibodies: α-cleaved caspase 3 (RRID:AB_2070042, CST; 1:200), α-MEK1 (RRID:AB_10693788, CST; 1:200), α-pMEK1/2 S218/222 (RRID:AB_331648, CST; 1:100), α-MEK1/2 (RRID:AB_823567, CST; 1:100), α-pMEK1 T292 (RRID:AB_568845, Upstate; 1:100), α-MEK2 (RRID:AB_2140641, CST; 1:100), α-ERK1/2 (RRID:AB_330744, CST; 1:200), α-pERK1/2 (RRID:AB_331646, CST; 1:200), α-AKT (RRID:AB_329827, CST; 1:200), α-pAKT S473 (RRID:AB_329825, CST; 1:100), α-FOXO3a (RRID:AB_836876, CST; 1:200), α-pFOXO3A S294 (RRID:AB_10696878, CST; 1:100), α-pFOXO3a S253 (RRID:AB_2106674, CST; 1:100), α-mTOR (RRID:AB_2105622, CST; 1:250), α-p-mTOR S2448 (RRID:AB_10691552, CST; 1:100), α-S6 (RRID:AB_331355, CST; 1:200), α-pS6 (RRID:AB_916156, CST; 1:100), α-PINK1 (RRID:AB_447627, Abcam; 1:200) or α-rabbit-mAb-IgG-isotype (RRID:AB_1550038, CST; concentration matched) and α-mouse-mAb-IgG-isotype (RRID:AB_10829607, CST; concentration matched). Cells were washed thrice in PermWash buffer and subsequently incubated with the conjugated secondary antibodies Alexa488-goat α-rabbit/-goat α-mouse or Alexa647-goat α-rabbit/-goat α-mouse (all from Thermo Fisher Scientific) for 1h at RT. After washing 3 times in PermWash buffer, cells were resuspended in 2%BSA/PBS and analyzed. All sorting and FACS analysis experiments were performed using a BD FACS Aria III or BD LSM Fortessa analyzers. Data were analyzed with the FlowJo (10.2) software. A validation of the antibodies used in this study is deposited in the Mendeley database (https://doi.org/10.17632/7rdg6mjk5h.1).
Immunofluorescence staining
HSCs (500-1000 cells) were FAC-sorted directly onto poly-L-lysine coated coverslips, incubated for 2h, and fixed in PBS/4% PFA for 20 min at RT. Cells were permeabilized in PBS/0.1% Triton X-100 for 10 min at RT prior to blocking in PBS/0.1% Triton/5% BSA for 1h at RT. Cells were incubated in blocking solution with the following primary Abs, either for 2h at RT or at 4°C overnight: rabbit α-FOXO3a (RRID:AB_836876; CST; 1:200), mouse α-TOMM20 (RRID:AB_945896, Abcam; 1:100,), rabbit α-LAMP1 (RRID:AB_775978, Abcam; 1:150) or mouse α-phospho-Histone H2A.X (S139 = anti-γH2A.X; RRID:AB_309864; 1:200). After incubation, cells were washed 3-5 times with PBS and incubated for 1h at RT in blocking solution with Alexa488-conjugated goat α-rabbit, Alexa488-conjugated goat α-mouse, or Alexa647-conjugated goat α-rabbit (all from Thermo Fisher Scientific). Cells were then washed thrice in PBS and mounted in VectaShield with DAPI (Vector Laboratories). Slides were imaged on an inverse point scanning confocal microscope (Zeiss LSM 710 Airyscan, 63X or 100X magnification), and images were processed using Zeiss imaging software. For statistics, 100 cells per condition were scored on a Zeiss Axio Imager Z2 microscope.
RNA-seq library preparation and data analysis
100 HSCs per sample were FAC-sorted in 96 well plates containing lysis buffer. cDNA synthesis and enrichment were performed according to the Smart-seq2 protocol as described previously (Picelli et al., 2014). ERCC spike-in RNA (Ambion) was added to the lysis buffer in a dilution of 1:1,000,000. Library preparation was conducted on 1 ng of cDNA using the Nextera XT library preparation kit (Illumina) as described. Sequencing was performed by the Biomedical Sequencing Facility at CeMM using the 50 bp single-read setup on the Illumina HiSeq 3000/4000 platform. To analyze the data, we first trimmed off sequencing adapters from the reads generated, and then aligned the reads using Bowtie v 1.1.1 (Langmead et al., 2009) parameters: -q -p 6 -a -m 100–minins 0–maxins 5000–fr–sam–chunkmbs 200) to the cDNA reference transcriptome (mm10 cDNA sequences from Ensembl v 77). Next, we removed duplicate reads before quantifying transcript levels with BitSeq v 1.12.0 (Glaus et al., 2012). The raw transcript counts were loaded into R and collapsed by taking the expression level of the most strongly expressed transcript per gene. For statistical analysis, we used these read counts as input for DESeq2 (Love et al., 2014) factoring in the date of library preparation and sequencing as a covariate to reduce the effect of technical variation. For visualization, we used variance-stabilized data.
The complete normalized RNA-seq data and results of the statistical analyzes are given in Table S1. Finally, we used the Enrichr API (Kuleshov et al., 2016) to extract the top 10 enriched terms (ranked by combined score) from the Gene Ontology (BP and CC) (Ashburner et al., 2000, The Gene Ontology Consortium, 2017)) and KEGG (Kanehisa et al., 2017, Kanehisa and Goto, 2000) databases.
qRT–PCR
cDNAs were synthesized and amplified from single cells using the same protocol as for RNA-seq, quantitative PCR was performed using GoTaq® qPCR Master Mix (Promega) according to the manufacturer’s instructions using a Mastercycler realplex real-time PCR system (Eppendorf). All primer sequences are listed in Table S2. mRNA levels were calculated and normalized to actin (Actb) housekeeping gene using the ΔCT method.
Lentiviral transduction protocol
For knockdown experiments, predicted mouse shMEK1 (5′-TGCTGTTGACAGTGAGCGA
CCAGGCTGAACTACAGTGAAATAGTGAAGCCACAGATGTATTTCACTGTAGTTCAGCCTGGCTGCCTACTGCCTCGGA-3′) and human shMEK1 (5′-GAAGGTATAT TGCTGTTGACAGTGAGCGAACTTCTGGTTGTATTTCTATATAGTGAAGCCACAGATGTATATAGAAATACAACCAGAAGTCTGCCTACTGCCTCGGACTTCAAGGGGCTAG-3′) sequences were subcloned into the lentiviral SGEP vector (containing GFP) using XhoI/EcoRI enzymes and Gibson cloning strategy (Fellmann et al., 2013). The same SGEP vector was used for the overexpression of the following constructs: MEK1 WT, MEK1 T292D, MEK1 T292A, FOXO3A WT, FOXO3A ERK-insensitive (S294A, S344A, S425A), FOXO3A AKT-insensitive (T32A, S253A, S315A), FOXO3A ERK/AKT-insensitive (combined ERK and AKT point mutations), and PINK1. The genes of interest were inserted into the lentiviral SGEP vector downstream of the PGK promoter using BveI/CsiI enzymes and thus replaced the selection marker puromycin. All MEK1 constructs have been described previously (Catalanotti et al., 2009, Zmajkovicova et al., 2013). FOXO3a WT and FOXO3a ERK-insensitive constructs were a kind gift of Mien-Chie Hung's Lab. FOXO3a AKT-insensitive and FOXO3a ERK/AKT-insensitive constructs were generated by site-directed mutagenesis. The PINK1 construct was purchased from Addgene (pGEX5X.1 PINK1 WT, Plasmid #13321).
The lentiviral constructs encode the gene of interest and a fluorescent marker for FAC-sorting. Lentiviral particles were produced in 293T cells using a standard protocol. HSCs were infected for 48 hours with lentivirus-containing medium supplemented with SCF (10ng/ml; PeproTech) and TPO (10ng/ml; PeproTech) in the presence of 8μg/ml polybrene. After 48 hours, positively transduced cells (GFP-positive) were sorted by FACS.
Quantification and Statistical Analyses
The experiment in Figures 2D, 2E, and S2 was performed once (n = 3-4 animals); the transplantation experiments in Figures 1B and 1C were performed twice; all other experiments were performed three times or more. In all Figs, n = number of biological replicates. In all LTC experiments n = 3, each assayed in three technical replicates. Unless otherwise specified, data are represented as mean ± standard deviation. Statistical analysis was performed using the two tailed Student's t test, with the exception of the survival data (Figures 1F and 3C), which were analyzed using the Mantel–Cox log-rank test. In all cases, ∗p < 0.05; ∗∗p < 0.01; and ∗∗∗p < 0.001 were considered statistically significant.
Data and Software Availability
The accession number for the preprocessed RNA-seq data of untreated and 5-FU stressed MEK1 F/F and cKO HSCs reported in this paper is GEO: GSE112842. A detailed description of data analysis and the software used can be found in Method Details. The analysis of the F/F and MEK1 cKO hematopoietic compartments in different stress models and the validation of the antibodies used in the phosflow experiments is deposited in the Mendeley database (https://doi.org/10.17632/7rdg6mjk5h.1).
Acknowledgments
We thank Johannes Zuber for the gift of the SGEP lentiviral vector; Mien-Chie Hung for the human Foxo3a constructs (wild-type and ERK site phospho-ablated mutant); Michaela Prchal-Murphy for help with the γ-irradiation of stromal feeder layers; and Karin Ehrenreiter, Karel Hanak, Clemens Bogner, and the animal facility for technical help. We also thank the team of the Biomedical Sequencing Facility at CeMM for support with next generation sequencing. This work was supported by grant P26303-B20 of the Austrian Science Fund (FWF) to M.B. M.F. is supported by the network grant FWF-SFB F061 (F6102). F.H. is supported by a postdoctoral fellowship of the German Research Council (DFG) (grant HA 7723/1-1). C. Bock is supported by a New Frontiers Group award of the Austrian Academy of Sciences and by an ERC Starting Grant (European Union’s Horizon 2020 research and innovation program; grant agreement no. 679146).
Author Contributions
Conceptualization, M.B., C. Bock, and V.S.; Methodology and Investigation, C. Baumgartner, S.T., I.F., M.F., and R.S.; Data Curation and Formal Analysis, F.H. and C. Bock; Writing and Visualization, C. Baumgartner and M.B.; Funding Acquisition and Supervision, M.B.
Declaration of Interests
The authors declare no competing interests.
Published: May 24, 2018
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.stem.2018.05.003.
Supplemental Information
References
- Akunuru S., Geiger H. Aging, clonality, and rejuvenation of hematopoietic stem cells. Trends Mol. Med. 2016;22:701–712. doi: 10.1016/j.molmed.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernitz J.M., Kim H.S., MacArthur B., Sieburg H., Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. 2016;167:1296–1309.e10. doi: 10.1016/j.cell.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigarella C.L., Liang R., Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141:4206–4218. doi: 10.1242/dev.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Busch K., Klapproth K., Barile M., Flossdorf M., Holland-Letz T., Schlenner S.M., Reth M., Höfer T., Rodewald H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N., Buettner F., Sommerkamp P., Klimmeck D., Ladel L., Thalheimer F.B., Pastor-Flores D., Roma L.P., Renders S., Zeisberger P. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. 2017;169:807–823.e19. doi: 10.1016/j.cell.2017.04.018. [DOI] [PubMed] [Google Scholar]
- Catalanotti F., Reyes G., Jesenberger V., Galabova-Kovacs G., de Matos Simoes R., Carugo O., Baccarini M. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat. Struct. Mol. Biol. 2009;16:294–303. doi: 10.1038/nsmb.1564. [DOI] [PubMed] [Google Scholar]
- Chandel N.S., Jasper H., Ho T.T., Passegué E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 2016;18:823–832. doi: 10.1038/ncb3385. [DOI] [PubMed] [Google Scholar]
- Chen C., Liu Y., Liu R., Ikenoue T., Guan K.L., Liu Y., Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang G.G., Abraham R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- Chung E., Kondo M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011;49:248–268. doi: 10.1007/s12026-010-8187-5. [DOI] [PubMed] [Google Scholar]
- Crane G.M., Jeffery E., Morrison S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017;17:573–590. doi: 10.1038/nri.2017.53. advance online publication. [DOI] [PubMed] [Google Scholar]
- Damnernsawad A., Kong G., Wen Z., Liu Y., Rajagopalan A., You X., Wang J., Zhou Y., Ranheim E.A., Luo H.R. Kras is required for adult hematopoiesis. Stem Cells. 2016;34:1859–1871. doi: 10.1002/stem.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J., Williams A., Skavdis G., Harker N., Coles M., Tolaini M., Norton T., Williams K., Roderick K., Potocnik A.J., Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- Desideri E., Cavallo A.L., Baccarini M. Alike but different: RAF paralogs and their signaling outputs. Cell. 2015;161:967–970. doi: 10.1016/j.cell.2015.04.045. [DOI] [PubMed] [Google Scholar]
- Dorard C., Vucak G., Baccarini M. Deciphering the RAS/ERK pathway in vivo. Biochem. Soc. Trans. 2017;45:27–36. doi: 10.1042/BST20160135. [DOI] [PubMed] [Google Scholar]
- Essers M.A., Offner S., Blanco-Bose W.E., Waibler Z., Kalinke U., Duchosal M.A., Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Fellmann C., Hoffmann T., Sridhar V., Hopfgartner B., Muhar M., Roth M., Lai D.Y., Barbosa I.A., Kwon J.S., Guan Y. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Foretz M., Guigas B., Bertrand L., Pollak M., Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Glaus P., Honkela A., Rattray M. Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics. 2012;28:1721–1728. doi: 10.1093/bioinformatics/bts260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F., Zhang S., Grogg M., Cancelas J.A., Varney M.E., Starczynowski D.T., Du W., Yang J.Q., Liu W., Thomas G. Mouse gene targeting reveals an essential role of mTOR in hematopoietic stem cell engraftment and hematopoiesis. Haematologica. 2013;98:1353–1358. doi: 10.3324/haematol.2012.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T.T., Warr M.R., Adelman E.R., Lansinger O.M., Flach J., Verovskaya E.V., Figueroa M.E., Passegué E. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshii T., Tadokoro Y., Naka K., Ooshio T., Muraguchi T., Sugiyama N., Soga T., Araki K., Yamamura K., Hirao A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Invest. 2012;122:2114–2129. doi: 10.1172/JCI62279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Carracedo A., Weiss D., Arai F., Ala U., Avigan D.E., Schafer Z.T., Evans R.M., Suda T., Lee C.-H., Pandolfi P.P. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Turcotte R., Cui J., Zimmerman S.E., Pinho S., Mizoguchi T., Arai F., Runnels J.M., Alt C., Teruya-Feldstein J. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354:1156–1160. doi: 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzidis D., Sykes S.M., Wang Z., Punt N., Tang Y., Ragu C., Sinha A.U., Lane S.W., Souza A.L., Clish C.B. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11:429–439. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T., Kang J., Lee T.H., Wojnowski L., Pritchard C.A., Leavitt A.D. A critical function for B-Raf at multiple stages of myelopoiesis. Blood. 2005;106:833–840. doi: 10.1182/blood-2004-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T., Dankort D., Kang J., Giblett S., Pritchard C.A., McMahon M., Leavitt A.D. Hematopoietic expression of oncogenic BRAF promotes aberrant growth of monocyte-lineage cells resistant to PLX4720. Mol. Cancer Res. 2013;11:1530–1541. doi: 10.1158/1541-7786.MCR-13-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharas M.G., Okabe R., Ganis J.J., Gozo M., Khandan T., Paktinat M., Gilliland D.G., Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbus A., Pilat S., Husak Z., Deiner E.M., Stengl G., Beug H., Baccarini M. Raf-1 antagonizes erythroid differentiation by restraining caspase activation. J. Exp. Med. 2002;196:1347–1353. doi: 10.1084/jem.20020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.Y., Nakada D., Yilmaz O.H., Tothova Z., Joseph N.M., Lim M.S., Gilliland D.G., Morrison S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell. 2010;7:593–605. doi: 10.1016/j.stem.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Bohin N., Wen T., Ng V., Magee J., Chen S.C., Shannon K., Morrison S.J. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504:143–147. doi: 10.1038/nature12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J.A., Ikenoue T., Nakada D., Lee J.Y., Guan K.L., Morrison S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11:415–428. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y., Zhang Y., Yamamoto K., Xie W., Mak T.W., You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl. Acad. Sci. USA. 2009;106:5153–5158. doi: 10.1073/pnas.0901104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K., Araki K.Y., Naka K., Arai F., Takubo K., Yamazaki S., Matsuoka S., Miyamoto T., Ito K., Ohmura M. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacka J.D., Baumgartner C., Pelorosso C., Roth M., Zuber J., Baccarini M. MEK1 is required for the development of NRAS-driven leukemia. Oncotarget. 2016;7:80113–80130. doi: 10.18632/oncotarget.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H., Ding L., Morrison S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.H., Ko J., Park Y.S., Park J., Hwang J., Koh H.C. Clearance of damaged mitochondria through PINK1 stabilization by JNK and ERK MAPK signaling in chlorpyrifos-treated neuroblastoma cells. Mol. Neurobiol. 2017;54:1844–1857. doi: 10.1007/s12035-016-9753-1. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Pickrell A.M., Youle R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak R., Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood. 2012;119:911–923. doi: 10.1182/blood-2011-07-366203. [DOI] [PubMed] [Google Scholar]
- Rimmelé P., Liang R., Bigarella C.L., Kocabas F., Xie J., Serasinghe M.N., Chipuk J., Sadek H., Zhang C.C., Ghaffari S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015;16:1164–1176. doi: 10.15252/embr.201439704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers J.T., King K.Y., Brett J.O., Cromie M.J., Charville G.W., Maguire K.K., Brunson C., Mastey N., Liu L., Tsai C.R. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubiolo C., Piazzolla D., Meissl K., Beug H., Huber J.C., Kolbus A., Baccarini M. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood. 2006;108:152–159. doi: 10.1182/blood-2005-09-3866. [DOI] [PubMed] [Google Scholar]
- Sanchez S., Tafforeau P., Ahlberg P.E. The humerus of Eusthenopteron: a puzzling organization presaging the establishment of tetrapod limb bone marrow. Proc. Biol. Sci. 2014;281:20140299. doi: 10.1098/rspb.2014.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai C.M., Babovic S., Upadhaya S., Knapp D.J.H.F., Lavin Y., Lau C.M., Goloborodko A., Feng J., Fujisaki J., Ding L. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity. 2016;45:597–609. doi: 10.1016/j.immuni.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M.B., Sinclair D.A. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 2016;143:3–14. doi: 10.1242/dev.130633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M., Hall M.N. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Staser K., Park S.-J., Rhodes S.D., Zeng Y., He Y.Z., Shew M.A., Gehlhausen J.R., Cerabona D., Menon K., Chen S. Normal hematopoiesis and neurofibromin-deficient myeloproliferative disease require Erk. J. Clin. Invest. 2013;123:329–334. doi: 10.1172/JCI66167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Ramos A., Chapman B., Johnnidis J.B., Le L., Ho Y.J., Klein A., Hofmann O., Camargo F.D. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H., Regoes R.R., Boddupalli C.S., Bonhoeffer S., Manz M.G. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J. Exp. Med. 2011;208:273–284. doi: 10.1084/jem.20101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z., Gilliland D.G. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1:140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Van Meter M.E., Díaz-Flores E., Archard J.A., Passegué E., Irish J.M., Kotecha N., Nolan G.P., Shannon K., Braun B.S. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109:3945–3952. doi: 10.1182/blood-2006-09-047530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga A., Ehrenreiter K., Aschenbrenner B., Kocieniewski P., Kochanczyk M., Lipniacki T., Baccarini M. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKα. Sci. Signal. 2017;10:eaai8482. doi: 10.1126/scisignal.aai8482. [DOI] [PubMed] [Google Scholar]
- Walter D., Lier A., Geiselhart A., Thalheimer F.B., Huntscha S., Sobotta M.C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- Wang J., Kong G., Liu Y., Du J., Chang Y.I., Tey S.R., Zhang X., Ranheim E.A., Saba-El-Leil M.K., Meloche S. Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood. 2013;121:5203–5207. doi: 10.1182/blood-2012-12-475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr M.R., Binnewies M., Flach J., Reynaud D., Garg T., Malhotra R., Debnath J., Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Yang J.Y., Zong C.S., Xia W., Yamaguchi H., Ding Q., Xie X., Lang J.Y., Lai C.C., Chang C.J., Huang W.C. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O.H., Valdez R., Theisen B.K., Guo W., Ferguson D.O., Wu H., Morrison S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Zhang J., Grindley J.C., Yin T., Jayasinghe S., He X.C., Ross J.T., Haug J.S., Rupp D., Porter-Westpfahl K.S., Wiedemann L.M. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zmajkovicova K., Jesenberger V., Catalanotti F., Baumgartner C., Reyes G., Baccarini M. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol. Cell. 2013;50:43–55. doi: 10.1016/j.molcel.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.