

Abstract
Background
The tropomodulins (TMODs) are a family of proteins that cap the pointed ends of actin filaments. Four TMODs have been identified in humans, with orthologs in mice. Mutations in actin or actin-binding proteins have been found to cause several human diseases, ranging from hypertrophic cardiomyopathy to immunodefiencies such as Wiskott-Aldrich syndrome. We had previously mapped Tropomodulin 2 (TMOD2) to the genomic region containing the gene for amyotrophic lateral sclerosis 5 (ALS5). We determined the genomic structure of Tmod2 in order to better analyze patient DNA for mutations; we also determined the genomic structure of Tropomodulin 4 (TMOD4).
Results
In this study, we determined the genomic structure of TMOD2 and TMOD4 and found the organization of both genes to be similar. Sequence analysis of TMOD2 revealed no mutations or polymorphisms in ALS5 patients or controls. Interestingly, we discovered that another gene, YL-1, intergenically splices into TMOD4. YL-1 encodes six exons, the last of which is 291 bp from a 5' untranslated exon of TMOD4. We used 5' RACE and RT-PCR from TMOD4 to identify several intergenic RACE products. YL-1 was also found to undergo unconventional splicing using non-canonical splice sites within exons (intraexonic splicing) to produce several alternative transcripts.
Conclusions
The genomic structure of TMOD2 and TMOD4 have been delineated. This should facilitate future mutational analysis of these genes. In addition, intergenic splicing at TMOD4/YL-1 was discovered, demonstrating yet another level of complexity of gene organization and regulation.
Background
Tropomodulins are the vertebrate proteins that cap the pointed ends of filamentous actin [1]. The first tropomodulin (TMOD) gene was cloned by Sung and colleagues (1992) from a fetal liver library. Three additional family members have been identified in humans, and TMOD homologs have been identified in mouse, rat, chick, Drosophila, and C. elegans[2-8]. Expression patterns vary extensively: Tropomodulin 3 (TMOD3) is ubiquitous; Tropomodulin 1 (TMOD1) is widespread in heart, muscle, brain, lens, erythrocytes, and arterioles; Tropomodulin 2 (TMOD2) is restricted to neuronal tissue; Tropomodulin 4 (TMOD4). is expressed in skeletal muscle, with a less abundant ~7 kb transcript expressed in both skeletal and cardiac muscle [8]. Whereas tropomodulins or the Arp 2/3 complex cap the pointed end of actin filaments, the barbed end can be capped by CapZ, α, β, and γ adducins as well as gelsolin [9-11]. Control of thin filament length is critical for maintaining proper sarcomere function and length[12]. Inhibition of Tmod1's capping activity – either by using an antibody to its C-terminal end or by decreasing expression using an antisense Tmod1 transcript – results in elongated thin filaments and decreased cardiac contractility [13,14]. Tmod1 overexpression in rat cardiomyocytes causes shortening of the thin filaments and sarcomere disorganization, resulting in myofibril degeneration [14]. Likewise, mice overexpressing TMOD1 in the heart show disrupted sarcomere organization with shortened thin filaments, leading to myofibril degeneration and dilated cardiomyopathy [15]. Recently, Littlefield and colleagues showed that overexpression of GFP-Tmod1 in chick cardiac myocytes results in shortening of thin filaments; the authors proposed that excess Tmod1 decreases the affinity between actin monomers and pointed ends, leading to monomer dissociation and filament shortening [42]. Mutations in many proteins making up the cardiac sarcomere have been shown to cause cardiac hypertrophy [16-19]:mutations in the TPM1 gene, for example, cause type 3 familial hypertrophic cardiomyopathy (CMH3), and a transgenic mouse expressing a CMH3 mutation develops ventricular myocyte disarray and hypertrophy [20]. Mutations in myosin heavy chain 7 are estimated to account for 40–50% of the cases of hypertrophic cardiomyopathy [21].
Given the importance of actin-binding proteins in human disease, we mapped TMOD2 and TMOD4 and found them to localize to 15q21.1 and 1q12, respectively [8]. These loci overlap with the genomic regions containing the genes for amyotrophic lateral sclerosis 5 (ALS5) and limb girdle muscular dystrophy 1B (LGMD1B), respectively [22,23]. We characterized these tropomodulin genomic loci for use in mutational analysis. During the course of this study, the gene mutated in LGMD1B was identified as Lamin A/C [24]. Our investigation, however, revealed that an unrelated gene, YL-1, intergenically spliced into TMOD4 . YL-1 is a putative transformation suppressor gene that localizes to the nucleus and binds to DNA [25,26]. YL-1 is organized into six exons and produces several alternatively spliced transcripts using both intergenic and cryptic splicing. This relationship between YL-1 and TMOD4 serves as yet another example of the complexity of genomic organization and gene regulation.
Results
Genomic organization of TMOD2, TMOD4, and YL-1
We determined the exon/intron boundaries of TMOD2 and TMOD4 by sequencing PCR products amplified from primers within adjacent exons, and sequencing short exon containing genomic fragments. We identified nine coding exons in TMOD2 and in TMOD4 which are spread over a 45 kb and 4.7 kb genomic region, respectively (Fig. 1). The exon/intron boundaries of the two genes are conserved, except that TMOD2 has more 5' untranslated region (UTR) in coding exon 1 and more 3' UTR in exon 9 than TMOD4. TMOD2 and TMOD4 also contain additional 5' UTR exons. Our reported cDNAs have 69 and 19 bp additional sequence 5' to what is contained in exon 1 for each gene, respectively [8]. A search of the EST database using the TMOD4 5' end extended this sequence by 8 bp (ESTs AA194560, AA192772, and AA178988). We were able to align this sequence and found that it lies 1006 bp from TMOD4 exon 1 and contained a consensus donor splice site. Using 5' rapid amplification of cDNA ends (RACE) from TMOD4 exon 2, we discovered that this exon was 93 bp in length and contained no consensus AG acceptor. 3' RACE from TMOD4 exon 2 extended our reported cDNA by an additional 9 bp, bringing the entire length of the TMOD4 transcript to 1.267 kb (Genbank Accession# AF393374).
Figure 1.

Genomic structure of TMOD2, TMOD4, and YL-1. Numbered rectangles represent coding exons, E0 represents 5' UTR exons. ATG represents the start codon of each gene and TAA or TAG represent the termination codon of each gene. Scale below is in kilobases (kb).
Using the 5' end of the TMOD2 cDNA, we searched the high throughput genomic sequences (HTGS) database and identified a sequence, NT 010204, that contained 100% identity to the 69 basepairs of TMOD2 5' UTR and contained a consensus GT donor splice site. The distance from this UTR exon to coding exon 1 is 14.6 kb, but the sequence is not completely ordered. Using nomenclature from previous tropomodulin genes we designated these 5' UTR exons as E0 [27].
We sought to determine the genomic structure of YL-1 when we discovered that it lies next to and intergenically splices into TMOD4 (see below). Using the same methods described above, we identified six coding exons of YL-1 spanning 14.2 kb (Fig. 1). The distance from the last exon of YL-1 to E0 TMOD4 is 291 bp. We sequenced the entire genomic region encompassing TMOD4 from the end of YL-1 exon 6 through TMOD4 exon 9 and deposited it in Genbank (Accession No. AF393375). The sizes of the exons and introns as well as the boundary sequences for TMOD2, TMOD4, and YL-1 are provided (See Table 1). Consensus splice sites were identified for all Tropomodulin exons, but YL-1 exon 1 contained a GC 5' splice site instead of the more common GT 5' splice site. This splice site also conforms well to other nonconsensus GC 5' splice sites as described in Mount (2000) with a G at the -1 and +5 positions [28]. We used the very 3' end of the YL-1 cDNA to look for additional 3' UTR. We identified three ESTs (ESTs AA128103, AW627363, and AI591193) that were identical at the 3' end and extended the reported UTR by an additional 150 bp.
Table 1.
| TMOD2 | ||||
| Exon | 5' intron (5'-3) | Exon (bp) | 3' intron (5'-3') | Intron (kb) |
| E0 | 5' UTR | 69 | GTGAGCGCCCCGGGGGGCGGGTGG | 14.6 |
| 1 | TATTGTGTTTCTTTTTTATTAACAG | 195 | GTTAGTGACATGGAACTGAAGCAGA | 1.7 |
| 2 | CCTGCTCACACCTCTTTCTTGTCAG | 157 | GTAAGGACCACAGGCAGAGCATCTT | 5 |
| 3 | TTTTTGTTTTTCTTCTGATTTAAAG | 123 | GTTAGTCCTGAACTCTGGGAACTTT | 3 |
| 4 | ATGCTCATTTGTTGACTGTTTTTAG | 87 | GTAAGTTGATGTGCAATCTGTGGTT | 3.6 |
| 5 | TTTAAAATGAAAACTTGTGTTTTAG | 131 | GTATTTCATTGTGATTATCATCAGT | 1.6 |
| 6 | ACTAGCCATGTCCTCTCTGTCTTAG | 108 | GTGAGTAAAATTCTTAGAAATATAA | 17 |
| 7 | GTTTTTTTTCTTTTTGCATCATAAG | 144 | GTAAGGAAGGCCAGAGAATAGGCAA | 8 |
| 8 | TGCATGTGTCTGCACCTGCAACCAG | 145 | GTAATTCAGCCATAATATTTGCTGT | 2 |
| 9 | TTTTGTTCCTTTCTTTCTTACCTAG | 1314 | 3' UTR | |
| TMOD4 | ||||
| Exon | 5' intron (5'-3') | Exon (bp) | 3' intron (5'-3') | Intron (bp) |
| E0 | 5' UTR | 93 | GTAGGTGGGGAAGACAGGGGGCTTA | 1006 |
| 1 | CATCCTTTTGTCCCATGTTGCCCAG | 165 | GTAGGGGCTCCAGCACAGGGAACCA | 205 |
| 2 | TTGCTCCCCAAAACTCATCCCTAAG | 157 | GTATGGGGACCCTGACATCCATGCC | 775 |
| 3 | GAGCCATCCTTCATCTCCCCATCAG | 117 | GTGGGTAGCTGTGGGAAGTTGAGGG | 952 |
| 4 | TGACTCCTCTTCTTACCCTCTCCAG | 90 | GTGAGTGTGTTTGGGGAGCATGGTC | 506 |
| 5 | TTCCCTTCCTGCCCTCATTCACCAG | 131 | GTATTTGACTTGCATCCTCCAAACC | 92 |
| 6 | ACCATCTCCTATTCCCCTCCGATAG | 108 | GTAAGGCTAGTTGACATCCCCAGCC | 368 |
| 7 | CCTAACCCAAGTCCCTACTACCCAG | 144 | GTCAGCATTCCCTTCCCCTGATGTT | 256 |
| 8 | CAGCTTTTGCCTCTCCCTCCCCCAG | 145 | GTGAGTAACTGCAGACATGATGTGT | 415 |
| 9 | TATGTTTAATATTGCTCCTTTACAG | 108 | 3' UTR | |
| YL-1 | ||||
| Exon | 5' intron (5'-3') | Exon (bp) | 3' intron (5'-3') | Intron (kb) |
| 1 | 5' UTR | 122 | GCAAGATCCGGGCCTGGGAAAAGGG | 4.5 |
| 2 | AACCCTTATCTCCCTGTGTCCTTAG | 153 | GTACAGGGGGAGTCCTAGTTGTCTT | 0.178 |
| 3 | GTATCTGATTCCAATCTGTCCTCAG | 115 | GTGAGTAGGGATGTTAGGGTCAATT | 1 |
| 4 | TTCTTCTTATCTCACTCTCTTCCAG | 177 | GTAAGTCTGTGGTTTCAGAAAAAGT | 6 |
| 5 | CTCATTTCTTCCTTTGACCCTAAAG | 145 | GTGAGTGAATCTCAAGAGGAAAAGA | 1.2 |
| 6 | GTTTTTCCTTGTCTTTTGTCCTCAG | 724 | 3' UTR | |
Mutational Analysis of TMOD2 in ALS5
Two patients and two controls from a family with an autosomal recessive form of familial ALS (ALS5) were analyzed for mutations in TMOD2. We amplified coding exons 1–9 using primers flanking each exon (See Table 2). These PCR products were sequenced to identify any potential point mutations or small deletions. We did not detect any mutations or polymorphisms in any of the patients or controls.
Table 2.
| TMOD2 | ||||
| Anneal | ||||
| Exon | (° C) | Forward Primer (5'-3') | Reverse Primer (5'-3') | Product size (bp) |
| 1 | 54 | TTCAATACAGTAGGCTCCA | CGAGCTAAGGGATTTCTAC | 393 |
| 2 | 55 | GGTATCCTTCCTGCTTCC | GGCTTATTTCCCATGTCT | 413 |
| 3 | 54 | TCTGCTCAGTCTACTTACCC | CCATAATGCCTTGTGTCA | 322 |
| 4 | 54 | TTCAAGGGACAAATTAACA | AAAGCCCAAGTGATCCTA | 289 |
| 5 | 54 | AGTGTTATTGGAATCAGCAT | CTCCAACTCAGTGACTACAAA | 358 |
| *GTAAAACGACGGCCAGTTGCTGAAGGCTA | **GGAAACAGCTATGACCATGACTATAAT | |||
| 6 | 57 | GGTAATGA | TTTGCTCACTAACTGT | 390 |
| 7 | 54 | TTAATTTGGGAGTGAGGG | ACAGGAAATCCAGTACATCA | 356 |
| 8 | 54 | CCTGTGCAAGTAACTGTTTC | AGTGACATCAGAGGCCAG | 323 |
| 9 | 54 | ATGTTGTTTATCTGGAATTTA | CCCATGAGCAAATAAAAC | 307 |
| *indicates a M13 universal sequence attached | **indicates a M13 reverse sequence | |||
| at 5' end | attached at 5' end | |||
Inter genic splicing of YL-1 into TMOD4
During our analysis of TMOD4's genomic structure we identified EST AI763406, which contains TMOD4 exon 9 and part of the coding region of another unrelated gene known as YL-1. Using 5' RACE we were able to confirm the presence of this transcript and identify other unique intergenic RACE products. Figure 2 summarizes our findings. There are two main intergenic products, each of which was represented multiple times in our RACE clones. The first intergenic splice products (RACE #9–11, and 13–15) are similar to EST AI763406. These variants consist of YL-1 exons 1–5, and TMOD4 exon 9. The ORF of this splice variant keeps the reading frame of YL-1 but changes the TMOD4 endogenous reading frame to make the reading frame of exon 9 completely open. Translation of this ORF would theoretically produce a protein of at least 271 amino acids containing the amino terminal half of YL-1 but a unique carboxy terminus. If, however, we extend the sequence to include intronic sequence downstream of exon 9, we discover that the frame is kept open an additional 45 bp, making this theoretical protein 286 amino acids in length. Using primers to exon 1 of YL-1 and exon 9 of TMOD4, we confirmed the presence of this intergenic product by RT-PCR on human heart RNA. RACE product #12 was similar to RACE products #9–11, 13–15 except that YL-1 splices from exon 5 to TMOD4 exon 8 which then splices to exon 9 (Fig. 2). This produces an ORF coding for a 252 amino acid protein that changes the frame of TMOD4, adding only 16 additional amino acids before encountering a stop codon. Another set of RACE products (RACE # 1–5) consists of YL-1 exons 4 and 5 and TMOD4 exons 1–9, except that exon 2 uses an internal consensus cryptic splice site. Using the YL-1 endogenous frame we find that this ORF terminates in TMOD4 exon 1. If we assume an ORF including YL-1 exons 1–3 along with the isolated RACE product, then it would theoretically produce a protein of 264 amino acids. We used the translated protein sequences from these intergenic ORFs and analyzed them using InterPro Scan via EXPASY. No additional sequence motifs or domains were identified. Another type of RACE product we identified (RACE# 26–31) is the incompletely spliced products of TMOD4. In no case did we observe the inclusion of YL-1 exon 6 or the use of the normal splice acceptor of TMOD4 exon 2 – the cryptic splice acceptor was always present in intergenic RACE products. Furthermore, the YL-1 alpha helical domain, which is thought to contain the DNA binding domain, is maintained in all intergenic splice products contained within YL-1 exons 4 and 5.
Figure 2.

Summary of RACE products: Hatched rectangles represent YL-1 exons. Diagonal striped rectangles represent TMOD4 exons. Numbers in parentheses represent the number of amino acids produced from each ORF. Black rectangles represent introns, and horizontally striped rectangles represent exons which used a cryptic splice site.
YL-1 intraexonic spliced transcripts and expression pattern
To better understand the nature of YL-1's intergenic splicing, we searched the EST database for other alternative and intergenic YL-1 transcripts. Using either the YL-1 or TMOD4 cDNAs in a BLAST search of the human EST database, we were unable to identify any additional intergenic transcripts, but we did uncover several YL-1 ESTs that had apparently spliced within exons in such a way as to not use the canonical consensus splice acceptors or donors, the rarer GC or AT donor sites or the AC acceptor site[29]. ESTs AI560960 and AI369608 were identical and contained YL-1 exons 1 and 2 but splice within exon 2 at position 276 (position is in bp relative to Genbank sequence D43642) to position 1107 (internal to exon 6) (Fig. 3). Translation of this ORF would produce a truncated protein of only 121 amino acids. EST AI560960 was obtained from the IMAGE consortium and sequenced. It is 476 bp in length and contains YL-1 exons 1 and up to position 276 in exon 2 at which it splices internal to exon 6 at position 1107. A second splice product was represented by ESTs AI366716 and AW005187, in which the sequence appears to splice from position 113 in exon 1 to exon 6 at position 936 (Fig. 3). This would produce another truncated protein that is 89 amino acids in length. Another unusual splice product was represented by ESTs AA443950 and AA653180. These ESTs demonstrate splicing within YL-1 exon six itself, from position 936 to 1062 (skipping over ~126 bp), changing the frame and making it open past the endogenous stop codon (Fig. 3). EST AA443950 was obtained from the image consortium and sequenced. It contained a 1199 bp insert which contained all of YL-1 exons 1–5 and exon six as described above and encodes an ORF for a 358 amino acid protein. However, EST AA653180 has an additional six base pairs at the splice junction site coding for leucine and proline, suggesting that either the site produces two different splice variants or there is some type of intermediate stage of splicing at this site. In either case, the protein produced from this ORF (if AA653180 contains the rest of the intact 5' YL-1 transcript) would be 360 amino acids. This truncates the reported YL-1 protein only by 6 or 4 amino acids. Using InterPro Scan via EXPASY, we determined that these few amino acids do not produce any potential phosphorylation sites or protein domains or motifs. Two of these three splice variants use the 936 position for splicing as a putative donor in one case and as an acceptor in the other. We cannot explain this finding, but it may suggest that YL-1 undergoes a great degree of intraexonic splicing. This high degree of splicing together with the lack of a GT donor site in the last exon, and the close proximity of YL-1 and TMOD4, may account for the intergenic splicing between these genes.
Figure 3.

Hatched rectangles represent YL-1 exons. Thick black lines represent the intraexonic splice events. The number of amino acids produced by each ORF is in parentheses. Asteriks represent the positions of intraexonic non-canonical splice sites used in multiple ESTs.
We also wanted to determine whether the YL-1 expression pattern overlapped with TMOD4. A human multiple tissue Northern blot was probed with a YL-1 RT-PCR product revealing a single 1.4–1.6 kb band in all lanes (Fig. 4). This is consistent with the reported ubiquitous expression of YL-1 in the rat [25].
Figure 4.
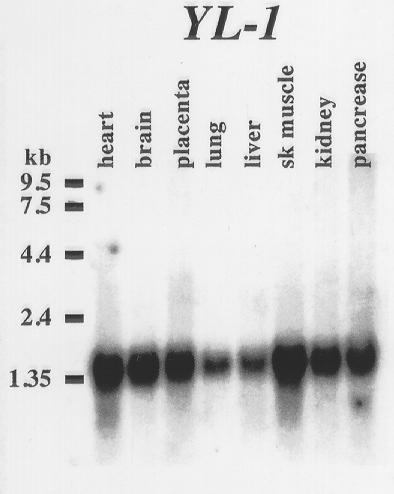
Human multiple tissue Northern blot probed with YL-1 RT-PCR product revealed a single highly expressed transcript between 1.4–1.6 kb in all tissues.
Discussion
Sequencing PCR fragments and genomic subclones from phage artificial chromosomes (PACs) and bacterial artificial chromosomes (BACs) that contain TMOD2 and TMOD4 allowed us to determine the genomic structure of these two Tropomodulin genes. Although mutational analysis of the coding region of TMOD2 in ALS5 and TMOD4 in LGMD1B patients failed to reveal any point mutations, small deletions, or polymorphisms, we did find that TMOD2 and TMOD4 consist of nine coding exons and at least one 5' untranslated exon (E0).
This is identical to the genomic structure reported for TMOD1 and Tmod1 by Chu and colleagues and Conley et al. [27,41]. In addition, they identified an 86 bp 5' UTR exon which was expressed in a tissue-specific manner. We performed 5' RACE for TMOD4 and identified only a single 93 bp 5' UTR exon (E0) which lies 1006 bp from coding exon 1. We did not examine whether the E0 exons of TMOD2 and TMOD4 were expressed in a tissue-specific manner. In addition, the TMOD2 transcript is approximately 9.5–10 kb in length and our reported cDNA only accounts for 2.47 kb [8]. Therefore, there are additional UTR exons or sequence that we have not identified in this study. This may not be the case for TMOD4, because our reported cDNA in addition to the 93 base pairs from the E0 sequence and the additional 9 bp from the 3' end produces a transcript of the same size as the major transcript seen by Northern. However, the larger transcripts (2.4 and 3.0 kb) seen on Northern may contain additional sequence that we were unable to identify in this study, or may represent incompletely spliced transcripts.
EST database searches in conjunction with the YL-1 genomic structure allowed us to identify three unique intraexonic splicing events. Two of the three events theoretically would produce truncated proteins with little similarity to the normal protein. These splicing events do not use the canonical AG and GT splice sites, nor do they use the rarer GC, AT, or AC splice sites. This may represent yet another form of splicing for the creation of unique transcripts and, or gene regulation.
We also identified two intergenic transcripts between YL-1 and TMOD4, both of which theoretically would code for truncated versions of YL-1. Intergenic splicing appears to be a rare event: apparently there have been only six other cases reported in mammals. Fears et. al (1996) reported that the Myelodysplasia syndrome 1(MDS1) gene spliced into exon 2 of the adjacent Ecotropic viral integration site 1 (EVI1) gene, producing a unique transcript containing an ORF that produces a GATA-binding transcriptional activator [30,31]. The human Galactose-1-phosphate uridylytransferase (GALT) gene produces an intergenic transcript by skipping its last exon and splicing into exon 2 of Interleukin-11 receptor α-chain (IL-11Rα)[32]. This produces an intergenic transcript that keeps the endogenous frame of both genes, thereby coding for a theoretical protein with domains from both proteins. A long terminal repeat from a human endogenous retrovirus (HERV) was shown to drive the expression of an intergenic transcript from an unknown gene named HERV-HLTR associating 1 (HHLA1) and human otoconin-90 (OC90) to produce a transcript that is highly expressed in teratocarcinoma cells but not in normal human tissues or cell lines [33]. In the mouse, a Prion-like downstream gene named doppel (Dpl) was found to have an intergenic transcript that is upregulated in Prion-deficient mice [34]. The deletion of the Prion gene in these mice removes the splice acceptor site of exon 3, causing the normal Prion transcript to skip the deleted exon and inadvertently upregulate the normally low-abundance intergenic transcript by forcing it to splice more often to the doppel gene. The human cytochrome P450 2C gene cluster maps to chromosome 10q24 and consists of four genes which are composed of nine exons with conserved exon/intron boundaries, and have been shown to produce intergenic transcripts [35-38]. These transcripts have been identified by RT-PCR for both neighboring genes such as CYP2C18 and CYP2C19, as well as CYP2C members separated by another gene, such as CYP2C18 and CYP2C8 [37,38]. The latter was shown by RT-PCR to include exons from both genes consisting of nine exons and therefore could code for a chimeric CYP2C protein [38]. Recently, the Translin-associated factor X gene (TRAX) was demonstrated to intergenically splice into Disrupted in schizophrenia gene 1 (DISCI)[39]. Several intergenic transcripts were described, the majority of which contain intervening non-coding exons that contain multiple stop codons.
At least half of the reported cases of intergenic splicing result from skipping the last exon of the upstream gene and the first exon of the downstream gene [32,33,39]. This is thought to be due to the lack of donor and acceptor splice sites in these exons, respectively. For YL-1 exon 6 there is no GT donor site at the end of the exon, and the E0 exon of TMOD4 contains no AG acceptor. Thus the same pattern seen in the other reported intergenic splicing events is born out in our studies. This idea is supported by the fact that we never identified any part of the E0 exon in our RACE products screened using the YL-1 cDNA. However, we did discover this product when we rescreened our RACE products using a 5' TMOD4 probe. Of the previously reported cases, the closest distance between adjacent genes is 4 kb [32]. YL-1 and TMOD4 are separated by 291 bp. Since this genomic region is so small, it raises the question as to the location of the TMOD4 promoter. One possibility is that it lies within the 1006 bp between the E0 exon and coding exon 1, or even within the 291 bp between the E0 exon and YL-1 exon 6. A more intriguing possibility is that the TMOD4 promoter lies further upstream, possibly within the intronic structure of YL-1, or as a shared promoter with YL-1.
Whether these intergenic splice products are biologically relevant is unclear. Due to the fact that the size of the transcripts for TMOD4 and YL-1 overlap (1.3 – 1.5 kb), we cannot demonstrate the co-existence of these intergenic variants by Northern blot analysis. The only reported case in which an intergenic transcript has been shown to produce a functional protein in vivo is the case of MDS1/EVI1[31]. In all other cases, the intergenic transcript or the theoretical protein produced from such transcripts has been difficult to demonstrate by Northern blot analysis, and none have been shown by Western analysis in normal tissues. However, all of these transcripts have been identified by RT-PCR and some by RNase protection assays. Thus, further analysis will be needed to determine if any of the YL-1/TMOD4 intergenic transcripts play an important biological role either at the RNA or protein levels.
Conclusion
Through our genomic analysis we were able to determine that TMOD2 and 4, like TMOD1/Tmod1, share conserved genomic structures consisting of nine coding exons and a single 5' UTR exon (E0). Using primers flanking the coding exons of TMOD2 we performed mutational analysis on ALS5 patients. No mutations or polymorphisms were detected, but our primer pairs and reaction conditions may prove useful for future mutational analyses. We determined that YL-1 comprises six exons, and its last exon lies 291 bp from TMOD4. We also identified six ESTs that show nonconventional splicing. Intergenic splice products between YL-1 and TMOD4 were identified in the EST database, by 5' RACE, and by RT-PCR. Although we did not determine whether this relationship between YL-1 and TMOD4 is conserved in the mouse, it may be an important possibility to consider when devising strategies for targeted deletion not only of YL-1 or TMOD4 but any gene pair in which intergenic splicing occurs. This is clearly illustrated by the Prion deficient mice produced by Sakaguchi et. al (1996) and Moore (1997). In these animals exon 3 and its splice acceptor were deleted, causing a rare intergenic transcript between Prion and Doppel to be highly upregulated and abnormally expressed in the brain, leading to late-onset ataxia and Purkinje cell degeneration[34].
The recent completion of the human genome by Ventner et. al (2001) has led to the new prediction that the human genome contains only ~30,000 genes, significantly lower than previous estimates of ~100,000 genes [40]. This estimate does not, however, include the possibility of additional proteins encoded by alternative or intergenic transcripts. The genomic organization between YL-1 and TMOD4 clearly raises the possibility of additional proteins being produced from intergenic transcripts. In addition, it highlights the difficulties that may arise when trying to understand gene regulation for genes that lie extremely close to one another or that form intergenic transcripts. YL-1's intraexonic splicing illustrates yet another level of genomic complexity in that alternative transcripts could be produced from the use of non-canonical splice sites that lie even within an individual exon. This, in conjunction with intergenic splicing, may significantly increase the number of predicted proteins encoded by the human genome and will certainly pose a challenge for anyone trying to decipher genomic organization and regulation.
Materials and Methods
Genomic organization and sequence analysis of TMOD2, TMOD4, and YL-1
The genomic organization of each gene was determined using two methods. The first involved PCR amplification from either PAC 61J10 (for TMOD2) or BAC HB342I3 (for TMOD4 and YL-1) using primers within each exon to amplify the exon and the intervening intron and use it as a substrate for sequencing the exon-intron boundaries. We also digested PAC 61J10 (from the RPCI-1 human PAC library, Roswell Park Cancer Institute) and BAC HB342I3 (from the human CITB BAC library. Research Genetics, Inc.) with RSAI and then subcloned the DNA fragments into pZero 2.0 according to the manufacturer's instructions. We screened colonies by hybridization using the cDNA for each gene as a probe. All labeling reactions for all procedures were performed using the Megaprime DNA labeling system (Amersham Pharmacia Biotech, Piscataway, NJ). Positive colonies were isolated and sequenced using a PRISM BigDye Terminator Cycle sequencing kit and an ABI 3700 automated DNA sequencer (Perkin-Elmer Applied Biosystems, Foster City, CA).
Mutational Analysis of TMOD2 for ALS5 and TMOD4 for LGMD1B
The coding exons of TMOD2 and TMOD4 were amplified from patient lymphoblast genomic DNA using primers flanking the exon (Table 2). Reaction conditions were as follows for a 50 μl PCR reaction : Perkin-Elmer Cetus PCR buffer; 1.5 mM MgCl2, 0.25 mM dNTPs, 1.5 μl of each primer (10 μM), 1% DMSO, 5 U of Taq polymerase (Life Technologies). Thermocycler conditions were as follows for a Perkin Elmer 9600 thermocycler: 94°C for 4 min, followed by 35 cycles of 94°C for 30 s, annealing temperature (see Table 2) for 30 s, 72°C for 1 min, and a final extension of 72°C for 10 min. The resulting PCR products were subjected to agarose gel electrophoresis and the DNA was recovered from excised bands using the Qiagen Quick Gel Extraction Kit (CAT# 28706). Each PCR product was then sequenced.
5' and 3' Rapid Amplification of cDNA ends for TMOD4
We used Clontech's Human Heart Marathon-Ready cDNA (Cat # 7404–1) to perform 5' and 3' RACE to the TMOD4 gene. Nested primers were designed within exon 9 and exon 2 of TMOD4, TMOD4EX9RACE5'.1 5'TACAGGAAAGGAGGACAGATGAGG3', and TMOD4EX9RACE5'.2 5'GATTTAAGTGTCCAGTGCTCCCAG3', and TMOD4EX2RACE5'.1 5'GGGCACCAAGTCATCACGCTCT3', TMOD4EX2RACE5' .2 5'CCAAGTACTGCAAAAGGGCCTC3', TMOD4EX2RACE3'.1 5'GGACTAAGACAACGTGACCAGACA3', and TMOD4EX2RACE3' .2 5'GAGGCCCTTTTGCAGTACTTGG3'. The initial RACE reaction to identify intergenic RACE products was performed using the gene specific primer, TMOD4RACE5'.1, and Clontech's AP1 primer in a 50 μl reaction using the Roche High Fidelity Kit (CAT# 1–732–641). Thermocycler conditions for a PE9600 were as follows: 94°C for 5 min, followed by 10 cycles of 94°C for 30 s, 71°C for 30 s, 72°C for 4 min; followed by one cycle of 94°C for 30 s, 68°C for 4 min 20 s, followed by 17 additional cycles each adding 20 s to each extension; final extension at 72°C for 10 min. 5 μl of a 1:50 dilution of the product from the first RACE reaction served as template for the second nested RACE reaction. Reaction and thermocycler conditions were the same as before except gene specific primer 2, TMOD4RACE5'.2, and Clontech's AP2 primer were used. RACE products were then subcloned into pbluescript KS II+ using TA vector cloning. Positive products were then identified by colony hybridization using the YL-1 cDNA as a probe. A secondary screening was then performed using exon 2 primers TMOD4EX2RACE5'.1, followed by a second round of race using TMOD4EX2RACE5'.2. Likewise the 3' RACE was performed using TMOD4EX2RACE3'.1, followed by TMOD4EX2RACE3'.2. RACE products were then subcloned and identified by using either a 5' or 3' probes made from the TMOD4 cDNA. Positive RACE products were sequenced.
RT-PCR of YL-1 and Expression Analysis of YL-1
For RT-PCR of YL-1, we performed the same procedure as described in Cox and Zoghbi (2000). Exceptions were the RNA source, human heart poly (A)+ RNA, (Clontech, 6533–1), primers were YL1-35, GTAGGCGGTATGAGTTTG, and YL1-1277, AAAACACAACGAAACCTG, respectively, and the annealing temperature was 53°C. The resulting RT-PCR product was subcloned using TA cloning into pbluescript KSII+. This product was then sequenced to confirm that it matched the reported cDNA. We examined the expression pattern of YL-1 using this RT-PCR product as a probe on a Clontech Human multiple tissue Northern blot (CAT# #7760–1).
Acknowledgments
Acknowledgements
This work was supported by the Howard Hughes Medical Institute (HYZ) and the Medical Scientist Training Program (PRC).
Contributor Information
Patrick R Cox, Email: pc992155@condor.bcm.tmc.edu.
Teepu Siddique, Email: t-siddique@nwu.edu.
Huda Y Zoghbi, Email: hzoghbi@bcm.tmc.edu.
References
- Weber A, Pennise CR, Babcock GG, Fowler VM. Tropomodulin caps the pointed ends of actin filaments. J Cell Biol. 1994;127:1627–1635. doi: 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung LA, Fowler VM, Lambert K, Sussman MA, Karr D, Chien S. Molecular cloning and characterization of human fetal liver tropomodulin. A tropomyosin-binding protein. J Biol Chem. 1992;267:2616–2621. [PubMed] [Google Scholar]
- Sussman MA, Sakhi S, Barrientos P, Ito M, Kedes L. Tropomodulin in rat cardiac muscle. Localization of protein is independent of messenger RNA distribution during myofibrillar development. Circ Res. 1994;75:221–232. doi: 10.1161/01.res.75.2.221. [DOI] [PubMed] [Google Scholar]
- Ito M, Swanson B, Sussman MA, Kedes L, Lyons G. Cloning of tropomodulin cDNA and localization of gene transcripts during mouse embryogenesis. Dev Biol. 1995;167:317–328. doi: 10.1006/dbio.1995.1026. [DOI] [PubMed] [Google Scholar]
- Watakabe A, Kobayashi R, Helfman DM. N-tropomodulin: a novel isoform of tropomodulin identified as the major binding protein to brain tropomyosin. J Cell Sci. 1996;109:2299–2310. doi: 10.1242/jcs.109.9.2299. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Ito M, Daniels MP, Flucher B, Buranen S, Kedes L. Chicken skeletal muscle tropomodulin: novel localization and characterization. Cell Tissue Res. 1996;285:287–296. doi: 10.1007/s004410050646. [DOI] [PubMed] [Google Scholar]
- Almenar-Queralt A, Lee A, Conley CA, Ribas de Pouplana L, Fowler VM. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J Biol Chem. 1999;274:28466–28475. doi: 10.1074/jbc.274.40.28466. [DOI] [PubMed] [Google Scholar]
- Cox PR, Zoghbi HY. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics. 2000;63:97–107. doi: 10.1006/geno.1999.6061. [DOI] [PubMed] [Google Scholar]
- Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein. J Biol Chem. 1999;274:33179–33182. doi: 10.1074/jbc.274.47.33179. [DOI] [PubMed] [Google Scholar]
- Caldwell JE, Heiss SG, Mermall V, Cooper JA. Effects of CapZ, an actin capping protein of muscle, on the polymerization of actin. Biochemistry. 1989;28:8506–8514. doi: 10.1021/bi00447a036. [DOI] [PubMed] [Google Scholar]
- Kuhlman PA, Hughes CA, Bennett V, Fowler VM. A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J Biol Chem. 1996;271:7986–7991. doi: 10.1074/jbc.271.14.7986. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Fowler VM. Defining actin filament length in striated muscle: rulers and caps or dynamic stability? Annu Rev Cell Dev Biol. 1998;14:487–525. doi: 10.1146/annurev.cellbio.14.1.487. [DOI] [PubMed] [Google Scholar]
- Gregorio CC, Weber A, Bondad M, Pennise CR, Fowler VM. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature. 1995;377:83–86. doi: 10.1038/377083a0. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Baque S, Uhm CS, Daniels MP, Price RL, Simpson D, Terracio L, Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res. 1998;82:94–105. doi: 10.1161/01.res.82.1.94. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Welch S, Cambon N, Klevitsky R, Hewett TE, Price R, Witt SA, Kimball TR. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J Clin Invest. 1998;101:51–61. doi: 10.1172/JCI1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Tanigawa G, Jarcho JA, Kass S, Solomon SD, Vosberg HP, Seidman JG, Seidman CE. A molecular basis for familial hypertrophic cardiomyopathy: an alpha/beta cardiac myosin heavy chain hybrid gene. Cell. 1990;62:991–998. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, Boivin GP, Wolska B, Evans C, Solaro RJ, Wieczorek DF. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- McKenna WJ. Hypertrophic cardiomyopathy: an update. Cardiologia. 1993;38:277–281. [PubMed] [Google Scholar]
- Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmad A, Yang Y, Rimmler J, Hung W, Schlotter B, Ahmed A, Ben Hamida M, Hentati F, Siddique T. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15ql5-q22 markers. Neurogenetics. 1998;2:55–60. doi: 10.1007/s100480050052. [DOI] [PubMed] [Google Scholar]
- van der Kooi AJ, van Meegen M, Ledderhof TM, McNally EM, de Visser M, Bolhuis PA. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11-21. Am J Hum Genet. 1997;60:891–895. [PMC free article] [PubMed] [Google Scholar]
- Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- Horikawa I, Tanaka H, Yuasa Y, Suzuki M, Oshimura M. Molecular cloning of a novel human cDNA on chromosome 1q21 and its mouse homolog encoding a nuclear protein with DNA-binding ability. Biochem Biophys Res Commun. 1995;208:999–1007. doi: 10.1006/bbrc.1995.1433. [DOI] [PubMed] [Google Scholar]
- Horikawa I, Tanaka H, Yuasa Y, Suzuki M, Shimizu M, Oshimura M. Forced expression of YL-1 protein suppresses the anchorage-independent growth of Kirsten sarcoma virus-transformed NIH3T3 cells. Exp Cell Res. 1995;220:11–17. doi: 10.1006/excr.1995.1286. [DOI] [PubMed] [Google Scholar]
- Chu X, Thompson D, Yee LJ, Sung LA. Genomic organization of mouse and human erythrocyte tropomodulin genes encoding the pointed end capping protein for the actin filaments. Gene. 2000;256:271–281. doi: 10.1016/S0378-1119(00)00327-9. [DOI] [PubMed] [Google Scholar]
- Mount SM. Genomic sequence, splicing, and gene annotation. Am J Hum Genet. 2000;67:788–792. doi: 10.1086/303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burset M, Seledtsov LA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fears S, Mathieu C, ZeIeznik-Le N, Huang S, Rowley JD, Nucifora G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc Natl Acad Sci USA. 1996;93:1642–1647. doi: 10.1073/pnas.93.4.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderholm J, Kobayashi H, Mathieu C, Rowley JD, Nucifora G. The leukemia-associated gene MDS1/EVI1 is a new type of GATA-binding transactivator. Leukemia. 1997;11:352–358. doi: 10.1038/sj.leu.2400584. [DOI] [PubMed] [Google Scholar]
- Magrangeas F, Pitiot G, Dubois S, Bragado-Nilsson E, Cherel M, Jobert S, Lebeau B, Boisteau O, Lethe B, Mallet J, Jacques Y, Minvielle S. Cotranscription and intergenic splicing of human galactose-1-phosphate uridylyltransferase and interleukin-11 receptor alpha-chain genes generate a fusion mRNA in normal cells. Implication for the production of multidomain proteins during evolution. J Biol Chem. 1998;273:16005–16010. doi: 10.1074/jbc.273.26.16005. [DOI] [PubMed] [Google Scholar]
- Kowalski PE, Freeman JD, Mager DL. Intergenic splicing between a HERV-H endogenous retrovirus and two adjacent human genes. Genomics. 1999;57:371–379. doi: 10.1006/geno.1999.5787. [DOI] [PubMed] [Google Scholar]
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- Gray IC, Nobile C, Muresu R, Ford S, Spurr NK. A 2.4-megabase physical map spanning the CYP2C gene cluster on chromosome 10q24. Genomics. 1995;28:328–332. doi: 10.1006/geno.1995.1149. [DOI] [PubMed] [Google Scholar]
- Romkes M, Faletto MB, Blaisdell JA, Raucy JL, Goldstein JA. Cloning and expression of complementary DNAs for multiple members of the human cytochrome P450IIC subfamily [published erratum appears in Biochemistry 1993 Feb 9;32(5):1390]. Biochemistry. 1991;30:3247–3255. doi: 10.1021/bi00227a012. [DOI] [PubMed] [Google Scholar]
- Zaphiropoulos PG. RNA molecules containing exons originating from different members of the cytochrome P450 2C gene subfamily (CYP2C) in human epidermis and liver. Nucleic Acids Res. 1999;27:2585–2590. doi: 10.1093/nar/27.13.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finta C, Zaphiropoulos PG. The human CYP2C locus: a prototype for intergenic and exon repetition splicing events. Genomics. 2000;63:433–438. doi: 10.1006/geno.1999.6063. [DOI] [PubMed] [Google Scholar]
- Millar JK, Christie S, Semple CA, Porteous DJ. Chromosomal location and genomic structure of the human translin-associated factor X gene (TRAX; TSNAX) revealed by intergenic splicing to DISCI, a gene disrupted by a translocation segregating with schizophrenia [In Process Citation]. Genomics. 2000;67:69–77. doi: 10.1006/geno.2000.6239. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, dark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Francesco VD, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang ZY, Wang A, Wang X, Wang J, Wei MH, Wides R, Xiao C, Yan C, et al. The Sequence of the Human Genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 2001;73:127–39. doi: 10.1006/geno.2000.6501. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Almenar_Queralt A, Fowler VM. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat Cell Biol. 2001;6:544–51. doi: 10.1038/35078517. [DOI] [PubMed] [Google Scholar]