

Abstract
Despite advances in prevention and treatment, vascular diseases, including atherosclerotic vascular disease, continues to account for significant morbidity and mortality in the developed world. It is exected to worsen with an increasing number of patients with common co-morbidities such as obesity and diabetes; conditions linked with atherosclerotic vascular disease, and reaching epidemic proportions. Atherosclerosis is a lipid-driven vascular inflammatory disease involving multiple cell types in various stages of inflammation, activation, apoptosis, and necrosis. One commonality between these cell types is that they are activated and communicate with each other in a paracrine fashion via a complex network of cytokines. Cytokines mediate atherogenesis by stimulating expression of numerous proteins necessary for induction of a host of cellular responses, including inflammation, extravasation, proliferation, apoptosis, and matrix production. Cytokine expression is regulated by a host of transcriptional and post-transcriptional mechanisms. In this context, proteins and mechanisms that control and fine-tune cytokine expression can be considered key players in development of atherosclerosis and also represent targets for rational drug therapy to combat this serious disease. This review will describe the cellular and molecular mechanisms that drive atherosclerotic plaque progression, and present some of the key cytokines that participate in this process. We will also describe some of the RNA binding proteins that mediate cytokine mRNA stability and regulate cytokine abundance. Identification and characterization of the cytokines and molecules that regulate their abundance are essential to our ability to identify therapeutic approaches to combat atherosclerotic vascular disease.
1. Introduction
Cardiovascular disease is the leading cause of death worldwide, with 1 in 5 deaths annually attributed to cardiovascular etiology. Despite increased awareness in the general public about how lifestyle choices affect vascular health, cardiovascular disease remains the #1 cause of death both nation and worldwide to date. It is widely recognized that atherosclerosis, the underlying cause of coronary heart disease, peripheral vascular disease, and stroke, is the primary contributor to the majority of all cardiovascular diseases [1]. Vascular disease can affect the entire body as arteries and veins are responsible for nourishing the body in its entirety, and roughly 92.1 million individuals are affected by at least one type of vascular disease in the United States alone [1]. As the developed world adopts a more sedentary, inactive lifestyle, atherosclerotic vascular disease itself accounts for significant morbidity and mortality. Controllable risk factors for vascular disease include obesity, smoking, physical inactivity, hypertension, stress, high LDL levels, and uncontrolled diabetes. Disease prevention focuses on healthy lifestyle choices that limit these risk factors. The incidence of atherosclerosis and other vascular diseases increases as the number of patients with co-morbidities such as obesity, metabolic syndrome, and Type 2 diabetes mellitus grows, along with an aging population in the developed world.
2. Development of atherosclerosis
Blood vessels are composed of three layers: the outermost layer or adventitia, the middle layer or media, which is composed of vascular smooth muscle cells (VSMCs), and the innermost layer, the intima, is formed by simple squamous endothelial cells (ECs) and contains the endothelium, which is the interface between the blood vessel wall and the circulating blood. Atherosclerotic lesions typically develop in large and medium-sized arteries and can lead to ischemia in areas including the heart and brain or development of peripheral arterial disease when affecting the extremities [2]. Numerous cells from each of these layers participate in the vascular inflammation indicative of atherosclerosis. ECs, VSMCs, and infiltrating immune cells communicate with each other bidirectionally through the production and secretion of cytokines and production of cytokine receptors [8]. Plaque rupture can result in myocardial infarction, stroke, or thrombosis, which can often be lethal. Atherosclerosis is a lipid-driven, chronic vascular inflammatory disease that develops in response to various insults to the vasculature such as oxidative stress in the form of oxidized excess low-density lipoprotein (oxLDL). The lipid theory of atherogenesis states that oxidation of low-density lipoprotein (LDL), one of the earliest initiation factors of atherosclerosis, becomes modified by oxidization as a normal metabolic consequence. Oxidation of LDL exposes numerous epitopes and subendothelial deposition of excess oxLDL acts as an antigenic, pro-inflammatory factor [3,4]. Internalization of oxLDL results in a series of dramatic events, including activation of Nuclear Factor Kappa B (NF-κB), a “master switch” for transactivation of many cytokine genes that drive the cellular atherogenic response. Several studies have shown that oxLDL stimulation of ECs, VSMCs, and leukocytes leads to expression of pro-inflammatory cytokines [3]. While the lipid theory has become both well established and accepted, unmodified or “native” LDL has been implicated as a trigger of inflammation and atherogenesis [4, 5]. Studies demonstrate that unmodified LDL, specifically Apolipoprotein B100 (ApoB100), the primary protein constituent of LDL, results in expression of pro-inflammatory cytokines as well as activate Mitogen Activated Protein Kinase (MAPK) and calcium signaling [6]. The majority of these experiments focus specifically on T cell activation and explore vaccination as a potential therapy, though there are additional studies investigating the effects of ApoB100 in additional cell types including macrophages, platelets, and dendritic cells [4, 7-9].
The endothelium in healthy individuals maintains a non-thrombogenic, non-adherent surface; it maintains vascular tone by synthesis of vascular dilatory and constricting molecules; acts as a permeability barrier for exchange of substances into artery wall; and regulates lipid modification as it is transported into the artery wall. Atherosclerosis begins with EC dysfunction, which is initiated by oxLDL induction of TNFα, a master pro-inflammatory cytokine which in turn induces expression of other pro-inflammatory cytokines and cell adhesion molecules (CAMs). Increased expression of cell adhesion molecules, leading to increased endothelial permeability and leukocyte extravasation gave rise to the inflammatory hypothesis of atherogenesis initially proposed by Ross which states that immune cell adhesion to activated, or “inflamed” endothelium is the earliest cellular event in development of atherosclerosis [7]. The earliest type of atherosclerotic lesion is termed the “fatty streak”, which is not uncommon in most children [10]. This initial lesion is purely inflammatory as it consists of only monocyte-derived macrophages and T cells [11]. As inflammation persists, additional monocytes are recruited into the subendothelial space, differentiate into macrophages, and rapidly uptake oxLDL via scavenger receptors. As macrophages continue to scavenge for oxLDL they can become overloaded and become “foam cells”, which are incapable of leaving the artery and continue to actively secrete pro-inflammatory cytokines, fueling the ongoing inflammation [2, 12]. As plaque development progresses to intermediate lesions, macrophages and foam cells become apoptotic. In early atherogenesis additional macrophages are able to clear the apoptotic cells, however in later pathogenesis, apoptotic macrophages and foam cells accumulate and form a necrotic core, which is a feature of complex atherosclerotic lesions.
Pro-inflammatory cytokines also cause VSMCs to undergo phenotypic switching during atherogenesis, specifically in the intermediate lesion stage. VSMCs transform from their normally differentiated, contractile phenotype to an active “synthetic” state. Synthetic VSMCs are able to migrate from the media into the intima and proliferate, phagocytize oxLDL, and secrete additional pro-inflammatory cytokines, which in turn, recruit additional VSMCs as well as immune cells. VSMCs also produce matrix, which retains lipids and the inflammatory cell types in the lesion. Eventually intermediate lesions can develop into fibrous plaques or advanced lesions, which typically lead to clinical manifestations. These advanced lesions are comprised of a necrotic core of apoptotic foam cells and T cells, covered in a thick layer of VSMCs that have formed a fibrous cap, consisting of collagen and connective tissue. Plaque vulnerability is dependent upon the thickness of the fibrous cap. As inflammation persists, pro-inflammatory cytokines are able to secrete matrix metalloproteinases (MMPs) that degrade the matrix, leading to thinning of the fibrous cap. Plaque rupture typically occurs as a consequence of decreased cap integrity, leading to myocardial infarction (MI) or stroke. Plaque rupture from a fractured cap may also lead to thrombus formation, which is the major cause of vascular mortality in humans.
3. Cytokines which participate in atherosclerosis
It is important to recognize that the microenvironment of the atherosclerotic plaque is a dynamic collection of multiple cell types including ECs, VSMCs, and infiltrating inflammatory cells, each in various stages of activation and synthesis of cytokine products. As such, most cytokines initiate a complex and varied repertoire of responses on their target cells and can initiate, promote, and potentially resolve plaque formation. Thus, this complex microenvironment results in a complex milieu consisting of multiple cytokines with redundant and opposing effects. To better describe the actions of cytokines on atherogenesis, it is convenient to classify them into different families, typically based on structure; we can also categorize them by function, specifically into one of two groups: pro-inflammatory and anti-inflammatory. Cytokines typically act in synergy with other cytokines of the same functional group and are able to induce simultaneous cellular processes as the previously mentioned cell types have extensive overlap in which cytokines they are able to produce and secrete and respond to. Cytokines are able to balance each other: proinflammatory cytokines initiate and sustain inflammation, while anti-inflammatory cytokines then limit the magnitude of inflammation, typically pushing it to resolve, limiting tissue damage. Skewing of this delicate balance can result in the body's inability to fight infection or chronic inflammation. Vascular diseases are often the consequence of such a shift and subsequent uncontrolled inflammation.
4. Pro-and anti-inflammatory cytokines
Pro- and anti-inflammatory cytokines are frequently associated with their effects on T helper cells, with pro-inflammatory cytokines being associated with Th1 and anti-inflammatory cytokines characterizing Th2. A polarization of T cells to Th1 in atherosclerosis has been established in mouse models and has been suggested in humans [13, 14]. Because atherosclerosis is primarily an inflammatory condition, Th1 cytokines are much more prevalent in human atherosclerotic lesions than Th2 cytokines. In addition to Th1 and Th2 are Th17 and T regulatory (Tregs) cells which have a similar but distinct dynamic. Th17 cells are characterized by expression of pro-inflammatory IL-17 and the ability to inhibit Tregs. Tregs, formerly called suppressor T cells, are immunosuppressive and are known to play a role in prevention of autoimmune disease as well as attenuate atherosclerosis in mice. Tregs lymphocytes are found in human atherosclerotic plaque, and currently the subject of intense study. This lymphocyte subset is considered to be atheroprotective, as adaptive transfer of Tregs reduces the production of Th1 cytokines, such as IFNγ and concurrently increases expression of the Th2 cytokine IL-10 [15]. Not surprisingly, depletion of these cells increase atherosclerosis in mice [16].
A paradigm similar to T helper cells exists among macrophages. Pro- and antiinflammatory cytokines are able to polarize macrophages to their classical M1 or alternative M2 phenotypes, respectively. Shifts in M1/M2 balance have been indicated in a number of inflammatory diseases, including atherosclerosis and peripheral arterial disease. Similar to the previously discussed cell types, macrophages secrete and respond to cytokines. Proinflammatory cytokines including TNFα and IFN-γ typically induce classically activated M1 macrophages, which in turn secrete high levels of additional pro-inflammatory cytokines including TNFα, IL-1β, IL-12, and IL-23, as well as low levels of anti-inflammatory IL-10 [17, 18]. We will broadly present cytokines in terms of their pro-atherogenic and anti-atherosclerotic properties, and briefly describe each.
Pro-atherogenic cytokines
Generally speaking, due to the pro-inflammatory environment of the atherosclerotic plaque, pro-inflammatory, Th1 cytokines are more prevalent in mouse and human lesions compared with anti-inflammatory, Th2 cytokines. We will begin with a brief discussion of reported pro-atherogenic cytokines, beginning with TNFα, as it is considered to be a global proinflammatory protagonist, and also because its post-transcriptional regulation is by far the best described. While TNFα is clearly a major player in atherogenesis, we will also briefly discuss other cytokines that participate in this process.
Tumor necrosis factor alpha
(TNFα) is a pleiotropic member of the tumor necrosis factor superfamily [19]. TNFα is considered to be a master pro-inflammatory cytokine as it induces expression of multiple pro-inflammatory genes, thus amplifying its potent inflammatory effects. Its induction of other pro-inflammatory cytokines has earned TNFα the moniker of a “master inflammatory cytokine”. While M1 macrophages are the primary source of TNFα in atherosclerosis, its expression can also be induced in ECs and VSMCs [20]. TNFα fuels the inflammatory cascade by recruiting additional T cells and macrophages to the atherosclerotic lesion by induction of several pro-inflammatory cytokines and CAMs. Consistent with its effects on CAM expression, TNFα promotes leukocyte/endothelial cell interaction and extravasation in vivo, an initial step in generation of the fatty streak leading to atherosclerosis [21, 22]. TNFα could potentially serve as a useful biomarker for coronary artery disease in the clinic as serum levels of TNFα correlate with early carotid atherosclerosis [20].
Our current understanding of cannonical TNFα receptor signaling points to Nuclear Factor kappa B, (NF-κB), p38 MAPK, and JUN N-terminal kinase (JNK) activation as essential participants of this pathway [23]. This leads to expression of NF-κB target genes which are crucial in inflammation, cell proliferation, and response to stress [19]. Included in this list are cytokines pertinent for progression of atherosclerosis, including, but not limited to IL-1β, IL-6, IL-8, MCP-1, as well as TNFα itself [24].
Long-term stimulation of macrophages by TNFα, via a MAPK-dependent pathway downregulates macrophage scavenger receptor gene expression and protein via transcriptional and post-transcriptional processes [25, 26]. These scavenger receptors play a vital role in the reverse cholesterol pathway, as macrophages uptake LDL using these receptors; exacerbated TNFa is therefore able to promote atherosclerosis by reducing the efficacy of this reverse cholesterol pathway. TNFα is also associated with plaque rupture as it stimulates production of MMPs as well as the thrombogenic protein, tissue factor, in VSMCs [27, 28].
Multiple studies completed in Tnfα-/-xApoe-/- double knockout mice have found decreases in atherosclerotic lesion size from 50 – 75% and increased plaque necrosis and apoptosis [29]. In studies using Apoe-/- mice treated with recombinant soluble TNFα p55 receptor, lesion size was reduced [30]. Blockade therapy targeting TNFα signaling with humanized antibody or recombinant fusion proteins has proven beneficial for patients with chronic inflammatory diseases, and incidence of acute cardiovascular events is lower in arthritis patients receiving this treatment [31-33]. However, adverse effects have limited the utility of this therapy. Considering its potent pro-inflammatory, pro-thrombogenic, and anti-cholesterol clearing effects, it is not surprising that TNFα is a major pro-atherogenic cytokine. The complex molecular mechanisms that modulate TNFα abundance by regulation of its mRNA will be discussed in detail in Section 5.
Interleukin-1 beta
(IL-1β) is a prototypic pro-inflammatory cytokine expressed by macrophages, ECs, and VSMCs [34]. IL-1β plays a key role in early atherogenesis. It is inflammation-responsive and induced by TNFα and it subsequently serves as a local paracrine and autocrine stimulator of several additional pro-inflammatory cytokines and CAMs, leading to immune cell extravasation and sustained local inflammation. IL-1β also promotes VSMC proliferation and migration, as well as release of MMPs [35, 36]. IL-1β has been established as a pro-atherogenic cytokine in a number of mouse models. Infusion of IL-1 receptor decoy reduced fatty-streak area in Apoe-/- mice [37]. Il1β-/-xApoe-/- double knockouts also have 30% less atherosclerotic lesion size compared to Apoe-/- mice [38]. Considering its potent proinflammatory effects on a variety of cell types, IL-1β is a major contributor to vascular inflammatory diseases including atherosclerosis.
Interleukin-2
(IL-2) is a T lymphocyte growth factor and as such, is considered to be a proinflammatory Th1 cytokine [39]. Studies have established IL-2 expression in atherosclerotic plaques, though its role has yet to be properly characterized. In vivo studies completed in mouse models have yielded conflicting results. One study reports Apoe-/- mice treated with IL-2 via i.p. injection while on high fat diet have increased atherosclerosis compared to controls [40]. Additionally, treatment with IL-2 antibody also decreased plaque burden in Apoe-/- mice [20]. However, another study demonstrates that IL-2 is able to attenuate atherosclerotic plaque development by activating Tregs [41]. Additional experimentation are required to further elucidate the role of IL-2 in atherogenesis.
Interleukin-6
(IL-6) Interleukin-6 is a pro-inflammatory cytokine and potent inducer of Th17 cells [42]. Patients with unstable angina have increased plasma levels of IL-6 [43]. IL-6 is expressed in a variety of cell types apropos to atherosclerosis, including macrophages, ECs, and VSMCs. IL-6 expression results in increased CAM expression in ECs, contributing to extravasation of leukocytes into the vessel wall [44]. Systemic treatment with IL-6 in Apoe-/-mice results in increased atherogenesis, supporting the notion that IL-6 is a pro-atherosclerotic cytokine [45]. Surprisingly, Il6-/-xApoe-/- double knockout mice have increased plaque burden compared to controls [46]. Double knockout mice also have decreased leukocyte infiltration into the lesions, as well as reduced expression of MMPs, suggesting increased plaque stability. These seemingly contradictory data may be explained by the compensatory increase in expression of anti-inflammatory IL-10, as well as IL-1Rα and TNFα receptors, which may neutralize plasma levels of these inflammatory cytokines. IL-6 participation in atherogenesis is complex and has yet to be completely understood.
Interleukin-12
(IL-12) is a pro-inflammatory cytokine expressed in atherosclerotic lesions and is known to induce signal transducer and activator of transcription 4 (STAT4) and subsequently activate the transcription factor T box expressed in T cells (TBet) [47]. TBet has been classified as a marker of the Th1 phenotype, as it transactivates expression of many proinflammatory genes. TBet results in expression of pro-inflammatory IFNγ, which acts as a major regulator of inflammation [48]. Importantly, TBet also inhibits expression of antiinflammatory, Th2 cytokine IL-4. oxLDL treatment of monocytes results in IL-12 expression. Additionally, Apoe-/- mice treated with IL-12 demonstrate exacerbated atherosclerotic plaques compared to controls [49]. Il12-/-xApoe-/- mice also have decreased plaque deposition compared to mice with baseline IL-12 expression. Inhibition of the IL-12 receptor via anti-IL-12 antibodies attenuates atherosclerosis in Ldlr-/- mice [50]. Studies reporting the atherogenic effects of IL-12 have focused on immune cells specifically; effects of IL-12 on vascular cells have not been reported, but it is clear that IL-12 is a major pro-atherogenic cytokine.
Interleukin-18
(IL-18) Pro-inflammatory cytokine IL-18 has been shown to polarize T cells to Th1 and induce IFNγ expression [51]. IL-18 levels are elevated in the sera of patients with coronary artery disease and IL-18 and its receptor have both been detected in human plaques [52]. IL-18 is primarily secreted by macrophages and its receptor is present on macrophages, as well as ECs and VSMCs, allowing it to possibly mediate crosstalk between immune cells and the vasculature. Il18-/-xApoe-/- double knockout mice have reduced atherosclerosis when compared to control mice [53, 54]. Additionally, Apoe-/- mice treated with IL-18 demonstrate increased plaque burden [55]. It is interesting to note that when treated with recombinant IL-18, Ifnγ-/-xApoe-/- double knockout mice have decreased plaque burden compared to IL-18 treated Apoe-/- mice, suggesting that IL-18 and IFNγ have a synergistic relationship [53, 54].
Interleukin-22
(IL-22) is a member of the IL-10 family, has been associated with Th17, and implicated in autoimmune diseases including lupus and rheumatoid arthritis [56, 57]. IL-22 expression has been found in human atherosclerotic plaques in carotid arteries and increased levels were found specifically in patients with unstable plaques [58]. IL-22 expression has also been confirmed in multiple inflammatory cell types, including macrophages and T cells, as well as in VSMCs, further implicating a role in atherosclerosis. Il22-/-xApoe-/- double knockout mice fed high fat diet for 14 weeks have decreased plaque deposition compared to Apoe-/- controls [59]. Interestingly, double knockout mice in these studies also had decreased collagen levels in their plaques, suggesting the smaller plaques were less stable and IL-22 may contribute to cap thickness and plaque stability. Double knockout mice also had increased expression of contractile VSMC markers, suggesting IL-22 influences VSMC phenotypic switching to a synthetic, proliferative state.
Interleukin-23 (IL-23) is a pro-inflammatory cytokine that also influences Th17 cells [60]. Macrophages express both IL-23 and the IL-23 receptor; IL-23 is able to induce expression of several pro-inflammatory cytokines including IL-17, IL-22, and TNFα in these cells [60]. Like IL-22, IL-23 has been detected in autoimmune diseases including psoriasis and rheumatoid arthritis, but has yet to be extensively researched in vascular disease. A recent study investigated IL-23 in patients with carotid atherosclerosis with a specific focus on stroke [61]. The study reports significantly increased levels of IL-23 in the plasma of patients with atherosclerosis compared to healthy controls. Furthermore, during follow-up, high levels of IL-23 in the plasma was associated risk of mortality. Expression of IL-23 and the IL-23 receptor genes were markedly increased in the carotid plaques when compared to healthy vessels [61]. IL-23/LPS co-treatment of monocytes from patients with carotid atherosclerosis also resulted in increased secretion of IL-17 and TNFα compared to monocytes from healthy controls. Although these reports suggest a pro-atherosclerotic role, further studies are necessary to define the exact role for IL-23 in development of atherosclerosis.
Interferon gamma
(IFNγ) is highly expressed by multiple cell types in atherosclerotic plaque and its role throughout atherogenesis continues to be extensively researched [62]. Studies focusing on IFNγ unanimously characterize it as a robust pro-atherosclerotic cytokine for several reasons. IFNγ production by Th1 cells is able to stimulate macrophages to further secrete pro-inflammatory cytokines [63]. IFNγ also promotes oxLDL uptake in both macrophages and VSMCs, promoting foam cell development [64]. Systemic treatment with IFNγ via i.p. injection exacerbates plaque deposition in Apoe-/- mice [65]. IFNγ treatment also results in decreased VSMC proliferation and subsequent collagen deposition in the plaque cap, suggesting it may compromise plaque stability. Reduced plaque size is observed in Ifnγ-/- xApoe-/- double knock out mice, and gene transfer of a secreted IFNγ receptor decoy in Apoe-/-mice attenuated development of atherosclerotic plaque [65, 66].
Antiatherogenic cytokines
Cytokine abundance in atherosclerotic plaque is overwhelmingly Th1 oriented [67, 68]. While expressed to a much lesser degree compared to pro-inflammatory cytokines, antiinflammatory cytokines associated with Th2 and M2 cells are present in atherosclerotic plaques. Overexpression of Th2 cytokines has been proposed to attenuate other inflammatory conditions such as rheumatoid arthritis, colitis, and asthma; therefore, they may also prove attractive an approach for attenuation of atherosclerosis. Polarization of inflammatory cells to anti-inflammatory or reparative phenotypes could serve as a promising therapeutic avenue for atherosclerosis. One therapeutic goal for regression of existing atherosclerotic plaque would be to tip the balance of these “opposing forces” from Th1, MI to an anti-inflammatory Th2, M2 plaque milieu. Increased Th2 cells have been linked in decreased risk of MI and stroke in women [69]. The majority of studies focusing on Th2 cytokines consider them to be indirectly anti-atherogenic by dampening adaptive immunity, with subsequent lesion formation [70, 71]. Additional studies exploring the potential protective effects of Th2 interleukins on resident vascular cells such as EC and VSMC would also aid in identifying possible therapeutic targets.
Interleukin-4
(IL-4) is an anti-inflammatory cytokine that is confirmed to polarize T cells to their Th2 phenotype [72]. Expression of IL-4 both at the mRNA and proteins levels has been confirmed in human atherosclerotic plaque [73]. IL-4 initiates a positive feedback loop as subsequent IL-4 induced Th2 cells produce additional IL-4, which can further potentiate antiinflammatory effects upon T cells, as well as macrophages [47, 74]. IL-4 polarizes T cells to Th2 by inducing expression of the transcription factor, GATA-binding protein 3 (GATA3) and inhibiting expression of pro-inflammatory IFNγ [75]. Other in vitro experiments demonstrate anti-inflammatory phenotypes including decreased monocyte/macrophage adhesion and decreased VSMC proliferation. The role of IL-4 in atherogenesis in mouse models remains unresolved, as several studies present conflicting experimental data. Despite being an antiinflammatory, Th2 cytokine, IL-4 can induce expression of CAMs in ECs, resulting in increased immune cell extravasation into the vasculature, promoting inflammation. Il4-/-xApoe-/- double knockout mice have decreased plaque formation compared to controls [73]. Bone marrow transplants from Il4-/- into Ldlr-/- mice also results in decreased atherosclerosis. Additionally, treatment with IL-4 in Apoe-/- mice does not ameliorate plaque burden [76]. Despite conflicting data, some reviews still report IL-4 as an atheroprotective cytokine. The case of IL-4 highlights the complexity of inflammatory vascular disease. An anti-inflammatory cytokine does not always have a clear-cut role in disease pathology.
Interleukin-10
(IL-10) is the archetypal anti-inflammatory cytokine that plays a key role in regulating Th1/Th2 and M1/M2 balance, pushing toward Th2 and M2 [77]. IL-10 has been extensively researched in atherosclerosis and has been established as an anti-atherosclerotic cytokine with multiple mechanisms in addition to Th2 and M2 polarization. IL-10 is able to dampen expression of inflammatory genes in various cell types and inhibit antigen presentation and T cell proliferation [47]. Ongoing studies continue to explore the complex molecular mechanisms of IL-10 mediated atheroprotection. IL-10 signals through the IL-10 receptor and subsequent Jak1 and STAT 1, 3, and 5, though primarily STAT3. IL-10 is able to block signaling cascades induced by master pro-inflammatory switches TNFα and NFkB in macrophages, ECs, and VSMCs. IL-10 is also able to dampen expression of pro-inflammatory transcripts by decreasing levels of the RNA binding protein (RBP), human antigen R (HuR) [78]. HuR stabilizes pro-inflammatory transcripts by binding “AU” rich regulatory elements, termed AREs, in the 3′untranscribed region (3′UTR). IL-10 mediated atheroprotection is considered to be immunomodulatory, as ECs and VSMCs do not express IL-10 [79]. Il10-/-xApoe-/- double knockout mice have significantly increased plaque burden as well an increased VSMC and inflammatory cell infiltration [80]. Similarly, Ldlr-/- and Apoe-/- mice administered AAV2 overexpressing IL-10 and transplant of bone marrow from IL-10 transgenic mice into Ldlr-/- mice all demonstrate decreased lesion sizes compared to controls [81, 82]. Taken together, several studies indicate that IL-10 is a potent immune modulator that reduces atherosclerosis at multiple levels, including gene expression, leukocyte extravasation, and polarization of adaptive immunity to the Th2 phenotype.
Interleukin-19
(IL-19) is an anti-inflammatory Th2 cytokine that is a member of the IL-10 family [83]. IL-19 signals through dimerization of the α- and β- subunits of the IL-20 receptor. IL-19 is expressed in macrophages and T cells, and unlike IL-10, can be induced in ECs and VSMCs by pro-inflammatory signals [84, 85]. IL-19 is able to polarize T cells and macrophages to Th2 and M2 phenotypes, promoting its role as anti-inflammatory [86, 87]. IL-19 is also able to decrease expression of CAMs and reduce leukocyte/EC interactions [88]. Immunohistochemical analysis of human coronary arteries with class 4 plaques show increased IL-19 expression compared to healthy controls, suggesting IL-19 may play a compensatory mechanism in atherogenesis [86]. IL-19 treatment decreases oxLDL lipid uptake in wild-type (WT) macrophages. Furthermore, Il19-/- macrophages demonstrate increased oxLDL lipid uptake compared to WT macrophages, suggesting that IL-19 may participate in reverse cholesterol transport, an emerging anti-atherosclerotic mechanism [87]. IL-19 is also able to influence activation of cultured VSMCs, reducing their proliferation, migration, and inflammatory gene expression [84, 89, 90]. Ldlr/- mice treated with IL-19 while simultaneously being fed high fat diet have decreased plaque burden compared to PBS injected controls [86]. Interestingly, IL-19 does not affect NF-κB; its strong anti-inflammatory effects are hypothesized to be through inhibition of mRNA stability mediated by the RBP HuR [89]. A potential mechanism is that IL-19 prevents HuR translocation from the nucleus to the cytoplasm, thereby blocking its ability to bind and stabilize pro-inflammatory transcripts. The emerging picture for IL-19 shows potent anti-atherosclerotic effects through its ability to engage multiple cell types and affect multiple mechanisms including reduction in inflammatory gene expression, macrophage polarization, reduction in extravasation, and increased reverse cholesterol transport.
Interleukin-33
(IL-33) is a member of the IL-1 family and is normally expressed in the healthy vasculature in multiple cell types [91]. It is able to polarize to Th2 and M2 by induction of IL-4, IL-5 and IL-13 and by decreasing expression of IFNγ [92]. IL-33 expression is increased in human atherosclerotic plaque, specifically in macrophages. IL-33 has been shown to reduce expression of scavenger receptors and enhance expression of proteins involved in cholesterol efflux, thereby reducing foam cell formation [93]. IL-33 treatment of Apoe-/- mice has resulted in decreased atherosclerosis as well as reduced macrophage infiltration in atherosclerotic plaques, though these data were from two independent studies and have not yet both been assessed together [91].
Transforming growth factor beta
(TGFβ) is an anti-atherosclerotic, Th2 cytokine that is part of the TGF super-family. TGFβ is the most investigated cytokine in the TGF family and is able to negatively regulate pro-inflammatory signaling. Global Tgfβ-/- mice are postnatally lethal with significant leukocyte infiltration in all organs, suggesting a vital immunoregulatory role. TGFβ is present in human atherosclerotic plaques and can be secreted by T cells, macrophages, ECs, and VSMCs. TGFβ has a number of effects including influencing T cell differentiation and immune cell modulation [47]. TGFβ is a known pro-fibrotic and wound-heali ng cytokine, and initial studies exploring the role of TGFβ in atherosclerosis focused on its effects on VSMCs. Importantly, TGFβ contributes to matrix deposition, which is crucial for maintaining plaque stability in humans. Abrogation of TGFβ in T cells results in exacerbated lesion sizes with decreased VSMCs and collagen in Apoe-/- mice. Since matrix deposition is a stability characteristic, this suggests a potential role for TGFβ in promotion of plaque stability in humans [94]. Additionally, global inhibition of TGFβ accelerates plaque deposition and also decreases collagen content, further implicating a role in plaque vulnerability in humans [95]. Patients with severe cases of atherosclerosis have decreased levels of circulating TGFβ in their serum, providing further clinical evidence of TGFβ as an anti-atherosclerotic cytokine [96].
5. Post-transcriptional control of cytokine expression
Cytokine expression is induced by numerous factors present in the plaque milieu. Many excellent reviews have described transcriptional activation of cytokine expression but fewer describe mechanisms involved in post-transcriptional modifications that regulate cytokine expression, particularly cytokine mRNA stability [97-99]. A major point of control for the cell to regulate cytokine abundance is regulation of cytokine mRNA stability. This section will describe what is known about RNA binding proteins (RBPs) that regulate cytokine abundance by modulation of mRNA. Although modulation of mRNA has been posited as a possible therapeutic strategy, surprisingly, there is very little literature exploring the concept that it could be directly regulated by inflammatory stimuli. In this regard, regulation of cytokine mRNA stability can be considered to be an overlooked therapeutic opportunity [100]. To this end, RBPs have emerged as critical regulators of cytokine expression, and potential targets of rational drug therapy. RBPs appear to play a major role in phenotypic switching of several cell types of cardiovascular disease and remain very understudied. While RNA stability will be predominantly discussed, it is important to note that in addition to influencing stability, RBPs are able to also alter mRNA composition and localization, and mediate translation.
Many cytokines have unstable mRNAs, often due to the presence of cis-acting AU-rich elements (AREs) in the 3′UTR, which promote degradation. Trans-acting factors, including RBPs, are able to bind the ARE elements and regulate mRNA fate. AREs were first identified in 1986 and subsequent studies established them as a mechanism of decay of inflammation related transcripts [101]. It is now known that RBPs are able to bind AREs and either stabilize or destabilize them, leading to translation or degradation respectively. Based on a database of human mRNAs, roughly 8% contain AREs, but the percentage of cytokines that contain them is much higher, attributing to their need for finely-controlled expression. Controlling mRNA decay allows the cell fine-tune mRNA abundance and translation for a quick adaptation to inflammatory conditions. It has been demonstrated that cytokines expressed early in inflammation have several AREs, compared to those expressed later. Multiple RBPs are able to bind the same transcript and influence protein levels by different mechanisms.
TNFα is an example of a potent, rapid-response pro-inflammatory cytokine whose mRNA is finely controlled by RBPs, allowing the cell to tightly regulate induction of other cytokines. TNFα is known to initiate and sustain pathogenesis of a number of inflammatory diseases including atherosclerosis, due to its central role in inflammation. TNFα is perhaps the most studied cytokine in terms of mechanisms of regulation. The TNFα 3′UTR is complex and offers the greatest opportunity to identify and understand RBPs. We will use TNFα as an example of how the 3′UTR of mRNA can be used by RBPs to regulate cytokine abundance. TNFα mRNA contains multiple class II AREs in the 3′UTR, which are overlapping copies of nonamer UUAUUUAUU within a U-rich region. Post-transcriptional control of TNFα by its ARE occurs at multiple points. The TNFα ARE is targeted to destabilize its transcripts and initiate mRNA decay, though at least one RBP that promotes stability has been identified. Spontaneous mouse models of autoimmune disease have decreased levels of TNFα associated with mutations in their TNFα ARE sequences [102]. Genetic deletion of TNFα AREs in mice (TNFARE) results in chronic TNFα protein overproduction, demonstrating the importance of these elements in regulation of TNFα abundance [103]. TNFARE mice also develop chronic inflammatory arthritis and inflammatory bowel disease. Compared to WT control cells, unstimulated thioglycollate-elicited peritoneal macrophages (TEPM) and bone marrow derived macrophages (BMDM) isolated from TNFΔRE mice spontaneously produce detectable levels of TNFα protein, and when stimulated with LPS exhibit a 3- to 5- fold increase in TNFα protein compared to controls. When stimulated with LPS, steady state levels of TNFα mRNA from TNFARE TEPM demonstrate a sustained, 148-fold increase over WT controls. Altogether, these data suggest the absence of AREs augments TNFα mRNA and subsequent protein abundance, and demonstrate the importance of these elements in regulation of cytokine levels. Effects of deletion of AREs in TNFα in the scope of atherosclerosis and other vascular diseases have yet to be investigated. Several ARE-binding RBPs that influence TNFα have been identified. The following section will briefly describe the function of each and its role in regulation of TNFα mRNA stability.
Human antigen R
(HuR) is a ubiquitously expressed member of a family of RBPs called the embryonic lethal abnormal vision (ELAV). HuR stability is linked to its translocation from the nucleus to the cytoplasm where mRNA is processed. HuR has been shown to recognize and bind AREs in the 3′UTR of many cytokine transcripts [104]. Hur-/- mice are embryonically lethal, demonstrating its importance in mRNA processing. Initial studies exploring HuR in RNA stability found HuR to bind unstable transcripts and suggested HuR was a destabilizing RBP. However, follow up in vitro studies in which HuR was silenced, vascular endothelial growth factor (VEGF) mRNA, which contains an ARE in its 3′UTR, was destabilized [105]. Furthermore, VEGF transcripts were stabilized in cells when HuR was overexpressed. Experiments performed in an LPS-sensitive macrophage line to identify proteins that bind AREs in TNFα recognized HuR as the RBP with the highest affinity. Overexpression of HuR in Tet-OFF HeLa cells stabilized reporter construct containing the human TNFα ARE [106]. HuR appears to be the only identified RBP confirmed to stabilize TNFα transcripts. It is likely that there is competition among RBPs to bind AREs and influence protein production. However, HuR and TIA-1 are known to cooperatively bind TNFα, which results in destabilization [104]. IL-10 and IL-19 have both been shown to dampen inflammation by decreasing HuR abundance and/or activity, resulting in destabilization of pro-inflammatory transcripts including TNFα. Though not yet reported, HuR activity would likely be a promising target in anti-atherosclerotic therapy.
Tristetraprolin
(TTP) is a Cys-Cys-Cys-His (CCCH) zinc finger protein. In contrast to HuR, TTP destabilizes TNFα transcripts by binding its AREs. Ttp-/- bone marrow and fetal liver derived macrophages exhibit increased TNFα mRNA and protein in response to LPS in vitro. Ttp-/- mice begin developing arthritis, dermatitis, autoimmunity, and myeloid hyperplasia shortly after birth. Virtually all these syndromes can be reversed by treatment with TNFα antibody, suggesting these phenotypes are due to uncontrolled TNFα expression. Ttp-/- mice also display increased TNFα at the mRNA and protein levels both with and without LPS stimulation [107]. Phosphorylation of TTP results in its sequestration and subsequent increased TNFα production as TTP can no longer bind the transcript.
ARE/poly(U)-binding/degradation factor
(AUF-1) is also known as heterogeneous nuclear ribonucleoprotein D0 (HNRNP D). AUF-1 is ubiquitously expressed and is able to bind poly-(U) sites on mRNAs, in addition to AREs [108]. Knockdown of AUF-1 in WT BMDM results in delayed degradation of TNFα transcripts, suggesting that like TTP, AUF-1 is an mRNA de-stability factor. Auf-/- mice have increased sensitivity to LPS-induced sepsis and also develop endotoxemia and chronic dermatitis with increased mortality and chronic systemic inflammation due to failure to effectively degrade TNFα as well as IL-1β transcripts. Experiments completed in transgenic mice have found confirmed destabilization of TNFα but interestingly have found stabilization of other ARE-containing transcripts. It has been suggested that AUF-1 has different effects on mRNAs depending on cell types. This is likely through interaction of AUF-1 with other cell type specific factors that modulate its activity, providing an additional level of control of cytokine abundance.
T-cell-restricted intracellular antigen-1
(TIA-1), TIA-1-related protein (TIAR) are both members of the RNA-recognition motif family of RBPs and have both been shown to bind the ARE on TNFα transcripts. TIA-1 and TIAR are able to repress TNFα protein production by blocking translation. Peritoneal macrophages isolated from Tia1-/- mice express the same amount of TNFα mRNA as macrophages isolated from WT mice, however Tia-/- macrophages produce excess TNFα protein compared to WT macrophages [109]. TNFα mRNA in Tia-/- and WT macrophages do not differ in half-life, suggesting it does not play a role in the stability of the transcript. Instead, TIA-1 and TIAF specifically silence translation [110]. Under stressed conditions TIA-1 and TIAR are able to prevent initiation of translation by recruiting and sequestering mRNAs to discrete cytoplasmic foci called stress granules. The role of TIAR in regulation of TNFα mRNA has not been as established as TIA-1 because Tiar-/- mice are embryonically lethal. The only identified functionally distinct feature of TIA-1 and TIAR at this point is their interactions with other RBPs.
CUG triplet repeat RNA binding protein 1
(CUGBP1) was originally found to bind CUG repeats in the 3′UTR of mRNAs in mytonic dystrophy [111]. CUGBP has been shown to affect alternative splicing, modulation of translation of mRNAs, and deadenylation or poly(A) shortening [112]. CUGBP1 is able to destabilize TNFα mRNA and is able to bind its 3′UTR at multiple sites [113]. CUGBP1 is able to bind a region called embryonic deadenylation elements (EDENs) found in the 3′UTR, in addition to AREs. EDENs are typically found in proximity to AREs and data suggests that neighboring AREs are involved in EDEN-dependent deadenylation [114]. Thus, CUGBP is able to destabilize TNFα by binding both AREs and EDENs, leading to deadenylation and mRNA decay; though data suggests CUGBP binds to AREs with greater affinity than EDENs. Knockdown of CUGBP in vitro results in stabilization and subsequent increased abundance of TNFα transcripts [113]
RBPs have yet to be extensively researched in the specific scope of cardiovascular disease. The few studies that have investigated RBPs in cardiovascular disease suggest an important role in phenotypic switching [115]. ARE-binding RBPs, as well as RBPs Quaking (QKI) and Roquin are able to influence the development of foam cells from monocytes and macrophages [116-118]. QKI is highly abundant in macrophages in advanced atherosclerotic lesions and abrogation of QKI is able to prevent monocyte extravasation and foam cell formation both in vitro and in vivo [118]. RNA-seq and microarray analysis suggest a role for QKI in mRNA abundance and alternative splicing of transcripts. One study has shown that Roquin is able to initiate TNFα degradation in macrophages by binding constitutive decay elements in its 3′UTR [116]. Additionally, HuR and QKI are able to affect dedifferentiation of VSMCs from contractile to synthetic [119, 120]. Stimulation of human VSMCs with platelet-derived growth factor results in greater levels of cytoplasmic HuR and subsequent changes in expression of genes influencing cell proliferation, structure, and metabolism and knockdown of HuR is able to reduce VSMC proliferation [120]. QKI is required for development of the vasculature and QKI global knockout mice are embryonic lethal [121]. One recently identified mechanism by which QKI is able to influence VSMCs phenotype is by influencing alternative splicing of Myocardin pre-RNA, resulting in an imbalance of splice variants [119]. Additional studies are necessary to determine the direct role QKI and other RBPs play in development of atherosclerosis. Based on our current understanding of their function, we can predict that those that stabilize ARE-containing transcripts would be pro-atherosclerotic, while those that destabilize them would be atheroprotective.
6. Summary and Conclusions
Cardiovascular disease and atherosclerosis in particular, is and will continue to be a significant socioeconomic burden in the developed world. As a dynamic and complex interaction of many cell types in various stages of pro-and anti-inflammatory states, atherosclerosis initiation and progression relies on cytokines for communication between the various cell types. The balance between pro-and anti-inflammatory cytokines can tip the balance between plaque progression and plaque regression. Post-transcriptional processes are a major point of regulation of cytokine synthesis, and several proteins participate in regulation of cytokine mRNA abundance. Understanding the mechanisms and molecules that regulate cytokine synthesis is key to our ability to identify therapeutic approaches to combat this significant disease.
Figure 1.
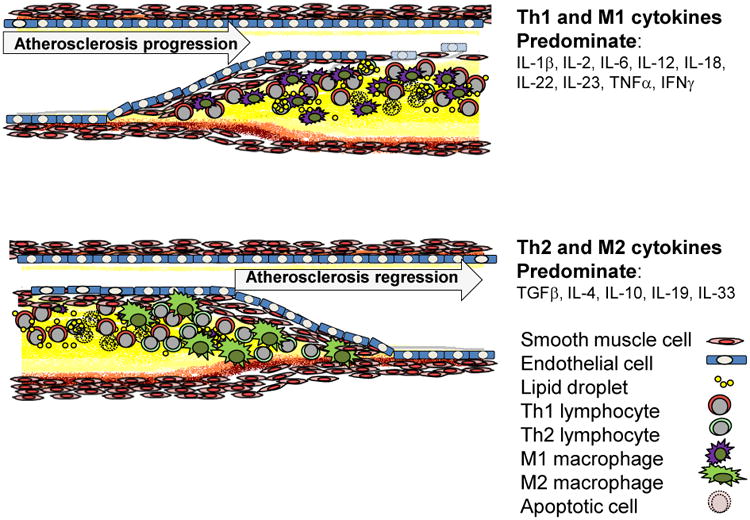
Cellular and cytokine profile in atherosclerotic plaque. Pro-inflammatory Th1 and M1 lymphocyte and macrophage derived cytokines dominate in atherogenesis, whereas anti-inflammatory Th2 and M2 cytokines would be expected to be expressed in resolving plaque. It is hypothesized that a therapeutic goal for atherosclerosis prevention, and potentially for regression would be to tip the balance of these “opposing forces” from a predominantly Th1 pro-inflammatory environment to a Th2, anti-inflammatory cytokine milieu.
Table 1.
Cytokines and predicted atherogenicity.
| Cytokine | Producer | T cell Phenotype | Macrophage Phenotype | Atherogenicity |
|---|---|---|---|---|
| IL-1β | ECs, Lymphocytes, Monocytes/Macrophages, VSMCs34 | Pro-atherogenic38 | ||
| IL-2 | Lymphocytes39 | Tregs41 | Conflicting data40,41,46 | |
| IL-6 | ECs, Lymphocytes, Monocytes/Macrophages, VSMCs42 | Conflicting data45,46 | ||
| IL-12 | Monocytes/Macrophages,Lymphocytes47 | Th147 | Pro-atherogenic49,50 | |
| IL-18 | Monocytes/Macrophages52 | Th152 | Pro-atherogenic53-55 | |
| IL-22 | Lymphocytes, Monocytes/Macrophages, VSMCs58 | Pro-atherogenic59 | ||
| IL-23 | Monocytes/Macrophages60 | Th1760 | Further Experiments Required | |
| TNFα | Lymphocytes, Monocytes/Macrophages, VSMCs20 | M120 | Pro-atherogenic29 | |
| IFNγ | ECs, Lymphocytes, Monocytes/Macrophages, VSMCs62 | Th163 | M164 | Pro-atherogenic65,66 |
| IL-4 | ECs, Lymphocytes, Monocytes/Macrophages,VSMCs47,74 | Th275 | Conflicting data73,76 | |
| IL-10 | Monocytes/Macrophages,Lymphocytes47,77 | Th247 | M279 | Atheroprotective79,81,82 |
| IL-19 | ECs, Lymphocytes, Monocytes/Macrophages,VSMCs84,85 | Th286 | M287 | Atheroprotective86 |
| IL-33 | Monocytes/Macrophages,Lymphocytes91-93 | Th292 | M292 | Atheroprotective91 |
| TGFβ | Lymphocytes, Monocytes/Macrophages, VSMCs47 | Th246 | Atheroprotective94 |
Table 2.
RNA binding proteins and predicted effects on atherogenicity.
| RNA-binding protein | Abbreviation | Effect on TNFα mRNA levels | Mechanism | Predicted Atherogenecity |
|---|---|---|---|---|
| Human antigen R | HuR | Increase105,106 | Stabilizes RNA105,106 | Pro-atherogenic |
| Tristetraprolin | TTP | Decrease107 | Destabilizes RNA107 | Anti-atherogenic |
| ARE/poly(U)-binding/degradation factor | AUF-1 | Decrease108 | Destabilizes RNA108 | Anti-atherogenic |
| T-cell-restricted intracellular antigen-1 | TIA-1 | Decrease109 | Inhibits translation110 | Anti-atherogenic |
| TIA-1-related protein | TIAR | Decrease109 | Inhibits translation110 | Anti-atherogenic |
| CUG triplet repeat RNA binding protein 1 | CUGBP1 | Decrease113 | Destabilizes RNA113,114 | Anti-atherogenic |
Acknowledgments
This work was supported by grants HL115575 and HL117724 from the National Heart Lung, and Blood Institute of the National Institutes of Health, and Grant 13GRNT1685003 from the American Heart Association to MVA. MR was supported by American Heart Association pre-doctoral fellowship 16PRE31220005.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benjamin EJ, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135(10):e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340(2):115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Chisolm GM, 3rd, Chai Y. Regulation of cell growth by oxidized LDL. Free Radic Biol Med. 2000;28(12):1697–707. doi: 10.1016/s0891-5849(00)00227-6. [DOI] [PubMed] [Google Scholar]
- 4.Tabas I, Weiland DA, Tall AR. Unmodified low density lipoprotein causes cholesteryl ester accumulation in J774 macrophages. Proc Natl Acad Sci U S A. 1985;82(2):416–20. doi: 10.1073/pnas.82.2.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye Q, et al. Inflammatory stress increases unmodified LDL uptake via LDL receptor: an alternative pathway for macrophage foam-cell formation. Inflamm Res. 2009;58(11):809–18. doi: 10.1007/s00011-009-0052-4. [DOI] [PubMed] [Google Scholar]
- 6.Ketelhuth DF, et al. Identification of a danger-associated peptide from apolipoprotein B100 (ApoBDS-1) that triggers innate proatherogenic responses. Circulation. 2011;124(22):2433–43. doi: 10.1161/CIRCULATIONAHA.111.051599. 1-7. [DOI] [PubMed] [Google Scholar]
- 7.Hermansson A, et al. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation. 2011;123(10):1083–91. doi: 10.1161/CIRCULATIONAHA.110.973222. [DOI] [PubMed] [Google Scholar]
- 8.Gistera A, et al. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med. 2017;281(4):383–397. doi: 10.1111/joim.12589. [DOI] [PubMed] [Google Scholar]
- 9.Assinger A, et al. Apolipoprotein B100 danger-associated signal 1 (ApoBDS-1) triggers platelet activation and boosts platelet-leukocyte proinflammatory responses. Thromb Haemost. 2014;112(2):332–41. doi: 10.1160/TH13-12-1026. [DOI] [PubMed] [Google Scholar]
- 10.Napoli C, et al. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100(11):2680–90. doi: 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stary HC, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89(5):2462–78. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- 12.Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37(3):208–22. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Tracy RP, et al. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2013;2(3):e000117. doi: 10.1161/JAHA.113.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Stemme S, Hansson GK. Evidence for a local immune response in atherosclerosis. CD4+ T cells infiltrate lesions of apolipoprotein-E-deficient mice. Am J Pathol. 1996;149(2):359–66. [PMC free article] [PubMed] [Google Scholar]
- 15.de Boer OJ, et al. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS One. 2007;2(8):e779. doi: 10.1371/journal.pone.0000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–80. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 17.De Duve C. The role of lysosomes in cellular pathology. Triangle. 1970;9(6):200–8. [PubMed] [Google Scholar]
- 18.Verreck FA, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A. 2004;101(13):4560–5. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119(3):651–65. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skoog T, et al. Plasma tumour necrosis factor-alpha and early carotid atherosclerosis in healthy middle-aged men. Eur Heart J. 2002;23(5):376–83. doi: 10.1053/euhj.2001.2805. [DOI] [PubMed] [Google Scholar]
- 21.Woollard KJ, et al. Pathophysiological levels of soluble P-selectin mediate adhesion of leukocytes to the endothelium through Mac-1 activation. Circ Res. 2008;103(10):1128–38. doi: 10.1161/CIRCRESAHA.108.180273. [DOI] [PubMed] [Google Scholar]
- 22.Zhou X, Hansson GK. Detection of B cells and proinflammatory cytokines in atherosclerotic plaques of hypercholesterolaemic apolipoprotein E knockout mice. Scand J Immunol. 1999;50(1):25–30. doi: 10.1046/j.1365-3083.1999.00559.x. [DOI] [PubMed] [Google Scholar]
- 23.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12(1):49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20(2):87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu HY, Nicholson AC, Hajjar DP. Inhibition of macrophage scavenger receptor activity by tumor necrosis factor-alpha is transcriptionally and post-transcriptionally regulated. J Biol Chem. 1996;271(13):7767–73. doi: 10.1074/jbc.271.13.7767. [DOI] [PubMed] [Google Scholar]
- 26.Hsu HY, Twu YC. Tumor necrosis factor-alpha -mediated protein kinases in regulation of scavenger receptor and foam cell formation on macrophage. J Biol Chem. 2000;275(52):41035–48. doi: 10.1074/jbc.M003464200. [DOI] [PubMed] [Google Scholar]
- 27.Galis ZS, et al. Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Ann N Y Acad Sci. 1995;748:501–7. doi: 10.1111/j.1749-6632.1994.tb17348.x. [DOI] [PubMed] [Google Scholar]
- 28.Taubman MB, et al. Tissue factor in the pathogenesis of atherosclerosis. Thromb Haemost. 1997;78(1):200–4. [PubMed] [Google Scholar]
- 29.Branen L, et al. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24(11):2137–42. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 30.Canault M, et al. Exclusive expression of transmembrane TNF-alpha in mice reduces the inflammatory response in early lipid lesions of aortic sinus. Atherosclerosis. 2004;172(2):211–8. doi: 10.1016/j.atherosclerosis.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):578–82. doi: 10.1038/nrrheum.2009.181. [DOI] [PubMed] [Google Scholar]
- 32.Jacobsson LT, et al. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005;32(7):1213–8. [PubMed] [Google Scholar]
- 33.Dixon WG, et al. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007;56(9):2905–12. doi: 10.1002/art.22809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, et al. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis. 1995;115(1):89–98. doi: 10.1016/0021-9150(94)05503-b. [DOI] [PubMed] [Google Scholar]
- 35.Kaden JJ, et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis. 2003;170(2):205–11. doi: 10.1016/s0021-9150(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 36.Eun SY, et al. IL-1beta enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vascul Pharmacol. 2015;72:108–17. doi: 10.1016/j.vph.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 37.Elhage R, et al. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97(3):242–4. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 38.Kamari Y, et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis. 2007;195(1):31–8. doi: 10.1016/j.atherosclerosis.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 39.Gajewski TF, Joyce J, Fitch FW. Antiproliferative effect of IFN-gamma in immune regulation. III. Differential selection of TH1 and TH2 murine helper T lymphocyte clones using recombinant IL-2 and recombinant IFN-gamma. J Immunol. 1989;143(1):15–22. [PubMed] [Google Scholar]
- 40.Upadhya S, et al. Atherogenic effect of interleukin-2 and antiatherogenic effect of interleukin-2 antibody in apo-E-deficient mice. Angiology. 2004;55(3):289–94. doi: 10.1177/000331970405500308. [DOI] [PubMed] [Google Scholar]
- 41.Dietrich T, et al. Local delivery of IL-2 reduces atherosclerosis via expansion of regulatory T cells. Atherosclerosis. 2012;220(2):329–36. doi: 10.1016/j.atherosclerosis.2011.09.050. [DOI] [PubMed] [Google Scholar]
- 42.Korn T, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105(47):18460–5. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng X, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127(1):89–97. doi: 10.1016/j.clim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Romano M, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315–25. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 45.Huber SA, et al. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19(10):2364–7. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 46.Schieffer B, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110(22):3493–500. doi: 10.1161/01.CIR.0000148135.08582.97. [DOI] [PubMed] [Google Scholar]
- 47.Tse K, et al. T cells in atherosclerosis. Int Immunol. 2013;25(11):615–22. doi: 10.1093/intimm/dxt043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buono C, et al. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. 2005;102(5):1596–601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee TS, et al. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19(3):734–42. doi: 10.1161/01.atv.19.3.734. [DOI] [PubMed] [Google Scholar]
- 50.Hauer AD, et al. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation. 2005;112(7):1054–62. doi: 10.1161/CIRCULATIONAHA.104.533463. [DOI] [PubMed] [Google Scholar]
- 51.Yoshimoto T, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161(7):3400–7. [PubMed] [Google Scholar]
- 52.Blankenberg S, et al. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106(1):24–30. doi: 10.1161/01.cir.0000020546.30940.92. [DOI] [PubMed] [Google Scholar]
- 53.Elhage R, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59(1):234–40. doi: 10.1016/s0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- 54.Mallat Z, et al. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89(7):E41–5. doi: 10.1161/hh1901.098735. [DOI] [PubMed] [Google Scholar]
- 55.Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ Res. 2002;90(2):E34–8. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- 56.Cheng F, et al. Decreased plasma IL22 levels, but not increased IL17 and IL23 levels, correlate with disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis. 2009;68(4):604–6. doi: 10.1136/ard.2008.097089. [DOI] [PubMed] [Google Scholar]
- 57.Ikeuchi H, et al. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum. 2005;52(4):1037–46. doi: 10.1002/art.20965. [DOI] [PubMed] [Google Scholar]
- 58.Xia Q, et al. Characterisation of IL-22 and interferon-gamma-inducible chemokines in human carotid plaque. Int J Cardiol. 2012;154(2):187–9. doi: 10.1016/j.ijcard.2011.10.093. [DOI] [PubMed] [Google Scholar]
- 59.Rattik S, et al. IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis. 2015;242(2):506–14. doi: 10.1016/j.atherosclerosis.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 60.Duvallet E, et al. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med. 2011;43(7):503–11. doi: 10.3109/07853890.2011.577093. [DOI] [PubMed] [Google Scholar]
- 61.Abbas A, et al. Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke. 2015;46(3):793–9. doi: 10.1161/STROKEAHA.114.006516. [DOI] [PubMed] [Google Scholar]
- 62.McLaren JE, Ramji DP. Interferon gamma: a master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009;20(2):125–35. doi: 10.1016/j.cytogfr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 63.Mallat Z, Ait-Oufella H, Tedgui A. Regulatory T cell responses: potential role in the control of atherosclerosis. Curr Opin Lipidol. 2005;16(5):518–24. doi: 10.1097/01.mol.0000182532.11512.90. [DOI] [PubMed] [Google Scholar]
- 64.Leon ML, Zuckerman SH. Gamma interferon: a central mediator in atherosclerosis. Inflamm Res. 2005;54(10):395–411. doi: 10.1007/s00011-005-1377-2. [DOI] [PubMed] [Google Scholar]
- 65.Gupta S, et al. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99(11):2752–61. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whitman SC, et al. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. 2000;157(6):1819–24. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172(6):1500–8. doi: 10.2353/ajpath.2008.070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frostegard J, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145(1):33–43. doi: 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 69.Engelbertsen D, et al. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler Thromb Vasc Biol. 2013;33(3):637–44. doi: 10.1161/ATVBAHA.112.300871. [DOI] [PubMed] [Google Scholar]
- 70.von der Thusen JH, et al. Interleukins in atherosclerosis: molecular pathways and therapeutic potential. Pharmacol Rev. 2003;55(1):133–66. doi: 10.1124/pr.55.1.5. [DOI] [PubMed] [Google Scholar]
- 71.Mallat Z, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85(8):e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 72.Swain SL, et al. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145(11):3796–806. [PubMed] [Google Scholar]
- 73.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163(3):1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–21. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wurtz O, Bajenoff M, Guerder S. IL-4-mediated inhibition of IFN-gamma production by CD4+ T cells proceeds by several developmentally regulated mechanisms. Int Immunol. 2004;16(3):501–8. doi: 10.1093/intimm/dxh050. [DOI] [PubMed] [Google Scholar]
- 76.King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol. 2007;171(6):2040–7. doi: 10.2353/ajpath.2007.060857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinez FO, et al. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 78.Krishnamurthy P, et al. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104(2):e9–18. doi: 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Von Der Thusen JH, et al. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr-/- mice. FASEB J. 2001;15(14):2730–2. doi: 10.1096/fj.01-0483fje. [DOI] [PubMed] [Google Scholar]
- 80.Caligiuri G, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9(1-2):10–7. [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Y, et al. Inhibition of atherogenesis in LDLR knockout mice by systemic delivery of adeno-associated virus type 2-hIL-10. Atherosclerosis. 2006;188(1):19–27. doi: 10.1016/j.atherosclerosis.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 82.Yoshioka T, et al. Adeno-associated virus vector-mediated interleukin-10 gene transfer inhibits atherosclerosis in apolipoprotein E-deficient mice. Gene Ther. 2004;11(24):1772–9. doi: 10.1038/sj.gt.3302348. [DOI] [PubMed] [Google Scholar]
- 83.Liao SC, et al. IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol. 2004;173(11):6712–8. doi: 10.4049/jimmunol.173.11.6712. [DOI] [PubMed] [Google Scholar]
- 84.Tian Y, et al. Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. Am J Pathol. 2008;173(3):901–9. doi: 10.2353/ajpath.2008.080163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jain S, et al. The anti-inflammatory cytokine interleukin 19 is expressed by and angiogenic for human endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31(1):167–75. doi: 10.1161/ATVBAHA.110.214916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ellison S, et al. Attenuation of experimental atherosclerosis by interleukin-19. Arterioscler Thromb Vasc Biol. 2013;33(10):2316–24. doi: 10.1161/ATVBAHA.113.301521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gabunia K, et al. IL-19 Halts Progression of Atherosclerotic Plaque, Polarizes, and Increases Cholesterol Uptake and Efflux in Macrophages. Am J Pathol. 2016;186(5):1361–74. doi: 10.1016/j.ajpath.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.England RN, et al. Interleukin-19 decreases leukocyte-endothelial cell interactions by reduction in endothelial cell adhesion molecule mRNA stability. Am J Physiol Cell Physiol. 2013;305(3):C255–65. doi: 10.1152/ajpcell.00069.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cuneo AA, Herrick D, Autieri MV. Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. J Mol Cell Cardiol. 2010;49(4):647–54. doi: 10.1016/j.yjmcc.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gabunia K, et al. Anti-inflammatory cytokine interleukin-19 inhibits smooth muscle cell migration and activation of cytoskeletal regulators of VSMC motility. Am J Physiol Cell Physiol. 2011;300(4):C896–906. doi: 10.1152/ajpcell.00439.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miller AM, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205(2):339–46. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16(11):676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 93.McLaren JE, et al. IL-33 reduces macrophage foam cell formation. J Immunol. 2010;185(2):1222–9. doi: 10.4049/jimmunol.1000520. [DOI] [PubMed] [Google Scholar]
- 94.Gojova A, et al. Specific abrogation of transforming growth factor-beta signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102(12):4052–8. doi: 10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 95.Mallat Z, et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89(10):930–4. doi: 10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 96.Grainger DJ, et al. The serum concentration of active transforming growth factor-beta is severely depressed in advanced atherosclerosis. Nat Med. 1995;1(1):74–9. doi: 10.1038/nm0195-74. [DOI] [PubMed] [Google Scholar]
- 97.Matsusaka T, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci U S A. 1993;90(21):10193–7. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84(3):331–4. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 99.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86(2):515–81. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 100.Eberhardt W, et al. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol Ther. 2007;114(1):56–73. doi: 10.1016/j.pharmthera.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 101.Caput D, et al. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci U S A. 1986;83(6):1670–4. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jacob CO, Lee SK, Strassmann G. Mutational analysis of TNF-alpha gene reveals a regulatory role for the 3′-untranslated region in the genetic predisposition to lupus-like autoimmune disease. J Immunol. 1996;156(8):3043–50. [PubMed] [Google Scholar]
- 103.Kontoyiannis D, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 104.Palanisamy V, et al. Control of cytokine mRNA expression by RNA-binding proteins and microRNAs. J Dent Res. 2012;91(7):651–8. doi: 10.1177/0022034512437372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Levy NS, et al. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273(11):6417–23. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 106.Dean JL, et al. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol. 2001;21(3):721–30. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Taylor GA, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4(5):445–54. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 108.Gratacos FM, Brewer G. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip Rev RNA. 2010;1(3):457–73. doi: 10.1002/wrna.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Piecyk M, et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000;19(15):4154–63. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kedersha NL, et al. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147(7):1431–42. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Timchenko LT, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24(22):4407–14. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moraes KC, Wilusz CJ, Wilusz J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA. 2006;12(6):1084–91. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang L, et al. The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. J Biol Chem. 2008;283(33):22457–63. doi: 10.1074/jbc.M802803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Audic Y, Omilli F, Osborne HB. Embryo deadenylation element-dependent deadenylation is enhanced by a cis element containing AUU repeats. Mol Cell Biol. 1998;18(12):6879–84. doi: 10.1128/mcb.18.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.de Bruin RG, et al. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. 2017;38(18):1380–1388. doi: 10.1093/eurheartj/ehw567. [DOI] [PubMed] [Google Scholar]
- 116.Leppek K, et al. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153(4):869–81. doi: 10.1016/j.cell.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 117.Sakurai S, Ohto U, Shimizu T. Structure of human Roquin-2 and its complex with constitutive-decay element RNA. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt 8):1048–54. doi: 10.1107/S2053230X15011887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.de Bruin RG, et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun. 2016;7:10846. doi: 10.1038/ncomms10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van der Veer EP, et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ Res. 2013;113(9):1065–75. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- 120.Pullmann R, Jr, et al. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J Biol Chem. 2005;280(24):22819–26. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noveroske JK, et al. Quaking is essential for blood vessel development. Genesis. 2002;32(3):218–30. doi: 10.1002/gene.10060. [DOI] [PubMed] [Google Scholar]