

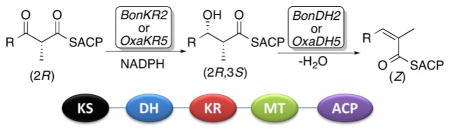
Abstract
Incubation of (±)-2-methyl-3-ketobutyryl-SNAC (3) and (±)-2-methyl-3-ketopentanoyl-SNAC (4) with BonKR2 or OxaKR5, ketoreductase domains from the bongkrekic acid (1) and oxazolomycin (2) polyketide synthases, in the presence of NADPH gave in each case the corresponding (2R,3S)-2-methyl-3-hydroxybutyryl-SNAC (5) or (2R,3S)-2-methyl-3-hydroxypentanoyl-SNAC (6) products, as established by chiral GC-MS analysis of the derived methyl esters. Identical results were obtained by BonKR2- and OxaKR5-catalyzed reduction of chemoenzymatically prepared (2R)-2-methyl-3-ketopentanoyl-EryACP6, (2R)-2-methyl-3-ketobutyryl-BonACP2 (12), and (2R)-2-methyl-3-ketopentanoyl-BonACP2 (13). The paired dehydratase domains, BonDH2 and OxaDH5, were then shown to catalyze the reversible syn dehydration of (2R,3S)-2-methyl-3-hydroxybutyryl-BonACP2 (14) to give the corresponding trisubstituted (Z)-2-methylbutenoyl-BonACP2 (16).
Graphical abstract
Double bonds are ubiquitous structural features found in thousands of known bacterial polyketide natural products. While the vast majority are disubstituted or trisubstituted E double bonds, numerous polyketides harbor isomeric Z double bonds, such as those found in bongkrekic acid (1)1 and oxazolomycin A (2)2 (Figure 1), as well as in fostriecin,3–5 phoslactomycin,6 borrellidin, 7, 8 bacillaene,9, 10 curacin,11,12 difficidin,13 chivosazol A,14 disorazol A,14 lactimidomycin,14 and macrolactin, 14 among many others. Modular polyketide synthases (PKSs) typically generate each double bond by the coupled action of a ketoreductase (KR) domain, which carries out the stereospecific, NADPH-dependent reduction of a 3-ketoacyl-acyl carrier protein (ACP) or a 2-methyl-3-ketoacyl-ACP chain elongation intermediate, and a paired dehydratase (DH) domain, which catalyzes the syn dehydration of the reduced 3-hydroxyacyl-ACP or 2-methyl-3-hydroxyacyl-ACP intermediate.
Figure 1.

Domain organization of the trans-AT polyketide synthase modules that generate (Z)-trisubstituted double bonds in bongkrekic acid (1) and oxazolomycin (2). The indicated methyl groups are introduced by C-methyl transferase (MT) domains.
A variety of DH domains from modular PKSs have been expressed as discrete proteins and their reactions characterized biochemically. Thus EryDH4, from module 4 of the erythromycin PKS,15 and NanDH2, from module 2 of the nanchangmycin PKS,16 each catalyze the syn dehydration of a (2R,3R)-2-methyl-3-hydroxyacyl-ACP substrate to the corresponding trisubstituted (E)-2-methylenoyl-ACP. By contrast, RifDH10, from module 10 of the rifamycin PKS, catalyzes the analogous syn dehydration of the diastereomeric (2S,3S)-2-methyl-3-hydroxyacyl-ACP to an (E)-2-methylenoyl-ACP product, in spite of the fact that the derived trisubstituted double bond in the ultimately formed rifamycin has the Z configuration, suggesting that the geometry must be altered subsequent to double bond formation.17 PicDH2, from module 2 of the picromycin PKS,18, 19 and FosKR1, from module 1 of the fostriecin PKS,5 each catalyze the dehydration of a (3R)-3-hydroxyacyl-ACP substrate to the corresponding disubstituted (E)-2-enoyl-ACP. The predicted syn stereochemistry of the latter two dehydration reactions has yet to be confirmed experimentally. We recently reported that FosDH2, from module 2 of the fostriecin PKS, catalyzes the reversible dehydration of a (3S)-3-hydroxyacyl-ACP to the corresponding disubstituted (Z)-2-enoyl-ACP product.5 This report was the first to document the in vitro DH-catalyzed formation of any (Z)-enoyl-thioester product. Indeed, for a variety of reasons, all prior attempts to demonstrate the predicted formation of disubstituted or trisubstituted (Z)-double bonds by recombinant PKS DH domains had been unsuccessful, resulting instead in the exclusive generation of the isomeric (E)-double bonds.7, 11, 12, 17, 20
We now report that BonDH2, from module 2 of the bongkrekic acid trans-AT PKS, and OxaDH5, from module 5 of the oxazolomycin trans-AT PKS (Figure 1), each catalyze the stereospecific syn dehydration of (2R,3S)-2-methyl-3-hydroxyacyl-ACP substrates, generated by the paired ketoreductases, BonKR2 and OxaKR5, respectively, to give the corresponding trisubstituted (Z)-2-methylenoyl-ACP products.
BonKR2 and BonDH2, from Burkholderia gladioli pathovar cocovenenans, and OxaKR5 and OxaDH5, from Streptomyces albus, were each expressed in Escherichia coli as N-terminal His6-tagged proteins using codon-optimized synthetic genes, based on consensus PKS domain boundaries (Figures S1–S4). Each of these four recombinant proteins was purified to homogeneity by immobilized Ni2+ affinity chromatography. The purity and Mr of each recombinant protein were assessed by SDS-PAGE and confirmed by LC–ESI(+)–MS analysis (Figure S5 and Table S1).
In common with the vast majority of polyketide synthase KR domains, all of which belong to the superfamily of short chain dehydrogenase/reductase (SDR) proteins, 21, 22 BonKR2 and OxaKR5 each harbor the highly conserved active site triad of Ser, Tyr, and Lys residues (Figure 2).23, 24 On the other hand, both KR domains, along with other KR domains from trans-AT PKSs such as BaeKR9, from module 9 of the bacilllaene PKS, and DifKR6, from module 6 of the difficidin PKS, lack the diagnostic sequence markers, such as the conserved Trp residue (A-Type KR domain) or Leu-Asp-Asp motif (B-Type KR domain), that are normally correlated with the respective (3S)- or (3R) configuration of the resultant 3-hdyroxy acyl thioester reduction product (Figure 2).23–26 Moreover, none of the additional conserved KR sequence features that are normally diagnostic of the (2R)- or (2S)-methyl configuration of the reduced 2-methyl-3-hydroxyacyl-thioester (KR subtypes 1 or 2) are evident in the amino acid sequences of these trans-AT KR domains. As a consequence, the intrinsic stereospecifity of BonKR2 or OxaKR5 cannot be deduced from simple consensus sequence alignments. (Although the Leu-Val-Asp triad of OxaKR5 might have suggested classification of OxaKR5 as a B-Type KR domain which would be predicted to generate a (3R)-3-hydroxyacyl group, the experiments described below establish firmly that OxaKR5 is an A1-Type KR.)
Figure 2.

Mega3.0 (http://www.megasoftware.net) sequence alignment of PKS ketoreductase domains that reduce 2-methyl-3-ketoacyl-ACP intermediates, showing the four different stereochemical classes of KR domains. Conserved K, S, and Y residues constitute the canonical active site catalytic triad of SDR proteins. The BaeKR9, BonKR2, DifKR6, and OxaKR5 domains lack the conserved sequence motifs diagnostic of established KR Types. PKS source: Amp, amphotericin; Ave, avermectin; Bae, bacillaene; Bon, bongkrekic acid; bor, borrelidin; Con, concanamycin A; Dif, difficidin; Ery, erythromycin; Lan, lankamycin; Lip, lipomycin; Meg, megalomicin; Mei, meilingmycin; Nys, nystatin; Oxa, oxazolomycin; Pic, picromycin; Pla, pladienolide; Tyl, tylactone.
The reductase activities of BonKR2 and OxaKR5 were each confirmed and the steady-state kinetic parameters were determined using the model N-acetylcysteamine thioester substrates (±)-2-methyl-3-ketobutyryl-SNAC (3) and (±)-2-methyl-3-ketopentanoyl-SNAC (4)27 and NADPH (Scheme 1a, Figures S6 and S7, Table S2). The stereochemistry of the resulting (2R,3S)-2-methyl-3-hydroxyacyl-SNAC products 5 and 6 was then established by chiral GC-MS analysis of the derived methyl esters 7-Me and 8-Me, including direct comparison with authentic synthetic standards of all four diastereomers of each product (Figures S8–S11, Tables S3 and S4).28 We also used ACP-bound substrates to confirm the stereospecificity of the BonKR2- and OxaKR5-catalyzed reductions. Thus, in one set of experiments, (2R)-2-methyl-3-ketopentanoyl-EryACP6, generated by incubation of propionyl-SNAC with Ery[KS6][AT6], the ketosynthase-acyltranferase didomain from module 6 of the erythromycin PKS, and EryACP6 plus methylmalonyl-CoA,28 was reduced in separate experiments with BonKR2 or OxaKR5 in the presence of NADPH to generate (2R,3S)-9 (Scheme 1b). After basic hydrolysis and methylation with TMSCHN2, chiral GC-MS analysis confirmed the exclusive formation of methyl (2R,3S)-2-methyl-3-hydroxypentanoate (8-Me) (Figures S12 and S13, Table S3).
Scheme 1.

Stereochemistry of BonKR2- and OxaKR5-Catalyzed Reduction of 2-Methyl-3-ketoacyl Thioesters.
Finally, in a complementary series of incubations, a mixture of BonKS2, the ketosynthase from module 2 of the bongkrekic acid PKS, and either acetyl-SNAC or propionyl-SNAC, was combined with malonyl-BonACP2, generated in situ as previously described by treatment of apo-BonACP2 with malonyl-CoA and the surfactin pantetheinyl transferase Sfp, so as to yield 3-ketobutyryl-BonACP2 (10) or 3-ketopentanoyl-BonACP2 (11) (Scheme 1c).29 Stereospecific methylation of 10 or 11 was achieved as previously described29 by treatment with a mixture of BonMT2, the C-methyl transferase from module 2 of the bongkrekic acid PKS, and S-adenosyl methionine (SAM), in the presence of S-adenosylhomocysteine (SAH) nucleosidase to prevent potent product inhibition by the co-product SAH, yielding (2R)-2-methyl-3-ketobutyryl-BonACP2 (12) or (2R)-2-methyl-3-ketopentanoyl-BonACP2 (13). In the presence of NADPH, incubation of 12 with either BonKR2 or OxaKR5 gave 14, while reduction of 13 with BonKR2 gave 15. Chiral GC-MS analysis of the derived methyl esters 7-Me and 8-Me, confirmed the exclusive formation of the corresponding (2R,3S)-2-methyl-3-hydroxybutyryl-BonACP2 (14) or (2R,3S)-2-methyl-3-hydroxypentanoyl-BonACP2 (15) (Figures S14–S16, Tables S3 and S4).
Having firmly established the stereochemistry of the BonKR2- and OxaKR5-catalyzed reductions, we next determined the stereospecificity of the paired dehydratase reactions. Chemoenzymatically prepared (2R,3S)-2-methyl-3-hydroxybutyryl-BonACP2 (14) was incubated in separate experiments with BonDH2 or OxaDH5 (Scheme 2a). Hydrolysis of the resultant acyl-ACP thioester (Z)-16, the product of syn dehydration, by treatment with PICS TE, the thioesterase from the picromycin PKS,30 gave exclusively (Z)-2-methylbutenoic acid (17), as established by GC-MS analysis and direct comparison with authentic standards of both (Z)- and (E)-2-methylbutenoic acid (Figures S17 and S18). In a negative control, NigDH1, from module 1 of the nigericin PKS, did not dehydrate 14.31 (Although NigDH1 naturally acts only as a 2-methyl-3-ketobutyryl-ACP epimerase, we have shown that it also harbors a cryptic dehydratase activity capable of converting (2R,3R)-2-methyl-3-hydroxypentanoyl-ACP to (E)-2-methyl-2-pentenoyl-ACP.)31 BonDH2 and OxaDH5 also catalyzed the reverse reaction, resulting in the stereospecific hydration of chemoenzymatically prepared (Z)-2-methylbutenoyl-BonACP2 (16) to yield exclusively the syn hydration product (2R,3S)-2-methyl-3-hydroxybutyryl-BonACP2 (14) (Scheme 2b). LC-ESI(+)-MS analysis after removal of the DH proteins confirmed the addition of water to (Z)-16, as established by the increase in mass of M+18 of each of the acyl-ACP products (Figure S19, Table S5). Finally, chiral GC-MS analysis established the exclusive formation of the derived methyl (2R,3S)-2-methyl-3-hydroxybutyrate (7-Me) (Figure S20). In the negative control, treatment of (Z)-16 with NigDH1 did not result in the formation of detectable hydration product.
Scheme 2.

Stereochemistry of BonDH2- and OxaDH5-Catalyzed Dehydration/Hydration Reactions
The above-described experiments establish conclusively that the dehydratase domains from the bongkrekic acid and oxazolomycin polyketide synthases, BonDH2 and OxaDH5, each catalyze the syn dehydration of (2R,3S)-2-methyl-3-hydroxyacyl-ACP substrates, which are exclusively generated by their respective paired BonKR2 and OxaKR5 domains, to give the corresponding (Z)-2-methylenoyl-ACP products. These findings constitute the first experimental demonstrations of the DH-catalyzed formation of a Z-trisubstituted double bond, in spite of a number of unsuccessful prior efforts, cited above, that have been directed toward this surprisingly elusive goal. Protein sequence alignments indicate that all of these dehydratases retain a conserved set of active site His and Asp residues but harbor no obvious DH sequence motifs that can be correlated with the E- or Z-configuration of their characteristic disubstituted or trisubstituted enoyl-ACP products (Figure S21).32 All PKS DH-catalyzed dehydration and/or hydration reactions for which the stereochemistry has been determined involve a reversible syn elimination/addition of water,15–17 in common with the established stereospecificity of the closely related FabA and FabZ dehydratase domains of E. coli fatty acid biosynthesis33, 34 as well as the analogous action of the yeast fatty acid synthase.35 The ultimate E or Z enoyl-ACP product geometry must reflect important differences in the active site conformation of the bound 3-hydroxyacyl-ACP substrates that are not obviously correlated with any conserved amino acid sequence motifs.
Supplementary Material
Acknowledgments
Funding
This work was supported by a grant from the U. S. National Institutes of Health, GM022172, to D.E.C.
Footnotes
Notes
The authors declare no competing financial interest.
Experimental methods, including sequence alignments, KR and DH design and expression, kinetic assays, and GC-MS and ESI-MS analysis of KR and DH incubations. This material is available free of charge on the ACS Publications website at DOI:10.1021/acs.biochem********
References
- 1.Moebius N, Ross C, Scherlach K, Rohm B, Roth M, Hertweck C. Chem Biol. 2012;19:1164–1174. doi: 10.1016/j.chembiol.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Zhao C, Coughlin JM, Ju J, Zhu D, Wendt-Pienkowski E, Zhou X, Wang Z, Shen B, Deng Z. J Biol Chem. 2010;285:20097–20108. doi: 10.1074/jbc.M109.090092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewy DS, Gauss CM, Soenen DR, Boger DL. Curr Med Chem. 2002;9:2005–2032. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- 4.Kong R, Liu X, Su C, Ma C, Qiu R, Tang L. Chem Biol. 2013;20:45–54. doi: 10.1016/j.chembiol.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 5.Shah DD, You YO, Cane DE. J Am Chem Soc. 2017;139:14322–14330. doi: 10.1021/jacs.7b08896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palaniappan N, Alhamadsheh MM, Reynolds KA. J Am Chem Soc. 2008;130:12236–12237. doi: 10.1021/ja8044162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vergnolle O, Hahn F, Baerga-Ortiz A, Leadlay PF, Andexer JN. Chembiochem. 2011;12:1011–1014. doi: 10.1002/cbic.201100011. [DOI] [PubMed] [Google Scholar]
- 8.Olano C, Wilkinson B, Sanchez C, Moss SJ, Sheridan R, Math V, Weston AJ, Brana AF, Martin CJ, Oliynyk M, Mendez C, Leadlay PF, Salas JA. Chem Biol. 2004;11:87–97. doi: 10.1016/j.chembiol.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 9.Moldenhauer J, Chen XH, Borriss R, Piel J. Angew Chem Int Ed Engl. 2007;46:8195–8197. doi: 10.1002/anie.200703386. [DOI] [PubMed] [Google Scholar]
- 10.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. Proc Natl Acad Sci U S A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Structure. 2010;18:94–105. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiers WD, Dodge GJ, Sherman DH, Smith JL, Aldrich CC. J Am Chem Soc. 2016;138:16024–16036. doi: 10.1021/jacs.6b09748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen XH, Vater J, Piel J, Franke P, Scholz R, Schneider K, Koumoutsi A, Hitzeroth G, Grammel N, Strittmatter AW, Gottschalk G, Sussmuth RD, Borriss R. J Bacteriol. 2006;188:4024–4036. doi: 10.1128/JB.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng YQ, Coughlin JM, Lim SK, Shen B. Chapter 8 Type I Polyketide Synthases That Require Discrete Acyltransferases. In Complex Enzymes in Microbial Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and Carbohydrates. 2009:165–186. [Google Scholar]
- 15.Valenzano CR, You YO, Garg A, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2010;132:14697–14699. doi: 10.1021/ja107344h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo X, Liu T, Valenzano CR, Deng Z, Cane DE. J Am Chem Soc. 2010;132:14694–14696. doi: 10.1021/ja1073432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gay D, You YO, Keatinge-Clay A, Cane DE. Biochemistry. 2013;52:8916–8928. doi: 10.1021/bi400988t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393–17404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Dodge GJ, Fiers WD, Fecik RA, Smith JL, Aldrich CC. J Am Chem Soc. 2015;137:7003–7006. doi: 10.1021/jacs.5b02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kandziora N, Andexer JN, Moss SJ, Wilkinson B, Leadlay PF, Hahn F. Chem Sci. 2014;5:3563–3567. [Google Scholar]
- 21.Kallberg Y, Oppermann U, Jornvall H, Persson B. Eur J Biochem. 2002;269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- 22.Kallberg Y, Oppermann U, Jornvall H, Persson B. Protein Sci. 2002;11:636–641. doi: 10.1110/ps.26902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keatinge-Clay AT. Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Zheng J, Keatinge-Clay AT. Med Chem Commun. 2013;4:34–40. [Google Scholar]
- 25.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 26.Caffrey P. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 27.Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Chem Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 28.Castonguay R, He W, Chen AY, Khosla C, Cane DE. J Am Chem Soc. 2007;129:13758–13769. doi: 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie X, Garg A, Khosla C, Cane DE. J Am Chem Soc. 2017;139:3283–3292. doi: 10.1021/jacs.7b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu H, Tsai SC, Khosla C, Cane DE. Biochemistry. 2002;41:12590–12597. doi: 10.1021/bi026006d. [DOI] [PubMed] [Google Scholar]
- 31.Xie X, Garg A, Khosla C, Cane DE. J Am Chem Soc. 2017;139:9507–9510. doi: 10.1021/jacs.7b05502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keatinge-Clay A. J Mol Biol. 2008;384:941–953. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwab JM, Habib A, Klassen JB. J Am Chem Soc. 1986;108:5304–5308. [Google Scholar]
- 34.Schwab JM, Henderson BS. Chem Rev. 1990;90:1203–1245. [Google Scholar]
- 35.Sedgwick B, Morris C, French SJ. J C S Chem Commun. 1978:193–194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.