

Abstract
A large number of mutations in genes that encode RNA binding proteins cause human disease. Many of these RNA binding proteins mediate key steps in post-transcriptional regulation of gene expression from mRNA processing to eventual decay in the cytoplasm. Surprisingly, these RNA binding proteins, which are ubiquitously expressed and play fundamental roles in gene expression, are often altered in tissue-specific disease. Mutations linked to disease impact nearly every post-transcriptional processing step and cause diverse disease phenotypes in a variety of specific tissues. This review summarizes steps in post-transcriptional regulation of gene expression that have been linked to disease providing specific examples of some of the many genes affected. Finally, recent advances that hold promise for treatment of some of these diseases are presented.
Keywords: RNA binding protein, tissue-specific disease, post-transcriptional regulation of gene expression, mRNA
Introduction
While all cells in the human body contain the same DNA blueprint, the differential expression of those genes confers distinct functional properties to each cell type. While much of this differential gene expression is achieved at the level of gene-specific transcription, there are numerous post-transcriptional events that also contribute to the cell-specific expression patterns, which dictate function. Much of this post-transcriptional regulation of gene expression occurs through the action of RNA binding proteins and processing factors that associate with RNAs from the initiation of transcription to the eventual death of the RNA in the cytoplasm [1]. Here we focus on RNA binding proteins/processing factors that mediate post-transcriptional regulation of gene expression and thus primarily interact with mRNA.
Given that the various steps in the production and eventual translation of mRNAs are critical in all cells types, the identification of a large number of mutations encoding these proteins that confer tissue-specific disease phenotypes and pathology is surprising [2]. In fact, analysis of gene expression data suggests that only 6% of RNA binding proteins are expressed in a tissue-specific manner [3] meaning that the vast majority of RNA binding proteins are ubiquitously expressed. As the majority of RNA binding proteins are not expressed in a tissue-specific manner, tissue-specific disease phenotypes that arise when RNA binding protein function is altered must reflect some critical function of that particular protein or process within that tissue [2]. Alternatively, there could be properties of that tissue that make it susceptible to altered RNA binding protein function.
Post-transcriptional steps in gene expression
Following transcription within the nucleus, a series of conserved processing steps is required to produce mature mRNA competent for translation in the cytoplasm (Figure 1). These nuclear processing steps include 5′-end capping to generate a 7-methylguanosine cap, splicing out of introns, and 3′-end cleavage/polyadenylation to create a mature, polyadenylated mRNA. Nuclear processing events are mediated by a myriad of RNA binding proteins that associate with the nascent mRNA as soon as the 5′-end emerges from RNA polymerase II [4]. These RNA binding proteins contribute to packaging the RNA into an mRNP complex competent for export to the cytoplasm [4]. Export is achieved through the action of specific mRNA export receptors that mediate interactions with the nuclear pore complex [5]. Remodeling of the mRNP complex at the cytoplasmic face of the nuclear pore provides directionality to this movement through the pore [6,7]. Once in the cytoplasm, the mRNA can be translated to protein, stored in cytoplasmic bodies for future translation, localized to specific regions of the cell, or targeted for decay [8–11]. All these steps are fundamental to achieve gene expression and support diverse cellular functions.
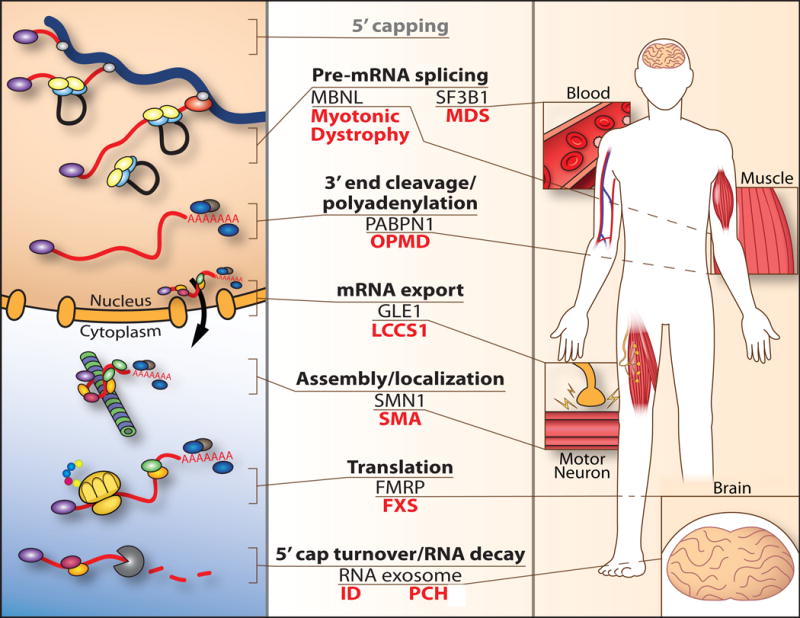
Figure 1.
Mutations in genes encoding components of post-transcriptional processing cause tissue-specific human disease. A schematic (left) illustrating steps (center) in post-transcriptional regulation of gene expression is shown highlighting the post-transcriptional regulatory factors that are altered in disease and described in this review (center). The right side of the schematic shows the diverse tissues that are affected when genes encoding these factors are mutated in disease. Left, processing of the mRNA (red) begins co-transcriptionally with the addition of an mRNA cap (5′ capping), splicing to remove introns (Pre-mRNA splicing), and 3′ end cleavage/polyadenylation. The simplified schematic shows a small number of RNA binding proteins that associate with the mRNA to form an export-competent mRNA ribonucleoprotein (mRNP) complex that is exported through the nuclear pore to the cytoplasm (mRNA export). In the cytoplasm, the mRNP can undergo remodeling but the RNA binding proteins assembled into the mRNA within the nucleus can also dictate the cytoplasmic fate of mRNA. The mRNA can undergo specific localization (Assembly/localization), can be translated (Translation), or can undergo regulated decay (5′ cap turnover/RNA decay). Here, a small subset of diseases that result from altered function of post-transcriptional regulators of gene expression is illustrated: Muscleblind (MBNL) is one alternative splicing factor sequestered in Myotonic Dystrophy type 1; SF3B1 is one of several splicing factors that is mutated in myelodysplastic syndrome (MDS), a cancer syndrome of the blood; the nuclear poly(A) RNA binding protein PABPN1 is altered in oculopharyngeal muscular dystrophy (OPMD); the mRNA export factor, GLE1 is altered in lethal congenital contracture syndrome 1 (LCCS1) causing loss of motor neurons; the mRNA chaperone protein SMN1 is lost in spinal muscular atrophy (SMA); a key regulator of translation, the fragile X mental retardation protein (FMRP) is lost in fragile X syndrome (FXS); and components of mRNA decay and processing including: the DCPS protein, which mediates turnover of the 5′ cap linked to an inherited form of intellectual disability (ID); and the RNA exosome, which mediates both RNA decay and precise RNA processing, has been linked to multiple diseases including pontocerebellar hypoplasia (PCH). See the text for details and references.
RNA binding proteins/processing factors linked to human disease
5′-end capping
The machinery required to produce the 7-methylguanosine cap and append this cap to the 5′-end of a nascent mRNA transcript is essential and highly conserved through evolution [12]. Thus far, mutations in genes that encode components of this machinery have not been linked to disease. However, mutation of the DCPS gene, which encodes a scavenger decapping enzyme that plays a critical role in turnover of the free m7GpppN cap produced when mRNAs are degraded in the cytoplasm [13] have been reported [14].
The mutation identified in the DCPS gene, which is described in a single family (OMIM 610534), alters the first splice donor site in exon 1 of the DCPS gene resulting in the absence of the major DCPS isoform [14]. The affected individuals are homozygous for this mutation and show no detectable DCPS enzymatic activity [14]. Clinical symptoms include congenital muscle hypotonia, craniofacial abnormalities, and developmental delays. Thus, this autosomal recessive disorder may represent a case where there is rather broad tissue involvement typical of what might be expected for the loss of critical post-transcriptional regulatory factors.
As DCPS functions at the step of mRNA turnover [15], the nuclear production of the 5′-end of a mature mRNA is one-step in post-transcriptional regulation of gene expression for which no mutations that directly impair critical steps in the pathway have been defined. Perhaps any perturbation of this initial step in the production of mRNA is incompatible with life or, alternatively, mutations that directly affect this process have simply not yet been identified.
Pre-mRNA splicing
The removal of introns to produce mature mRNA transcripts is mediated by a large macromolecular complex termed the spliceosome in cooperation with a number of additional factors required for alternative splicing and regulation [16]. The spliceosome coordinates a series of steps that are mediated by small nuclear ribonucleoprotein complexes termed the U1, U2, U4, U5, and U6 snRNPs [17]. Mutations in a number of genes that encode splicing machinery have been linked to human disease [2]. Some of these mutations occur in components of the primary splicing machinery while others impact alternative splicing factors.
Recently, mutations in the SF3B1 gene, which encodes subunit 1 of the splicing factor 3b protein complex [18,19], have been linked to a cancer syndrome termed myelodysplastic syndrome [20](OMIM 605590) as well as some other solid tumors [21,22]. Splicing factor 3b, together with splicing factor 3a and a 12S RNA unit, form the U2 snRNP [23]. Splicing factor 3b is also a component of the minor U12-type spliceosome [24]. In addition to mutations in SF3B1, mutations in other splicing factor genes including SRSF2, U2AF1 and ZRSR2 have also been identified in myelodysplastic syndrome [25] and some other cancers [22].
Myelodysplastic syndromes (OMIM # 614286) are a group of cancers in which immature blood cells in the bone marrow do not mature into healthy blood cells [26]. Studies have been performed to identify target RNAs that are aberrantly spliced that could explain the pathophysiology of these syndromes [22]. Recently resolved structures of the spliceosome complex bound to target RNAs [27,28] allow speculation that the mutational hot spots associated with disease could be in regions critical for interactions with target RNA [29]. An emerging model for disease suggests that some of the altered splicing factors could favor use of alternative splice sites generating aberrant transcripts that contribute to pathology [22]. Why these changes would preferentially impact a subset of cell types and tissues is not yet clear.
In addition to mutations within core splicing components, there are additional disease mechanisms that can contribute to alterations in splicing or alternative splicing. One such example is the case of myotonic dystrophy [30]. While myotonic dystrophy is a multi-system disease, the primary symptom is muscle atrophy with weakness of skeletal and respiratory muscles [31]. Myotonic dystrophy type 1 (DM1; OMIM #160900) arises from an expansion of CTG repeats in the 3′UTR of the dystrophia myotonica protein kinase (DMPK) gene (Figure 2). The disease phenotypes arises not from an effect of these repeats on the function of DMPK but rather because the repeats encode a toxic RNA that forms a hairpin structure [31], which sequesters key splicing factors [32]. Among these splicing factors, is the muscleblind (MBNL) family of proteins [33]. Studies have defined a number of genes that are critical for proper muscle function that show aberrant splicing when muscleblind function is impaired including the skeletal muscle chloride channel CIC-1 [34], which could provide some insight into why muscle is preferentially affected in this disease.
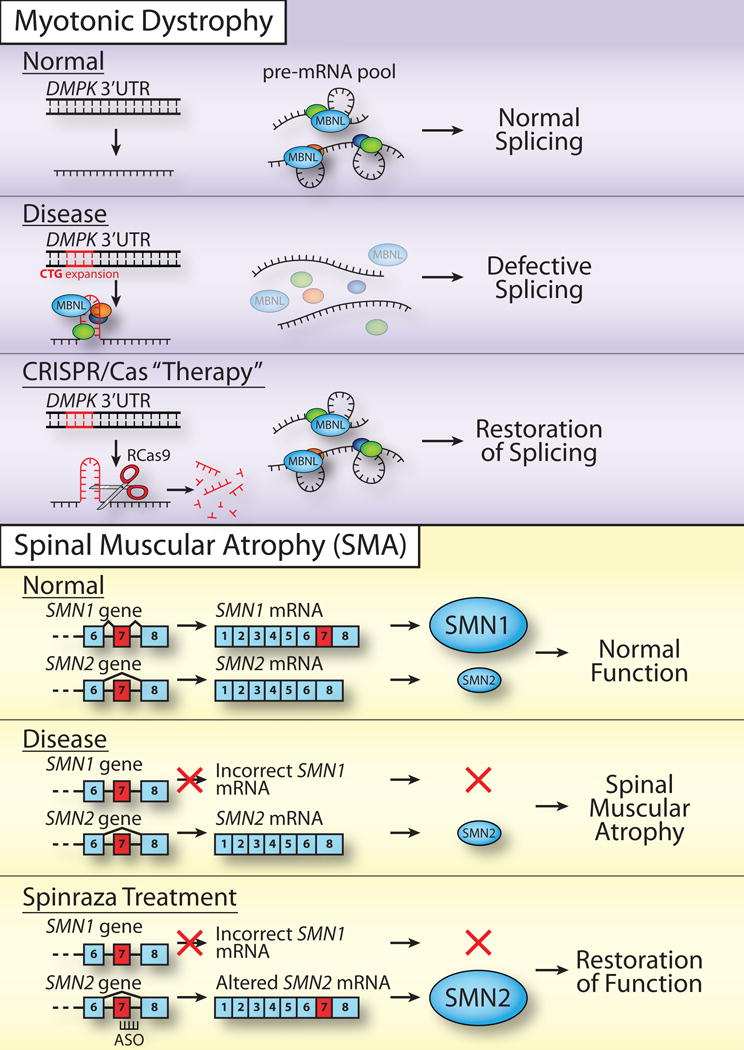
Figure 2.
Targeting post-transcriptional processing for therapy. Myotonic Dystrophy (top). As described in the text, several approaches have been employed to target the triplet expansion that encodes a toxic mRNA species in myotonic dystrophy. In this Disease, the Normal 3′UTR of the DMPK gene undergoes a CTG triplet expansion that produces a toxic RNA, which sequesters multiple alternative splicing factors including muscleblind (MBNL) causing Defective Splicing. In one of several recent approaches to restore function, Batra et al. [40] adapted Cas9 for cleavage of RNA to produce an RCas9 that could directly target the toxic RNA species and liberate the sequestered alternative splicing factors to allow Restoration of Splicing. This proof of principle result, together with others that involve genome editing described in more detail in the text, provides potential for future therapeutic approaches. However, muscle cells, which are very large and multi-nucleated, may represent a particularly challenging target tissue for delivery and implementation. Spinal Muscular Atrophy (bottom). Many years of important fundamental studies to define the function of alternative splicing factors together with more recent translational work have culminated in the development of and FDA approval of nusinersen/Spinraza® to treat SMA. As described in the text, the human genome encodes two nearly identical SMN genes SMN1 and SMN2. In Normal individuals, the SMN1 gene is highly expressed providing the majority of the SMN protein, which is required to support proper motor neuron function. In the SMA Disease state, a mutation in the SMN1 gene leads to loss of expression of SMN1 and individuals who live with this disease depend on the pool of SMN2 protein, which is not sufficient to support proper motor neuron function (Spinal Muscular Atrophy). The nusinersen/Spinraza® treatment relies on the fact that the low levels of SMN2 result from an alternative splicing event that normally causes exclusion of exon 7 in SMN2 that underlies the low steady-state amount of SMN2 protein produced. An antisense oligonucleotide (ASO) was developed and weaponized [71] to block this alternative splicing event and increase the steady-state level of SMN2 protein. The increase in SMN2 protein allows a Restoration of Function and thus hope for individuals who live with SMA.
As the toxic species in DM1 is the RNA produced, researchers have been creative in developing potential therapies to treat this disease [35]. There have been efforts to identify small molecule therapeutics to target the toxic RNA using a variety of approaches [36] including disrupting the RNA hairpin that forms and decreasing expression of the DMPK gene. Given that the expansion of the CTG repeats in DMPK occur in a region of the genome that does not appear to have a critical function, a number of studies have deployed CRISPR/Cas9 editing to eliminate the encoded toxic RNA [37–39] or even adapted Cas9 (RCas9) to directly target and eliminate the toxic RNA [40]. As illustrated in Figure 2 (Myotonic Dystrophy), the approach of targeting the toxic RNA directly has the advantage of not altering the genome thus eliminating some concerns about off target effects. Importantly, this study demonstrated that a variety of triplet repeat encoded RNAs could be targeted by RCas9 [40] opening the door to a general treatment strategy. With respect to treatment of DM1, how these methods, thus far validated in isolated patient cells as well as primary cells and cell lines, could be extended into the large, multinucleated muscle cells that comprise the effected tissue in DM1 patients remains to be determined. At this juncture, AAV-mediated delivery appears to be the most feasible option but challenges remain particularly for delivery into multinucleated muscle cells.
3′-end cleavage and polyadenylation
Generation of a mature eukaryotic mRNA includes a 3′-end cleavage event to generate the end of the RNA followed by coupled polyadenylation to produce a poly(A) tail that is critical for export, translation, and stability [41]. A number of mutations that cause disease have been mapped to genes that encode critical components of 3′-end cleavage/polyadenylation [2,42,43]. As with other post-transcriptional processing factors, these diseases often present with tissue-specific pathology.
Mutations in the PABPN1 gene, which encodes a ubiquitously expressed, nuclear poly(A) RNA binding protein [42], cause a form of muscular dystrophy termed oculopharyngeal muscular dystrophy (OPMD) (OMIM # 164300) [44]. OPMD is a late onset disease where symptoms most commonly manifest in the 5th or 6th decade of life [45]. A subset of skeletal muscles including the pharyngeal and eyelid muscles are most impacted with weakness also noted in proximal limb muscles [46]. Thus, not only does the disease predominantly affect a single tissue type, muscle, a subset of muscles is affected.
The mutation in the PABPN1 gene that causes OPMD consists of a very modest expansion of a GCG triplet repeat in the first exon of the gene [44]. The most common disease-causing mutation is a 9-base expansion that increases an existing ten-alanine tract to 13 alanines meaning that the addition of a mere three additional alanines to an existing stretch of ten alanines causes disease [42]. Furthermore, OPMD is typically inherited in an autosomal dominant pattern [45] so this expansion occurs within a single PABPN1 allele causing pathology in a dominant manner.
Several mechanisms by which alanine-expanded PABPN1 could confer pathology in skeletal muscle have been proposed [42,47,48]. As a hallmark of OPMD is the presence of nuclear aggregates of the alanine-expanded PABPN1, most models have suggested that toxic aggregates underlie pathology [47]. However, recent studies that show that PABPN1 levels are very low in skeletal muscle as compared to other unaffected tissues [49] would seem counter to this model. If toxic aggregates were the sole cause of disease, then tissues with higher levels of PABPN1 expression would seem to be most vulnerable to disease. Furthermore, there is a report that PABPN1 levels decline with age [50]. As OPMD is a late onset disease [45], this observation would be consistent with a model where low levels of PABPN1 confer disease susceptibility. Potentially, a complex mechanism where aggregation of PABPN1 leads to a functional depletion of PABPN1 could explain the tissue-specific nature of the disease. In fact, muscles most impacted in OPMD have even lower levels of PABPN1 than muscles that are not impacted in disease [49]. These studies emphasize the importance of studies in the tissue impacted in disease to develop models [51] that could explain the tissue-specific nature of disease.
Export of mRNA to the cytoplasm
Once mRNA processing is completed in the nucleus, the mature mRNP complex is targeted to the nuclear pore for export. While a number of conditions have been linked to mutations in the genes encoding the nuclear pore proteins that comprise the nuclear pore [52], there are less clear cases of genetic disease links specifically to the mRNA export machinery [53]. However, mutations in human GLE1 cause a very severe condition termed lethal congenital contracture syndrome 1 (LCCS1) (OMIM # 253310) that results in death of the fetus [54]. This condition causes lack of anterior horn motoneurons as well as severe atrophy of the ventral spinal cord [55,56]. The disease is inherited in an autosomal recessive manner and was identified in a Finnish population where the most common mutation is an A->G base change located in the 3rd intron of GLE1, which creates an aberrant splice acceptor site introducing an insertion of three amino acids (PFQ) within a coiled-coil region of the protein [54].
The GLE1 gene encodes an evolutionarily conserved mRNA export factor that was originally identified and characterized in budding yeast [57]. Studies in budding yeast have identified a key role for Gle1 in mediating a critical remodeling step at the cytoplasmic face of the nuclear pore to deliver mRNA to the cytoplasm [58]. Subsequent studies exploiting a zebrafish model to assess the tissues impacted by mutations in GLE1 suggest that the defects in mRNA export have broad affects across organs that manifest as the loss of motor neurons [59]. Mutations in GLE1 are highly pleotropic with effects observed in multiple tissues and organs [56]. Thus, mRNA export may be one of the steps in post-transcriptional regulation of gene expression that is most sensitive to alterations.
mRNP assembly/mRNA localization
Once mRNA is exported from the nucleus to the cytoplasm, the mRNA can be translated, stored, targeted for decay, or localized to specific subcellular regions [9]. Which of these fates awaits an exported mRNA is dictated by the complement of associated RNA binding proteins starting from the moment of transcription in the nucleus until the mRNA reaches the cytoplasm. The composition of the mRNP complex is dynamic over the life of the mRNA facilitating distinct steps in the gene expression pathway [4]. Among the most critical steps to achieve proper cellular function is the localization of a subset of mRNAs. In particular, neurons require very precise spatial and temporal gene expression to support neuronal activity [60–62].
Among the proteins critical for the proper assembly and hence fate of mRNPs is the Survival of Motor Neuron (SMN) protein [63,64], which in humans is encoded by both SMN1 and SMN2 [65,66]. Mutations that lead to loss of function of the SMN1 gene cause Spinal Muscular Atrophy (SMA) (OMIM 253300) [66], which is inherited in an autosomal recessive manner. SMA is a neuromuscular disease where patients lose the function of motor neurons and suffer from muscle wasting that can result in early death. A single C->T transition within exon 7 of SMN2 compared to SMN1 leads to exclusion of exon 7 in the resulting SMN2 transcript, which results in a much lower steady-state level of SMN2 protein relative to SMN1 [67]. In normal individuals, the SMN1 gene provides sufficient function; however, in SMA, where SMN1 is lost, the level of functional SMN2 is not sufficient to support the survival of motor neurons [68].
The SMN protein localizes to sub-nuclear bodies called gems, which are proximal to coiled bodies that contain high concentrations of small ribonucleoproteins (snRNPs) [69]. Based on interactions with the gemin proteins that comprise gems as well as interactions with snRNPs, the SMN proteins were first implicated in assembly of spliceosome components [64]. However, why the defects in splicing that would seem likely to result from defective assembly of the spliceosome would cause pathology primarily in motor neurons was unclear [70]. Recent studies have uncovered more general roles for SMN in both mRNP assembly and mRNP localization that could explain the loss of motor neurons [63]. As the cells that may depend most on proper transport of mRNA transcripts to distal locations, this cell type could be most susceptible to loss of SMN function. While dissecting the possible contributions of defects in splicing and mRNP assembly/localization to SMA remains an area of intense focus [70], there have been major advances in using antisense oligonucleotide-based technology [71,72] to modulate splicing of the SMN2 gene in SMA patients (Figure 2) and thus raise the level of functional SMN protein. This treatment, called nusinersen or Spinraza®, relies on many years of important fundamental studies to define pathways for splicing and alternative splicing that have now been harnessed to improve the quality of life for those individuals living with SMA.
Translation
Once mRNAs are transported to the cytoplasm and properly localized, they interface with ribosomes to direct protein synthesis.
While the process of translation is essential for all tissues, there are some cell types where spatial and temporal control of translation is particularly critical [62,73]. This control can be achieved by specific localization of RNAs and/or through regulated, local translation.
The Fragile X Mental Retardation Protein (FMRP), which is encoded by the FMR1 gene [74], is an RNA binding protein that regulates translation through multiple mechanisms [75]. Although FMRP is ubiquitously expressed [76], loss of expression of FMR1 causes Fragile X Syndrome (FXS) (OMIM # 300624), which is the most common form of inherited intellectual disability as well as the most prevalent single-gene cause of autism spectrum disorder (ASD). The most common mechanism leading to loss of FMRP expression is expansion of a CGG repeat in the promoter of FMR1, which causes DNA methylation and silencing [77]. FMRP typically functions as a translational repressor [78]; however, studies suggest that pathophysiology in FXS results from not from the modest increase in steady-state levels of FMRP-regulated targets but rather from an inability of neurons to achieve regulated local translation particularly in response to stimuli [75]. This need for exquisite regulation of local translation for proper neuronal function likely explains why loss of FMRP causes neurological disease.
There is evidence to support several modes by which FMRP can regulate translation. The most heavily regulated step in translation is initiation where the mRNA cap is recognized by translation initiation factors that recruit the 40S ribosomal subunit [73]. Indeed, FMRP interacts with mRNA cap binding protein and other factors to regulate translation initiation [75]. However, recent studies mapped FMRP binding within gene bodies and identified interactions with translating ribosomes [79], consistent with studies that identified FMRP in polysome fractions [75]. These data support a model where FMRP functions as a negative regulator of elongation. This model was recently tested in elegant experiments that show that mouse forebrain lysates lacking FMRP show a 40–50% increase in the rate of elongation compared to control lysate [80]. To further complicate matters, FMRP can also regulate translation through interaction with miRNAs [75].
A major challenge for developing treatments for FXS is the identification of the mis-regulated FMRP target RNAs that contribute to pathology [75]. A number of pathways have been identified that show altered function in FXS models [81]. However, various attempts to define the spectrum of FMRP-regulated RNAs have not yet yielded a consensus set of target transcripts or particular insight into what defines an FMRP target RNA. An in vitro RNA selection approach identified G-quartet-containing RNAs as potential FMRP targets [75], but attempts to validate these findings via UV-crosslinking approaches have not been successful. Thus, many questions persist about how FMRP recognizes and regulates the translation of key target mRNAs.
mRNA decay/processing
At the end of their useful lifespan, mRNAs are turned over through specific decay pathways [11]. These decay pathways can also play critical regulatory roles. Indeed, some of the RNA decay machinery also contributes to essential steps in precise RNA processing. Cells have a variety of evolutionarily conserved RNA decay pathways and mechanisms [11]. One of the RNA decay machines that contributes to much of cellular RNA decay is the RNA exosome complex. The RNA exosome is a multi-subunit 3′-5′ exonuclease that both processes and destroys numerous classes of RNAs including mRNAs [82].
The RNA exosome complex consists of ten highly conserved subunits that were originally identified and have been studied most extensively in budding yeast [82]. The subunits are arranged into a three subunit cap (EXOSC1, EXOSC2, EXOSC3), a six subunit central core (EXOSC4, EXOSC5, EXOSC6, EXOSC7, EXOSC8, EXOSC9), and an active 3′-5′ exonuclease (DIS3) located at the base of the complex [82]. The corresponding genes in budding yeast, which are termed RRP genes based on their original identification in screens for ribosomal RNA processing mutants [83], are all essential for cell viability [84]. Given that these genes are all essential in yeast, it was surprising when the cause of an autosomal recessive, neurodevelopmental disorder, Pontocerebellar Hypoplasia Type 1b (PCH1b) (OMIM # 614678) was mapped to the EXOSC3 gene [85]. In fact, disease mutations have now been mapped to genes encoding four of the nine structural exosome subunits (EXOSC2, 3, 8, and 9) [84].
While the structure and function of the RNA exosome have been defined in elegant studies of this large complex [82,86], there is little information about any tissue-specific functions of this machine, which plays critical roles in both RNA decay and RNA processing. Thus, why mutations in several RNA exosome subunit genes cause pathology in specific regions of the brain is not at all clear. While the number of patients with mutations in EXOSC genes remain relatively small [84], EXOSC3, EXOSC8 (OMIM # 616081), and EXOSC9 mutations all cause cerebellar atrophy [85,87,88] suggesting that the cerebellum could be particularly vulnerable to altered RNA exosome function. Surprisingly, mutations in the EXOSC2 gene cause a distinct syndrome (OMIM # 617763) with pleiotropic effects in a variety of tissues where pathology includes retinitis pigmentosa, progressive hearing loss, premature ageing and mild intellectual disability [89]. How changes in specific subunits of the RNA exosome could lead to distinct consequences is not at all clear. Notably, the RNA exosome gene mutations that cause disease do not cause complete loss of function but rather single amino acid substitutions [84]. A combined approach of defining how these disease-causing amino acid substitutions alter the function of the RNA exosome and exploring requirements for RNA exosome function in specific tissues will be required to define the mechanisms that underlie pathophysiology in RNA exosome-linked disease.
Conclusions
The specific examples described here represent a small fraction of the RNA binding proteins that mediate critical steps in post-transcriptional regulation of gene expression, which are implicated in human disease [3]. Defining the complex functions of such proteins is challenging because many play multiple roles within the post-transcriptional processing pathway. To understand the nature of tissue-specific pathology, studies are required both to define basic mechanisms and to understand the requirements to support normal function in different tissues and cell types. As with the development of nusinersen/Spinraza® to treat SMA [71,72], there are exciting examples where years of fundamental research have now culminated in treatment regimens that are transforming the lives of patients.
Acknowledgments
Members of the Corbett laboratory and colleagues in the field are acknowledged for helpful discussions and comments. Ms. Stephanie Jones provided the artwork presented in the figures. Funding: This work was supported by the National Institutes of Health [grant numbers R21AG054206, R01AR061987, R01GM058728, R01MH107305] and the Muscular Dystrophy Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author declares that there are no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
*of special interest
** of outstanding interest
- 1.Dassi E. Handshakes and Fights: The Regulatory Interplay of RNA-Binding Proteins. Front Mol Biosci. 2017;4:67. doi: 10.3389/fmolb.2017.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carey KT, Wickramasinghe VO. Regulatory Potential of the RNA Processing Machinery: Implications for Human Disease. Trends Genet. 2018 doi: 10.1016/j.tig.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 3**.Gerstberger S, Hafner M, Ascano M, Tuschl T. Evolutionary conservation and expression of human RNA-binding proteins and their role in human genetic disease. Adv Exp Med Biol. 2014;825:1–55. doi: 10.1007/978-1-4939-1221-6_1. Remarkable cataloguing of RNA binding proteins using various data-mining and analysis approaches. A very complete table of those RNA binding proteins that have been linked to human disease is presented. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh G, Pratt G, Yeo GW, Moore MJ. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu Rev Biochem. 2015;84:325–354. doi: 10.1146/annurev-biochem-080111-092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Natalizio BJ, Wente SR. Postage for the messenger: designating routes for nuclear mRNA export. Trends Cell Biol. 2013;23:365–373. doi: 10.1016/j.tcb.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oeffinger M, Zenklusen D. To the pore and through the pore: a story of mRNA export kinetics. Biochim Biophys Acta. 2012;1819:494–506. doi: 10.1016/j.bbagrm.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stewart M. Ratcheting mRNA out of the nucleus. Mol Cell. 2007;25:327–330. doi: 10.1016/j.molcel.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Buxbaum AR, Haimovich G, Singer RH. In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol. 2015;16:95–109. doi: 10.1038/nrm3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing L, Bassell GJ. mRNA localization: an orchestration of assembly, traffic and synthesis. Traffic. 2013;14:2–14. doi: 10.1111/tra.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 12.Ramanathan A, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44:7511–7526. doi: 10.1093/nar/gkw551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Ng CK, Shboul M, Taverniti V, Bonnard C, Lee H, Eskin A, Nelson SF, Al-Raqad M, Altawalbeh S, Seraphin B, et al. Loss of the scavenger mRNA decapping enzyme DCPS causes syndromic intellectual disability with neuromuscular defects. Hum Mol Genet. 2015;24:3163–3171. doi: 10.1093/hmg/ddv067. Identified a family with mutations in the DCPS gene with intellectual disability providing the first example of a genetic disease associated with decay/recycling of the mRNA cap. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grudzien-Nogalska E, Kiledjian M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip Rev RNA. 2017;8 doi: 10.1002/wrna.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat Rev Mol Cell Biol. 2017;18:655–670. doi: 10.1038/nrm.2017.86. [DOI] [PubMed] [Google Scholar]
- 17.Papasaikas P, Valcarcel J. The Spliceosome: The Ultimate RNA Chaperone and Sculptor. Trends Biochem Sci. 2016;41:33–45. doi: 10.1016/j.tibs.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Chua K, Seghezzi W, Lees E, Gozani O, Reed R. Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev. 1998;12:1409–1414. doi: 10.1101/gad.12.10.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer A, Mulhauser F, Wersig C, Groning K, Bilbe G. Mammalian splicing factor SF3a120 represents a new member of the SURP family of proteins and is homologous to the essential splicing factor PRP21p of Saccharomyces cerevisiae. RNA. 1995;1:260–272. [PMC free article] [PubMed] [Google Scholar]
- 20.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bejar R. Splicing Factor Mutations in Cancer. Adv Exp Med Biol. 2016;907:215–228. doi: 10.1007/978-3-319-29073-7_9. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong RN, Steeples V, Singh S, Sanchi A, Boultwood J, Pellagatti A. Splicing factor mutations in the myelodysplastic syndromes: target genes and therapeutic approaches. Adv Biol Regul. 2017 doi: 10.1016/j.jbior.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem. 2015;84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verma B, Akinyi MV, Norppa AJ, Frilander MJ. Minor spliceosome and disease. Semin Cell Dev Biol. 2017 doi: 10.1016/j.semcdb.2017.09.036. [DOI] [PubMed] [Google Scholar]
- 25.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Germing U, Kobbe G, Haas R, Gattermann N. Myelodysplastic syndromes: diagnosis, prognosis, and treatment. Dtsch Arztebl Int. 2013;110:783–790. doi: 10.3238/arztebl.2013.0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Zhan X, Yan C, Zhang X, Lei J, Shi Y. Structure of a human catalytic step I spliceosome. Science. 2018 doi: 10.1126/science.aar6401. Shows cryo-EM structures of the first catalytic step in splicing for the human spliceosome providing insight into the mechanism by which the human splicesome mediates the initial steps in mRNA splicing. Builds on recent advances in the structural analysis of the spliceosome. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y. The Spliceosome: A Protein-Directed Metalloribozyme. J Mol Biol. 2017;429:2640–2653. doi: 10.1016/j.jmb.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins JL, Kielkopf CL. Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet. 2017;33:336–348. doi: 10.1016/j.tig.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev. 2017;44:30–37. doi: 10.1016/j.gde.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 32.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 33.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 34.Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev. 2017;44:135–140. doi: 10.1016/j.gde.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konieczny P, Selma-Soriano E, Rapisarda AS, Fernandez-Costa JM, Perez-Alonso M, Artero R. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov Today. 2017;22:1740–1748. doi: 10.1016/j.drudis.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 37*.Provenzano C, Cappella M, Valaperta R, Cardani R, Meola G, Martelli F, Cardinali B, Falcone G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol Ther Nucleic Acids. 2017;9:337–348. doi: 10.1016/j.omtn.2017.10.006. Shows that CRISPR/Cas9 genome editing can be employed to remove a triplet repeat expansion in the DMPK locus, which is altered in Myotonic Dystrophy 1 patient to restore function. This provides proof of principle for this approach as a potential therapeutic application for triplet repeats diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Agtmaal EL, Andre LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek W, Gourdon G, Furling D, Mouly V, Monckton DG, et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol Ther. 2017;25:24–43. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Pinto BS, Saxena T, Oliveira R, Mendez-Gomez HR, Cleary JD, Denes LT, McConnell O, Arboleda J, Xia G, Swanson MS, et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol Cell. 2017;68:479–490 e475. doi: 10.1016/j.molcel.2017.09.033. Uses inactivated Cas9 to block transcription of the locus that encodes the toxic mRNA in Myotonic Dystrophy providing proof of principle for using such an approach for future therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell. 2017;170:899–912 e810. doi: 10.1016/j.cell.2017.07.010. Develops an RNA-directed version of Cas9 (Rcas9) and uses RCas9 to directly target the toxic mRNA that is produced in Myotonic Dystrophy. This approach provides a potential application of modified CRISPR/Cas9 that avoids the need for genome editing and could be broadly applied to disease caused by toxic RNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neve J, Patel R, Wang Z, Louey A, Furger AM. Cleavage and polyadenylation: Ending the message expands gene regulation. RNA Biol. 2017;14:865–890. doi: 10.1080/15476286.2017.1306171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banerjee A, Apponi LH, Pavlath GK, Corbett AH. PABPN1: molecular function and muscle disease. FEBS J. 2013 doi: 10.1111/febs.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curinha A, Oliveira Braz S, Pereira-Castro I, Cruz A, Moreira A. Implications of polyadenylation in health and disease. Nucleus. 2014;5:508–519. doi: 10.4161/nucl.36360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 45.Brais B. Oculopharyngeal muscular dystrophy: a polyalanine myopathy. Curr Neurol Neurosci Rep. 2009;9:76–82. doi: 10.1007/s11910-009-0012-y. [DOI] [PubMed] [Google Scholar]
- 46.Luigetti M, Lo Monaco M, Mirabella M, Primiano G, Lucchini M, Monforte M, Servidei S. Oculopharyngeal muscular dystrophy: Clinical and neurophysiological features. Clin Neurophysiol. 2015;126:2406–2408. doi: 10.1016/j.clinph.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 47.van der Sluijs BM, Raz V, Lammens M, van den Heuvel LP, Voermans NC, van Engelen BG. Intranuclear Aggregates Precede Clinical Onset in Oculopharyngeal Muscular Dystrophy. J Neuromuscul Dis. 2016;3:101–109. doi: 10.3233/JND-150118. [DOI] [PubMed] [Google Scholar]
- 48.Raz Y, Raz V. Oculopharyngeal muscular dystrophy as a paradigm for muscle aging. Front Aging Neurosci. 2014;6:317. doi: 10.3389/fnagi.2014.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apponi LH, Corbett AH, Pavlath GK. Control of mRNA stability contributes to low levels of nuclear poly(A) binding protein 1 (PABPN1) in skeletal muscle. Skelet Muscle. 2013;3:23. doi: 10.1186/2044-5040-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anvar SY, Raz Y, Verway N, van der Sluijs B, Venema A, Goeman JJ, Vissing J, van der Maarel SM, t Hoen PA, van Engelen BG, et al. A decline in PABPN1 induces progressive muscle weakness in Oculopharyngeal muscle dystrophy and in muscle aging. Aging (Albany NY) 2013 doi: 10.18632/aging.100567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vest KE, Phillips BL, Banerjee A, Apponi LH, Dammer EB, Xu W, Zheng D, Yu J, Tian B, Pavlath GK, et al. Novel mouse models of oculopharyngeal muscular dystrophy (OPMD) reveal early onset mitochondrial defects and suggest loss of PABPN1 may contribute to pathology. Hum Mol Genet. 2017;26:3235–3252. doi: 10.1093/hmg/ddx206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakuma S, D’Angelo MA. The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin Cell Dev Biol. 2017;68:72–84. doi: 10.1016/j.semcdb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hautbergue GM. RNA Nuclear Export: From Neurological Disorders to Cancer. Adv Exp Med Biol. 2017;1007:89–109. doi: 10.1007/978-3-319-60733-7_6. [DOI] [PubMed] [Google Scholar]
- 54.Nousiainen HO, Kestila M, Pakkasjarvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vuopala K, Herva R. Lethal congenital contracture syndrome: further delineation and genetic aspects. J Med Genet. 1994;31:521–527. doi: 10.1136/jmg.31.7.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herva R, Leisti J, Kirkinen P, Seppanen U. A lethal autosomal recessive syndrome of multiple congenital contractures. Am J Med Genet. 1985;20:431–439. doi: 10.1002/ajmg.1320200303. [DOI] [PubMed] [Google Scholar]
- 57.Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- 58.Alcazar-Roman AR, Tran EJ, Guo S, Wente SR. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol. 2006;8:711–716. doi: 10.1038/ncb1427. [DOI] [PubMed] [Google Scholar]
- 59.Jao LE, Appel B, Wente SR. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Development. 2012;139:1316–1326. doi: 10.1242/dev.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gumy LF, Katrukha EA, Kapitein LC, Hoogenraad CC. New insights into mRNA trafficking in axons. Dev Neurobiol. 2014;74:233–244. doi: 10.1002/dneu.22121. [DOI] [PubMed] [Google Scholar]
- 61**.Zappulo A, van den Bruck D, Ciolli Mattioli C, Franke V, Imami K, McShane E, Moreno-Estelles M, Calviello L, Filipchyk A, Peguero-Sanchez E, et al. RNA localization is a key determinant of neurite-enriched proteome. Nat Commun. 2017;8:583. doi: 10.1038/s41467-017-00690-6. Differentiates mouse embryonic stem cells into neurons, and analyzes the local transcriptome, proteome, and translated transcriptome comparing cell bodies and neurites, providing insight into mechanisms that control neuronal polarity. The authors conclude that mRNA localization is a major driver to achieve neuronal polarity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Driesche SJ, Martin KC. New Frontiers in RNA Transport and Local Translation in Neurons. Dev Neurobiol. 2018 doi: 10.1002/dneu.22574. [DOI] [PubMed] [Google Scholar]
- 63*.Donlin-Asp PG, Fallini C, Campos J, Chou CC, Merritt ME, Phan HC, Bassell GJ, Rossoll W. The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for mRNP Assembly. Cell Rep. 2017;18:1660–1673. doi: 10.1016/j.celrep.2017.01.059. Expands the role of SMN to a more general mRNP chaperone required to package mRNP for transport providing insight into critical functions of SMN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, Zhang Z, Han K, Wan L, Dreyfuss G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- 65.Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet. 2001;108:255–266. doi: 10.1007/s004390100473. [DOI] [PubMed] [Google Scholar]
- 66.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 67.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowerman M, Becker CG, Yanez-Munoz RJ, Ning K, Wood MJA, Gillingwater TH, Talbot K, Consortium USR Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis Model Mech. 2017;10:943–954. doi: 10.1242/dmm.030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng W, Gubitz AK, Wan L, Battle DJ, Dostie J, Golembe TJ, Dreyfuss G. Gemins modulate the expression and activity of the SMN complex. Hum Mol Genet. 2005;14:1605–1611. doi: 10.1093/hmg/ddi168. [DOI] [PubMed] [Google Scholar]
- 70.Lanfranco M, Vassallo N, Cauchi RJ. Spinal Muscular Atrophy: From Defective Chaperoning of snRNP Assembly to Neuromuscular Dysfunction. Front Mol Biosci. 2017;4:41. doi: 10.3389/fmolb.2017.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71**.Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J, Fey RA, Gaus H, Hua Y, Grundy JS, et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350:46–55. doi: 10.1124/jpet.113.212407. Describes oligonucleotide chemistry that serves as the basis for the development of nusinersen/Spinraza® to treat spinal muscular atrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wood MJA, Talbot K, Bowerman M. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum Mol Genet. 2017;26:R151–R159. doi: 10.1093/hmg/ddx215. [DOI] [PubMed] [Google Scholar]
- 73.Kapur M, Monaghan CE, Ackerman SL. Regulation of mRNA Translation in Neurons-A Matter of Life and Death. Neuron. 2017;96:616–637. doi: 10.1016/j.neuron.2017.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 75.Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.LaFauci G, Adayev T, Kascsak R, Brown WT. Detection and Quantification of the Fragile X Mental Retardation Protein 1 (FMRP) Genes (Basel) 2016:7. doi: 10.3390/genes7120121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 78.Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med. 2013;19:1473–1477. doi: 10.1038/nm.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Castagnola S, Bardoni B, Maurin T. The Search for an Effective Therapy to Treat Fragile X Syndrome: Dream or Reality? Front Synaptic Neurosci. 2017;9:15. doi: 10.3389/fnsyn.2017.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zinder JC, Lima CD. Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 2017;31:88–100. doi: 10.1101/gad.294769.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mitchell P, Petfalski E, Tollervey D. The 3′ end of yeast 5.8S rRNA is generated by an exonuclease processing mechanism. Genes Dev. 1996;10:502–513. doi: 10.1101/gad.10.4.502. [DOI] [PubMed] [Google Scholar]
- 84.Morton DJ, Kuiper EG, Jones SK, Leung SW, Corbett AH, Fasken MB. The RNA Exosome and RNA Exosome-linked Disease. RNA. 2017 doi: 10.1261/rna.064626.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wan J, Yourshaw M, Mamsa H, Rudnik-Schoneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Makino DL, Halbach F, Conti E. The RNA exosome and proteasome: common principles of degradation control. Nat Rev Mol Cell Biol. 2013;14:654–660. doi: 10.1038/nrm3657. [DOI] [PubMed] [Google Scholar]
- 87.Boczonadi V, Muller JS, Pyle A, Munkley J, Dor T, Quartararo J, Ferrero I, Karcagi V, Giunta M, Polvikoski T, et al. EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nat Commun. 2014;5:4287. doi: 10.1038/ncomms5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burns DT, Donkervoort DT, Bharucha-Goebel DX, G M, Munro B, Scavina M, Foley R, Müller JS, Bönnemann CG, Horvath R. A recessive mutation in EXOSC9 causes abnormal RNA metabolism resulting in a novel form of cerebellar hypoplasia/atrophy with early motor neuronopathy. Neuromuscul Disord. 2017;27:S38. [Google Scholar]
- 89.Di Donato N, Neuhann T, Kahlert AK, Klink B, Hackmann K, Neuhann I, Novotna B, Schallner J, Krause C, Glass IA, et al. Mutations in EXOSC2 are associated with a novel syndrome characterised by retinitis pigmentosa, progressive hearing loss, premature ageing, short stature, mild intellectual disability and distinctive gestalt. J Med Genet. 2016;53:419–425. doi: 10.1136/jmedgenet-2015-103511. [DOI] [PubMed] [Google Scholar]