

Abstract
Objective
The extent to which major depression is the outcome of a single biological mechanism or represents a final common pathway of multiple disease processes remains uncertain. Genetic approaches can potentially identify etiologic heterogeneity in major depression by dividing patients on their experience of major adverse events.
Method
Data are from China, Oxford, and VCU Experimental Research on Genetic Epidemiology (CONVERGE), a study of Han Chinese women with recurrent major depression aimed at identifying genetic risk factors for major depression in a rigorously ascertained cohort carefully assessed for key environmental risk factors (n = 9599). To detect etiologic heterogeneity, genome-wide association studies (GWAS), heritability analyses, and gene-by-environment interaction analyses were performed.
Results
GWAS stratified by exposure to adversity revealed three novel loci associated with major depression only in subjects with no history of adversity. Significant GxE interactions were seen between adversity and genotype at all three loci and 13.2% of major depression liability can be attributed to genome-wide interaction with adversity exposure. The genetic risk in major depression for samples who reported major adverse life events (27%) was partially shared with that in samples who did not (73%) (genetic correlation = +0.64). Together with results from simulation studies, these findings suggest etiologic heterogeneity within major depression as a function of environmental exposures.
Conclusions
The genetic contributions to major depression in women may differ in those with and without major adverse life events. These results have implications for the molecular dissection of major depression and other complex psychiatric and biomedical diseases.
Keywords: major depressive disorder, childhood sexual abuse, stressful life events, genome-wide association, etiologic heterogeneity, clinical heterogeneity, gene-by-environment interaction
Introduction
The heterogeneity of major depression, demonstrated by variable symptom presentation, course of illness, and treatment response, has hindered our understanding of its etiology (1,2). To counter this, researchers have attempted to study homogeneous subtypes (e.g., atypical depression, early age-of-onset) (3,4). Indeed, the decades-long debate about the number of distinct depressive subtypes remains unresolved (1). Of particular interest, a large literature suggests that major depression can be usefully divided into a stress-responsive subtype (e.g., “reactive” depression) and a subtype with no apparent environmental precipitants (e.g., “endogenous”) (5–9).
In this paper, we examine whether genetic approaches can identify etiologically heterogeneous depressive subtypes. We explore whether the two main classes of known causal factors for major depression, genes and environment, represent partially distinct pathways to major depression. Genetic effects on major depression are well established from twin studies (10) and genome-wide association data (11–13). Molecular genetic analysis reveals that major depression, like other complex diseases, has a polygenic architecture with multiple loci of small effect (14, 15).
Adversity exposure increases risk for major depression (16) with a dose-response relationship between severity of stressors and disease risk (17–19). Results of co-twin control studies suggest that this association is largely causal (18, 20). However, adversity is neither necessary nor sufficient to produce major depression and it has been difficult to identify clinical features distinguishing cases of major depression with versus without environmental precipitants (21–23).
Genetic risk factors for major depression not only alter average risk but also influence sensitivity to depressogenic effects of environmental adversities, particularly childhood maltreatment and adult life events (24–26). For example, exposure to severe stressful life events increases risk to major depression more strongly in those with high versus low genetic liability (27). Despite strong effects of environmental exposures on major depression risk, there have been limited efforts incorporating these factors into large-scale molecular genetic studies. Furthermore, the major depression-associated genetic loci identified to-date only account for a small portion of the variance in disease liability (12–14), underscoring the importance of continued research on detecting heterogeneity as a mechanism to identify etiologically relevant determinants.
Therefore, we investigate whether genetic approaches can demonstrate etiologic heterogeneity among major depression cases by classifying individuals on the basis of adversity exposure. Using data from the China, Oxford, and VCU Experimental Research on Genetic Epidemiology (CONVERGE) (13), a study of Han Chinese women with recurrent major depression (n=9599) aimed at identifying genetic risk factors in a rigorously ascertained cohort assessed for key environmental risk factors, we explore whether major depression with and without major environmental adversities may represent, from a genetic perspective, partially distinct subtypes.
Methods
Sample collection
Recurrent major depression cases were recruited from 58 provincial mental health centers and psychiatric departments of medical hospitals in 45 cities and 23 provinces of China. Controls were recruited from multiple locations including general hospitals and local community centers. Subjects were Han Chinese women with four Han grandparents. Cases were ages 30–60 and had ≥ two episodes of major depression meeting DSM-IV criteria with the first episode at ages 14–50. The study was approved by the Ethical Review Boards of Oxford University and participating hospitals. All participants provided written informed consent. Details on sample collection, phenotypes, and sequencing are reported elsewhere (13,14,28).
Adversity measures
A binary measure of adversity was derived from self-reported stressful life events and childhood sexual abuse (Supplemental Methods). The stressful life events questionnaire was adapted from a prior study (29) and assessed 16 traumatic events and their age at occurrence (Supplemental Table 1). The childhood sexual abuse questionnaire was a shortened version of a scale (30) that queried whether, before aged 16, any older person involved them in unwanted sexual incidents including sexual invitation, fondling, and intercourse. Subjects were considered “adversity-exposed” if they a) had data on stressful life events and childhood sexual abuse and b) endorsed any childhood sexual abuse and/or had high aggregate stressful life event scores (+3SD). Since life events vary in severity, our score was constructed by weighting each event by its estimated effect-size on major depression and summing across events. Stressful life events for cases were only included if they preceded depression onset. We thereby grouped subjects into “adversity-exposed” and “unexposed” subgroups.
Genome-wide association
Genome-wide associations between 4,313,801 imputed autosomal single nucleotide polymorphisms (SNPs) (with minor allele frequency (MAF) >5%, imputation information >0.95, P-value for violation of Hardy-Weinberg equilibrium >10−6) and major depression was performed in: a) whole cohort, b) reduced cohort unexposed to adversity (‘unexposed’), and c) only those exposed to adversity (‘exposed’) using linear mixed modeling (BOLT-LMM v.2.2) (31). To calibrate the BOLT-LMM statistic we calculated linkage disequilibrium (LD) scores of each SNP using LDSC (v.1.0.0) (32). The kinship matrix used was constructed from 413,669 LD pruned SNPs (LD <0.8). PLINK v.1.9 (33,34) was used for logistic regression to obtain odds ratios for top variants identified from BOLT. SNPs with P-values smaller than 5×10−8 were selected for gene-by-environment interaction tests. Regional association plots were constructed using LocusZoom v.0.4.8 (35).
Polygenic risk scores
Polygenic risk scores (PRS) for CONVERGE have been previously constructed by two methods (14,36). First, using a random independent 50–50 split (Sample-1, Sample-2) we estimated Sample-1 SNP effects using the best linear unbiased prediction (BLUP) method implemented in GCTA and tested PRS constructed using the profile option in PLINK using SNP BLUP-solutions as weights in Sample-2 and vice versa (Conv-PRS) (14). Second, using summary statistics from the Psychiatric Genomics Consortium (PGC) meta-analysis of European studies of major depression, recurrent depression PRS were constructed from SNP weights based on P-value threshold <0.2 (36).
Interaction test
Gene-by-environment interaction effects were tested at loci identified from GWAS. Interaction was tested on both the multiplicative (logistic regression) and additive scales (blm package in R) (37–39) including 10 principal components as covariates to control for population stratification.
Random-effect meta-analysis
Heterogeneity of SNP effects on depression in the adversity-exposed and unexposed cohorts was tested using random-effect models that identify both main and heterogeneity effects and Cochran’s Q-test, all implemented in METASOFT v.2.0.1 (40,41).
Heritability estimation
SNP-based heritability (h2SNP) was estimated using GCTA v.1.26.0 (42) with a genetic relatedness matrix (GRM) constructed from 413,669 LD pruned SNPs and 10 principal components used as covariates in a) the full cohort, b) the adversity-exposed cohort, and c) the unexposed cohort. Using the bivariate option (43) the genetic correlation (ρ) was estimated for major depression between the adversity-exposed and unexposed subgroups, for adversity between major depression-cases and controls, and between major depression and adversity. GCTA was used to estimate the proportion of variance in major depression due to aggregate additive gene-by-environment interaction between adversity and all GRM SNPs. Details for ascertainment adjustment (K) and alternative h2SNP estimation using LDAK (44) and PCGC-s are in the Supplemental Methods.
Simulations of etiologic heterogeneity
We used simulations to mirror genetic approaches to discern features of heterogeneity and to demonstrate stratification of samples by adversity is an appropriate means for uncovering heterogenous genetic effects (Supplemental Methods). Three scenarios were applied: a) SNP effect and adversity exposure contribute additively to liability (no etiologic heterogeneity), b) SNP effect is only present in the adversity unexposed (reflecting etiologic heterogeneity), and c) h2SNP estimates under the presence and absence of etiologic heterogeneity by replacing the single causal SNP with polygenic contributions. For each simulation, SNP effects were tested under four logistic regression models: I) ignoring effects of adversity, II) controlling for effects of adversity by incorporating it as a covariate, III) including an interaction between SNP and adversity, and IV) analyzing adversity-exposed and unexposed cohorts separately. For scenarios a) and b), we simulated 1,000 independent replicates of a SNP effect (matching SNPs associated in the unexposed) on a disease with prevalence of 5% in a cohort with adversity exposure, prevalence, and sample sizes matching CONVERGE. For scenario c), the single causal SNP was replaced with polygenic contributions from 10,000 simulated independent SNPs.
Results
Association between adversity and major depression
Adversity was significantly associated with major depression, confirming prior analyses (45,46). Depression cases experienced significantly more life events than controls (P=2.66×10−81, Supplemental Figure 1, Supplemental Table 1). Childhood sexual abuse was significantly associated with major depression (10.3% cases vs. 2.5% controls, odds ratio=2.98, P=2.6×10−19) with effects increasing with greater abuse severity (Supplemental Figure 2). Together, stressful life events and childhood sexual abuse accounted for 11.6% of the phenotypic variance in major depression. Twenty-seven percent of the sample (1,646 cases, 982 controls) was adversity-exposed and 73% (3,139 cases, 3,832 controls) was not. Supplemental Table 2 shows rates of key clinical features by adversity exposure. Adversity-exposed individuals endorsed higher levels of neuroticism, younger age of onset, and were more likely to have comorbid dysthymia and anxiety disorders.
Genome-wide association of major depression in cohorts with and without adversity
Figure 1 shows Manhattan plots for the GWAS of major depression in: a) CONVERGE subjects with complete information on adversity (n=9,599), b) those who reported adversity (n=2628), and c) those without adversity (n=6971). The genomic control factors (λ) were 1.047, 1, and 1.047; the adjusted measure to that of 1,000 cases and 1,000 controls (λ1000) were 1.01, 1, and 1.014 respectively (Supplemental Figure 3).
Figure 1.
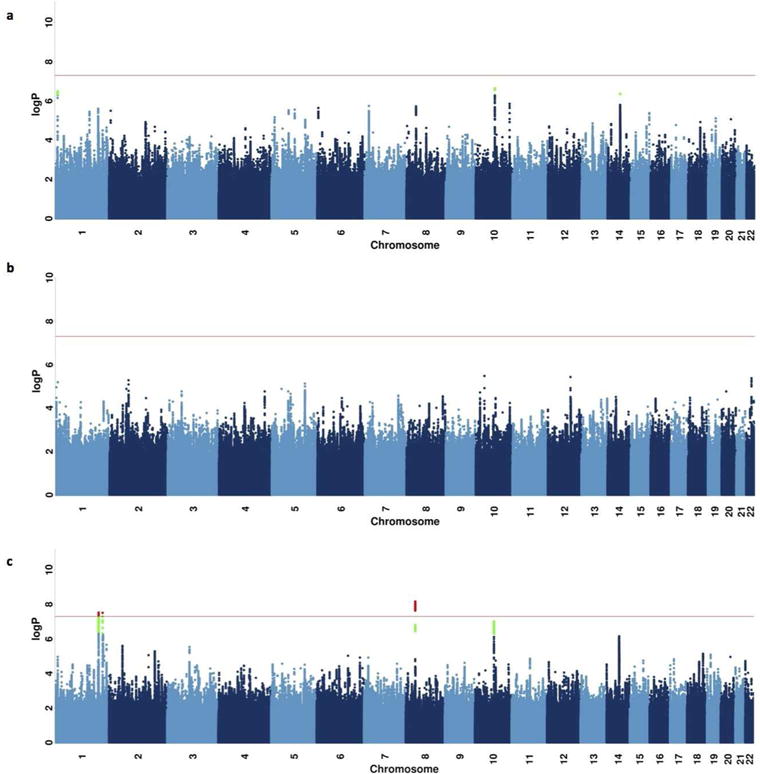
Manhattan plots of loci associated with major depression.
(a) Manhattan plot of major depression for all subjects for whom information on exposure to adversity is available; (b) Manhattan plot of major depression in a subgroup reporting exposure to adversity; (c) Manhattan plot of major depression in a subgroup reporting no exposure to adversity. In each plot, the -log10 P-values of imputed SNPs associated with major depression by leave-one-chromosome-out linear mixed model association in BOLT-LMM are shown on the left y axes. The horizontal axis gives the position on each chromosome; chromosomes are numbered below the axis.
In the adversity-exposed subset (Figure 1b), no locus exceeded P<5.0×10−8. In the subset without adversity, neither of the two previously reported loci on chromosome 10 exceeded P<5.0×10−8 (rs12415800: P=3.2×10−6, rs35936514: P=8.7×10−5), likely due to reduced power as odds ratios were not significantly different from the full cohort odds ratios (Supplemental Figure 6, Supplemental Table 10). However, three novel loci were detected (Table 1, Figure 1c): i) on chromosome 1 near LPGAT1 (lysophosphatidylglycerol acyltransferase 1) (rs7526682, chr1:211973950, MAF=13.3%, P=3.0×10−8, odds ratio=1.31, Figure 2a), ii) on chromosome 1 in C1ORF95 (rs11577545, chr1:226799083, MAF=21.5%, P=3.1×10−8, odds ratio=1.25, Figure 2b), and iii) on chromosome 8 at the 5′ end of SLC25A37 (Mitoferrin-1) (rs950893, chr8:23450510, MAF=28.0%, P=6.9×10−9, odds ratio=0.79, Figure 2c).
Table 1.
Top SNP associations with major depression in full cohort and subgroups.
| rs ID | rs7526682 | rs11577545 | rs950893 | rs12415800 | rs35936514 | ||
|---|---|---|---|---|---|---|---|
| Chr | 1 | 1 | 8 | 10 | 10 | ||
| Position | 211973950 | 226799083 | 23450510 | 69624180 | 126244970 | ||
| Major/Mino r Alleles | C/G | C/T | A/G | G/A | C/T | ||
| Linear mixed model (BOLT-LMM) | Full cohort | MAF | 0.13 | 0.22 | 0.28 | 0.46 | 0.26 |
| P-value | 4.1×10−5 | 3.2×10−5 | 1.9×10−6 | 5.1×10−7 | 1.8×10−6 | ||
| z score | 4.10 | 4.16 | −4.76 | 5.02 | −4.78 | ||
|
| |||||||
| No adversity | MAF | 0.13 | 0.21 | 0.28 | 0.46 | 0.26 | |
| P-value | 3.0×10−8 | 3.1×10−8 | 6.9×10−9 | 3.2×10−6 | 8.7×10−5 | ||
| z score | 5.54 | 5.54 | −5.79 | 4.66 | −3.93 | ||
|
| |||||||
| Adversity | MAF | 0.13 | 0.23 | 0.27 | 0.45 | 0.27 | |
| P-value | 3.8×10−1 | 9.4×10−2 | 8.2×10−2 | 6.0×10−2 | 2.50×10−3 | ||
| z score | −0.89 | −1.67 | 0.82 | 1.88 | −3.03 | ||
|
| |||||||
| Logistic regression (PLINK) | Full cohort | P-value | 3.7×10−5 | 9.0×10−5 | 6.9×10−7 | 9.6×10−7 | 1.5×10−6 |
| odds ratio | 1.19 | 1.15 | 0.85 | 1.15 | 0.85 | ||
| CI | 1.10–1.30 | 1.07–1.23 | 0.80–0.91 | 1.09–1.22 | 0.80–0.91 | ||
|
| |||||||
| No adversity | P-value | 4.6×10−8 | 8.0×10−8 | 2.1×10−9 | 2.9×10−6 | 8.8×10−5 | |
| odds ratio | 1.31 | 1.25 | 0.79 | 1.17 | 0.86 | ||
| CI | 1.19–1.45 | 1.15–1.36 | 0.74–0.86 | 1.10–1.26 | 0.80–0.93 | ||
|
| |||||||
| Adversity | P-value | 3.7×10−1 | 9.7×10−2 | 4.6×10−1 | 5.1×10−2 | 2.0×10−3 | |
| odds ratio | 0.93 | 0.90 | 1.05 | 1.12 | 0.82 | ||
| CI | 0.78–1.10 | 0.79–1.02 | 0.92–1.19 | 1.00–1.25 | 0.72–0.93 | ||
This table reports the test statistics at the SNPs associated with major depression in the subgroup unexposed to adversity a) in the full cohort, b) in the subgroup unexposed to adversity (No Adversity) and c) adversity-exposed (Adversity); the minor allele at each SNP is the tested allele. Results from leave-one-chromosome-out linear mixed model association testing in BOLT-LMM and logistic regression with 10 principal components as covariates in PLINK are shown. SNPs showing genome-wide significant associations are shown in bold. Two SNPs (rs12415800 and rs35936514) showing genome wide significant association with major depression in our previous analysis of all samples in CONVERGE (including those without the self-reported adversity measure) are included for comparison.
Figure 2.
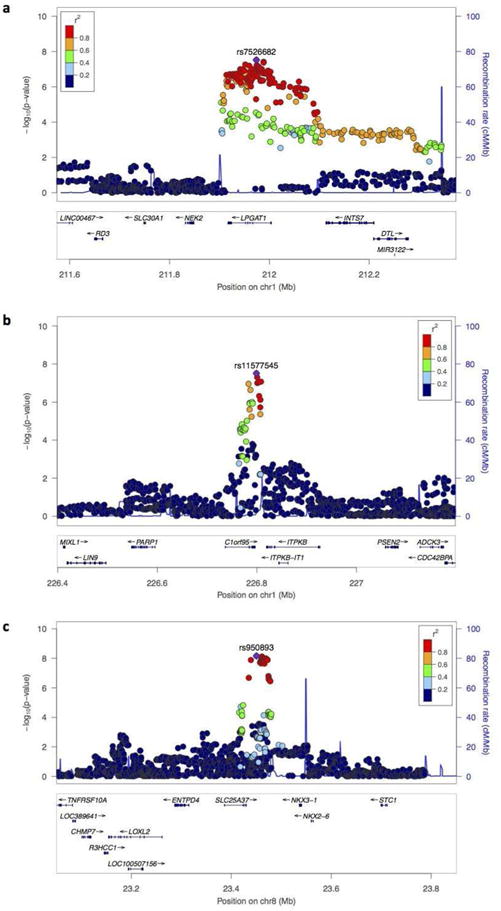
Genes at three loci associated with major depression in the subgroup reporting no adversity.
(a) Locus on chromosome 1, 212 Mb, over the gene encoding lysophosphatidylglycerol acyltransferase 1 (LPGAT1) (peak SNP = rs7526682,); (b) Locus on chromosome 1, 226 Mb, over the gene C1ORF95 (peak SNP = rs11577545); (c) Locus on chromosome 8 at 23.4Mb, over the 3′ end of the mitoferrin gene SLC25A37 (peak SNP = rs950893). The -log10 P-values of imputed SNPs associated with major depression by logistic regression are shown on the left y axes together the recombination rates (NCBI Build GRCh37), represented by light-blue lines, with scales on the right y axes. Genes within the regions are shown in the bottom panels. The horizontal axis gives the chromosomal position in megabases (Mb). The index SNPs are shown as larger purple diamonds labeled by their marker names; linkage disequilibrium (hg19 1000 Genomes ASN panel Nov 2014) with the remaining SNPs is indicated by different colors.
Comparison of these newly identified loci with the PGC mega-analysis of European studies (11) revealed an association between rs950893 on chromosome 8 and major depression (P=0.009), in the same direction as observed in CONVERGE. In contrast, the chromosome 1 loci (rs7526682, rs11577545) were not associated in the PGC study (P=0.37, P=0.81), although results were in the same direction (Supplemental Figure 4).
We performed four further analyses on the three newly identified SNPs in the unexposed group. First, to determine if results were due to stochastic effects, we randomly removed samples equal in size to the adversity-exposed group 10,000 times and obtained empirical distributions of odds ratios at these SNPs for major depression (Supplemental Methods). Our results were unlikely to have arisen by chance as all SNPs showed significant deviation in odds ratio from the full cohort (rs7526682 99.9th percentile of the empirical distribution of odds ratios, rs11577545 100th percentile, rs950893 0.2th percentile) (Supplemental Figure 5). In comparison, the two previously reported SNPs on chromosome 10 (rs12415800, rs35936514) were not significant (Supplemental Figure 6).
Second, we tested for statistical interaction between adversity and the minor allele at each locus and compared findings to results including adversity as a covariate. For the two previously reported SNPs (rs12415800, rs35936514), the interaction terms were not significant. The three newly identified SNPs, however, all had significant multiplicative and additive interaction terms (Table 2, Supplemental Table 3, Supplemental Figure 7).
Table 2.
Tests for gene-by-environment interaction between adversity and genetic variants predicting major depression.
| Test | Model 1: Interaction | Model 2: Covariate | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| OR | 95%CI | P-value | R2 | OR | 95%CI | P-value | |
| chr1:rs7526682_G | 1.29 | 1.17–1.43 | 1.94×10−7 | 0.0024 | 1.20 | 1.10–1.31 | 2.50×10−5 |
| Adversity | 2.21 | 1.99–2.46 | 2.22×10−48 | 0.0316 | 2.03 | 1.85–2.23 | 5.21×10−51 |
| adversity:rs7526682 | 0.73 | 0.60–0.89 | 0.0016 | 0.0013 | – | – | – |
|
| |||||||
| chr1:rs11577545_T | 1.28 | 1.17–1.39 | 1.48×10−8 | 0.0018 | 1.14 | 1.06–1.23 | 0.0003 |
| Adversity | 2.41 | 2.15–2.71 | 4.38×10−49 | 0.0311 | 2.02 | 1.84–2.22 | 3.11×10−50 |
| adversity:rs11577545 | 0.67 | 0.57–0.79 | 9.31×10−7 | 0.0032 | – | – | – |
|
| |||||||
| chr8:rs950893_G | 0.80 | 0.74–0.86 | 5.05×10−9 | 0.0029 | 0.86 | 0.81–0.92 | 3.28×10−6 |
| Adversity | 1.74 | 1.54–1.97 | 6.47×10−19 | 0.0311 | 2.02 | 1.84–2.22 | 3.04×10−50 |
| adversity:rs950893 | 1.31 | 1.13–1.52 | 0.0003 | 0.0018 | – | – | – |
|
| |||||||
| chr10:rs12415800_A | 1.17 | 1.10–1.26 | 2.77×10−6 | 0.0035 | 1.16 | 1.10–1.23 | 2.87×10−7 |
| Adversity | 2.10 | 1.81–2.45 | 2.76×10−22 | 0.0315 | 2.03 | 1.85–2.23 | 1.00×10−50 |
| adversity:rs12415800 | 0.96 | 0.84–1.10 | 0.5630 | <0.0001 | – | – | – |
|
| |||||||
| chr10:rs35936514_T | 0.85 | 0.79–0.92 | 3.61×10−5 | 0.0037 | 0.84 | 0.79–0.90 | 1.61×10−7 |
| Adversity | 2.09 | 1.85–2.36 | 2.80×10−32 | 0.0315 | 2.03 | 1.85–2.23 | 7.43×10−51 |
| adversity:rs35936514 | 0.95 | 0.82–1.10 | 0.5010 | <0.0001 | – | – | – |
This table shows the odds ratio (OR), 95% confidence interval of OR, and P-value of the minor allele of each single nucleotide polymorphism (SNP) association with major depression in the full cohort in logistic regression, with an interaction term between SNP and self-reported adversity (adversity:SNP) term included in Model 1, and without it in Model 2. All analyses performed using 10 principal components as covariates, bold font indicates significant genetic effect (P < 5.0×10−8) or gene by environment interaction (P < 0.005).
Third, we investigated differences in variant effects in the adversity-exposed and unexposed groups using random-effect meta-analysis. Supplemental Table 4 shows significant effect-size heterogeneity at the three new loci (Q-tests: rs7526682 P=3.13×10−4, rs11577545 P=9.42×10−6, rs950893 P=1.82×10−4) and significant random effect tests for heterogeneity (P=1.02×10−7, P=1.07×10−7, and P=2.34×10−8, respectively). This method detected significant heterogeneity of SNP effects across the adversity exposure groups for the three newly identified loci. Major depression case-only and control-only association of adversity also demonstrated effect-size differences at these variants (Supplemental Table 8).
Fourth, we performed simulations to determine whether the difference in the estimated SNP effects between the adversity-exposed and unexposed groups implicates heterogeneity. The average logistic regression results for scenario a) (no heterogeneity) are displayed in the left panel of Table 3. Three results are noteworthy. First, in model IV (analyzing adversity groups separately), the P-values are orders of magnitude less significant than in models I (adversity ignored) and II (adversity as covariate). Second, as no heterogeneity is simulated, the P-value difference between the two groups in model IV must only reflect power differences, not heterogeneity. Crucially, this shows that disparate P-values between cohorts alone does not indicate heterogeneity. Third, the G×E interaction test in model III is well calibrated and shows no evidence of (false) inflation. These features of homogeneous SNP effects are all evident for both loci on chromosome 10.
Table 3.
Average test output from four types of logistic regression on 1,000 simulated datasets: one ignoring adversity (model I); one controlling for the additive effect of adversity (model II); one additionally incorporating an interaction between genotype and adversity (model III); and finally a model which analyzes adversity-exposed and unexposed cohorts separately (model IV).
| Regression Model | Without Heterogeneity | With Heterogeneity | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Z Stat | odds ratio | P-value | Z Stat | odds ratio | P-value | |
| Model I, g | 5.36 | 1.18 | 8.22×10−8 | 4.67 | 1.16 | 3.06×10−6 |
| Model II, g | 5.36 | 1.18 | 8.15×10−8 | 4.99 | 1.17 | 6.05×10−7 |
| Model II, s | 13.95 | 1.90 | 2.94×10−44 | 11.14 | 1.68 | 7.77×10−29 |
| Model III, g | 4.64 | 1.19 | 3.34×10−6 | 5.85 | 1.24 | 4.80×10−9 |
| Model III, s | 10.2 | 1.92 | 1.93×10−24 | 10.22 | 1.92 | 1.64×10−24 |
| Model III, g:s | −0.15 | 0.99 | 0.877 | −3.08 | 0.8 | 2.10×10−3 |
| Model IV, g, no adversity | 4.64 | 1.19 | 3.44×10−6 | 5.85 | 1.24 | 4.80×10−9 |
| Model IV, g, adversity | 2.69 | 1.18 | 7.14×10−3 | −0.08 | 1.00 | 0.94 |
For each row, Z statistic (Z Stat), odds ratio, and P-value are shown for SNP effect (g), adversity effect (s), or an interaction effect (g:s). The three columns on the left (Without Etiologic Heterogeneity) show results for simulations of no etiologic heterogeneity between simulated phenotype in samples with and without adversity; the three columns on the right show that for simulations with heterogeneity. Data was simulated using a liability threshold model with realistic ascertainment, effect-sizes and allele frequencies (Supplemental Methods).
Next, we modified the baseline simulation by making the SNP causal only in the adversity-unexposed group (scenario b) and performed the same tests (Table 3, right panel). The presence of heterogeneity induces three novel features: the genetic effect-sizes for each cohort estimated in model IV are now different; the unexposed cohort test in model IV is more powerful than the test in model I (ignoring adversity), despite an attendant reduction in sample size; and the G×E interaction test in model III is statistically significant. These simulation results all distinguish the loci on chromosomes 1 and 8 from those on chromosome 10.
The genetic basis for major depression in adversity-exposed and unexposed groups may differ
To determine the presence of heterogeneity on aggregate genetic effects, estimates of the additive SNP contribution (h2SNP) on the liability scale, after correction for sample ascertainment, were compared between adversity-exposed and unexposed depressive subgroups. Without etiologic heterogeneity, h2SNP in subgroups should be similar to that in the entire sample. However, given genetic heterogeneity, h2SNP may be larger in both subgroups than in the entire sample.
Although the h2SNP estimate of major depression in the adversity unexposed cohort (h2SNP =38.0%, SE=4.8%, P=1.11×10−16) was higher than the exposed cohort (h2SNP=34.2%, SE=15.9%, P=0.013), and the overall combined major depression sample (h2SNP =30.5%, SE=3.7%, P<10−16), they were not statistically different. Because differences in h2SNP estimation methods may impact estimates and their interpretations (44,47–49), we accounted for LD in dense, imputed data using LDAK (44) and assessed underestimation from restricted maximum likelihood using PCGC-s. These results were consistent with results from GCTA (Supplemental Table 5).
Second, the proportions of variance in major depression due to aggregate additive G×E interaction between adversity and all GRM SNPs, Conv-PRS, or PGC-PRS were estimated. The interaction component for the GRMxAdversity term was significant (P=0.038), with the proportion of variance attributable to additive genetic (h2SNP) and G×E interaction components estimated at 23.3% (SE=5.8%) and 13.2% (SE=7.4%) respectively. However, none of the PRSxAdversity interactions were significant (Supplemental Table 6) perhaps due to limitations of a PRS-based approach (Supplemental Material).
Third, the genetic correlation of major depression between the adversity-exposed and unexposed cohorts was estimated at +0.64 (SE=0.23). While less than unity, this is known so imprecisely that it is not significantly different from 1 (95%CI=[0.19,1.0]).
Finally, we consider which models of genetic architecture are consistent with observed trends. The resulting h2SNP estimates from the overall cohort with and without adversity exposure are shown in Supplemental Figure 9. The two within-cohort heritabilities, along with genetic correlation and G×E estimates, are shown in Supplemental Figure 10. The results confirm our prior intuition: without heterogeneity, within-group heritabilities coincide with the overall average heritability, though the reduced sample sizes induce larger variance in the within-group estimators; however, as heterogeneity increases (or causal variant sharing decreases) the overall heritability decreases while the within-cohort heritabilities remain constant.
Exposure to adversity may have a heterogeneous genetic basis
One interpretation of our findings is that the presence of adversity in one group attenuates the contribution of genetic effects. However, self-reported environmental measures are moderately heritable (reviewed in (50)) and ~29% of the variance in the number of stressful life events has been attributed to SNPs (51). Here, the h2SNP of adversity was 18.2% in the overall sample (SE=6.2%, P=0.001, K=0.215), 25.7% (SE=12.0%, P=0.013, K=0.344) among major depression cases, and 44.2% for controls (SE=14.7%, P=0.001, K=0.20). The genetic correlation of adversity between major depression cases and controls was +0.34 (SE=0.31) but was not statistically significant.
Assessment of G-E correlation
Since G–E correlation can bias G×E results, we tested for G–E correlation by three methods. The estimated SNP-based genetic correlation between major depression and self-reported adversity was negligible as ρ=−0.02 (SE=0.15) and not significantly different from 0 (P=0.45). Additional tests of G–E correlation examined association of major depression-PRS with adversity and were not significant (Supplemental Table 7). An exploratory test of G–E correlation examined by genome-wide correlation of SNP odds ratios for adversity between major depression-cases and controls was also small (r=0.008, Supplemental Table 9, Supplemental Figure 8). These results do not support significant systematic G–E correlation in our sample between major depression and adversity exposure.
Discussion
We applied molecular genetic methods to a large sample of carefully characterized depressed women to evaluate etiologic heterogeneity between those exposed versus unexposed to severe environmental adversities. These efforts yielded three major findings.
First, classifying samples based on adversity exposure identified genetic loci with heterogeneous effects. We identified three novel loci on chromosomes 1 and 8 that confer risk of major depression only among individuals unexposed to adversity. The newly discovered locus on chromosome 8 is at the 5′ end of the SLC25A37 gene which encodes an iron carrier localized in the mitochondrial inner membrane (52) adding further support for a mitochondrial role in major depression (13,14,53). Second, we found evidence for interaction between adversity and genotype at all three loci. Third, we provide modest evidence for heterogeneity at the whole-genome level: i) 13.2% of the variance in major depression liability arises from interaction between genome-wide SNP effects and adversity, ii) genetic correlation for major depression between subgroups with and without adversity exposure was +0.64, and iii) although confidence intervals overlapped, SNP-based heritability estimates of major depression in the unexposed subgroup was higher (~39%) than the overall sample (30%). Furthermore, simulations reflecting etiologic heterogeneity are consistent with our results.
These results have several implications. First, they provide support for long-debated typology that major depression patients can be meaningfully divided into those whose illness arises in reaction to environmental stressors and those whose disorder emerges “from within” (5–8). The genetic substrates of these two forms of major depression appear to be correlated but not identical, and some genetic factors may have subtype specific effects.
Second, these findings provide insight into how effects of genes and environment combine to give rise to major depression. A leading hypothesis consistent with prior studies (24,25,27) is that certain genes have a stronger impact on risk for major depression in adversity-exposed than unexposed individuals. We unexpectedly find, that for three loci, an opposite pattern in which effects were stronger in cases without adversity exposure. While the CONVERGE sample may contain loci with an increased impact on adversity-exposed individuals, power to detect these is low as only 27% of our sample reported severe adversity.
An appealing interpretation of our findings is that absent environmental stressors, a higher genetic loading is required to cause depression. This cannot, however, explain our findings as it would predict a graded response at the three identified loci in exposed and unexposed individuals. Rather, our results suggest at least two classes of molecular variants that predispose to major depression: those whose effects are present in all cases and those whose effects depend on the history of adversity. In contrast to the three SNPs discovered by stratifying on adversity, results for the two previously reported SNPs on chromosome 10 (13) are consistent with a liability threshold model. Major depression may be a syndrome arising from several partially distinct etiologies. The design of CONVERGE enabled the combination of “deep” phenotypes and genotypes to detect differences in the genetic architecture of those with and without adversity. Other subtypes may be detectable in a similar manner.
Our findings counter the dominant paradigm in psychiatric molecular genetics research that increasing sample size should be the primary method for detecting more genetic loci (12). Here, the newly detected loci were discovered in a sample size 30% smaller than the cohort that yielded the two previously reported loci (13). These results support the value of more detailed phenotyping, especially the assessment of environmental adversities. To characterize major depression etiology, future efforts may need both careful assessment of the phenotype and environmental exposures in large samples.
Three limitations to the study should be noted. First, our power to detect genetic variants with the expected small effect-sizes is limited (Supplemental Table 10). Second, we are unaware of Asian replication cohorts with genetic information and environmental adversities. Therefore, these results should be considered tentative, although their plausibility is supported by simulations. Third, our assessments of age at onset of depression and adversity exposure were retrospective. Although interviewers encouraged effortful responding, we cannot rule out recall biases. Despite these limitations, our results highlight the value of empirically driven approaches to address heterogeneity and provide a framework applicable to other complex psychiatric diseases to identify putative subtypes and etiologically relevant genetic variation.
Supplementary Material
Acknowledgments
This work was funded by the Wellcome Trust (WT090532/Z/09/Z, WT083573/Z/07/Z, WT089269/Z/09/Z) and by NIH grant MH100549. Roseann E. Peterson is supported by NIH T32 grant MH020030; Na Cai by the ESPOD Fellowship from the European Molecular Biology Laboratory, European Bioinformatics Institute, and Wellcome Trust Sanger Institute; Alexis C. Edwards by K01 grant AA021399; and Silviu-Alin Bacanu by R21MH100560 and R21AA022717. Authors are part of the CONVERGE consortium (China, Oxford and Virginia Commonwealth University Experimental Research on Genetic Epidemiology) and gratefully acknowledge the support of all partners in hospitals across China. Special thanks to all the CONVERGE collaborators and patients who made this work possible.
Footnotes
Supplemental Material: Supplementary information is available at AJP’s website and includes Supplemental Methods, 11 Supplemental Tables, and 10 Supplemental Figures.
Disclosures: There are no conflicts of interest to report.
References
- 1.Flint J, Kendler KS. The genetics of major depression. Neuron. 2014 Feb 5;81(3):484–503. doi: 10.1016/j.neuron.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg D. The heterogeneity of “major depression”. World psychiatry: official journal of the World Psychiatric Association (WPA) 2011 Oct;10(3):226–228. doi: 10.1002/j.2051-5545.2011.tb00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milaneschi Y, Lamers F, Peyrot WJ, Abdellaoui A, Willemsen G, Hottenga J, et al. Polygenic dissection of major depression clinical heterogeneity. Molecular psychiatry. 2015 doi: 10.1038/mp.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Power RA, Tansey KE, Buttenschøn HN, Cohen-Woods S, Bigdeli T, Hall LS, et al. Genome-wide Association for Major Depression Through Age at Onset Stratification: Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Biol Psychiatry. 2016 May 24; doi: 10.1016/j.biopsych.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendels J, Cochrane C. The nosology of depression: the endogenous-reactive concept. Am J Psychiatry. 1968 May;124(11):11. doi: 10.1176/ajp.124.11S.1. [DOI] [PubMed] [Google Scholar]
- 6.Coryell W, Winokur G, Maser JD, Akiskal HS, Keller MB, Endicott J. Recurrently situational (reactive) depression: a study of course, phenomenology and familial psychopathology. J Affect Disord. 1994 Jul;31(3):203–210. doi: 10.1016/0165-0327(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 7.Nelson JC, Charney DS. Primary affective disorder criteria and the endogenous-reactive distinction. Arch Gen Psychiatry. 1980 Jul;37(7):787–793. doi: 10.1001/archpsyc.1980.01780200065007. [DOI] [PubMed] [Google Scholar]
- 8.Nelson JC, Charney DS. The symptoms of major depressive illness. Am J Psychiatry. 1981 Jan;138(1):1–13. doi: 10.1176/ajp.138.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Pollitt J. The relationship between genetic and precipitating factors in depressive illness. Br J Psychiatry. 1972 Jul;121(560):67–70. doi: 10.1192/bjp.121.1.67. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 11.Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, et al. A mega-alysis of genome-wide association studies for major depressive disorder. Molecular psychiatry. 2013;18(4):497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016 Sep;48(9):1031–1036. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Converge Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523(7562):588–591. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson RE, Cai N, Bigdeli TB, Li Y, Reimers M, Nikulova A, et al. The Genetic Architecture of Major Depressive Disorder in Han Chinese Women. JAMA Psychiatry. 2017 Feb 01;74(2):162–168. doi: 10.1001/jamapsychiatry.2016.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lubke GH, Hottenga JJ, Walters R, Laurin C, de Geus Eco JC, Willemsen G, et al. Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biological psychiatry. 2012 Oct 15;72(8):707–709. doi: 10.1016/j.biopsych.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- 17.Brown GW, Harris T. Social origins of depression: a study of psychiatric disorder in women. London: Tavistock; 1978. [Google Scholar]
- 18.Kendler KS, Bulik CM, Silberg J, Hettema JM, Myers J, Prescott CA. Childhood sexual abuse and adult psychiatric and substance use disorders in women: an epidemiological and cotwin control analysis. Arch Gen Psychiatry. 2000 Oct;57(10):953–959. doi: 10.1001/archpsyc.57.10.953. [DOI] [PubMed] [Google Scholar]
- 19.Kendler KS, Karkowski LM, Prescott CA. Stressful life events and major depression: risk period, long-term contextual threat, and diagnostic specificity. J Nerv Ment Dis. 1998 Nov;186(11):661–669. doi: 10.1097/00005053-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999 Jun;156(6):837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 21.Cole J, McGuffin P, Farmer AE. The classification of depression: are we still confused? Br J Psychiatry. 2008 Feb;192(2):83–85. doi: 10.1192/bjp.bp.107.039826. [DOI] [PubMed] [Google Scholar]
- 22.Farmer A, McGuffin P. The classification of the depressions. Contemporary confusion revisited. Br J Psychiatry. 1989 Oct;155:437–443. doi: 10.1192/bjp.155.4.437. [DOI] [PubMed] [Google Scholar]
- 23.Kendell RE. The classification of depressions: a review of contemporary confusion. Br J Psychiatry. 1976 Jul;129:15–28. doi: 10.1192/bjp.129.1.15. [DOI] [PubMed] [Google Scholar]
- 24.Kendler KS, Kuhn J, Prescott CA. The interrelationship of neuroticism, sex, and stressful life events in the prediction of episodes of major depression. Am J Psychiatry. 2004 Apr;161(4):631–636. doi: 10.1176/appi.ajp.161.4.631. [DOI] [PubMed] [Google Scholar]
- 25.Heath AC, Eaves LJ, Martin NG. Interaction of marital status and genetic risk for symptoms of depression. Twin Res. 1998 Sep;1(3):119–122. doi: 10.1375/136905298320566249. [DOI] [PubMed] [Google Scholar]
- 26.Phelan J, Schwartz JE, Bromet EJ, Dew MA, Parkinson DK, Schulberg HC, et al. Work stress, family stress and depression in professional and managerial employees. Psychol Med. 1991 Nov;21(4):999–1012. doi: 10.1017/s0033291700029998. [DOI] [PubMed] [Google Scholar]
- 27.Kendler KS, Kessler RC, Walters EE, MacLean C, Neale MC, Heath AC, et al. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry. 1995 Jun;152(6):833–842. doi: 10.1176/ajp.152.6.833. [DOI] [PubMed] [Google Scholar]
- 28.Cai N, Bigdeli TB, Kretzschmar WW, Li Y, Liang J, Hu J, et al. 11,670 whole-genome sequences representative of the Han Chinese population from the CONVERGE project. Sci Data. 2017 Feb 14;4:170011. doi: 10.1038/sdata.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Kendler K, Prescott C. Genes, Environment, and Psychopathology: Understanding the Causes of Psychiatric and Substance Use Disorders. New York, NY: Guilford Press; 2006. [Google Scholar]
- 30.Martin J, Anderson J, Romans S, Mullen P, O’Shea M. Asking about child sexual abuse: methodological implications of a two stage survey. Child Abuse Negl. 1993 May-Jun;17(3):383–392. doi: 10.1016/0145-2134(93)90061-9. [DOI] [PubMed] [Google Scholar]
- 31.Loh P, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015 Mar;47(3):284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulik-Sullivan B, Loh P, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature genetics. 2015;47(3):291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007 Sep;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010 Sep 15;26(18):2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bigdeli TB, Ripke S, Peterson RE, Trzaskowski M, Bacanu S, Abdellaoui A, et al. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl Psychiatry. 2017 Mar 28;7(3):e1074. doi: 10.1038/tp.2016.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kendler KS, Gardner CO. Interpretation of interactions: guide for the perplexed. Br J Psychiatry. 2010 Sep;197(3):170–171. doi: 10.1192/bjp.bp.110.081331. [DOI] [PubMed] [Google Scholar]
- 38.R Development Core Team. R: A language and environment for statistical computing. 2009 2.9.2. [Google Scholar]
- 39.Kovalchik S, Varadhan R. Fitting Additive Binomial Regression Models with the R Package blm. Journal of Statistical Software. 2013 Sep 3;:54. [Google Scholar]
- 40.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011 May 13;88(5):586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang EY, Han B, Furlotte N, Joo JWJ, Shih D, Davis RC, et al. Meta-analysis identifies gene-by-environment interactions as demonstrated in a study of 4,965 mice. PLoS Genet. 2014 Jan;10(1):e1004022. doi: 10.1371/journal.pgen.1004022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A Tool for Genome-wide Complex Trait Analysis. The American Journal of Human Genetics. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28(19):2540–2542. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet. 2012 Dec 7;91(6):1011–1021. doi: 10.1016/j.ajhg.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Cai Y, Cong E, Liu Y, Gao J, Li Y, et al. Childhood sexual abuse and the development of recurrent major depression in Chinese women. PLoS ONE. 2014;9(1):e87569. doi: 10.1371/journal.pone.0087569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tao M, Li Y, Xie D, Wang Z, Qiu J, Wu W, et al. Examining the relationship between lifetime stressful life events and the onset of major depression in Chinese women. J Affect Disord. 2011 Dec;135(1-3):95–99. doi: 10.1016/j.jad.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SH. Implications of simplified linkage equilibrium SNP simulation. Proc Natl Acad Sci U S A. 2015 Oct 06;112(40):5449. doi: 10.1073/pnas.1502868112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golan D, Rosset S, Lander ES. Reply to Lee: Downward bias in heritability estimation is not due to simplified linkage equilibrium SNP simulation. Proc Natl Acad Sci U S A. 2015 Oct 06;112(40):5452. doi: 10.1073/pnas.1511370112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Golan D, Lander ES, Rosset S. Measuring missing heritability: inferring the contribution of common variants. Proc Natl Acad Sci U S A. 2014 Dec 9;111(49):5272. doi: 10.1073/pnas.1419064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kendler KS, Baker JH. Genetic influences on measures of the environment: a systematic review. Psychol Med. 2007 May;37(5):615–626. doi: 10.1017/S0033291706009524. [DOI] [PubMed] [Google Scholar]
- 51.Power RA, Wingenbach T, Cohen-Woods S, Uher R, Ng MY, Butler AW, et al. Estimating the heritability of reporting stressful life events captured by common genetic variants. Psychol Med. 2013 Sep;43(9):1965–1971. doi: 10.1017/S0033291712002589. [DOI] [PubMed] [Google Scholar]
- 52.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci U S A. 2009 Sep 22;106(38):16263–16268. doi: 10.1073/pnas.0904519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, et al. Molecular signatures of major depression. Curr Biol. 2015 May 4;25(9):1146–1156. doi: 10.1016/j.cub.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.