

Abstract
The nuclear envelope (NE), which is a critical barrier between the DNA and the cytosol, is capable of extensive dynamic membrane remodeling events in interphase. One of these events, interphase NE rupture and repair, can occur in both normal and disease states and results in the loss of nucleus compartmentalization. NE rupture is not lethal, but new research indicates that it could have broad impacts on genome stability and activate innate immune responses. These observations suggest a new model for how changes in NE structure could be pathogenic in cancer, laminopathies, and autoinflammatory syndromes, and redefine the functions of nucleus compartmentalization.
Introduction
The nuclear envelope (NE) surrounds the nucleus and is comprised of two membrane sheets fused at the nuclear pore complexes enclosing a lumen that is contiguous with the endoplasmic reticulum. The other main components of the NE are the nuclear pore complexes (NPCs) and the underlying nuclear lamina, a meshwork of lamin intermediate filament and transmembrane proteins that connect the chromatin to the inner nuclear membrane. The structure and composition of the NE regulates many aspects of nucleus biology, including nucleus morphology, response to mechanical stress, heterochromatin binding, gene expression, and nuclear functions, such as DNA damage repair, which led to a model of the NE as a scaffold [1,2]. Recently, a new model of NE structure has emerged that highlights an essential requirement for interphase NE remodeling in many cell processes and behaviors [3]. Most of these dynamics preserve nucleus compartmentalization [2]. The exception is NE rupture, which results in the loss of nucleus compartmentalization and can be corrected by NE repair or lead to persistent chromatin mislocalization [4–7].
Analysis of the current examples of NE rupture indicates that this process can have both protective and pathogenic consequences, depending on the biological context. Transient NE rupture during cell migration is thought to release intranuclear pressure [8], which could facilitate nuclear deformation and migration through small pores. However, it is also correlated with increased genome instability and nucleus fragmentation [9–11]. Similarly, NE rupture in micronuclei can generate highly rearranged chromosomes [12], but may also stimulate senescence and clearance of aneuploid cells from tissues [13,14]. Finally, induction of pro-inflammatory responses after NE rupture may have a dual nature, triggering autoinflammatory disease in some contexts [15], but also able to cause systemic anti-tumor responses after irradiation [13,16]. These observations raise new questions about the function of nucleus compartmentalization and the importance of regulating NE remodeling to prevent disease.
Interphase NE rupture and repair
Interphase NE rupture is defined as the loss of nucleus integrity due to membrane rupture in interphase and is characterized by rapid mislocalization of nuclear and cytoplasmic proteins in the absence of chromatin condensation [4–6]. Ruptures in the nuclear membranes typically occur at a single spot, often at sites of chromatin herniation [4–6]. Transient NE rupture, where membrane repair occurs within a few minutes to a few hours, has been observed in several conditions in vitro [9,10,17–21] (Figure 1). NE rupture without repair occurs frequently in micronuclei [7], small nuclei that form in addition to the primary nucleus as a result of chromosome missegregation. Micronuclei are distinct from nuclear buds, which often resemble micronuclei in shape but are connected to the primary nucleus via a thin chromatin bridge [22]. Micronuclei arise from many causes, including unrepaired DNA damage, defects in spindle assembly, and chromatin bridge breakage [23]. Persistent NE rupture in micronuclei has been observed in cultured cells after missegregation of whole chromosomes or acentric fragments, due to spindle misassembly or DNA damage, and in tumor cells and embryos in vivo [7,13,15,24,25]. In cycling cells the chromatin from disrupted micronuclei is not lost, but persists in the cytosol and frequently reincorporates into the primary nucleus in the next cell cycle [7,26].
Figure 1. Mechanism of nuclear envelope rupture.
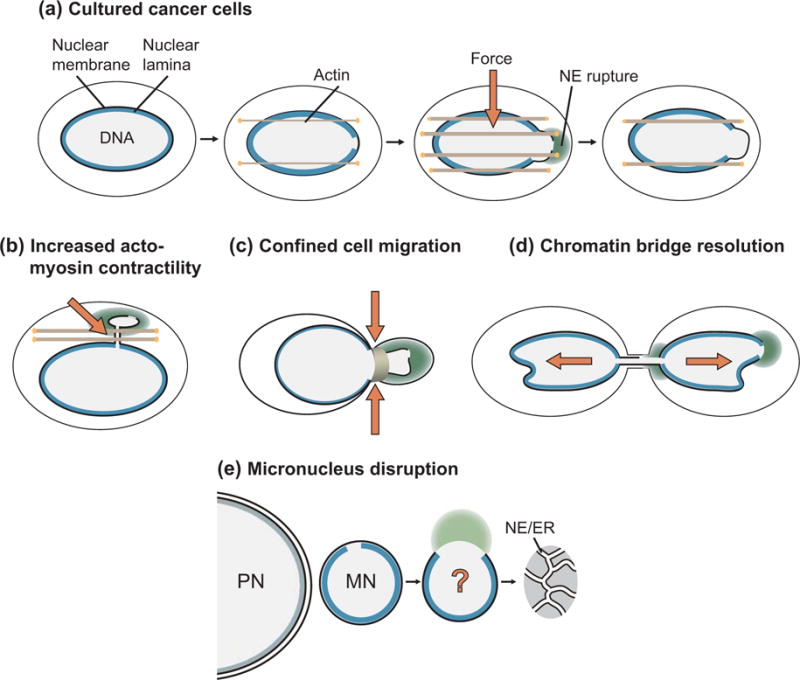
(a) Nuclear envelope rupture in cultured cancer cells occurs as a result of defects in nuclear lamina organization that give rise to gaps in the lamina meshwork. Confinement by actin bundles increases stress on the nuclear membrane and results in chromatin herniation and rupture of the nuclear membranes. After rupture, the nuclear membrane is resealed. Nuclear lamina defects and increased membrane stress are also associated with NE rupture in laminopathy mutations (not pictured) (b) increased actomyosin contractility, (c) cell migration through narrow channels, and (d) chromatin bridge resolution. In contrast, NE rupture in (e) micronucleus disruption requires nuclear lamina defects, but the role of membrane stress is unknown.
Analysis of NE rupture has identified two significant contributors to NE stability – nuclear lamina organization and mechanical stress (Figure 1). Membrane rupture occurs at the site of gaps in the nuclear lamina, which can appear after mitosis or form during mechanical stress [4,5,7,9,10,18–20,27]. In addition, membrane rupture can be inhibited by overexpressing lamin proteins [7,18,27], and occurs more frequently in cell types characterized by altered lamina structure [20] [28], indicating that nuclear lamina disorganization is required for NE rupture. NE rupture is frequently observed in nuclei experiencing significant mechanical stress [9,10,18–20,28,29], and a current model is that external force on the nucleus triggers chromatin herniation and membrane rupture at sites of nuclear lamina breaks. One exception to this model is micronucleus disruption, where membrane rupture frequently occurs hours after the appearance of lamina gaps and does not depend on actin-based forces [7,19]. The ESCRT-III membrane remodeling complex, which seals the NE after mitosis and in response to nuclear pore defects [30–32], is transiently recruited to sites of NE rupture and increases the efficiency of NE repair [9,10]. Nuclear lamina proteins are also recruited to the rupture site and persist after membrane resealing [10,33]. Although significant progress has been made on the mechanism of NE rupture and repair, many questions remain about the molecular and mechanical details of these processes, including how the membrane ruptures, how repair proteins are targeted to the membrane breaks, and whether membrane stability can be fully restored.
NE rupture and genome instability
The loss of nucleus integrity can have several impacts on genome stability, including altered nucleotide sequence, chromosome structure, and chromosome number (Figure 2). Several of these consequences were first described in micronucleus disruption. NE rupture without repair terminates many nuclear processes, including transcription and DNA replication [7,34], which can result in both temporary and heritable aneuploidy [12]. In addition, NE rupture during DNA replication in micronuclei is thought to cause extensive DNA damage, specifically double-stranded DNA breaks (DSBs), that cannot be repaired due to the loss of DNA damage repair proteins [7,12,26,34–36]. Consistent with multiple DNA breaks, micronucleated chromatin frequently appears fragmented in mitotic spreads [26,37,38]. However, NE disruption may not be required for this fragmentation. Cells containing intact micronuclei frequently enter mitosis before DNA replication has finished [26], which could cause chromosome fragmentation by a process called premature chromatin condensation [39]. In support of a mitotic damage mechanism, missegregation of the Y chromosome into micronuclei results in a high frequency of DNA fragmentation in mitosis, even though NE disruption frequency in interphase is low [38].
Figure 2. Nuclear envelope rupture and genome instability.
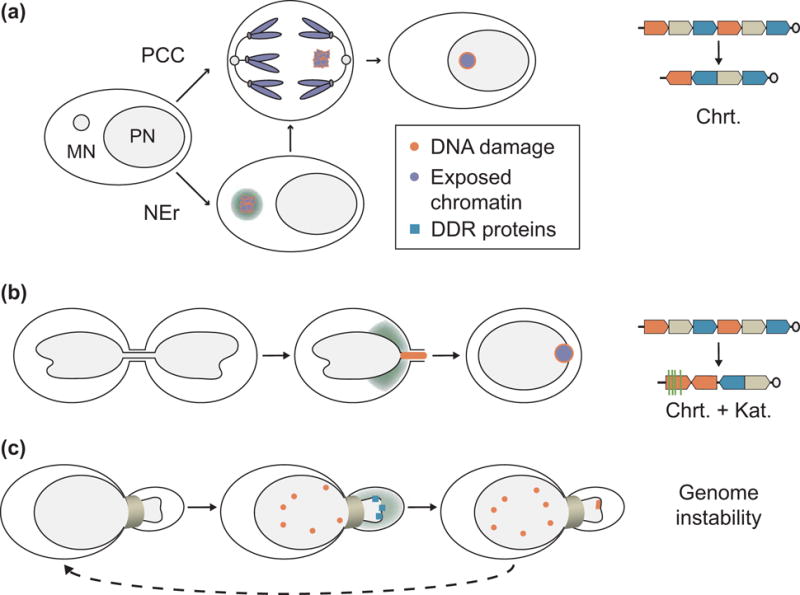
(a) Micronucleus mechanism of chromothripsis. Micronucleation of a chromosome leads to DNA damage and fragmentation after DNA replication initiation, either by NE rupture in interphase, or by premature chromatin condensation in mitosis. Chromothriptic are visible in the next cell cycle, likely as a result of the chromatin being reincorporated in the nucleus and exposed to DNA damage repair proteins. (b) Chromothripsis and kataegis after chromatin bridge resolution. Chromatin bridge breakage is preceded by NE rupture and results in DNA damage and both chromothriptic chromatin rearrangements and APOBEC-associated kataegis. (c) Transient NE rupture in cells migrating through narrow channels is associated with increased DNA damage and mislocalization of DNA damage repair proteins. Repeated migration leads to increased genetic diversity. APOBEC = apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like. Chrt = chromothriptic chromosome rearrangements; DDR = DNA damage repair; Kat = kataegis; NEr = nuclear envelope rupture; PPC = premature chromosome condensation.
Transient NE rupture in the primary nucleus can also cause DNA damage. Direct evidence of this process comes from analysis of chromatin bridge resolution after telomere fusion. Bridge breakage in this system is facilitated by DNA fragmentation by cytoplasmic nucleases, including TREX1, that gain access to the chromatin through NE rupture and cause DSBs [18]. Inhibiting NE rupture decreases the amount of ssDNA in the bridge, which is likely a key step in this process [18]. NE rupture and repair frequently precedes DSBs in migrating cells [9,10], but it is unclear whether this is the primary cause of DNA damage in these cells as nucleus deformation is also sufficient to induce DSBs [10]. Similar to persistent NE rupture in micronuclei, NE rupture in migrating cells may inhibit DNA damage repair by causing mislocalization of repair proteins to the cytoplasm [11]. These models of DNA damage after transient NE rupture raise several interesting questions. First, TREX1 is not required for bridge resolution, it only accelerates the timing of resolution [18], indicating that additional mechanisms for chromatin bridge resolution exist and may occur independently of NE rupture. In addition, DNA damage appears to be restricted to chromatin in the bridge in cells with telomere fusions [18], but frequently occurs all over the nucleus during confined migration [9,10]. This suggests two interesting possibilities: that the ability of NE rupture to cause DNA damage depends on pre-existing conditions in the nucleus, or that the consequences of NE rupture can be mitigated by undefined protective mechanisms.
DNA damage due to NE rupture is thought to be a critical initiating event for chromothripsis and kataegis, two “all at-once” complex genome alteration mechanisms frequently found together in broad array of cancer types [40–42]. In chromothripsis a subset of chromatin, e.g. a single chromosome arm up to a few chromosomes, becomes fragmented and is randomly religated [43]. In addition to DNA rearrangement, chromothripsis causes gene amplification by fragment circularization and gene loss by fragment deletion [44]. NE rupture in micronuclei and chromatin bridges is highly correlated with chromothripsis in human cells [12,18]. Chromothripsis also occurs after micronucleus induction in Arabidopsis thaliana [45], suggesting that chromosome rearrangement is a conserved consequence of micronucleation. Chromatin bridge resolution is also correlated with kataegis [18], a type of clustered hypermutation associated with APOBEC3 cytidine deaminase activity [46], suggesting that bridge breakage could be a mechanism linking chromothripsis and kataegis in cancer. Breakpoint analysis and DNA repair enzyme depletion experiments indicate that both non-homologous end-joining and microhomology-mediated pathways can contribute to chromosome reassembly [12,18,38,43,45] and further analysis of these pathways in chromothripsis is the subject of an excellent recent review [47]. One major question about these models is whether or not NE rupture is required for chromothripsis or kataegis. Analysis of genome stability after repeated rounds of confined migration suggest a link between conditions that increase NE rupture and changes in chromosome copy number [11], but additional studies will be important to define a causal connection between NE rupture, DNA damage, chromothripsis, and kataegis.
NE rupture and innate immunity
Several conditions, including DNA damage and expression of autoinflammatory disease mutations, activate the DNA sensor cGAS (cyclic GMP-AMP (cGAMP) Synthase) and cause an inflammatory response [13,15,16,48]. Recent studies suggest that NE rupture in micronuclei may be the critical trigger of cGAS activation in both these situations [13,15]. Ionizing radiation and loss of RNAseH2b activity, a model for the autoinflammatory disease Aicardi-Goutières Syndrome, both cause an increase in micronucleus frequency and induce a cGAS-STING (stimulator of interferon genes) dependent interferon and inflammatory response specifically in micronucleated cells [13,15]. A current model is that NE rupture leads to cGAS accumulation on micronucleated chromatin, which activates cGAS to produce cGAMP and signal to the STING adaptor protein, leading to expression of interferons and pro-inflammatory cytokines [13,15,49] (Figure 3a). In support of the micronucleation model of innate immune induction, DNA damage alone is insufficient to activate the cGAS-STING pathway, but inducing micronucleation on its own is sufficient to induce a pro-inflammatory response [13,15]. Recent analysis of cGAS activity in senescent cells also supports this model. The secretion of pro-inflammatory cytokines by senescent cells, called the SASP (senescence-associated secretory phenotype), requires the formation of cytosolic chromosome fragments (CCFs), which bind cGAS and activate the cGAS-STING pathway [49–51]. CCFs share many hallmarks of NE disruption with disrupted micronuclei, including γ-H2AX accumulation, loss of H3K9 acetylation, and loss of nuclear lamina proteins [7,52], but accumulate after the cells have stopped cycling and are thought to arise from NE budding [52]. Together, these results suggest that exposure of self-DNA to the cytoplasm by NE instability could be a widespread trigger of inflammation. cGAS also accumulates on exposed chromatin during transient NE rupture in the main nucleus [9,10,15,18], but whether this is sufficient to activate an inflammatory response is unknown.
Figure 3. Nuclear envelope rupture and innate immunity.
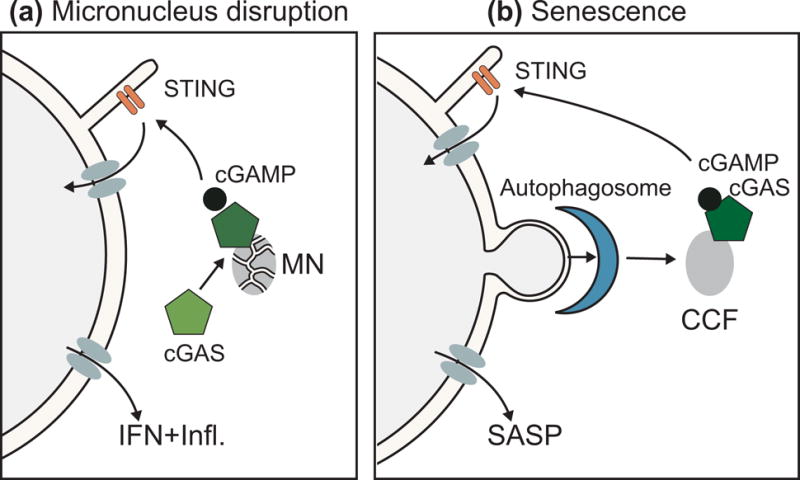
(a) Micronucleus model of cGAS-STING activation. Disruption of the micronucleus NE causes accumulation and activation of cGAS, which initiates interferon and pro-inflammatory responses via cGAMP production and STING activation. (b) Senescence model of cGAS-STING activation. Increased NE instability during senescence leads to budding off of nucleus material, association of chromatin with autophagosomes, and generation of cytoplasmic chromatin fragments that accumulate and activate cGAS, which initiates the senescence-associated secretory phenotype via STING. CCF = cytoplasmic chromatin fragments; cGAMP = cyclic GMP-AMP; cGAS = cyclic GMP-AMP Synthase; IFN = interferon response; Infl = pro inflammatory pathway activation; MN = micronuclei; NE = nuclear envelope; SASP = senescence-associated secretory phenotype; STING = stimulator of interferon genes.
The current studies identify a strong correlation between micronucleus formation and innate immune responses, but additional work will help determine whether cGAS accumulation on disrupted micronuclei is sufficient for cGAS-STING pathway activation. One unresolved question is the contribution of other cGAS-STING activating conditions to the immune response in micronucleated cells. Micronuclei frequently co-occur with aneuploidy because they form as a result of chromosome missegregation, and micronucleus disruption contributes to aneuploidy by interrupting DNA replication and transcription [7,12]. Recently, aneuploidy has been shown to cause upregulation of the cGAS-STING pathway [14], which suggests an alternative mechanism for the pro-inflammatory response in micronucleated cells. In addition, ionizing radiation causes senescence, which could activate cGAS-STING through CCF formation [49,50]. Thus, it will be important for future studies to tease apart the relative contributions of aneuploidy, DNA damage, senescence, and micronucleus disruption in these models.
Another outstanding question is whether negative regulators of auto-inflammation, such as TREX1 or autophagy, affect cGAS-STING pathway activation after micronucleus disruption. Digestion of cytosolic DNA by TREX1 is thought to be a critical mechanism to prevent cGAS activation by self-DNA [48]. TREX1 localizes to chromatin bridges and disrupted micronuclei [18], but its effect on cGAS activation from exposed chromatin is unclear. In the presence of TREX1, the interferon response occurs several days after irradiation and is linked to the presence of micronuclei [13]. However, irradiation of cells lacking TREX1 increases the amount of cytosolic DNA and leads to an interferon response within one day [16], which complicates assessment of the role of TREX1 activity in the slower micronucleus-mediated cGAS activation. Targeting of self-DNA to the lysosome by autophagy also prevents cGAS activation after DNA damage [53]. Whether autophagy targets disrupted micronuclei is likely to be dependent on the cellular environment. Disrupted micronuclei in cycling cells have little association with autophagy markers [26,54], but in senescent cells similar material in CCFs frequently co-localizes with them [55]. Autophagy is thought be required to activate cGAS-STING in senescent cells, via generation of CCFs [55], but stimulating autophagy in RNAseH2b knockout cells inhibits cGAS activation, via depletion of cytosolic DNA [25]. Thus, TREX1 and autophagy proteins appear to interact with chromatin uncovered by NE rupture, but further studies are needed to identify what conditions enable them to inhibit auto-inflammation.
Conclusions
The recognition that the NE is capable of extensive membrane remodeling in interphase, and that this behavior is linked to changes in nuclear lamina organization, has opened up a new model for how altered NE structure can cause disease. The recent studies linking NE rupture to increased genome instability and innate immune responses identify new potential molecular mechanisms for cancer evolution, radiotherapy efficacy, laminopathy symptoms, and autoinflammatory syndromes. At the same time, we are just beginning to see whether NE rupture can facilitate normal cell behaviors, such as migration of immune cells through dense tissues [9]. In addition, there are likely additional consequences of NE rupture that have yet to be defined. Loss of membrane integrity causes removal of histone acetyl groups in micronuclei [7] and chromatin compaction in both micronuclei and the primary nucleus [7,28]. These events could affect genome stability, gene regulation, and chromosome organization, but currently their consequences are unknown. Going forward, the challenge will be to determine which of the many consequences of nucleus integrity loss is responsible for persistent changes in the chromatin, the cell, and the organism. A second challenge will be to untangle the consequences of NE rupture from those of concurrent conditions, including karyotype changes and altered nuclear lamina structure. New studies on the mechanisms of NE rupture and repair will be critical to develop new tools to address these challenges. These types of studies will also add new information to fundamental questions in nuclear lamina biology, including how the lamin protein meshwork assembles, and how changes in lamina protein sequences can have multifaceted effects on the cell.
Acknowledgments
I thank Susan Parkhurst for helpful suggestions and comments on this manuscript. EMH is supported by a grant from the National Institutes of Health [1R35GM124766-01].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Added note
A recently published paper (Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018)) suggests that cGAS-STING activation by micronucleus formation and rupture may be critical for the initiation of metastasis, through STING-dependent upregulation of pro-metastatic genes. Activation of specific cell signaling pathways, i.e. non-cannonical NFκB, is thus a third consequence of nuclear membrane disruption, in addition to increased genome instability and induction of pro-inflammatory responses.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
• of outstanding interest
- 1.Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–164. doi: 10.1146/annurev-biochem-060614-034115. [DOI] [PubMed] [Google Scholar]
- 2.King MC, Lusk CP. A model for coordinating nuclear mechanics and membrane remodeling to support nuclear integrity. Curr Opin Cell Biol. 2016;41:9–17. doi: 10.1016/j.ceb.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ungricht R, Kutay U. Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2016.153. [DOI] [PubMed] [Google Scholar]
- 4.de Noronha CM, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, Greene WC. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294:1105–1108. doi: 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- 5.De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, Manders EMM, Verstraeten VLRM, van Steensel MAM, Marcelis CLM, et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet. 2011;20:4175–4186. doi: 10.1093/hmg/ddr344. [DOI] [PubMed] [Google Scholar]
- 6.Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus. 2012;3:88–100. doi: 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60. doi: 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah P, Wolf K, Lammerding J. Bursting the Bubble – Nuclear Envelope Rupture as a Path to Genomic Instability? Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil AM, Manel N, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016 doi: 10.1126/science.aad7611. With Denais et al. 2017, this study was the first to characterize nuclear envelope rupture and repair in cells migrating through small pores. [DOI] [PubMed] [Google Scholar]
- 10••.Denais CM, Gilbert RM, Isermann P, McGregor AL, Lindert Te M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016 doi: 10.1126/science.aad7297. This work, along with Raab et al demonstrated that nuclei transiently rupture when during migration through small pores, that NE rupture is associated with DNA damage, that cGAS accumulates at NE rupture sites, and that ESCRTIII increased membrane repair efficiency. In addition, these authors were the first to demonstrate that NE rupture and repair could occur in migrating cells in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, et al. DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Curr Biol. 2017;27:210–223. doi: 10.1016/j.cub.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13••.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. This study, along with Mackenzie et al 2017, was the first to suggest that micronuclei are critical for cGAS-STING activation after DNA damage and, along with Vanpouille-Box et al 2017, was the first to connect the cGAS-STING pathway to the abscopal tumor effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, Amon A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell. 2017;41:638–651.e5. doi: 10.1016/j.devcel.2017.05.022. This study carefully analyzes the effect of chromosome missegregation on genomic instability, cell cycle arrest, senescence, and immune responses. Their data suggest that cells do not count chromosomes, but that stress from aneuploidy leads to increased genomic instability, which triggers cell cycle arrest and clearance by natural killer cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;2:1199. doi: 10.1038/nature23449. This study, along with Harding et al 2017, was the first to show that disrupted micronuclei accumulate cGAS and that interferon and inflammatory responses after DNA damage are limited to micronucleated cells. The authors also demonstrated that cGAS is activated by chromatin in vitro and that a mouse model of inflammatory disease has an increased frequency of micronucleus formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618. doi: 10.1038/ncomms15618. This study, along with Harding et al 2017, connects cGAS-STING activation to the abscopal tumor effect. These authors also demonstrated that high levels of DNA damage induce TREX1 and dampen this effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatch EM, Hetzer MW. Breaching the nuclear envelope in development and disease. J Cell Biol. 2014;205:133–141. doi: 10.1083/jcb.201402003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Hatch EM, Hetzer MW. Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol. 2016;114 doi: 10.1083/jcb.201603053. jcb.201603053. This study demonstrated that NE rupture requires membrane tension, and that this tension in 2D culture is induced by formation of perinuclear actin bundles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Takaki T, Montagner M, Serres MP, Le Berre M, Russell M, Collinson L, Szuhai K, Howell M, Boulton SJ, Sahai E, et al. Actomyosin drives cancer cell nuclear dysmorphia and threatens genome stability. Nat Commun. 2017;8:16013. doi: 10.1038/ncomms16013. This study demonstrated that overactivation of actomyosin contractility in cultured cells causes the formation of nuclear buds that rupture frequently and induces DNA damage. These authors also linked proper nuclear lamina organization to decreased sensitivity to increased actomyosin contractility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z, Maciejowski J, de Lange T. Nuclear envelope rupture is enhanced by loss of p53 or Rb. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-17-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montero-Montoya R, Serrano L, Ostrosky-Wegman P. In vitro induction of micronuclei in lymphocytes: the use of bromodeoxyuridine as a proliferation marker. Mutat Res. 1997;391:135–141. doi: 10.1016/s1383-5718(97)00060-0. [DOI] [PubMed] [Google Scholar]
- 23.Fenech M, Knasmueller S, Bolognesi C, Bonassi S, Holland N, Migliore L, Palitti F, Natarajan AT, Kirsch-Volders M. Molecular mechanisms by which in vivo exposure to exogenous chemical genotoxic agents can lead to micronucleus formation in lymphocytes in vivo and ex vivo in humans. Mutat Res. 2016;770:12–25. doi: 10.1016/j.mrrev.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 24.Vázquez-Diez C, Yamagata K, Trivedi S, Haverfield J, FitzHarris G. Micronucleus formation causes perpetual unilateral chromosome inheritance in mouse embryos. Proc Natl Acad Sci U S A. 2016;113:626–631. doi: 10.1073/pnas.1517628112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A, et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet. 2017;26:3960–3972. doi: 10.1093/hmg/ddx283. [DOI] [PubMed] [Google Scholar]
- 26.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus. 2012;3:88–100. doi: 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robijns J, Molenberghs F, Sieprath T, Corne TDJ, Verschuuren M, De Vos WH. In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. [Internet] Scientific Reports. 2016;6:30325. doi: 10.1038/srep30325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamiello C, Kamps MAF, van den Wijngaard A, Verstraeten VLRM, Baaijens FPT, Broers JLV, Bouten CCV. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus. 2013;4:61–73. doi: 10.4161/nucl.23388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522:236–239. doi: 10.1038/nature14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, Stenmark H. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522:231–235. doi: 10.1038/nature14408. [DOI] [PubMed] [Google Scholar]
- 32.Webster BM, Thaller DJ, Jäger J, Ochmann SE, Borah S, Lusk CP. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016;35:2447–2467. doi: 10.15252/embj.201694574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Berre M, Aubertin J, Piel M. Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr Biol (Camb) 2012;4:1406–1414. doi: 10.1039/c2ib20056b. [DOI] [PubMed] [Google Scholar]
- 34.Terradas M, Martin M, Hernandez L, Tusell L, Genesca A. Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat Res. 2012;729:35–40. doi: 10.1016/j.mrfmmm.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Medvedeva NG, Panyutin IV, Panyutin IG, Neumann RD. Phosphorylation of histone H2AX in radiation-induced micronuclei. Radiat Res. 2007;168:493–498. doi: 10.1667/RR0788.1. [DOI] [PubMed] [Google Scholar]
- 36.Yoshikawa T, Kashino G, Ono K, Watanabe M. Phosphorylated H2AX foci in tumor cells have no correlation with their radiation sensitivities. J Radiat Res. 2009;50:151–160. doi: 10.1269/jrr.08109. [DOI] [PubMed] [Google Scholar]
- 37.Sandberg AA, Aya T, Ikeuchi T, Weinfeld H. Definition and morphologic features of chromosome pulverization: a hypothesis to explain the phenomenon. JNCI J Natl Cancer Inst. 1970;45:615–623. [PubMed] [Google Scholar]
- 38•.Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol. 2016 doi: 10.1038/ncb3450. This study devised an elegant method to inducibly micronucleate the Y chromosome and demonstrate a role for non-homologous end joining in repairing shattered chromatin fragments after mitosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson RT, Rao PN. Mammalian Cell Fusion: Induction of Premature Chromosome Condensation in Interphase Nuclei. Nature. 1970;226:717–722. doi: 10.1038/226717a0. [DOI] [PubMed] [Google Scholar]
- 40.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg Å, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. doi: 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maciejowski J, Imielinski M. Modeling cancer rearrangement landscapes. Current Opinion in Systems Biology. 2017;1:54–61. doi: 10.1016/j.coisb.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rode A, Maass KK, Willmund KV, Lichter P, Ernst A. Chromothripsis in cancer cells: An update. Int J Cancer. 2016;138:2322–2333. doi: 10.1002/ijc.29888. [DOI] [PubMed] [Google Scholar]
- 45.Tan EH, Henry IM, Ravi M, Bradnam KR, Mandakova T, Marimuthu MP, Korf I, Lysak MA, Comai L, Chan SW. Catastrophic chromosomal restructuring during genome elimination in plants. eLife Sciences. 2015;4:82. doi: 10.7554/eLife.06516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikkilä J, Kumar R, Campbell J, Brandsma I, Pemberton HN, Wallberg F, Nagy K, Scheer I, Vertessy BG, Serebrenik AA, et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br J Cancer. 2017;117:113–123. doi: 10.1038/bjc.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ly P, Cleveland DW. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol. 2017;35:313–336. doi: 10.1146/annurev-immunol-051116-052331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. This study, along with Gluck et al 2017 and Yang et al 2017, was the first to connect the SASP response in senescent cells to cGAS-STING pathway activation. The authors demonstrated that cGAS accumulates on CCFs and that CCFs are required for cGAS-STING dependent activation of the SASP and maintenance of senescence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–1070. doi: 10.1038/ncb3586. Along with Dou et al 2017, this study connected cGAS accumulation on senescent chromatin to the cGAS-STING-dependent SASP response in vitro and in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013;202:129–143. doi: 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lan YY, Londoño D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014;9:180–192. doi: 10.1016/j.celrep.2014.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Mariño G, Vitale I, Jemaá M, Harper F, Pierron G, et al. Autophagic removal of micronuclei. Cell Cycle. 2012;11:170–176. doi: 10.4161/cc.11.1.18564. [DOI] [PubMed] [Google Scholar]
- 55.Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109. doi: 10.1038/nature15548. [DOI] [PMC free article] [PubMed] [Google Scholar]