

Abstract
A protein-derived cofactor is a catalytic or redox-active site in a protein that is formed by post-translational modification of one or more amino acid residues. These post-translational modifications are irreversible and endow the modified amino acid residues with new functional properties. This Perspective focusses on the following advances in this area that have occurred during recent years. The biosynthesis of the tryptophan tryptophylquinone (TTQ) cofactor is catalyzed by a di-heme enzyme, MauG. A bis-FeIV redox state of the hemes performs three two-electron oxidations of specific Trp residues via long-range electron transfer. In contrast, a flavoenzyme catalyzes the biosynthesis of the cysteine tryptophylquinone (CTQ) cofactor present in a newly discovered family of CTQ-dependent oxidases. Another carbonyl cofactor, the pyruvoyl cofactor found in classes of decarboxylases and reductases, is formed during an apparently autocatalytic cleavage of a precursor protein at the N-terminus of the cleavage product. It has been show that in at least some cases, the cleavage is facilitated by binding to an accessory protein. Tyrosylquinonine cofactors, topaquinone (TPQ) and lysine tyrosylquinone (LTQ) are found in copper-containing amine oxidases and lysyl oxidases, respectively. The physiological roles of different families of these enzymes in humans has been more clearly defined and shown to have significant implications towards human health. There has also been continued characterization of the roles of covalently cross-linked amino acid side-chains that influence the reactivity of redox-active metal centers in proteins. These include Cys-Tyr species in galactose oxidase and cysteine dioxygenase, and the Met-Tyr-Trp species in the catalase-peroxidase KatG.
Graphical Abstract
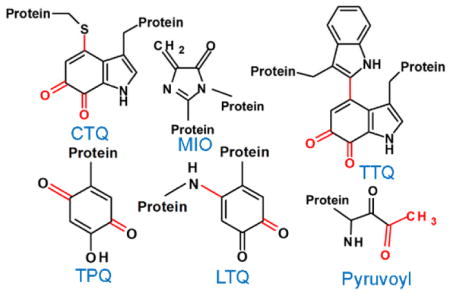
The range of chemical reactions that can be catalyzed by enzymes and the diversity of mechanisms used for catalysis continue to expand. This trend will likely continue given the large number of open reading frames in the databases of sequenced genomes that are annotated as proteins of unknown function. In many cases, enzymes require cofactors to perform these remarkable tasks. Typically, cofactors are organic molecules, metals or organometallic compounds that are acquired from the environment. In some cases, the need for an external cofactor is circumvented by the construction of a protein-derived cofactor. A protein-derived cofactor is a catalytic or redox-active site in a protein that is formed by post-translational modification of one or more amino acid residues. These post-translational modifications are irreversible and endow the modified residues with new functional properties.
In 2007, I reviewed the status of the field of protein-derived cofactors in a “Current Topics” published in Biochemistry1. This Perspective focusses on advances that have occurred since that time. Significant advances have been made in elucidating the mechanism of biosynthesis of protein-derived cofactors, especially in cases where the post-translational modifications that generate the cofactor are catalyzed by another enzyme, rather than being autocatalytic. This is best evidenced by studies of the biosynthesis of the tryptophylquinone cofactors, tryptophan tryptophylquinone (TTQ) and cysteine tryptophylquinone (CTQ). There have been surprising new findings related to the mechanism of biosynthesis of the pyruvoyl cofactor, one of the first such protein-derived cofactors to be described. There is now evidence that the biogenesis of the cofactor is not an autocatalytic event, as was long believed, but requires an accessory protein to facilitate the event. There has also been a clear definition that there are two discrete classes of these enzymes; decarboxylases that possess only the pyruvoyl cofactor and reductases that also possess a catalytic selenocysteine. There has been progress in identifying previously unrecognized biological roles for enzymes that used protein-derived cofactors. This has been especially true for the human enzymes that use tyrosylquinone cofactors, topaquinone (TPQ) and lysine tyrosylquinone (LTQ). There has also been continued characterization of the roles of covalently cross-linked amino acid side-chains that influence the reactivity of redox-active metal centers in proteins.
TRYPTOPHYLQUINONE COFACTORS
Tryptophan tryptophylquinone (TTQ)
In recent years, the complex mechanism of the enzyme-catalyzed formation of TTQ in methylamine dehydrogenase (MADH) from Paracoccus denitrificans has been described in detail. Biosynthesis of TTQ from two specific Trp residues requires insertion of two oxygen atoms into the indole ring of one of the Trp residues and formation of a covalent crosslink to the indole ring of the other2 (Figure 1). It had been known for many years that this process required a modifying enzyme, from work done by van Spanning and coworkers3, 4. The genes that encode the two MADH subunits, together with nine other genes related to MADH, reside in the methylamine utilization (mau) locus in P. denitrificans. A role in TTQ biosynthesis was demonstrated for mauG, as when this gene was knocked out, the MADH subunits produced by the bacterium lacked both activity and the absorbance spectrum characteristic of TTQ.4 Its gene product, MauG, was recombinantly expressed, purified, and shown to be di-heme enzyme with c-type hemes.5 It was also possible to express a precursor protein of MADH in the absence of mauG. The precursor had a Trp which was mono-hydroxylated, but lacked the additional post-translational modifications required for TTQ maturation.6 From these results, the physiologic role of MauG was known, but questions remained. What redox state of MauG would be generated with a sufficiently high redox potential to perform these oxidation reactions? How could the activated hemes interact with the substrate amino acid residues located within another protein?
Figure 1.

Tryptophylquinone cofactors
The reactive intermediate of MauG that oxidizes the protein substrate was shown to be a previously undetected high-valent iron species, a bis-FeIV species7 in which both heme irons are FeIV. The crystal structure of MauG revealed that in the oxidized state, one heme is five coordinate with an axial His ligand and the other is six-coordinate with axial His and Tyr ligands.8 The heme irons are separated by 21 Å with a Trp residue between the hemes (Figure 2). Spectroscopic studies showed that in the diferric state the five coordinate heme is low-spin and the six-coordinate heme is high-spin.5 In the bis-FeIV state, the two ligands to the six-coordinate heme are retained and oxygen binds to the open site on the five-coordinate heme (Figure 2). The two FeIV hemes are stabilized by a charge-resonance transfer process that is mediated by the intervening Trp.7, 9 Site-directed mutagenesis studies identified roles of several amino acid residues in controlling the redox properties and reactivity of this di-heme redox center.10–14
Figure 2.

The hemes of MauG. The structure of MauG (PDB entry 3L4M) is in gray with the two hemes and intervening Trp93 in stick. In the inset is a representation of the bis-FeIV diheme system.
A clue as to how the hemes oxidize the specific residues on the precursor protein was provided from a crystal structure of a complex of MauG and the TTQ-lacking precursor of MADH (preMADH).8 Surprisingly, that structure revealed that the modified residues, βTrp57 and βTrp108, do not make direct contact with either heme of MauG. In fact, the distance separating the βTrp108 of preMADH and the iron of the oxygen-binding five-coordinate heme is 40.1 Å, and the closest distance to the iron of the other heme is 19.4 Å (Figure 3). While this might suggest that the complex that crystallized was not physiologically relevant, it was shown that addition of H2O2 to the crystal resulted in formation of the mature TTQ cofactor.8, 15 Thus, the reaction does occur over long distance in this complex in this orientation. Site-directed mutagenesis and kinetic studies16, 17 described a process in which the bis-FeIV hemes oxidize the residues that form TTQ on precursor protein via long-range electron transfer, which proceeds via a hole-hopping mechanism16, 17 (Figure 4). The hopping is mediated by another Trp residue on the surface of MauG at the interface with preMADH.
Figure 3.

The structure of the complex of preMADH and MauG. The structure is composed of two MADH α subunits (blue), two MADH β subunits (green) and two MauG molecules (red). The hemes and residues βTrp57 and βTrp108 (preTTQ) are black and in stick. This was drawn using the coordinates in PDB entry 3L4M.
Figure 4.

Electron transfer required for oxidation of preMADH by bis-FeIV MauG. The orientation of the residues are as they are in the structure (PDB entry 3L4M). Residues βTrp57 and βTrp108 that form TTQ (preTTQ) are shown on the left and all other residues are from preMADH. The proposed hole-hopping segments are indicated by the curved arrows. The charge resonance stabilization of the two FeIV hemes is indicated by the double arrow line.
The overall TTQ biosynthetic reaction requires multiple two-electron oxidations of the precursor by MauG. It was possible to characterize the intermediate radical species on the MADH precursor that are generated during this process, including a novel di-Trp radical species15 (Figure 5) and observe reaction intermediates in crystallo15. The order of reaction steps is cross-link formation between the mono-hydroxylated and unmodified Trp residues, followed by the second hydroxylation of the cross-linked intermediate, followed by oxidation of the quinol to the TTQ.
Figure 5.

The overall reaction of TTQ formation from preTTQ. The initial two-electron oxidation proceeds through a di-radical intermediate as shown. After cross-link formation, two additional two-electron oxidations occur. The post-translational modifications are shown in red.
An unexpected finding during the studies of MauG-dependent TTQ biosynthesis was that when the bis-FeIV state was generated in the absence of the precursor protein substrate, MauG damaged itself by oxidizing specific Met residues.18 This occurs through an unusual mechanism of proton-coupled electron transfer19, 20 where the source of the electron is a Met residue and the source of the proton is solvent. The results of these studies described how the protein controls the reactivity of radical intermediates that are centered on amino acid residues and directs them towards catalysis rather than oxidative damage.
Cysteine tryptophylquinone (CTQ)
The biosynthesis of CTQ requires insertion of two oxygen atoms into the indole ring of a Trp residue, and formation of a covalent cross-link between the indole ring and the sulfur of a Cys residue21 (Figure 1). Given the remarkable and complex mechanism of TTQ biosynthesis that was described for MADH, one might assume that a similar mechanism would be common to other tryptophylquinone-dependent enzymes. Recent studies of LodA-like proteins, however, have shown this not to be the case. These proteins comprise a recently identified family of enzymes22 predicted from their sequence to possess CTQ. These enzymes are interesting in light of their function. The members of this class that have been thus far functionally characterized are amino acid oxidases. This is unusual because all previously characterized TTQ- and CTQ-dependent enzymes are dehydrogenases. Furthermore, these are the first amino acid oxidases to be described that use something other than a flavin as their cofactor.23 LodA-like proteins are named after the first member of this group to be described, LodA from Marinomonas mediterranea,24 the gene product of lodA. In contrast to the mau gene cluster, which contains several genes including mauG, only one other gene is present in the same operon with lodA. That gene, lodB, was predicted to encode a flavoprotein. It was shown that lodA expression in the absence of lodB produces an inactive precursor protein with no chromophore, both in the native bacterium25 and in a recombinant E. coli expression system26. Active LodA was only obtained when lodA was co-expressed with lodB. Interestingly, the precursor of LodA that was expressed in the absence of lodB had a mono-hydroxlated Trp residue26, as was observed for the precursor of MADH that was expressed in the absence of MauG6. Thus, while the modifying enzyme required for CTQ biosynthesis in LodA is a flavoenzyme, rather than the di-heme enzyme required for TTQ biosynthesis, the mono-hydroxylation of the precursor protein in the absence of its modifying enzyme is observed in both systems.
Genes predicted to encode LodA-like proteins have been identified in several classes of bacteria and fungi. In each case, a gene similar to lodB is present in the operon. Phylogenetic analysis using the Integrated Microbial Genomes database performed in 2015 revealed 168 LodA-like proteins that could be clustered in five different major groups, I-V22. Group I LodA-like proteins are all believed to be lysine ε-oxidases, as this was shown for the proteins synthesized by M. mediterranea27 and Pseudoalteromonas tunicate28. A LodA-like protein from Group II is GoxA from M. mediterranea, which is a glycine oxidase29. It was shown for GoxA that production of the active enzyme with the mature CTQ cofactor required co-expression of the goxA gene with goxB, which is analogous to lodB.26, 29 Co-expression of goxA and goxB from Pseudoalteromonas luteoviolacea also produced a GoxA with glycine oxidase activity30, suggesting that all Group II LodA-like proteins may be glycine oxidases. Despite their similarities, it was shown that lodB and goxB cannot substitute for each other, and each is specific for maturation of LodA and GoxA, respectively.26
Of particular interest is the multi-functionality of the LodA-like proteins. The protein must provide a scaffold for CTQ biosynthesis from specific Cys and Trp residues to form the active site, and provide a pathway for electron transfer between the flavin on the modifying enzyme and the residues that are oxidized during CTQ formation. The protein must also provide a site for controlled oxygen-dependent reactivity of the mature CTQ. How such versatility evolved within a single enzyme active site is of considerable interest from a protein structure/function perspective. Each of the four LodA-like proteins characterized thus far is an amino acid oxidase. This is unusual because all previously characterized amino acid oxidases utilize a flavin cofactor. In fact, enzymes that function as oxidases in general, typically require either a bound metal, organometallic cofactor or a flavin cofactor.31, 32 The crystal structures of LodA from M. mediterranea (PDB entry 3WEU)33 and GoxA from P. luteoviolacea (PDB entry 6BYW)30 revealed that they possess none of these cofactors or metals, but only the protein-derived CTQ cofactor. Furthermore, all previously characterized TTQ and CTQ enzymes that are not LodA-like proteins are dehydrogenases, and are very stable in their quinol states in the presence of O2.
The results obtained thus far for LodA-like proteins raise interesting questions. What features of the active sites of the LodA-like proteins allow them to function as oxidases rather than dehydrogenases? What exactly is the mechanism of oxidation of the quinol by O2? The overall crystal structure of LodA33 (Figure 6A) looks nothing like that of the TTQ-bearing MADH34 (Figure 3). In LodA, there is much less interaction between the identical subunits, which appear to be interacting via long antiparallel beta strand extensions. Despite the differences in the overall structures, features of the active site structure are conserved. The relative positions of the TTQ/CTQ and an Asp residue that is conserved in all tryptophylquinone enzymes are structurally conserved (Figure 6B). The other potentially reactive residue, Asp in MADH and Cys in LodA, also are conserved in position. Thus far, steady-state kinetic studies of the reactions catalyzed by LodA from M. mediterranea27 and GoxAs from M. mediterranea29 and P. luteoviolacea30 have been reported and site-directed mutagenesis studies have identified roles for specific residues in influencing CTQ biosynthesis, kinetic parameters of the mature enzymes and the extent of cooperativity toward the amino acid substrate29, 35.
Figure 6.

Structure of LodA. A. The structure of LodA (PDB entry 3WEU) with two identical subunits colored red and blue. B. The tryptophylquinone cofactor and nearby residues in the active sites of LodA and MADH (PDB entry 2BBK) are superimposed.
The post-translational modifications to form CTQ in LodA and GoxA each require a flavoenzyme (LodB or GoxB). For each of these it has been shown that the initial hydroxylation of the Trp occurs prior to the post-translational modifications that are catalyzed by the LodB and GoxB.26 Despite the many distinctions between the LodA-like proteins and TTQ-dependent enzymes, they share this feature. A long-standing question initially raised during studies of the biosynthesis of TTQ in MADH36 is how this seemingly impossible reaction occurs; the auto-catalytic hydroxylation of a specific tryptophan residue of a precursor protein at a specific position on the indole side chain. Study of an D512A variant of LodA provided a clue as to how this may happen.37 The majority of the purified protein was the completely unmodified form without the initial hydroxylation, but a small fraction contained active protein with CTQ. Metal analysis revealed that the active form contained weakly-bound copper. The unmodified protein contained no copper but a variety of other metals. This led to the hypothesis that copper is required for the initial hydroxylation of the unmodified precursor, and is expelled from the active site after mature CTQ is formed. The presence of Asp512 likely dictates the selectivity for Cu binding in the unmodified protein. In the D512A LodA, the selectivity is lost so many different metals could bind, and only in the small fraction that randomly bound Cu did the hydroxylation occur allowing the subsequent LodB-dependent post-translational modifications during expression of the protein. The position of this Asp is conserved in all TTQ and CTQ enzymes that have been structurally characterized.21, 30, 33, 34, 38, 39 The proposed mechanism involves a Cu3+-OH intermediate that oxidizes the Trp. While this proposed organometallic intermediate mechanism is uncommon, it has been reported that Cu3+-OH can stabilize intermediates with an aromatic ring via such an organometallic complex40. The intermediate was proposed to be stabilized by a Cys sulfur, which could be the Cys that later becomes part of CTQ, as it was shown for LodA that mutation of this residue yielded a precursor that lacked any post-translational modifications.26
PROTEIN-DERIVED CARBONYL COFACTORS
Pyruvoyl cofactor
Contrary to its name, the pyruvoyl cofactor41 is not derived from external pyruvate. It is formed by post-translational modification of an internal amino acid residue that occurs during cleavage of an inactive precursor protein that yields a two-subunit active enzyme. Enzymes that possess a pyruvoyl cofactor may be divided into two classes. One class is comprised of decarboxylases from bacterial and eukaryotic sources. This includes a variety of metabolic enzymes, such as aspartate decarboxylase,42 phosphatidylserine decarboxylases,43, 44 S-adenosylmethionine decarboxylase,45 and histidine decarboxylase.46 The other class contains reductases such as glycine reductase47 and D-proline reductase,48 which are found in Clostridium difficile, as well as other pathogenic bacteria. While each class uses the pyruvoyl cofactor for catalysis, they have major structural distinctions. The decarboxylases are comprised of two subunits that are derived from the single precursor protein, and require only the pyruvoyl cofactor for catalysis. The reductases have two analogous subunits derived from a precursor, but also have an additional subunit encoded by another gene. This additional subunit possesses a selenocysteine residue, which is also required for catalysis by the reductases.
It has long been assumed that processing of the pre-proteins of pyruvoyl enzymes to generate the α and β subunits with the pyruvoyl cofactor at the N-terminus of the α subunit was an autocatalytic event.49 The reaction mechanism for the cleavage of the pre-protein and formation of the pyruvoyl cofactor is well established (Figure 7). The side-chain hydroxyl group of a specific serine residue supplies an oxygen that becomes part of the C-terminus of the β-chain. Ammonia is released and the remainder of the Ser is transformed into the pyruvoyl group at the N terminus of the α-chain. The initial reaction step forms a cyclic oxyoxazolidine intermediate, which then forms an ester intermediate.50 The ester undergoes β-elimination releasing the β-chain and leaving dehydroalanine at the N-terminus of the α-chain. After addition of water and loss of ammonia, the pyruvoyl cofactor is formed at the end of the α-chain in the mature enzyme. The importance of this specific Ser was confirmed by site-directed mutagenesis of this residue in the S-adenosylmethionine decarboxylase pro-enzyme. Conversion of this Ser to Ala prevented the processing and the formation of active enzyme.51 This made it possible to determine the crystal structure of the unprocessed pro-enzyme.52
Figure 7.

Reaction mechanism of pre-protein cleavage and pyruvoyl cofactor generation.
Recently, multiple groups presented evidence for the requirement of a previously unrecognized accessory protein for processing of the pre-proteins of the decarboxylase members of this class of enzymes.53–56 The most compelling evidence was obtained for PanD, an aspartate α-decarboxylase that is present in the pantothenate biosynthesis pathway.53, 57 A crystal structure was presented of a complex of the uncleaved precursor of PanD with its accessory protein, PanZ. Activation of this complex also requires Coenzyme A, the cofactor that is derived from pantothenate. Formation of this complex was shown to stabilize a conformation of PanD in which the serine hydroxyl group is in close proximity to the adjacent carbonyl group, thus facilitating the formation of the initial oxyoxazolidine intermediate.
In contrast to the pyruvoyl-dependent decarboxylases, the site of internal cleavage and modification of these reductases is believed to be a Cys rather than a Ser.47, 48 A chemical reaction mechanism similar to that in Figure 7 could be imagined with a Cys side chain, but this has not yet been proven. Further distinction of the reductases is the presence of another selenocysteine-bearing residue in the mature active reductase. The physiological roles of the reductases are also quite different from the decarboxylases. C. difficile has been classified as a Stickland fermentor.58 This refers to bacteria that catalyze the coupled oxidation and reduction of amino acids (Stickland reactions) during their fermentation. D-proline reductase and glycine reductase are key enzymes in this metabolic process for their respective amino acid substrate.58, 59 It was shown that C. difficile thrives in culture using glycine or D-proline as an electron acceptor.60 Thus, these enzymes likely allow C. difficile to thrive in the colon, an abundant source of these amino acids.61 A study of C. difficile gene expression in animals revealed that the genes encoding D-proline reductase are differentially up-regulated during colonization.62 Recently, it was shown that if incorporation of selenium into selenoproteins in C. difficile is blocked, this leads to defects in growth in proline-rich medium.63
4-Methylideneimidazole-5-one (MIO)
MIO was first discovered in the crystal structures of two ammonia lyases; phenylalanine ammonia-lyase64 and histidine ammonia lyase.65 MIO was subsequently found in the structure of SgcC4 L-tyrosine 2,3-aminomutase.66 MIO is formed through autocatalytic post-translational modification of a sequence of three consecutive amino acid residues: Ala, Ser and Gly (Figure 8).67, 68 The process involves peptide cyclization and dehydration reactions. Alternative mechanisms have been proposed for catalysis by MIO-dependent enzymes.69 In each mechanism, the reactions are initiated by covalent adduct formation between MIO and the amino acid substrate. These enzymes have attracted a great deal of interest for applications in biocatalysis in biotechnology and medicine.70
Figure 8.

Formation of MIO.
TYROSYLQUINONE COFACTORS
2,4,5-Trihydroxyphenylalanine-quinone (topaquinone or TPQ)
TPQ has been found in copper-containing amine oxidases from bacterial, mammalian and plant sources.71 The formation of TPQ requires the insertion of two oxygen atoms into the phenyl ring of a Tyr residue72 (Figure 9). The mechanism of TPQ biosynthesis, which is summarized in Figure 10, was established some time ago as an autocatlytic event that requires copper and O2.73–77 Copper binds near the Tyr that becomes TPQ. Deprotonation of the tyrosine yields a charge-transfer complex between the tyrosinate and Cu2+, which is in resonance stabilization with a Cu+-tyrosyl radical complex. Reaction of this species with O2 yields superoxide, which forms a copper-coordinated peroxide intermediate that breaks down to form a dopaquinone (4,5-dihydroxyphenylalanine quinone) intermediate. It is then believed that the side-chain of the dopaquinone rotates 180° around its Cβ–Cγ bond,75, 78 followed by addition of hydroxide. Then oxidation by another O2 yields TPQ. An interesting feature of the TPQ-dependent enzymes is that the copper required for TPQ biosynthesis remains bound at the active site and participates in catalysis. The role in catalysis appears to be stabilization of transient intermediates in the oxidative half-reaction.79 Thus, copper serves dual roles in cofactor biosynthesis and catalysis.
Figure 9.

Tyrosylquinone cofactors
Figure 10.

Autocatalytic biosynthesis of TPQ
In recent years, the physiological roles of TPQ-containing copper amine oxidases in humans have been more clearly defined.80 Three classes of these enzymes have been described. The class 1 enzymes are referred to as diamine oxidases, and are responsible for the metabolism of histamine.81 They are expressed in and secreted from the kidney, and are sometimes referred to as kidney amine oxidases. They are present in high concentrations in the digestive tract and are highly expressed in the placenta. Low levels in the placenta have been associated with high-risk pregnancies.82 Decreased metabolic capacity of this oxidase has also been associated with hypersensitivity to non-steroidal anti-inflamatory drugs.83 The class 2 enzyme are sometimes called retina-specific amine oxidases. That is because activity has only been reported in the retina, although the gene appears to be expressed in other issues. The class 3 enzymes have held many names including, plasma amine oxidase and semicarbazide sensitive amine oxidase. They are now referred to as vascular adhesion protein or VAP-1.84 These are membrane-bound proteins that are expressed in many tissues. They are released into the serum after cleavage of the protein by a protease. Elevated serum levels of this soluble form have been associated with a variety of medical conditions, including type I85 and type II86 diabetes, hemorrhagic stroke,87 liver disease88 and kidney disease.89 These and other such findings have implicated these TPQ-dependent enzymes as potential biomarkers and therapeutic targets for a variety of disorders.
Lysine Tyrosylquinone (LTQ)
LTQ was originally described as the protein-derived cofactor of mammalian lysyl oxidase (LOX), which is required for the formation of connective tissue. LOX catalyzes the post-translational modification of elastin and collagen.90 It oxidizes peptidyl Lys residues to peptidyl α-aminoadipic δ-semialdehyde, the first step in the formation of cross-links that insolubilize these proteins. As with the TPQ enzymes, copper is also present in the active site.
LTQ is formed by post-translational modifications in which one atom of oxygen is incorporated into the phenyl ring of a Tyr, and a covalent bond is formed between the C5 carbon of the tyrosine ring and the side-chain nitrogen of a Lys residue91 (Figure 9). This bond is formed at the same position at which the second oxygen atom is added to complete the biosynthesis of TPQ. The exact mechanism of LTQ biogenesis has not yet been characterized, but there is evidence92 to suggest that the LTQ mechanism biosynthesis is similar to that described above for TPQ. In this case, the dopaquinone intermediate reacts with the amino nitrogen of a Lys side-chain rather than hydroxide.
In recent years it has been determined that LOX is just one of a family of human proteins that includes four additional lysyl oxidase-like proteins (LOXL1, LOXL2, LOXL3 and LOXL4).80 Although these are secreted proteins, intracellular functions of the LOX family have been identified, notably as transcription regulators. Some of these enzymes have been shown to localize in the nucleus and use histones as substrates suggesting an epigenetic role.93 Altered levels of expression of LOX family proteins have been linked to multiple types of cancers as well as other adverse medical conditions.80 Stiffening of the extracellular matrix caused by the activity of LOX proteins has been associated with cancer94, 95 and medical conditions including pulmonary fibrosis.96
CROSS-LINKED AMINO ACIDS IN METALLOPROTEINS
Cys-Tyr cofactor
The crystal structure of galactose oxidase97 revealed that the active site contained Cys and Tyr which were cross-linked by a thioether bond between the side chains. A copper, which was coordinated by two His residues, was also coordinated to the oxygen on the Tyr side chain. Formation of this cross-link was shown to be autocatalytic, requiring only copper and O2. Furthermore, the reaction was far more efficient when Cu1+ was used.98 A proposed mechanism is summarized in Figure 11. It begins with Cu1+ initially coordinated with the unmodified Tyr. Reaction of Cu1+ with O2 yields Cu2+ coordinated to superoxide, which abstracts a hydrogen atom from a nearby Cys to generate a thiyl free radical. This adds to the aromatic ring of Tyr to form the cross-link while the copper remains coordinated with the modified Tyr. This species may also be converted to a Cu2+-phenoxy radical. An analogous Cys-Tyr-copper cofactor is also present in glyoxal oxidase99 from fungi and yeast. A similar Cys-Tyr cross-linked species has been identified in the active site of the mammalian enzyme, cysteine dioxygenase. In contrast to galactose oxidase, iron rather than copper is present in the active site.100 The non-cross-linked enzyme has low initial activity and that enzyme activity increases with crosslink formation.101 It has been proposed that the Cys–Tyr cross-link formation properly positions the Tyr–OH for binding the cysteine substrate for catalysis.102 The cross-link may also increase activity by controlling deleterious interactions involving the thiolate of the un-cross-linked Cys.103
Figure 11.

Autocatalytic biosynthesis of the Cys-Tyr cofactor
Met-Tyr-Trp cofactor
KatG is a bi-functional catalase-peroxidase found in Mycobacterium tuberculosis, as well as other bacteria. The crystal structures of KatGs from M. tuberculosis,104 and other bacteria,105, 106 revealed the presence of three covalently cross-linked amino acid residues located on the distal side of the active-site heme. The covalent bonds are between Trp, Tyr and Met residues, with both cross-links to the aromatic rings of the tyrosine. The overall structure of KatG is similar to those of the large family of Class I peroxidases.107 However, the Met-Tyr-Trp crosslink is only seen in KatG and site-directed mutagenesis studies demonstrated that the cross-link is needed for the catalatic activity, but not for peroxidatic activity.108 Furthermore, the structure of KatG contains a large loop, not present in the peroxidase, which is essential for catalase activity.109, 110 It is believed that heme is required to generate the cross-linked amino acids during the maturation of the active site.111 The cross-linked amino acids in addition to the heme are then for its catalatic activity.
Tyr-His cross-link
A His that is cross-linked to a Tyr provides one of the copper ligands in the bi-nuclear CuB of the CuB-hemea3 center of cytochrome c oxidase.112, 113 There are three classes of proton-pumping heme-copper oxygen reductases in nature; aa3-type oxidases, ba3-type oxidases, and cbb3-type oxidases.114 The Tyr-His cross-link is believed to be common to each of the three distinct families.115
Cys-His cross-link
The crystal structure of catechol oxidase revealed that one of the His ligands coordinating the coppers of the di-nuclear CuA-CuB metal site is covalently cross-linked to a Cys via a thioether bond116 . Similarly, a Cys-His cross-link is observed in the di-nuclear metal site of Octopus hemocyanin.117 An interesting aspect of these findings is that the functions of the metal sites are different in these two proteins. Unlike the oxidase, hemocyanin functions as an O2 transporter.
CONCLUSIONS
The characterization of protein-derived cofactors challenges the tenets of protein evolution. In the cases where modifying enzymes and accessory proteins are required to generate the protein-derived cofactor, it seems unlikely that the enzyme we see today is not the product of a linear evolutionary process. It is more likely that the protein initially evolved for some other role, until it acquired a structure susceptible to the observed post-translational modifications. The modifications then endowed the protein with a new functionality, which was the first step in a new line of evolution to produce the enzyme that we see today. This also raises questions as to the evolution of the modifying enzymes, such as MauG and LodB. It seems likely that they originally evolved for some function other than the irreversible modification of specific amino acid residues within another protein. Given the huge number of open reading frames with no known function in sequenced genomes, it is expected that some of these encode enzymes with protein-derived cofactors and modifying enzymes that catalyze their formation. Uncovering them will require expression of the proteins to discover what they actually look like and do. Genome mining and modeling of protein structure from predicted sequences have become valuable tools for analysis of genome sequences. However, there is no way predict the presence of a yet uncharacterized protein-derived cofactor from a protein sequence. This will require discovery-based science, which has the potential to yield unimaginable results. Prior to the characterization of the protein-derived cofactors described herein, and their mechanisms of biosynthesis, no one would have predicted their existence. The characterization of protein-derived cofactors also has implications for protein engineering. This knowledge could provide strategies for the design and modification of sequences of existing proteins to generate protein-derived cofactors that create a new redox or catalytic center within a protein. This would introduce a new function as well as circumvent the need for an exogenous cofactor. Continued characterization of protein-derived cofactors promises to unveil novel mechanisms of catalysis by both the cofactors and the enzymes that catalyze their formation.
Acknowledgments
Funding
Work from the author’s laboratory was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R37GM41574 (V.L.D.).
ABBREVIATIONS
- CTQ
cysteine tryptophylquinone
- QHNDH
quinohemoprotein amine dehydrogenase
- LOX
lysyl oxidase
- LTQ
lysine tyrosylquinone MADH, methylamine dehydrogenase
- MIO
4-methylideneimidazole-5-one
- TPQ
topaquinone
- TTQ
tryptophan tryptophylquinone
Footnotes
Notes
The author declares no competing financial interest.
References
- 1.Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 2.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 3.van der Palen CJ, Reijnders WN, de Vries S, Duine JA, van Spanning RJ. MauE and MauD proteins are essential in methylamine metabolism of Paracoccus denitrificans. Antonie van Leeuwenhoek. 1997;72:219–228. doi: 10.1023/a:1000441925796. [DOI] [PubMed] [Google Scholar]
- 4.van der Palen CJ, Slotboom DJ, Jongejan L, Reijnders WN, Harms N, Duine JA, van Spanning RJ. Mutational analysis of mau genes involved in methylamine metabolism in Paracoccus denitrificans. Eur J Biochem. 1995;230:860–871. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CW, Davidson VL. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 6.Pearson AR, de la Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Aggar SA, Grimsrud PA, Davidson VL, Wilmot CW. Further insights into quinone cofactor biogenesis: Probing the role of mauG in methylamine dehydrogenase TTQ formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. A catalytic di-heme bis-Fe(IV) intermediate, alternative to an Fe(IV)=O porphyrin radical. Proc Natl Acad Sci USA. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. In crystallo posttranslational modification within a MauG/pre-methylamine dehydrogenase complex. Science. 2010;327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geng J, Dornevil K, Davidson VL, Liu A. Tryptophan-mediated charge-resonance stabilization in the bis-Fe(IV) redox state of MauG. Proc Natl Acad Sci U S A. 2013;110:9639–9644. doi: 10.1073/pnas.1301544110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu Tarboush N, Jensen LM, Feng M, Tachikawa H, Wilmot CM, Davidson VL. Functional importance of tyrosine 294 and the catalytic selectivity for the bis-Fe(IV) state of MauG revealed by replacement of this axial heme ligand with histidine. Biochemistry. 2010;49:9783–9791. doi: 10.1021/bi101254p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng M, Jensen LM, Yukl ET, Wei X, Liu A, Wilmot CM, Davidson VL. Proline 107 is a major determinant in maintaining the structure of the distal pocket and reactivity of the high-spin heme of MauG. Biochemistry. 2012;51:1598–1606. doi: 10.1021/bi201882e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin S, Feng M, Davidson VL. Mutation of Trp(93) of MauG to tyrosine causes loss of bound Ca(2+) and alters the kinetic mechanism of tryptophan tryptophylquinone cofactor biosynthesis. Biochem J. 2013;456:129–137. doi: 10.1042/BJ20130981. [DOI] [PubMed] [Google Scholar]
- 13.Shin S, Yukl ET, Sehanobish E, Wilmot CM, Davidson VL. Site-directed mutagenesis of Gln103 reveals the influence of this residue on the redox properties and stability of MauG. Biochemistry. 2014;53:1342–1349. doi: 10.1021/bi5000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin S, Feng M, Li C, Williamson HR, Choi M, Wilmot CM, Davidson VL. A T67A mutation in the proximal pocket of the high-spin heme of MauG stabilizes formation of a mixed-valent FeII/FeIII state and enhances charge resonance stabilization of the bis-FeIV state. Biochim Biophys Acta. 2015;1847:709–716. doi: 10.1016/j.bbabio.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yukl ET, Liu F, Krzystek J, Shin S, Jensen LM, Davidson VL, Wilmot CM, Liu A. Diradical intermediate within the context of tryptophan tryptophylquinone biosynthesis. Proc Natl Acad Sci U S A. 2013;110:4569–4573. doi: 10.1073/pnas.1215011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abu Tarboush N, Jensen LMR, Yukl ET, Geng J, Liu A, Wilmot CM, Davidson VL. Mutagenesis of tryptophan199 suggets that hopping is required for MauG-dependent tryptophan tryptophylquinone biosynthesis. Proc Natl Acad Sci USA. 2011;108:16956–16961. doi: 10.1073/pnas.1109423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi M, Shin S, Davidson VL. Characterization of electron tunneling and hole hopping reactions between different forms of MauG and methylamine dehydrogenase within a natural protein complex. Biochemistry. 2012;51:6942–6949. doi: 10.1021/bi300817d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yukl ET, Williamson HR, Higgins L, Davidson VL, Wilmot CM. Oxidative damage in MauG: implications for the control of high-valent iron species and radical propagation pathways. Biochemistry. 2013;52:9447–9455. doi: 10.1021/bi401441h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma Z, Williamson HR, Davidson VL. Roles of multiple-proton transfer pathways and proton-coupled electron transfer in the reactivity of the bis-FeIV state of MauG. Proc Natl Acad Sci U S A. 2015;112:10896–10901. doi: 10.1073/pnas.1510986112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Z, Williamson HR, Davidson VL. A suicide mutation affecting proton transfers to high-valent hemes causes inactivation of MauG during catalysis. Biochemistry. 2016;55:5738–5745. doi: 10.1021/acs.biochem.6b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc Natl Acad Sci U S A. 2001;98:14268–14273. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campillo-Brocal JC, Chacon-Verdu MD, Lucas-Elio P, Sanchez-Amat A. Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity. BMC Genomics. 2015;16:231. doi: 10.1186/s12864-015-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campillo-Brocal JC, Lucas-Elio P, Sanchez-Amat A. Distribution in different organisms of amino acid oxidases with FAD or a quinone as cofactor and their role as antimicrobial proteins in marine bacteria. Mar Drugs. 2015;13:7403–7418. doi: 10.3390/md13127073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez D, Lucas-Elio P, Sanchez-Amat A, Solano F. A novel type of lysine oxidase: L-lysine-epsilon-oxidase. Biochim Biophys Acta. 2006;1764:1577–1585. doi: 10.1016/j.bbapap.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 25.Gomez D, Lucas-Elio P, Solano F, Sanchez-Amat A. Both genes in the Marinomonas mediterranea lodAB operon are required for the expression of the antimicrobial protein lysine oxidase. Mol Microbiol. 2010;75:462–473. doi: 10.1111/j.1365-2958.2009.07000.x. [DOI] [PubMed] [Google Scholar]
- 26.Chacon-Verdu MD, Campillo-Brocal JC, Lucas-Elio P, Davidson VL, Sanchez-Amat A. Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-epsilon-oxidase and glycine oxidase from Marinomonas mediterranea. Biochim Biophys Acta. 2015;1854:1123–1131. doi: 10.1016/j.bbapap.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 27.Sehanobish E, Shin S, Sanchez-Amat A, Davidson VL. Steady-state kinetic mechanism of LodA, a novel cysteine tryptophylquinone-dependent oxidase. FEBS Lett. 2014;588:752–756. doi: 10.1016/j.febslet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mai-Prochnow A, Lucas-Elio P, Egan S, Thomas T, Webb JS, Sanchez-Amat A, Kjelleberg S. Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several gram-negative bacteria. J Bacteriol. 2008;190:5493–5501. doi: 10.1128/JB.00549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sehanobish E, Williamson HR, Davidson VL. Roles of conserved residues of the glycine oxidase GoxA in controlling activity, cooperativity, subunit composition, and cysteine tryptophylquinone biosynthesis. J Biol Chem. 2016;291:23199–23207. doi: 10.1074/jbc.M116.741835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreo-Vidal A, Mamounis K, Sehanobish E, Avalos D, Campillo-Brocal JC, Sanchez-Amat A, Yukl ET, Davidson VL. Structure and enzymatic properties of an unusual cysteine tryptophylquinone-dependent glycine oxidase from Pseudoalteromonas luteoviolacea. Biochemistry. 2018;57:1155–1165. doi: 10.1021/acs.biochem.8b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadda G. Oxygen activation in flavoprotein oxidases: the importance of being positive. Biochemistry. 2012;51:2662–2669. doi: 10.1021/bi300227d. [DOI] [PubMed] [Google Scholar]
- 32.Malmstrom BG. Enzymology of Oxygen. Ann Rev Biochem. 1982;51:21–29. doi: 10.1146/annurev.bi.51.070182.000321. [DOI] [PubMed] [Google Scholar]
- 33.Okazaki S, Nakano S, Matsui D, Akaji S, Inagaki K, Asano Y. X-Ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in L-lysine epsilon-oxidase from Marinomonas mediterranea. J Biochem. 2013;154:233–236. doi: 10.1093/jb/mvt070. [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Doi M, Durley RC, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 A resolution. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 35.Sehanobish E, Chacon-Verdu MD, Sanchez-Amat A, Davidson VL. Roles of active site residues in LodA, a cysteine tryptophylquinone dependent epsilon-lysine oxidase. Arch Biochem Biophys. 2015;579:26–32. doi: 10.1016/j.abb.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. MauG-dependent in vitro biosynthesis of tryptophan tryptophylquinone in methylamine dehydrogenase. J Am Chem Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 37.Williamson HR, Sehanobish E, Shiller AM, Sanchez-Amat A, Davidson VL. Roles of copper and a conserved aspartic acid in the autocatalytic hydroxylation of a specific tryptophan residue during cysteine tryptophylquinone biogenesis. Biochemistry. 2017;56:997–1004. doi: 10.1021/acs.biochem.6b01137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sukumar N, Chen ZW, Ferrari D, Merli A, Rossi GL, Bellamy HD, Chistoserdov A, Davidson VL, Mathews FS. Crystal structure of an electron transfer complex between aromatic amine dehydrogenase and azurin from Alcaligenes faecalis. Biochemistry. 2006;45:13500–13510. doi: 10.1021/bi0612972. [DOI] [PubMed] [Google Scholar]
- 39.Roujeinikova A, Scrutton NS, Leys D. Atomic level insight into the oxidative half-reaction of aromatic amine dehydrogenase. J Biol Chem. 2006;281:40264–40272. doi: 10.1074/jbc.M605559200. [DOI] [PubMed] [Google Scholar]
- 40.McCann SD, Stahl SS. Copper-catalyzed aerobic oxidations of organic molecules: Pathways for two-electron oxidation with a four-eectron oxidant and a one-eectron redox-active catalyst. Acc Chem Res. 2015;48:1756–1766. doi: 10.1021/acs.accounts.5b00060. [DOI] [PubMed] [Google Scholar]
- 41.van Poelje PD, Snell EE. Pyruvoyl-dependent enzymes. Ann Rev Biochem. 1990;59:29–59. doi: 10.1146/annurev.bi.59.070190.000333. [DOI] [PubMed] [Google Scholar]
- 42.Albert A, Dhanaraj V, Genschel U, Khan G, Ramjee MK, Pulido R, Sibanda BL, von Delft F, Witty M, Blundell TL, Smith AG, Abell C. Crystal structure of aspartate decarboxylase at 2.2 A resolution provides evidence for an ester in protein self-processing. Nat Struct Biol. 1998;5:289–293. doi: 10.1038/nsb0498-289. [DOI] [PubMed] [Google Scholar]
- 43.Li QX, Dowhan W. Structural characterization of Escherichia coli phosphatidylserine decarboxylase. J Biol Chem. 1988;263:11516–11522. [PubMed] [Google Scholar]
- 44.Schuiki I, Daum G. Phosphatidylserine decarboxylases, key enzymes of lipid metabolism. IUBMB Life. 2009;61:151–162. doi: 10.1002/iub.159. [DOI] [PubMed] [Google Scholar]
- 45.Stanley BA, Pegg AE, Holm I. Site of pyruvate formation and processing of mammalian S-adenosylmethionine decarboxylase proenzyme. J Biol Chem. 1989;264:21073–21079. [PubMed] [Google Scholar]
- 46.Gallagher T, Rozwarski DA, Ernst SR, Hackert ML. Refined structure of the pyruvoyl-dependent histidine decarboxylase from Lactobacillus 30a. J Mol Biol. 1993;230:516–528. doi: 10.1006/jmbi.1993.1168. [DOI] [PubMed] [Google Scholar]
- 47.Wagner M, Sonntag D, Grimm R, Pich A, Eckerskorn C, Sohling B, Andreesen JR. Substrate-specific selenoprotein B of glycine reductase from Eubacterium acidaminophilum Biochemical and molecular analysis. Eur J Biochem. 1999;260:38–49. doi: 10.1046/j.1432-1327.1999.00107.x. [DOI] [PubMed] [Google Scholar]
- 48.Kabisch UC, Grantzdorffer A, Schierhorn A, Rucknagel KP, Andreesen JR, Pich A. Identification of D-proline reductase from Clostridium sticklandii as a selenoenzyme and indications for a catalytically active pyruvoyl group derived from a cysteine residue by cleavage of a proprotein. J Biol Chem. 1999;274:8445–8454. doi: 10.1074/jbc.274.13.8445. [DOI] [PubMed] [Google Scholar]
- 49.Ramjee MK, Genschel U, Abell C, Smith AG. Escherichia coli L-aspartate-alpha-decarboxylase: preprotein processing and observation of reaction intermediates by electrospray mass spectrometry. Biochem J. 1997;323:661–669. doi: 10.1042/bj3230661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ekstrom JL, Tolbert WD, Xiong H, Pegg AE, Ealick SE. Structure of a human S-adenosylmethionine decarboxylase self-processing ester intermediate and mechanism of putrescine stimulation of processing as revealed by the H243A mutant. Biochemistry. 2001;40:9495–9504. doi: 10.1021/bi010736o. [DOI] [PubMed] [Google Scholar]
- 51.Xiong H, Pegg AE. Mechanistic studies of the processing of human S-adenosylmethionine decarboxylase proenzyme. Isolation of an ester intermediate. J Biol Chem. 1999;274:35059–35066. doi: 10.1074/jbc.274.49.35059. [DOI] [PubMed] [Google Scholar]
- 52.Tolbert WD, Zhang Y, Cottet SE, Bennett EM, Ekstrom JL, Pegg AE, Ealick SE. Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry. 2003;42:2386–2395. doi: 10.1021/bi0268854. [DOI] [PubMed] [Google Scholar]
- 53.Monteiro DC, Patel V, Bartlett CP, Nozaki S, Grant TD, Gowdy JA, Thompson GS, Kalverda AP, Snell EH, Niki H, Pearson AR, Webb ME. The structure of the PanD/PanZ protein complex reveals negative feedback regulation of pantothenate biosynthesis by coenzyme A. Chem Biol. 2015;22:492–503. doi: 10.1016/j.chembiol.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nozaki S, Webb ME, Niki H. An activator for pyruvoyl-dependent L-aspartate alpha-decarboxylase is conserved in a small group of the gamma-proteobacteria including Escherichia coli. Microbiologyopen. 2012;1:298–310. doi: 10.1002/mbo3.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trip H, Mulder NL, Rattray FP, Lolkema JS. HdcB, a novel enzyme catalysing maturation of pyruvoyl-dependent histidine decarboxylase. Mol Microbiol. 2011;79:861–871. doi: 10.1111/j.1365-2958.2010.07492.x. [DOI] [PubMed] [Google Scholar]
- 56.Stuecker TN, Hodge KM, Escalante-Semerena JC. The missing link in coenzyme A biosynthesis: PanM (formerly YhhK), a yeast GCN5 acetyltransferase homologue triggers aspartate decarboxylase (PanD) maturation in Salmonella enterica. Mol Microbiol. 2012;84:608–619. doi: 10.1111/j.1365-2958.2012.08046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnott ZLP, Nozaki S, Monteiro DCF, Morgan HE, Pearson AR, Niki H, Webb ME. The mechanism of regulation of pantothenate biosynthesis by the PanD-PanZ.AcCoA complex reveals an additional mode of action for the antimetabolite N-pentyl pantothenamide (N5-Pan) Biochemistry. 2017;56:4931–4939. doi: 10.1021/acs.biochem.7b00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mead GC. The amino acid-fermenting clostridia. J Gen Microbiol. 1971;67:47–56. doi: 10.1099/00221287-67-1-47. [DOI] [PubMed] [Google Scholar]
- 59.Seto B, Stadtman TC. Purification and properties of proline reductase from Clostridium sticklandii. J Biol Chem. 1976;251:2435–2439. [PubMed] [Google Scholar]
- 60.Jackson S, Calos M, Myers A, Self WT. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol. 2006;188:8487–8495. doi: 10.1128/JB.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jackson-Rosario S, Cowart D, Myers A, Tarrien R, Levine RL, Scott RA, Self WT. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J Biol Inorg Chem. 2009 doi: 10.1007/s00775-009-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun. 2014;82:914. doi: 10.1128/IAI.00515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McAllister KN, Bouillaut L, Kahn JN, Self WT, Sorg JA. Using CRISPR-Cas9-mediated genome editing to generate C difficile mutants defective in selenoproteins synthesis. Sci Rep. 2017;7:14672. doi: 10.1038/s41598-017-15236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calabrese JC, Jordan DB, Boodhoo A, Sariaslani S, Vannelli T. Crystal structure of phenylalanine ammonia lyase: multiple helix dipoles implicated in catalysis. Biochemistry. 2004;43:11403–11416. doi: 10.1021/bi049053+. [DOI] [PubMed] [Google Scholar]
- 65.Schwede TF, Retey J, Schulz GE. Crystal structure of histidine ammonia-lyase revealing a novel polypeptide modification as the catalytic electrophile. Biochemistry. 1999;38:5355–5361. doi: 10.1021/bi982929q. [DOI] [PubMed] [Google Scholar]
- 66.Christianson CV, Montavon TJ, VanLanen SG, Shen B, Bruner SD. The structure of l-tyrosine 2,3-aminomutase from the c-1027 enediyne antitumor antibiotic biosynthetic pathway. Biochemistry. 2007;46:7205–7214. doi: 10.1021/bi7003685. [DOI] [PubMed] [Google Scholar]
- 67.Retey J. Discovery and role of methylidene imidazolone, a highly electrophilic prosthetic group. Biochim Biophys Acta. 2003;1647:179–184. doi: 10.1016/s1570-9639(03)00091-8. [DOI] [PubMed] [Google Scholar]
- 68.Poppe L. Methylidene-imidazolone: a novel electrophile for substrate activation. Curr Opin Chem Biol. 2001;5:512–524. doi: 10.1016/s1367-5931(00)00253-2. [DOI] [PubMed] [Google Scholar]
- 69.Turner NJ. Ammonia lyases and aminomutases as biocatalysts for the synthesis of alpha-amino and beta-amino acids. Curr Opin Chem Biol. 2011;15:234–240. doi: 10.1016/j.cbpa.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 70.Parmeggiani F, Weise NJ, Ahmed ST, Turner NJ. Synthetic and therapeutic applications of ammonia-lyases and aminomutases. Chem Rev. 2018;118:73–118. doi: 10.1021/acs.chemrev.6b00824. [DOI] [PubMed] [Google Scholar]
- 71.Mure M. Tyrosine-derived quinone cofactors. Acc Chem Res. 2004;37:131–139. doi: 10.1021/ar9703342. [DOI] [PubMed] [Google Scholar]
- 72.Janes SM, Mu D, Wemmer D, Smith AJ, Kaur S, Maltby D, Burlingame AL, Klinman JP. A new redox cofactor in eukaryotic enzymes: 6-hydroxydopa at the active site of bovine serum amine oxidase. Science. 1990;248:981–987. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 73.Brazeau BJ, Johnson BJ, Wilmot CM. Copper-containing amine oxidases. Biogenesis and catalysis; a structural perspective. Arch Biochem Biophys. 2004;428:22–31. doi: 10.1016/j.abb.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 74.Wilce MC, Dooley DM, Freeman HC, Guss JM, Matsunami H, McIntire WS, Ruggiero CE, Tanizawa K, Yamaguchi H. Crystal structures of the copper-containing amine oxidase from Arthrobacter globiformis in the holo and apo forms: implications for the biogenesis of topaquinone. Biochemistry. 1997;36:16116–16133. doi: 10.1021/bi971797i. [DOI] [PubMed] [Google Scholar]
- 75.Kim M, Okajima T, Kishishita S, Yoshimura M, Kawamori A, Tanizawa K, Yamaguchi H. X-ray snapshots of quinone cofactor biogenesis in bacterial copper amine oxidase. Nat Struct Biol. 2002;9:591–596. doi: 10.1038/nsb824. [DOI] [PubMed] [Google Scholar]
- 76.Dove JE, Klinman JP. Trihydroxyphenylalanine quinone (TPQ) from copper amine oxidases and lysyl tyrosylquinone (LTQ) from lysyl oxidase. Adv Protein Chem. 2001;58:141–174. doi: 10.1016/s0065-3233(01)58004-3. [DOI] [PubMed] [Google Scholar]
- 77.DuBois JL, Klinman JP. Role of a strictly conserved active site tyrosine in cofactor genesis in the copper amine oxidase from Hansenula polymorpha. Biochemistry. 2006;45:3178–3188. doi: 10.1021/bi052025m. [DOI] [PubMed] [Google Scholar]
- 78.Chen Z, Schwartz B, Williams NK, Li R, Klinman JP, Mathews FS. Crystal structure at 2.5 A resolution of zinc-substituted copper amine oxidase of Hansenula polymorpha expressed in Escherichia coli. Biochemistry. 2000;39:9709–9717. doi: 10.1021/bi000639f. [DOI] [PubMed] [Google Scholar]
- 79.Dooley DM, McGuirl MA, Brown DE, Turowski PN, McIntire WS, Knowles PF. A Cu(I)-semiquinone state in substrate-reduced amine oxidases. Nature. 1991;349:262–264. doi: 10.1038/349262a0. [DOI] [PubMed] [Google Scholar]
- 80.Finney J, Moon HJ, Ronnebaum T, Lantz M, Mure M. Human copper-dependent amine oxidases. Arch Biochem Biophys. 2014;546:19–32. doi: 10.1016/j.abb.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McGrath AP, Hilmer KM, Collyer CA, Shepard EM, Elmore BO, Brown DE, Dooley DM, Guss JM. Structure and inhibition of human diamine oxidase. Biochemistry. 2009;48:9810–9822. doi: 10.1021/bi9014192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maintz L, Schwarzer V, Bieber T, van der Ven K, Novak N. Effects of histamine and diamine oxidase activities on pregnancy: a critical review. Hum Reprod Update. 2008;14:485–495. doi: 10.1093/humupd/dmn014. [DOI] [PubMed] [Google Scholar]
- 83.Agundez JA, Ayuso P, Cornejo-Garcia JA, Blanca M, Torres MJ, Dona I, Salas M, Blanca-Lopez N, Canto G, Rondon C, Campo P, Laguna JJ, Fernandez J, Martinez C, Garcia-Martin E. The diamine oxidase gene is associated with hypersensitivity response to non-steroidal anti-inflammatory drugs. PLoS One. 2012;7:e47571. doi: 10.1371/journal.pone.0047571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Airenne TT, Nymalm Y, Kidron H, Smith DJ, Pihlavisto M, Salmi M, Jalkanen S, Johnson MS, Salminen TA. Crystal structure of the human vascular adhesion protein-1: unique structural features with functional implications. Protein Sci. 2005;14:1964–1974. doi: 10.1110/ps.051438105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Januszewski AS, Mason N, Karschimkus CS, Rowley KG, Best JD, O’Neal DN, Jenkins AJ. Plasma semicarbazide-sensitive amine oxidase activity in type 1 diabetes is related to vascular and renal function but not to glycaemia. Diabetes Vasc Dis Res. 2014;11:262–269. doi: 10.1177/1479164114532963. [DOI] [PubMed] [Google Scholar]
- 86.Li H-Y, Jiang Y-D, Chang T-J, Wei J-N, Lin M-S, Lin C-H, Chiang F-T, Shih S-R, Hung CS, Hua C-H, Smith DJ, Vanio J, Chuang L-M. Serum vascular adhesion protein-1 predicts 10-year cardiovascular and cancer mortality in individuals with type 2 diabetes. Diabetes. 2011;60:993–999. doi: 10.2337/db10-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hernandez-Guillamon M, Sole M, Delgado P, Garcia-Bonilla L, Giralt D, Boada C, Penalba A, Garcia S, Flores A, Ribo M, Alvarez-Sabin J, Ortega-Aznar A, Unzeta M, Montaner J. VAP-1/SSAO plasma activity and brain expression in human hemorrhagic stroke. Cerebrovasc Dis. 2012;33:55–63. doi: 10.1159/000333370. [DOI] [PubMed] [Google Scholar]
- 88.Weston CJ, Adams DH. Hepatic consequences of vascular adhesion protein-1 expression. J Neural Transm. 2011;118:1055–1064. doi: 10.1007/s00702-011-0647-0. [DOI] [PubMed] [Google Scholar]
- 89.Lin M-S, Li H-Y, Wei J-N, Lin C-H, Smith DJ, Vainio J, Shih S-R, Chen Y-H, Lin L-C, Kao H-L, Chuang L-M, Chen M-F. Serum vascular adhesion protein-1 is higher in subjects with early stages of chronic kidney disease. Clin Biochem. 2008;41:1362–1367. doi: 10.1016/j.clinbiochem.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 90.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 91.Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP. A crosslinked cofactor in lysyl oxidase: redox function for amino acid side chains. Science. 1996;273:1078–1084. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 92.Moore RH, Spies MA, Culpepper MB, Murakawa T, Hirota S, Okajima T, Tanizawa K, Mure M. Trapping of a dopaquinone intermediate in the TPQ cofactor biogenesis in a copper-containing amine oxidase from Arthrobacter globiformis. J Am Chem Soc. 2007;129:11524–11534. doi: 10.1021/ja0731165. [DOI] [PubMed] [Google Scholar]
- 93.Iturbide A, Garcia de Herreros A, Peiro S. A new role for LOX and LOXL2 proteins in transcription regulation. FEBS J. 2015;282:1768–1773. doi: 10.1111/febs.12961. [DOI] [PubMed] [Google Scholar]
- 94.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ng MR, Brugge JS. A stiff blow from the stroma: collagen crosslinking drives tumor progression. Cancer Cell. 2009;16:455–457. doi: 10.1016/j.ccr.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 96.Aumiller V, Strobel B, Romeike M, Schuler M, Stierstorfer BE, Kreuz S. Comparative analysis of lysyl oxidase (like) family members in pulmonary fibrosis. Sci Rep. 2017;7:149. doi: 10.1038/s41598-017-00270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ito N, Phillips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KD, Knowles PF. Novel thioether bond revealed by a 1.7 A crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 98.Whittaker MM, Whittaker JW. Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor. J Biol Chem. 2003;278:22090–22101. doi: 10.1074/jbc.M300112200. [DOI] [PubMed] [Google Scholar]
- 99.Whittaker MM, Kersten PJ, Cullen D, Whittaker JW. Identification of catalytic residues in glyoxal oxidase by targeted mutagenesis. J Biol Chem. 1999;274:36226–36232. doi: 10.1074/jbc.274.51.36226. [DOI] [PubMed] [Google Scholar]
- 100.Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase. A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 101.Siakkou E, Rutledge MT, Wilbanks SM, Jameson GNL. Correlating crosslink formation with enzymatic activity in cysteine dioxygenase. Biochim Biophys Acta. 2011;1814:2003–2009. doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 102.Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, Karplus PA. Structure-based insights into the role of the Cys–Tyr crosslink and inhibitor recognition by mammalian cysteine dioxygenase. J Mol Biol. 2016;428:3999–4012. doi: 10.1016/j.jmb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, Jameson GNL. The Cys-Tyr cross-Link of cysteine dioxygenase changes the optimal pH of the reaction without a structural change. Biochemistry. 2014;53:7961–7968. doi: 10.1021/bi501277a. [DOI] [PubMed] [Google Scholar]
- 104.Bertrand T, Eady NA, Jones JN, Jesmin Nagy JM, Jamart-Gregoire B, Raven EL, Brown KA. Crystal structure of Mycobacterium tuberculosis catalase-peroxidase. J Biol Chem. 2004;279:38991–38999. doi: 10.1074/jbc.M402382200. [DOI] [PubMed] [Google Scholar]
- 105.Yamada Y, Fujiwara T, Sato T, Igarashi N, Tanaka N. The 2.0 A crystal structure of catalase-peroxidase from Haloarcula marismortui. Nat Struct Biol. 2002;9:691–695. doi: 10.1038/nsb834. [DOI] [PubMed] [Google Scholar]
- 106.Carpena X, Loprasert S, Mongkolsuk S, Switala J, Loewen PC, Fita I. Catalase-peroxidase KatG of Burkholderia pseudomallei at 1.7A resolution. J Mol Biol. 2003;327:475–489. doi: 10.1016/s0022-2836(03)00122-0. [DOI] [PubMed] [Google Scholar]
- 107.Zamocky M, Furtmuller PG, Obinger C. Evolution of structure and function of Class I peroxidases. Arch Biochem Biophys. 2010;500:45–57. doi: 10.1016/j.abb.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 108.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. Probing the structure and bifunctionality of catalase-peroxidase (KatG) J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 109.Kudalkar SN, Njuma OJ, Li Y, Muldowney M, Fuanta NR, Goodwin DC. A role for catalase-peroxidase large loop 2 revealed by deletion mutagenesis: control of active site water and ferric enzyme reactivity. Biochemistry. 2015;54:1648–1662. doi: 10.1021/bi501221a. [DOI] [PubMed] [Google Scholar]
- 110.Jakopitsch C, Droghetti E, Schmuckenschlager F, Furtmuller PG, Smulevich G, Obinger C. Role of the main access channel of catalase-peroxidase in catalysis. J Biol Chem. 2005;280:42411–42422. doi: 10.1074/jbc.M508009200. [DOI] [PubMed] [Google Scholar]
- 111.Ghiladi RA, Knudsen GM, Medzihradszky KF, de Montellano PR. The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG): autocatalytic formation and effect on enzyme catalysis and spectroscopic properties. J Biol Chem. 2005;280:22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- 112.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 A resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc Natl Acad Sci U S A. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 114.Pereira MM, Santana M, Teixeira M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim Biophys Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 115.Hemp J, Robinson DE, Ganesan KB, Martinez TJ, Kelleher NL, Gennis RB. Evolutionary migration of a post-translationally modified active-site residue in the proton-pumping heme-copper oxygen reductases. Biochemistry. 2006;45:15405–15410. doi: 10.1021/bi062026u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klabunde T, Eicken C, Sacchettini JC, Krebs B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat Struct Biol. 1998;5:1084–1090. doi: 10.1038/4193. [DOI] [PubMed] [Google Scholar]
- 117.Cuff ME, Miller KI, van Holde KE, Hendrickson WA. Crystal structure of a functional unit from Octopus hemocyanin. J Mol Biol. 1998;278:855–870. doi: 10.1006/jmbi.1998.1647. [DOI] [PubMed] [Google Scholar]