

Abstract

Bacterial identification is of great importance in clinical diagnosis, environmental monitoring, and food safety control. Among various strategies, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) has drawn significant interest and has been clinically used. Nevertheless, current bioinformatics solutions use spectral libraries for the identification of bacterial strains. Spectral library generation requires acquisition of MALDI-TOF spectra from monoculture bacterial colonies, which is time-consuming and not possible for many species and strains. We propose a strategy for bacterial typing by MALDI-TOF using protein sequences from public database, that is, UniProt. Ten genes were identified to encode proteins most often observed by MALD-TOF from bacteria through 500 times repeated a 10-fold double cross-validation procedure, using 403 MALDI-TOF spectra corresponding to 14 genera, 81 species, and 403 strains, and the protein sequences of 1276 species in UniProt. The 10 genes were then used to annotate peaks on MALDI-TOF spectra of bacteria for bacterial identification. With the approach, bacteria can be identified at the genus level by searching against a database containing the protein sequences of 42 genera of bacteria from UniProt. Our approach identified 84.1% of the 403 spectra correctly at the genus level. Source code of the algorithm is available at https://github.com/dipcarbon/BacteriaMSLF.
Keywords: bacterial identification, proteomics, MALDI-TOF, library-free, double-cross validation, ribosomal proteins, UniProt, MALDI-TOF peak annotation, parameter optimization, data filtering
1. Introduction
Bacterial infections are one of the most common threats to public health worldwide. According to the Review on Antimicrobial Resistance,1 global deaths attributed to antimicrobial resistant bacteria are estimated to reach 10 million by 2050. Appropriate and authentic information on the type of bacteria involved in infection is crucial in ensuring to select proper antibiotic therapy. Traditional methods in bacterial identification usually include morphological observations of bacteria cultured from samples,2 physiological and biochemical reactions with bacterial proteins,3 and enzymatic tests.4 The aforementioned methods take several days to carry out, and further delay in starting necessary antibiotic treatments, as well as incur high labor and patient care costs.5 Furthermore, many pathogenic bacteria cannot be successfully cultured due to inappropriate sample collection or the unavailability of selective bacterial culture media.
Recently, genomic methods have been used for the identification of bacterial species such as 16S rRNA identification6 with polymerase chain reaction (PCR)7,8 and the next generation high-throughput sequencing.9 These techniques hold certain advantages such as high sensitivity and strong accuracy. However, the genome-based techniques are still limited in several aspects. PCR is an analysis method that needs prior knowledge of the target sequences, and it is easily affected by contaminations.10 The resolving power of 16S rRNA identification at the level of species is not satisfying.11 Next-generation high-throughput sequencing is the technique with highest specificity in the differentiation of strains, but is not suitable for routine testing because the analysis takes several days.12
Since the 1980s, matrix-assisted laser desorption/ionization (MALDI)5 and electrospray ionization (ESI)13 as part of mass spectrometers have been used for the analysis of proteins. Specifically, MALDI time-of-flight (TOF) mass spectrometry (MS) has been suggested as an alternative for microbial identification by analyzing monoculture bacteria.14 This technique has several desirable features, such as a minimal requirement for sample preparation and consumables, and short analysis time. The principle is based on the high specificity of MALDI-TOF spectra for different bacteria, which are considered as the fingerprint of bacteria and can be used for bacterial identification. It was reported that peaks in MALDI-TOF spectra of bacteria mainly originate from ribosomal proteins, which are enriched during the sample preparation steps of protein extraction from bacterial cells using formic acid.15 However, when using rotating culture, strain media culture, and ethanol extraction, it is possible to detect other types of proteins.16 Therefore, sample preparation methods have great influence on protein profiles and mass spectra and hence on identification results.
Another prerequisite for MALDI-TOF based bacterial identification is the availability of accurate spectral library acquired from known monoculture bacterial samples, which limits the method only to cultivable bacteria. There are several commercial companies that manufacture MALDI-TOF instruments, while only a handful of commercial systems, such as Biotyper from Bruker Daltonics17 and VITEK MS from BioMérieux,18 are optimized for high-throughput bacterial identification. These companies integrate mass spectrometry platforms, spectral libraries, and software to identify and classify microbial organisms. These systems typically apply robust algorithms associated with multivariate statistical approaches for spectral matching. However, these algorithms and spectra database are not open source and are expensive, which certainly hinder the wide application of MALDI-TOF technology for bacterial identification. Furthermore, the spectral libraries are normally associated with specific instruments and sample preparation methods, limiting the share and process of data from different instruments and laboratories.
An alternative approach to this method is to annotate MALDI-TOF spectral peaks with genomic or proteomic sequences available in public repositories such as UniProt or Ensembl. In 2001, Demirev et al.19 established a microbiological rapid identification method called “Non-Protein-Based Approaches”. This method used the information contained in protein sequences database, that is, SwissProt. They chose 35 biomarkers, for example, P56058, P56056, O25662, O25451, etc., to identify Helicobacter pylori. In 2003, the same research team20 extended this approach to four genera, that is, Bacillus, Escherichia, Pseudomonas, and Haemophilus. They selected different biomarkers for the characterization of different microorganisms (5 species from 4 genera) and achieved an accurate identification rate of 95%. Since this research team’s endeavor, there has been little progress on the bacterial identification approach using genomic or proteomic sequences, despite the rapid development of public sequences databases such as UniProt and Ensembl.
In this study, we have developed an approach that allows for the generic identification of a wide range of bacterial strains by directly matching their MALDI-TOF spectra to protein sequences in UniProt. We identified ten genes that encode proteins most often observed in MALDI-TOF spectra using a double cross-validation procedure with a data set consisting of 403 MALDI-TOF spectra corresponding to 14 genera, 81 species, and 403 strains and the protein sequences of 1276 species in UniProt. The 10 genes were then used to annotate peaks on MALDI-TOF spectra of bacteria for bacterial identification, resulting in a global identification accuracy of 84.1% at the genus level.
Common genera, for example, Bacillus, Pseudomona, Escherichia, Brucella, Listeria, etc., can be well identified with accuracy >80%, while genera with little protein sequence information in UniProt can hardly be identified. Because of the narrow distribution of the molecular weights of the proteins encoded by the genes within the same genera, it is hard to resolve species within a genus through this method. Comparing to the previous spectral library-free MALDI-TOF bacterial identification approaches, it is the first attempt to implement and test the idea in a systematic way, and expands the strategy to a much wider range of bacterial species. Although this spectral library-free method is still not as accurate as the spectral library based approaches, it provides a possibility to estimate the genus of unknown bacteria or the bacteria that can hardly be cultured. With the increasing amount of microbes’ proteomic and genomic data, protein sequences in public databases will become more complete and more accurate including information such as PTMs and sequence variations, leading to a better future performance of the method. The source code of the algorithm is available at https://github.com/dipcarbon/BacteriaMSLF. It can be easily rerun on new MALDI-TOF data sets and new proteomic sequence databases to update the identification model.
2. Methods
2.1. Protein Sequences Database and Spectra
Proteomic sequence database including 86 452 protein entries, which has been uploaded to Github (https://github.com/dipcarbon/BacteriaMSLF), from 42 genera and 1276 species, as detailed in S1 part of the Supporting Information, was downloaded from UniProt (http://www.uniprot.org). The protein sequences were filtered with the following rules: (1) entries with a PE value >3 were removed; (2) entries without a specific gene name were removed; (3) entries from strains without a specific strain name were removed.
MALDI-TOF MS spectra (645) of 24 genera, 103 species, and 645 strains were downloaded from various publicly available databases, as specified in the S2 part of the Supporting Information. Intensities were converted to relative intensities between 0% and 100% with respect to the strongest peak in each spectrum. Spectra with <70 peaks with signal-to-noise ratio (S/N) ≥ 3 were eliminated. The 70 most abundant peaks with S/N ≥ 3 of each spectrum were used for building the model and for bacterial identification.
2.2. Peak Labeling
Peak labeling was achieved by comparing the mass-to-charge ratio (m/z) of peaks reduced with the weight of proton (1 Da) to the molecular weight of proteins with a tolerance of 2000 ppm. The large tolerance was used because of the relatively low resolving power of linear TOF at high m/z.21 When multiple proteins could be matched to one peak, the protein with the smallest difference between measured m/z and theoretical molecular weight was selected.
2.3. Identification of Most Informative Genes by Double Cross-Validation Procedure
Statistical approaches with a double cross-validation procedure were applied to identify the most informative genes for bacterial identification as shown in Figure 1. The double cross-validation process consisted of two nested cross-validation loops, the inner loop and the outer loop. In the outer loop of the double cross-validation, the 403 bacterial spectra were divided equally into 10 subsets. During each outer training, nine subsets were selected as the outer training set, and one was selected as the outer test set. In the inner loop, the outer training set was again divided equally into 10 subsets, where nine of them were selected as the inner training set and one was selected as the inner test set. For each outer training loop, 10 inner training were performed; both the inner and outer loops during the full process tested the complete data available within their loops.
Figure 1.

Identification of the most informative genes for bacterial identification using a double cross-validation approach. In the inner loop, parameters such as intensity threshold (THR), gene weight table (W), R-score threshold (R0), and constant k were optimized and the spectra of the test outer loop were used to assess the performance of the method using the optimal parameters.
During the inner loop training, peaks in each mass spectrum were annotated with the gene names of the proteins of its corresponding species using the peak labeling strategy described above. After labeling, gene weight (Wj) was calculated for each gene during each inner loop training as follows:
| 1 |
where Wj is the weight of gene j; P is the molecular mass in Da–1 of matched peaks in the spectra; I is the unit vector; Mij is the molecular weight of the protein expressed by the gene j in species i; k is a constant optimized during the double cross-validation training; and m is the number of species assessed in each inner loop training. A larger value for Wj indicated that the gene j was more frequently observed as a matched peak within the relative mass difference tolerance in various species by MALDI-TOF. The differences between strains within a species were ignored because of the low number of protein sequences available in UniProt at strain level. Ten genes with the largest averaged Wj values were chosen for the identification of pseudounknown samples during tests in inner loop and outer loop. Herein, we chose the 10 most informative genes, where the number of 10 was selected to keep short the computation time, while maintaining high identification accuracy, as detailed in Supporting Information S3.
A molecular weight comparison matrix (M) was created with the 1276 species from UniProt database as rows and the 10 most informative genes as columns. Each cell contained the molecular weight of the protein encoded by the corresponding gene in the corresponding species. Certain species have no protein sequence information in UniProt for some genes. In this case, the median molecular weights of the proteins encoded by the genes in the same genus was used to fill the missing values. A gene weight table W was obtained after normalization of gene weights (Wj) against the largest gene weight resulting in a relative value between 0 and 1. The identification of test spectra as pseudounknown samples was performed by matrix calculation. For each spectrum, a difference matrix (C) was calculated based on the molecular weight comparison matrix (M), where each cell in the molecular weight comparison matrix was filled with the values calculated using eq 2:
| 2 |
where k is the same constant used in eq 1. The C matrix was then multiplied by the gene weight table W to obtain a list of species and their corresponding scores R:
| 3 |
where n is the number of the most informative genes (n = 10) for spectra identification; m is the number of species in database (m = 1276). The species with the largest R-score value and larger than R0 was considered as positive identification result. Meanwhile, the R-score for the best match must be larger than R̅ + SD, where R̅ is the averaged R-score value for all the 1276 species; SD is the standard deviation of all the R-score values. Otherwise the spectra were reported to be not identified. An example of the calculation for the identification of a mass spectrum is given in S4 section of the Supporting Information.
Spectra identification accuracy was calculated during each inner loop testing. R0, Wj, k, THR, and the most informative genes were optimized using genetic algorithm, a method to solve the problem of constraint optimization by simulating evolution.22 Using R0 as an example, the R0 value was converted to a binary value. For instance, the value of 0.8125 in the decimal system can be converted to 0.1101 in the binary system. Then a Python open source framework, Pyevolve,23 was applied for the optimization of the R0 value. In an ideal situation, the value could be the optimal after several generations of evolution by the genetic algorithm to reach the highest identification accuracy of the test sets. Detailed description of the usage of the open source framework is given in Github at https://github.com/dipcarbon/BacteriaMSLF#gapy. The medians of of R0, Wj, THR, and k parameters from 10 inner loop trainings were selected to enter the outer loop. Identification accuracy was calculated using the outer tests in the outer loop. The medians of R0, Wj, and k parameters from 10 outer loop training were selected for the final model of that particular double cross-validation loop. Herein, we repeated 500 times the double cross-validation procedure, and the average value of R0, Wj, THR, and k parameters from each double cross-validation run were chosen as the final parameter values for bacterial identification.
3. Results and Discussion
3.1. Database Filtering
The current strategy for spectra matching against protein sequences database is based on the hypothesis that more peaks in a MALDI-TOF spectrum can be labeled when using the protein sequences of the corresponding genus (or species) than using protein sequences from any other genera (or species). The hypothesis has been demonstrated on the 14 genera, as illustrated in the Figures S2 and S5 part of Supporting Information. However, we found that many peaks in mass spectra could not be labeled even with the corresponding species’ protein sequences from UniProt. There can be three reasons for this phenomenon: (i) the current protein sequences database for many bacteria is still far from complete in UniProt; (ii) post-translational modifications (PTMs) and sequence variations should be considered for peak matching, and (iii) some peaks in mass spectra are not from the intact bacterial proteins, but underwent to post-translational modifications or fragmentations during sample preparation or MALDI procedure. It is noteworthy that some of protein mass variances such as due to PTMs and sequence variability is taken into account by the 2000 ppm protein mass matching tolerance. As shown in Figure 2, the averaged numbers of matched peaks in mass spectra from a genus is positively correlated with the number of reviewed protein entries in UniProt of the corresponding genus. The peak labeling was performed using the corresponding species’ protein sequences. When the number of reviewed proteins in a genus is less than 2000, the average number of matched peaks in mass spectra from the genus is ≤5, which is too small to be considered during model training. Therefore, we used only the mass spectra from genera with at least 2000 reviewed proteins in UniProt for the double cross-validation procedure. In the current UniProt database, there are 42 genera of bacteria, which meet this requirement, corresponding to 1276 species, as detailed in the S1 part of Supporting Information. From various publicly available databases, we have obtained 403 spectra corresponding to 14 genera, 81 species, and 403 strains, which are subset of the 42 genera, as detailed in the S2 part of Supporting Information.
Figure 2.
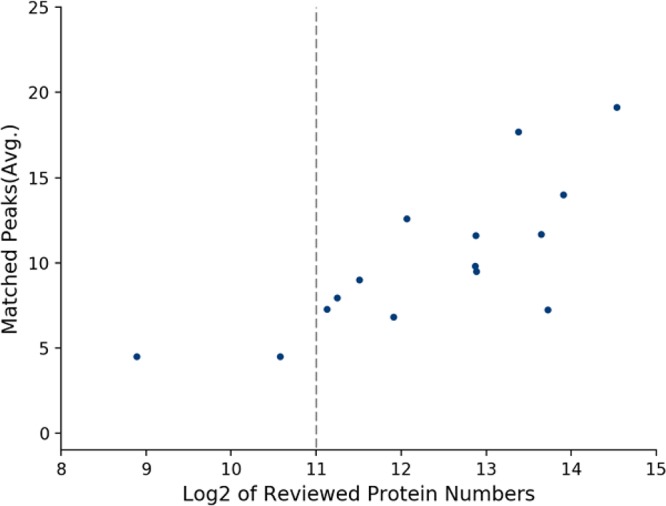
Correlation between the averaged numbers of matched peaks in mass spectra from each genus and the number of reviewed proteins in UniProt of the corresponding genus. Protein sequences of the genera of bacillus, bordetella, brucella, burkholderia, escherichia, flavobacterium, francisella, klebsiella, lactobacillus, listeria, pseudomonas, salmonella, staphylococcus, vibrio, and yersinia, and the mass spectra corresponding to the genera as listed in S2 were used to plot the figure.
3.2. Assessment of Optimal Model Performance
This work employed a double cross-validation24 method to obtain the optimal gene weight table W for spectral matching. In the model, the outer test group was independent from the data used in the inner loop for the optimization of parameters, and therefore overfitting of model parameters can be avoided when assessing the statistical validity of the library-free MALDI-TOF bacterial identification.25 Ten genes were identified as the most informative genes to annotate peaks on MALDI-TOF spectra for bacterial identification (Figure S1 in section S3 of the Supporting Information). The 10 genes together with their gene weights are shown on Figure 3. The obtained genes corresponded to ribosomal subunit proteins and DNA-binding proteins. Ribosomal proteins in conjunction with rRNA form the ribosomal subunits and are involved in protein translation.26 DNA-binding proteins are responsible for binding to DNA regions essential for DNA replication, recombination, and repair.27 Ribosomal proteins are highly abundant in bacteria. By statistics,28 the weight of ribosomal proteins accounts for one-fifth of the total weight of all proteins in a bacterial cell. Figure 3 shows that three genes, rpmC, rpmJ, and rpmH, have relative much higher gene weights compared to the other genes, which correspond to 50S ribosomal protein L34, 50S ribosomal protein L36, and 50S ribosomal protein L29, respectively. The expression levels of the three proteins in microorganism are high and their molecular weights are ranging from 4000 to 8000 Da. Mass spectra of bacteria by MALDI-TOF MS normally show strongest peaks within the mass range of 2000 to 20000 Da.14 The ribosomal proteins are conserved proteins but specific to bacteria at genera level, which make them as potential markers for bacterial identification. By only considering ribosomal protein sequences for spectral matching, we have obtained another gene weight table as shown in the S6 part of Supporting Information. The top eight genes are the same as the ones in Figure 3 and with similar gene weights.
Figure 3.
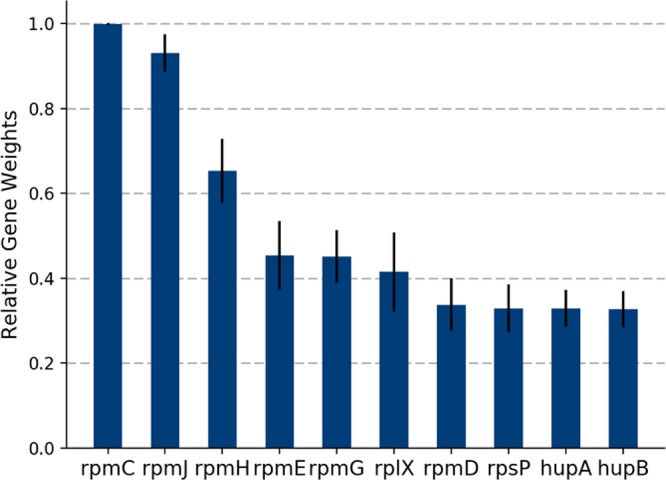
Bar plot showing the relative gene weight W for the 10 most informative genes that can be used for spectral identification. The gene weights were obtained with the double cross-validation procedure using 403 MALDI-TOF spectra and protein sequences of 1276 species in Unirpot. The relative gene weights were averaged from 500 trials of the double cross-validation procedure.
A previous study on bacterial identification by MALDI-TOF also supports the results in Figure 3.29 Arnold and Reilly characterized the proteins isolated from Escherichia coli ribosomes by MALDI-TOF. They detected 58 ribosomal proteins, and about half of the peaks in the MALDI-TOF spectra were attributed to ribosomal proteins. Arnold and Reilly found out that the MALDI-TOF spectra of proteins isolated from Escherichia coli ribosomes were rather similar to the MALDI-TOF spectra of the whole bacterial cells. In their study, some of the peaks were assigned to ribosomal proteins with post-translational modifications. Despite the increasing interests in bacterial identification by MALDI-TOF, there is still little understanding of the impact of protein sequence variations and the effect of PTMs on bacterial identification using MALDI-TOF spectra of whole bacterial cells. Knowing the exact PTMs of proteins and any protein sequence variations would lead to more accurate calculation of protein molecular weights and would improve identification accuracy of bacteria using the presented library-free approach.
3.3. Identification of Bacteria Using 10 Most Informative Genes
Identification of bacteria was performed by using the proteins encoded by the 10 most informative genes in different species to match the peaks in mass spectra. Protein sequences from 1276 species of 42 genera (S1 section of the Supporting Information) were considered during the calculation of R-score, and the species with the largest R-score and exceeding the thresholds of R-score (R0) and R̅ + SD was considered as positive identification result. The value of R0 was determined as 1.103, averaged from 500 trials of the double cross-validation procedure. To reduce the negative effects from low abundant peaks in MALDI-TOF spectra for bacterial identification, which showed normally large variations in peak m/z and intensity values during repeated measurements, only peaks with relative intensity larger than a specific value were considered for peak labeling during bacterial identification. The threshold for peak relative intensity (THR) was found as 1.13%, averaged from 500 trials of the double cross-validation procedure. The R0 and THR were optimized during each inner training to achieve the highest identification accuracy using the corresponding testing sets.
With the presented approach, the identification accuracy at the genus level was 84.1% and at the species level was 31.2% for the 403 mass spectra. Specifically, the identification results at the genus level for mass spectra from Bacillius, Yersinia, Pseudomonas, Escherichia, Lactobacillus, Vibrio, Brucella, and Francisella, which had at least 10 spectra for identification, are presented in Figure 4. When considering only ribosomal protein sequences for peak matching, that is, using the gene weights as shown in Figure S3 in the S6 section of Supporting Information, similar identification accuracies were obtained. Among the genera, Vibrio and Lactobacillus showed low identification accuracies, that is, 68% and 65%. The genomes of Lactobacillus and Vibrio are highly variable, ranging in size from 1.8 to 3.3 Mbp (mega bases pairs) for Lactobacillus(30) and 4.1 to 5.2 Mb for Vibrio,31,32 respectively. Accordingly, low identification accuracy of spectra from Lactobacillus and Vibrio at the genus level was observed due to the broad diversity within the genera.
Figure 4.
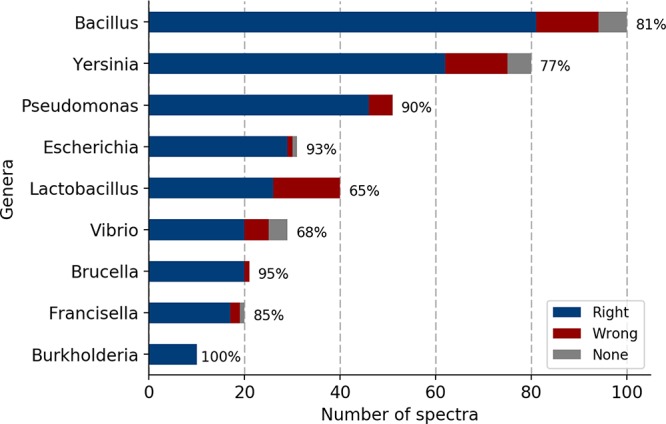
Identification accuracy for mass spectra from different genera using the 10 most informative genes. Blue, correctly identified; red, incorrectly identified; gray, spectra without identification result.
The identification accuracy at the genus level is acceptable for certain applications, but the identification accuracy of our approach at the species level is still too low. Figure 5 shows variance in molecular weight of the proteins encoded by the gene rpmC in different genera. Many genera, such as Bordetella, Brucella, Escherichia, Francisella, Pseudomonas, and Vibrio, show very small variance of the molecular weights of the proteins encoded by the gene between species within the same genera. Similar results were obtained using the other genes of the 10 most informative genes, as shown in Figure S4a,b in section S7 of the Supporting Information. Because of the small variability of molecular mass of the proteins encoded by the 10 most informative genes, our approach cannot provide sufficient resolving power in bacterial identification at the species level.
Figure 5.
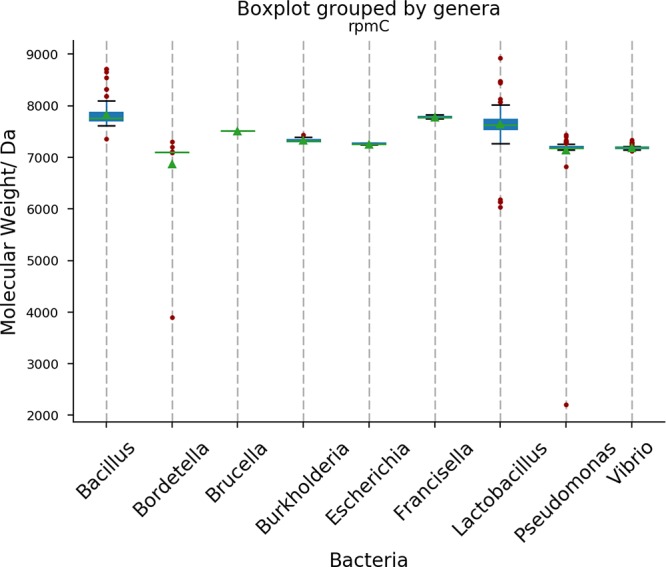
Variation of the molecular weights of the proteins encoded by rpmC in different genera using the protein sequences database from UniProt. Red dots shows outliers, the green triangle indicates mean value, the green line shows the median value, and the blue box shows values between inter quartile range (25%–75%).
4. Conclusion and Perspectives
In this work, we developed a bacterial MALDI-TOF spectra identification approach without using standard spectra library, but only protein sequences from UniProt database. Genetic algorithm with double cross-validation was performed to get the proteins encoded by 10 most informative genes as protein panels to be considered for bacterial identification. At the genus level, the identification accuracy exceeded 80%. Although the performance is still behind the performance of spectral library search based approach, it is the first attempt to implement and test the idea of library-free MALDI-TOF MS bacterial identification in a systematic way and provides possibility to identify bacteria that cannot be obtained in monoculture colonies.
The bacteria identification model can be improved in the future by updating the model with increasing reliable protein sequence annotations, including information such as PTMs and sequence variations, available in UniProt or other public proteomic database. The framework includes an automatic model update options, which can be used to update model and test its performance with new annotated MALDI-TOF spectra and new protein sequences database.
Acknowledgments
L.Q. and D.C. would like to thank the National Natural Science Foundation of China NSFC (81671849), Science and Technology Commission of Shanghai Municipality 17JC1400900, Ministry of Science and Technology of China MOST (2016YFE0132400), and Chinese Thousand Talents Program for funding support. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7, 2007-2013), Research Infrastructures action, under Grant Agreement No. FP7-228310 (EMbaRC project).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00065.
Proteomic database; spectra details; optimization of number of most informative genes for bacterial identification; example of identification; chance of random matches; bacterial identification by considering only ribosomal proteins; variation in molecular weights of proteins encoded by most informative genes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- O’Neill J.Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Review on Antimicrobial Resistance; HM Government, 2014; pp 13.
- Hallez R.; Bellefontaine A.-F.; Letesson J.-J.; De Bolle X. Morphological and functional asymmetry in alpha-proteobacteria. Trends Microbiol. 2004, 12 (8), 361–365. 10.1016/j.tim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Hilger A. E.; Lancaster M. V. Spectral analysis of biochemical reactions used for identification of bacteria. Eur. J. Clin. Microbiol. 1984, 3 (4), 310–315. 10.1007/BF01977479. [DOI] [PubMed] [Google Scholar]
- Bascomb S.; Manafi M. Use of enzyme tests in characterization and identification of aerobic and facultatively anaerobic gram-positive cocci. Clin. Microbiol. Rev. 1998, 318–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuddhe S. B.; Maier T.; Schwarz G.; Kostrzewa M.; Hof H.; Domann E.; Chakraborty T.; Hain T. Rapid identification and typing of listeria species by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 2008, 74 (17), 5402–5407. 10.1128/AEM.02689-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarridge J. E.; Alerts C. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17 (4), 840–862. 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Mishra A.; Puri N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. 10.1016/j.jbiotec.2017.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y.; Lau S. K. P.; Teng J. L. L.; Tse H.; Yuen K. Y. Then and now: Use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin. Microbiol. Infect. 2008, 14, 908–934. 10.1111/j.1469-0691.2008.02070.x. [DOI] [PubMed] [Google Scholar]
- Loman N. J.; Constantinidou C.; Chan J. Z. M.; Halachev M.; Sergeant M.; Penn C. W.; Robinson E. R.; Pallen M. J. High-throughput bacterial genome sequencing: An embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 2012, 10 (9), 599–606. 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- Kemp M.; Jensen K. H.; Dargis R.; Christensen J. J. Routine ribosomal PCR and DNA sequencing for detection and identification of bacteria. Future Microbiol. 2010, 5 (7), 1101–1107. 10.2217/fmb.10.59. [DOI] [PubMed] [Google Scholar]
- Mellmann A.; Cloud J.; Maier T.; Keckevoet U.; Ramminger I.; Iwen P.; Dunn J.; Hall G.; Wilson D.; LaSala P.; et al. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J. Clin. Microbiol. 2008, 46 (6), 1946–1954. 10.1128/JCM.00157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumaz S.; Stevens P.; Grumaz C.; Decker S. O.; Weigand M. A.; Hofer S.; Brenner T.; von Haeseler A.; Sohn K. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016, 8, 73. 10.1186/s13073-016-0326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Mazumdar S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 282574. 10.1155/2012/282574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonnelle E.; Mesquita C.; Bille E.; Day N.; Dauphin B.; Beretti J. L.; Ferroni A.; Gutmann L.; Nassif X. MALDI-TOF mass spectrometry tools for bacterial identification in clinical microbiology laboratory. Clin. Biochem. 2011, 44, 104–109. 10.1016/j.clinbiochem.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Ryzhov V.; Fenselau C. Characterization of the protein subset desorbed by MALDI from whole bacterial cells. Anal. Chem. 2001, 73 (4), 746–750. 10.1021/ac0008791. [DOI] [PubMed] [Google Scholar]
- Williams T. L.; Andrzejewski D.; Lay J. O.; Musser S. M. Experimental factors affecting the quality and reproducibility of MALDI TOF mass spectra obtained from whole bacteria cells. J. Am. Soc. Mass Spectrom. 2003, 14 (4), 342–351. 10.1016/S1044-0305(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Pusch W. Bruker Daltonics: leading the way from basic research to mass-spectrometry-based clinical applications. Pharmacogenomics 2007, 8 (6), 663–668. 10.2217/14622416.8.6.663. [DOI] [PubMed] [Google Scholar]
- Pincus D. H.Microbial identification using the bioMérieux VITEK® 2 system. In Encycl. Rapid Microbiol. Methods; Michael J. M., Ed.; PDA/DHI, 2010; pp 1–32. [Google Scholar]
- Demirev P. A.; Lin J. S.; Pineda F. J.; Fenselau C. Bioinformatics and mass spectrometry for microorganism identification: Proteome-wide post-translational modifications and database search algorithms for characterization of intact H. pylori. Anal. Chem. 2001, 73 (19), 4566–4573. 10.1021/ac010466f. [DOI] [PubMed] [Google Scholar]
- Pineda F. J.; Antoine M. D.; Demirev P. A.; Feldman A. B.; Jackman J.; Longenecker M.; Lin J. S. Microorganism Identification by Matrix-Assisted Laser/Desorption Ionization Mass Spectrometry and Model-Derived Ribosomal Protein Biomarkers. Anal. Chem. 2003, 75 (15), 3817–3822. 10.1021/ac034069b. [DOI] [PubMed] [Google Scholar]
- Fagerquist C. K. Top-down proteomic identification of bacterial protein biomarkers and toxins using MALDI-TOF-TOF-MS/MS and post-source decay. Rev. Anal. Chem. 2013, 32 (2), 127–133. 10.1515/revac-2012-0035. [DOI] [Google Scholar]
- Kumar M.; Husian M.; Upreti N.; Gupta D. Genetic Algorithm: Review and Application. Int. J. Inf. Technol. Knowl. Manag. 2010, 2 (2), 451–454. [Google Scholar]
- Perone C. S. Pyevolve: a Python open-source framework for genetic algorithms. SIGEVOlution 2009, 4, 12–20. 10.1145/1656395.1656397. [DOI] [Google Scholar]
- Roy K.; Ambure P. The “double cross-validation” software tool for MLR QSAR model development. Chemom. Intell. Lab. Syst. 2016, 159, 108–126. 10.1016/j.chemolab.2016.10.009. [DOI] [Google Scholar]
- Smit S.; van Breemen M. J.; Hoefsloot H. C. J.; Smilde A. K.; Aerts J. M. F. G.; de Koster C. G. Assessing the statistical validity of proteomics based biomarkers. Anal. Chim. Acta 2007, 592 (2), 210–217. 10.1016/j.aca.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Wilson D. N.; Nierhaus K. H. Ribosomal proteins in the spotlight. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 243–267. 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- Nelson H. C. Structure and function of DNA-binding proteins. Curr. Opin. Genet. Dev. 1995, 5 (2), 180–189. 10.1016/0959-437X(95)80006-9. [DOI] [PubMed] [Google Scholar]
- Melnikov S.; Ben-Shem A.; Garreau De Loubresse N.; Jenner L.; Yusupova G.; Yusupov M. One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. 10.1038/nsmb.2313. [DOI] [PubMed] [Google Scholar]
- Arnold R. J.; Reilly J. P. Observation of Escherichia coli ribosomal proteins and their posttranslational modifications by mass spectrometry. Anal. Biochem. 1999, 269, 105–112. 10.1006/abio.1998.3077. [DOI] [PubMed] [Google Scholar]
- Boekhorst J.; Siezen R. J.; Zwahlen M. C.; Vilanova D.; Pridmore R. D.; Mercenier A.; Kleerebezem M.; de Vos W. M.; Brüssow H.; Desiere F. The complete genomes of Lactobacillus plantarum and Lactobacillus johnsonii reveal extensive differences in chromosome organization and gene content. Microbiology 2004, 150 (11), 3601–3611. 10.1099/mic.0.27392-0. [DOI] [PubMed] [Google Scholar]
- Reimer A. R.; Van G. H.; Stroika S.; Walker M.; Kent H.; Tarr C.; Talkington D.; Rowe L.; Olsen-Rasmussen M.; Frace M. Comparative genomics of Vibrio cholerae from Haiti and recent clinical cases with travel to Asia and Africa. Emerg. Infect. Dis. 2011, 1. 10.3201/eid1711.110794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.; Chung H.; Lim S.; Kim K.; Kim S.; Na E.; Caetano-Anolles K.; Lee J.; Ryu S.; Choi S.; Kim H. Complete genome of Vibrio parahaemolyticus FORC014 isolated from the toothfish. Gut Pathog. 2016, 8, 59. 10.1186/s13099-016-0134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.