

Abstract
The ratoon stunting disease (RSD), caused by the bacterium Leifsonia xyli subsp. xyli (Lxx), is one of the most economically devastating diseases impacting sugarcane. RSD causes significant yield losses and variety degradation. Diagnosis of RSD is challenging because it does not exhibit any discernible internal and external symptoms. Moreover, the Lxx bacteria are very small and difficult to isolate, cultivate, and detect. In this study, conventional polymerase chain reaction (PCR), real-time quantitative PCR (RT-qPCR), and Lxx-loop-mediated isothermal amplification (Lxx-LAMP) were utilized to specifically detect the presence of Lxx pathogens in the juice from Lxx-infected sugarcane stalks and an Lxx-pMD18-T recombinant plasmid. The results showed that Lxx was a highly specific causal pathogen for RSD. All three techniques provided great reproducibility, while Lxx-LAMP had the highest sensitivity. When the DNA extract from Lxx-infected sugarcane juice was used as a template, Lxx-LAMP was 10 and 100 times more sensitive than RT-qPCR and conventional PCR, respectively. When the Lxx-pMD18-T recombinant plasmid was used as a template, Lxx-LAMP was as sensitive as RT-qPCR but was 10 times more sensitive than conventional PCR. Based on the Lxx-LAMP detection system established, adding 0.4 μM loop primers (LF/LP) can accelerate the reaction and reduce the total time required. In addition, the optimal amount of Bst DNA polymerase for Lxx-LAMP reactions was determined to be 6.0 U. The results provide technical support for the detection of RSD Lxx pathogen that will help manage sugarcane RSD.
1. Introduction
The ratoon stunting disease (RSD), first detected in 1944–1945 from sugarcane cultivar Q 28 in Queensland, Australia, is now recognized as one of the most devastating sugarcane diseases worldwide [1]. The disease is caused by a bacterium that colonizes in the xylem vessels of the sugarcane plant. Davis et al. initially named the infectious agent Clavibacter xyli subsp. xyli (Cxx) based on the morphology of the bacterium [2]; however, Evtushenko et al. renamed it Leifsonia xyli subsp. xyli (Lxx) after evaluation of its rRNA gene characteristics [3]. Sugarcane plants with RSD infection usually show a reduction in stalk height (stunting), stalk diameter, and number of tillers. These symptoms may become worse as the perennial roots age. However, these symptoms are very similar to the stunted growth caused by drought or inefficient field management. As a result, diagnosis of RSD based on visual inspection is very difficult. As a result, transmission of the Lxx pathogen from field to field by propagating cuttings from infected plants is common [4]. RSD can cause yield losses of 12%–37% under normal conditions and up to 60% under drought conditions. Moreover, RSD may also lead to variety degradation [5–7].
The Lxx bacteria are very small and difficult to isolate, cultivate, and detect [8]. Current techniques for RSD diagnosis mainly include microscope inspection, serological tests, and DNA-based molecular detection. Damann discovered a host response to the presence of the causal bacterium in the metaxylem of sugarcane with RSD [9]. This response, alkaline-induced metaxylem autofluorescence (AIMA), can be used to detect Lxx under dark-field microscopy for RSD diagnosis. However, this method is not sensitive or accurate. Later, Roach and Hoy et al. detected RSD causal pathogen directly from sugarcane juice using phase contrast microscopy (PCM) [10, 11]. This method is more accurate than the AIMA method and can determine the number of pathogens quantitatively, but its sensitivity is still not satisfactory and the procedure is tedious and complicated. Enzyme-linked immunosorbent assay based techniques include dot blot enzyme immunoassays (DB-EIA) [12], evaporative-binding enzyme immunoassays (EB-EIA) [13], and tissue blot enzyme immunoassays (TB-EIA) [14]. In 1980, the successful isolation and cultivation of sugarcane Lxx bacteria enabled application of immunoassays [15]. Since 1984, many researchers have applied immunological techniques for the diagnosis of RSD. Matthews used ELISA for detection of the causal pathogen of sugarcane RSD and it was able to test 700 samples per day, while phase contrast microscopy can only test 50–100 samples per day [16]. Shen et al. compared the diagnostic accuracy of the internal symptoms inspection technique and DB-EIA and found that the former method was less reliable, while DB-EIA was more accurate and had higher sensitivity [17]. Later, Shen et al. utilized a DB-EIA assay and detected 28.4% RSD incidence from 232 sugarcane stalk samples randomly collected from the Weng-yuan sugarcane production region [18]. Li et al. (2010a) developed a simple, rapid, accurate, and effective TB-EIA assay for RSD detection that was suitable for high-throughput diagnosis in the field [19]. Hoy et al. compared the accuracy, false positive rate, and false negative rate of five diagnosis techniques including AIMA, microscopic inspection, DB-EIA, EB-EIA, and TB-EIA. The results demonstrated that TB-EIA provides the highest sensitivity and accuracy [11].
PCR is a more accurate method of detection over microscopic inspection and serological tests. Pan et al. pioneered the development of a PCR protocol for the specific detection of Lxx. Based on the intergenic transcribed spacer (ITS) region of the 16S–23S ribosomal DNA (NCBI nucleotide database number: AF056003), two Lxx-specific primers that amplified a 438 bp PCR product were designed [20]. In the same year, Fegan et al. reported another two Lxx-specific primers that amplified a 278 bp PCR product. Since then, these two sets of primers have been widely used to detect sugarcane RSD [21]. For example, Deng et al. reported PCR detection of RSD in sugarcane samples from Guangxi Province, China [22, 23]. Shen et al. detected an Lxx isolate from Guangdong Province of China that shared almost 100% nucleotide sequence identity with those from Australia, Brazil, and USA [24]. Dan et al. were able to detect Lxx through PCR in virus-free seeds of sugarcane [25]. Zhou et al. further improved the detection accuracy using nested-PCR [26]. Kazeem et al. conducted PCR analysis of DNA extracted from sugarcane sap of 76 cultivars in Nigeria. Although internal symptoms of RSD were observed in samples of cultivar Co 510, none of the sugarcane samples, including those from Co 510, yielded the 438 bp band expected for PCR detection of Lxx [27].
Real-time quantitative PCR (RT-qPCR) provides higher accuracy and sensitivity than conventional PCR [28–30]. As a result, RT-PCR is gaining increasing applications in the diagnosis and quantification of causal pathogen in plants [30–32]. In 2007, Grisham et al. developed an RT-PCR protocol for early Lxx detection in sugarcane. Because of its quantitative capability, real-time PCR was used to rank cultivars for susceptibility to Lxx infection [33].
In 2000, Notomi et al. reported a novel PCR technique known as loop-mediated isothermal amplification (LAMP) [34]. This technique employs a set of four specifically designed primers that recognize a total of six distinct sequences on the target gene. The reaction mixture contains a strand displacement DNA polymerase (Bst) and is kept under isothermal condition (65°C) for a period of time to obtain the final PCR product. The LAMP technique does not require heat denaturation of the template, thermal cycling, or gel electrophoresis of the final product. Instead, the amplified DNA product can be analyzed by staining of fluorescence dye or measuring of the turbidity of a byproduct, magnesium pyrophosphate. The LAMP technique is simple, quick, and highly specific. The technique has been utilized for the detection of genetically modified crops [35–38], as well as viruses [39–41], fungi [42], or bacteria [43–45] in the plants. In 2013, Liu et al. successfully developed an Lxx-LAMP protocol for the detection of Lxx in RSD-infected sugarcane. When the total DNA extracted from sugarcane juice was used as a template, LAMP detection of Lxx was 10 times more sensitive than conventional PCR [43]. Su et al. developed a LAMP protocol targeting the core effector pep1 gene of the sugarcane smut pathogen, Sporisorium scitamineum. Although the LAMP method was equally sensitive to conventional PCR in amplifying the pep1 gene, it was 100 times more sensitive amplifying the bE gene of S. scitamineum [42].
In the present study, three molecular diagnostic techniques, namely, conventional PCR, RT-qPCR, and Lxx-LAMP, were used to specifically detect Lxx DNA in a dilution series of both DNA samples extracted from the juice of Lxx-infected stalks of sugarcane cultivar Yue-gan 18 and Lxx-pMD18-T recombinant plasmids. The sensitivities of these techniques in terms of the lowest detection limit of Lxx were determined. Loop primers and the amount of Bst DNA polymerase were optimized to improve the Lxx-LAMP technique. The results from this study will provide a scientific basis for selecting the best molecular diagnostic method to detect RSD infection in sugarcane.
2. Materials and Methods
2.1. Materials
Two sugarcane varieties, ROC 22 and Yue-gan 18 (also known as Guangdong sugar 00-236), were from the Key Laboratory of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture, Fuzhou, Fujian Province, China. ROC 22 was free of RSD and negative for Lxx, while Yue-gan 18 was naturally Lxx-infected, from which Lxx bacteria were isolated. Two model bacteria, Leifsonia ginseng and Leifsonia poae, were purchased from the Agricultural Culture Collection of China (Beijing, China). The Lxx-pMD18-T recombinant plasmid was constructed by inserting the 438 bp Cxx1/Cxx2-PCR amplicon [20] into the pMD18-T Vector.
2.2. Methods
2.2.1. Extraction of DNA from Sugarcane Juice
Material pretreatment: sugarcane xylem sap was collected into a 2.0 ml sterile centrifuge tube and centrifuged at 3,000 × g for 5 min at room temperature. The supernatant was then transferred to a new sterile centrifuge tube and centrifuged at 12,000 × g for 10 min at room temperature. The resulting supernatant was discarded and the pellet was kept.
The DNA was extracted from the pellets based on the CTAB method reported by Pan et al. with a minor modification [20]. The collected pellets were transferred into the cold mortar; after grinding the samples with liquid nitrogen, the ground samples were transferred into the 1.5 ml centrifuge tube. 1.0 ml CTAB extraction buffer (with 1μL mercaptoethanol) was added and then incubated for about 60 min at 65°C, mixed occasionally by hand. 0.5 ml phenol: chloroform: isoamyl alcohol (25: 24: 1) was added to each sample; the tubes were gently inverted, rocked back and forth to mix well, and then centrifuged at 12,000 × g for 10 min at room temperature. 500μL upper aqueous phase was transferred to a new 1.5 ml centrifuge tube; then, 500μL cold isopropyl alcohol was added, mixed well by inverting the tube several times, and then incubated at -20°C for at least 2 hours. The DNA pellet was obtained by centrifuging at 12,000 × g and 4°C for 15 min, and then the supernatant was removed and washed twice with 500 μL 75% ethanol. Following removal of the ethanol, the DNA pellet was centrifuged at 4,000 × g for 2 min at 4°C, after which the remaining ethanol was removed by pipette. The resultant DNA pellet was air-dried in a clean hood for about 20 min until transparent.
2.2.2. Preparation of Lxx-Infected Sugarcane Juice and Plasmid DNA Samples
The DNA concentrations of Lxx-infected juice DNA and Lxx-pMD18-T recombinant plasmid were found to be 100 ng/μL and 25 ng/μL, respectively, using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). After the initial concentration was determined, the two DNA samples were subjected to 10-fold serial dilutions. For Lxx-infected juice DNA sample, eight dilutions were prepared, namely, 100, 10−1, 10−2, 10−3, 10−4, 10−5, 10−6, and 10−7. For Lxx-pMD18-T plasmid sample, twelve dilutions were prepared, 100, 10−1, 10−2, 10−3, 10−4, 10−5, 10−6, 10−7, 10−8, 10−9, 1010, and 10−11. Next, 1.0 μl from each dilution was subjected to PCR reaction. Sterile water was used as a blank control and DNA extracted from Lxx-free ROC 22 sugarcane juice was used as a negative control.
2.2.3. Conventional PCR Detection of Lxx
The PCR was conducted on a Veriti 96-well thermal cycler (ABI, Foster City, CA, USA), and the reaction system and thermal cycles were according to Pan et al. with minor modification [20]. The PCR reaction mixture was composed of 2.5 μL 10 × Ex Taq PCR buffer, 2.5 μL 1.0% BSA, 2.0 μL dNTPs (2.5 mM each), 0.5 μL each of primers Cxx1 (10 μM) and Cxx2 (10 μM) (Table 1), 0.125 μL Ex Taq polymerase (5 U/μL), 1.0 μL DNA template, and ddH2O to a final volume of 25 μL. The thermal cycling program was 95°C for 10 min; 35 cycles (of 95°C for 30 s, 56°C for 30 s, and 72°C for 40 s); and 72°C for 5 min. PCR reactions were then held at 4°C until subsequent analysis. 5.0 μL of the PCR product was separated by 1.5% agarose gel electrophoresis photographed using a multifunctional gel imaging system and subsequently analyzed.
Table 1.
Sequences of PCR, LAMP, and RT-qPCR primers used for Lxx detection.
| Primer | Sequence (5′to 3′) |
|---|---|
| Cxx1a | CCGAAGTGAGCAGATTGACC |
| Cxx2 | ACCCTGTGTTGTTTTCAACG |
| F3b | ACATCGGTACGACTGGGT |
| B3 | TGGCCGACCAAAAAAGGT |
| FIP | GGCGTACTAAGTTCGAGCCGTT-GGTCAGCTCATGGGTGGA |
| BIP | CCTCGCACATGCACGCTGTT-CTCAGCGTCTTGAAGACACA |
| LF | CTCCGCACCAATGTCAATGT |
| LP | CTGAGGGACCGGACCTCATC |
| Lxx82Fc | TTCAACGCAGAAATTGTCCAGG |
| Lxx22R | CAAGCAGGCGTACTAAGTTCGA |
2.2.4. Real-Time Quantitative PCR
The RT-qPCR reaction mixture consisted of 12.5 μL FastStart Universal SYBR Green Master (ROX) (Roche, Shanghai, China), 0.75 μL each of Lxx82F (10 μM) and Lxx22R (10 μM) (Table 1), 1.0 μL DNA template, and 10.0 μL sterile ddH2O to give a final volume of 25 μL.
The RT-qPCR program was conducted according to Grisham et al. with minor modification [33]. Briefly, samples were subjected to 95°C for 10 s, followed by 35 cycles of 94°C for 40 s, 64°C for 45 s, and a melting process composed of 94°C for 15 s, 64°C for 1 min, and 94°C for 15 s. Each sample was performed in triplicate at each repeat. RT-qPCR was conducted on an Applied Biosystems 7500 Real-Time PCR System (ABI, Foster City, CA, USA). At the end of the reaction, the Ct value for each dilution was analyzed. Lxx was considered to be present if a positive result was observed in less than 35 cycles.
The amplification efficiency of the Lxx82F/Lxx22R primer pair was determined using a real-time PCR standard curve that was represented as a semi-log regression line plot of Ct values versus (−log) of the input DNA template amount. The efficiency (E) of the real-time PCR assay was calculated using E = (10−1/slope) − 1. Theoretically, when 0.99 < R2 < 1.0, the slope of standard curve was considered valid. The results of RT-qPCR using the specific primer pair were validated when 0.9 < E < 1.1, with an E value closer to 1.0 indicating higher amplification efficiency.
2.2.5. Lxx-LAMP Assay
The Lxx-LAMP reaction mixture was set up according to Liu et al. (2013). Briefly, the mixture consisted of 10 mM KCl, 20 mM Tris-HCl (pH 8.8), 10 mM (NH4)2SO4, 5.75 mM MgSO4, 0.1% Triton X-100, 0.2 μM each of external primers F3 and B3, 0.8 μM each of internal primers FIP and BIP (Table 1), 8 U Bst DNA polymerase, 1.4 mM dNTPs, and ddH2O to a final volume of 25 μL. The mixture was incubated at 65°C for 60 min and then heated at 80°C for 3 min to terminate the reaction. LAMP products were kept at 4°C until further analysis by adding 1.0 μL SYBR Green I (1,000X) (New England Biolabs, USA). Samples that turned green were considered to be Lxx-positive, while those that remained orange were considered to be Lxx-negative.
Two Loop primers, namely, LF and LP (Table 1), were designed according to the DNA sequence of Lxx reported by Pan et al. (1998) using the Primer Explorer 4.0 software (http://primerexplorer.jp/e/).
3. Results
3.1. Detection Specificity of the RSD Causal Agent Lxx
The RSD causal agent Lxx belongs to Leifsonia spp. In this study, two model strains of Leifsonia spp. were utilized, namely, Leifsonia ginseng and Leifsonia poae, to test the Lxx detection specificity by different methods. DNA samples prepared from xylem juice of RSD diseased Yue-gan 18 and recombinant plasmid Lxx-pMD18-T containing an Lxx-specific gene fragment were used as positive controls, while sterile water was used as a blank and DNA prepared from xylem juice of Lxx-free ROC 22 was used as a negative control. As shown in Figure 1(a), no Lxx-specific product was amplified from the blank and negative control. However, DNA sample from Lxx-infected xylem juice and recombinant Lxx-pMD18-T plasmid yielded a 438 bp Lxx-specific amplification product. In addition, no Lxx-specific product was amplified from Leifsonia ginseng and Leifsonia poae. Also shown in Figure 1(b), only the LAMP sample tubes containing Lxx-infected xylem juice DNA or recombinant Lxx-pMD18-T plasmid emitted green fluorescence. These results showed that the reaction mixtures were not contaminated and the RSD causal agent Lxx was highly specific to the molecular diagnostic techniques.
Figure 1.
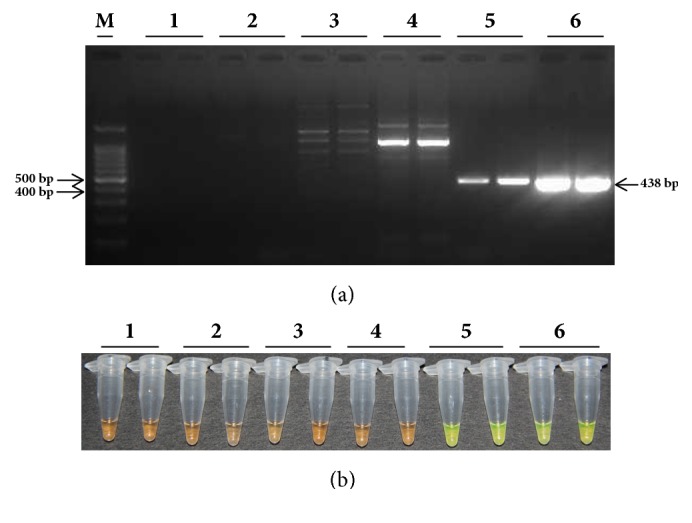
The specificity of detecting Leifsonia xyli subsp. xyli, the causal pathogen of sugarcane ratoon disease. (a) Agarose gel electropherograms of PCR amplification products. (b) Fluorescent color change of PCR amplification products. Lane M: 100 bp marker; Lane 1: ddH2O; Lane 2: 20 ng/μL DNA sample extracted from Lxx-free sugarcane juice; Lane 3: 20 ng/μL Leifsonia poae plasmid; Lane 4: 20 ng/μL Leifsonia ginsengi plasmid; Lane 5: 20 ng/μL DNA sample from Lxx-infected sugarcane juice; Lane 6: 20 ng/μL Lxx-pMD18-T plasmid.
3.2. Detection of Lxx by Conventional PCR
The initial concentration of total DNA extracted from Lxx-infected juice of Yue-gan 18 and the Lxx-pMD18-T recombinant plasmid was 100 ng/μL and 25 ng/μL, respectively. Accordingly, in a 25 μL reaction mixture, the concentration of the DNA template should be 4 ng/μL and 1 ng/μL for 100 dilution, 0.4 ng/μL and 0.1 ng/μL for 10−1 dilution, and so on, for Lxx-infected juice DNA and Lxx-pMD18-T recombinant plasmid, respectively. In our study, the lowest level of Lxx-infected juice DNA detected by the conventional PCR using Cxx1/Cxx2 primers (Pan et al., 1998) was 0.4 ng/μL (10−1 dilution) (Figure 2) and the lowest amount of Lxx-pMD18-T plasmid DNA by the conventional PCR using Cxx1/Cxx2 primers was 0.1 fg/μL or 10−7 ng/μL (10−7 dilution) (Figure 3).
Figure 2.

Agarose gel electropherograms of conventional PCR products of DNA extract from Lxx-infected sugarcane juice. Lane M: 100 bp molecular marker; Lane 1: ddH2O; Lane 2: 20 ng/μL negative DNA extracted from Lxx-free juice; Lanes 3 to 10: 10-fold serial dilutions of DNA (100 to 10−7, 4.0 ng/μL to 4.0 × 10−7 ng/μL) extracted from Lxx-infected sugarcane juice. The red arrow points to the limiting detection concentration of Lxx-infected sugarcane juice DNA.
Figure 3.
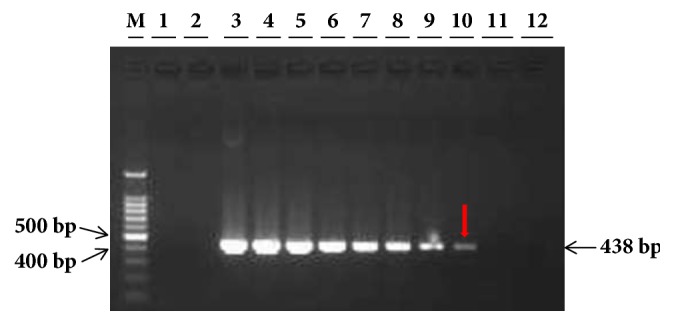
Agarose gel electropherograms of conventional PCR products of Lxx-pMD18-T plasmids. Lane M: 100 bp molecular marker; Lane 1: ddH2O; Lane 2: 20 ng/μL negative DNA extracted from Lxx-free juice; Lanes 3 to 12: 10-fold serial dilutions of Lxx-pMD18-T plasmid (100 to 10−10, 1.0 ng/μL to 1.0 × 10−10 ng/μL). The red arrow points to the limiting detection concentration of Lxx-pMD18-T plasmid.
3.3. Detection of Lxx by RT-qPCR
3.3.1. Melt-Curve and Amplification Efficiency of the Lxx82F/Lxx22R Primers
The melt-curve plots of amplification products using primer pair Lxx82F/Lxx22R are shown in Figure 4(a). A single peak melting profile representing a specific amplification product was observed, indicating that the Lxx82F/Lxx22R primers were highly specific to Lxx and could be used for the further detection of Lxx. As shown in Figure 4(b), the RT-qPCR amplification efficiency of the Lxx82F/Lxx22R primer pair was 1.01, which demonstrated that the primer pair was highly effective.
Figure 4.

Sensitivity assay of Lxx82F/Lxx22R primers set for Lxx-infected sugarcane juice DNA and Lxx-pMD18-T plasmid using RT-qPCR. (a) Melt-curve analysis; (b) standard curve; (c) amplification plot of serial dilutions of DNA sample extracted from Lxx-infected sugarcane juice; templates 1-3 were 10-fold serial dilutions of Lxx-infected sugarcane juice DNA (100 to 10−2, 4.0 ng/μL to 0.04 ng/μL); and (d) amplification plot of serial dilutions of Lxx-pMD18-T plasmid; templates 1-9 were 10-fold serial dilutions of Lxx-pMD18-T plasmid (100 to 10−8, 1.0 ng/μL to 1.0 × 10−8 ng/μL). The red arrow points to the limiting detection concentration.
Each RT-qPCR amplification reaction was performed three times that showed reproducibility. The lowest amount of Lxx-infected juice DNA detected was 0.04 ng/μL (10−2 dilution) (Table 2, Figure 4(c)) and the lowest limit of Lxx-pMD18-T plasmid detection was 10−8 ng/μL (10−8 dilution) (Table 3, Figure 4(d))
Table 2.
Ct values of RT-qPCR using serial dilutions of DNA extracted from Lxx-infected sugarcane juice as templates.
| Sample | 2013.5.6a | 2013.6.26b | ||
|---|---|---|---|---|
| Ctc | Mean Ct | Ct | Mean Ct | |
| Black (ddH2O) | — d | — | — | — |
| Negative juice DNA | — | — | — | — |
| 100 positive juice DNA | 25.6029; 25.1696; 25.139 | 25.304 | 24.6533; 23.8346; 24.3586 | 24.2822 |
| 10−1 positive juice DNA | 29.1754; 28.7072; 28.249 | 28.711 | 27.7062; 27.4855; 27.4397 | 27.5438 |
| 10−2 positive juice DNA | 32.437; 32.138; 32.4321 | 32.336 | 31.3557; 32.5417; — | 31.9487 |
| 10−3 positive juice DNA | — | — | — | — |
| 10−4 positive juice DNA | — | — | — | — |
| 10−5 positive juice DNA | — | — | — | — |
| 10−6 positive juice DNA | — | — | — | — |
| 10−7 positive juice DNA | — | — | — | — |
a Results of experiments conducted on 04/25/2013.
b Results of experiments conducted on 06/26/2013.
c “Ct” means cycle threshold.
d “—” means absence detection of Lxx.
Table 3.
Ct values of RT-qPCR using serial dilution of Lxx-pMD18-T plasmid as templates.
| Sample | 2013.5.6a | 2013.6.26b | ||
|---|---|---|---|---|
| Ctc | Mean Ct | Ct | Mean Ct | |
| Blank (ddH2O) | — d | — | — | — |
| Negative plasmid | — | — | — | — |
| 100 Lxx-pMD18-T plasmid | 8.3205; 8.4208; 8.2091 | 8.3168 | 8.5207; 7.8205; 7.947 | 8.096 |
| 10−1 Lxx-pMD18-T plasmid | 11.5595; 11.4471; 11.3005 | 11.4357 | 11.2019; 11.4752; 10.9927 | 11.223 |
| 10−2 Lxx-pMD18-T plasmid | 13.515; 13.4772; 13.5749 | 13.5224 | 13.62; 13.7713; 13.5248 | 13.639 |
| 10−3 Lxx-pMD18-T plasmid | 14.7635; 14.6707; 14.6183 | 14.6842 | 16.1848; 16.1488; 16.2099 | 16.121 |
| 10−4 Lxx-pMD18-T plasmid | 18.0854; 18.1271; 18.0325 | 18.0817 | 20.161; 20.0562; 19.7764 | 19.998 |
| 10−5 Lxx-pMD18-T plasmid | 23.0183; 23.3196; — | 23.169 | 24.3614; 24.4379; 23.9269 | 24.242 |
| 10−6 Lxx-pMD18-T plasmid | 25.0384; 24.8664; 24.7652 | 24.8900 | 28.0877; 28.1149; 27.6601 | 27.955 |
| 10−7 Lxx-pMD18-T plasmid | 29.1153; 19.8728; 29.2228 | 29.0703 | 30.9799; 30.3203; — | 30.6501 |
| 10−8 Lxx-pMD18-T plasmid | 33.412; 32.8918; — | 33.1519 | 34.906; 32.892; 33.412 | 33.737 |
| 10−9 Lxx-pMD18-T plasmid | — | — | — | — |
| 10−10 Lxx-pMD18-T plasmid | — | — | — | — |
a Results of experiments conducted on 04/25/2013.
b Results of experiments conducted on 06/26/2013.
c “Ct” means cycle threshold.
d “—” means absence detection of Lxx.
3.4. Detection of Lxx by Lxx-LAMP
Each Lxx-LAMP amplification reaction was conducted in two tubes. The experiment was repeated on two different dates, one on 04/25/2013 and one on 06/28/2013. The amplification results were reproducible. The lowest detection limit was 0.004 ng/μL (10−3 dilution) for Lxx-infected juice DNA (Figures 5(a) and 5(b)) and 10−8 ng/μL (10−8 dilution) for Lxx-pMD18-T plasmid (Figures 6(a) and 6(b)).
Figure 5.
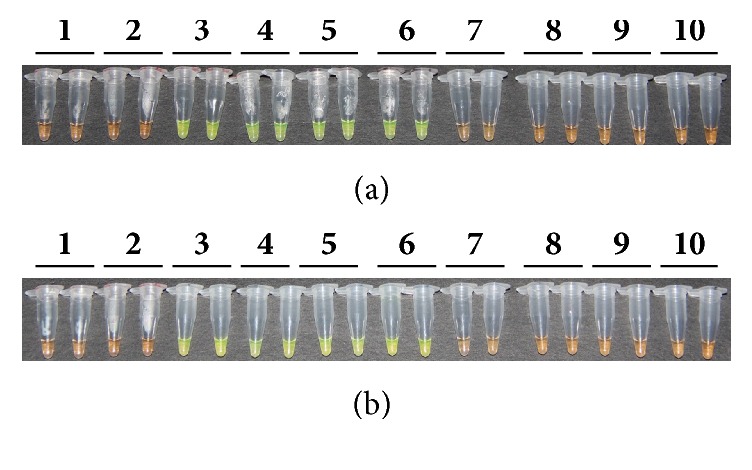
Detection of products amplified by LAMP reactions with DNA extracted from Lxx-infected sugarcane juice based on color changes. (a) Results of experiments conducted on 04/25/2013; (b) results of experiments conducted on 06/28/2013. Lane 1: ddH2O; Lane 2: 20 ng/μL DNA sample extracted from Lxx-free sugarcane juice; Lanes 3 to 10: 10-fold serial dilutions of DNA (100 to 10−7, 4.0 ng/μL to 4.0 × 10−7 ng/μL) extracted from Lxx-infected sugarcane juice.
Figure 6.
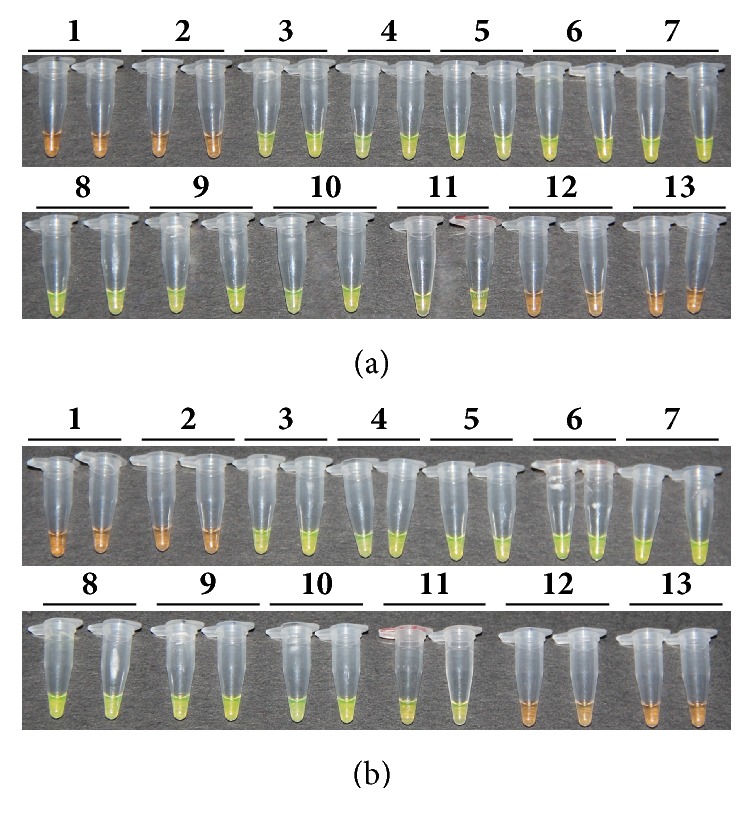
Detection of products amplified by LAMP reactions with Lxx-pMD18-T plasmids. (a) Results of experiments conducted on 04/25/2013; (b) results of experiments conducted on 06/28/2013. Lane 1: ddH2O; Lane 2: 20 ng/μL negative plasmid DNA; Lanes 3 to 13: 10-fold serial dilutions of Lxx-pMD18-T plasmid (100 to 10−11, 1.0 ng/μL to 1.0 × 10−11 ng/μL).
3.5. Optimization of Lxx-LAMP Reaction Rate
To further reduce the reaction time of Lxx-LAMP, 0.4 μM each of two additional loop primers, LF and LP, were added to an established Lxx-LAMP reaction mixture containing 20 ng Lxx-pMD18-T plasmid. The reaction system was then incubated at 65°C for 60 min; during this time an aliquot was taken out from the reaction system every 10 min and incubated at 80°C for 3 min to terminate the reaction. As shown in Figure 7(a), green fluorescence started to present after 20 min of incubation at 65°C. The results obtained from 2% agarose gel electrophoresis were similar, with ladder-like DNA bands starting to present after 20 min of incubation at 65°C (Figure 7(b)). These results demonstrated that addition of two more loop primers to the established Lxx-LAMP reaction mixture could accelerate the reaction rate.
Figure 7.

Product amplified by Lxx-LAMP reaction in the presence of loop primers LF/LP. (a) Detection of Lxx-LAMP products based on color change; (b) agarose gel electropherograms of Lxx-LAMP products. Lane M: 15,000 + 2,000 bp molecular marker; Lane NG: 20 ng/μL Lxx-negative plasmid; Lanes 10 min to 60 min: the incubation time of LAMP reaction mixture containing 20 ng/μL Lxx-positive plasmid.
3.6. Optimization of the Amount of Bst DNA Polymerase in Lxx-LAMP
To find out the optimal amount of Bst DNA polymerase, five different enzyme concentrations, namely, 0 U, 2.0 U, 4.0 U, 6.0 U, and 8.0 U, were tested in the Lxx-LAMP reaction mixture (containing 20 ng/μL Lxx-pMD18-T plasmid and 0.4 μM loop primers). The reaction mixture was incubated at 65°C for 30 min, followed by 85°C for 5 min to terminate the reaction. The amplification product was analyzed by observing the color change upon SYBR Green I staining (Figure 8(a)) and 2% agarose gel electrophoresis (Figure 8(b)).
Figure 8.
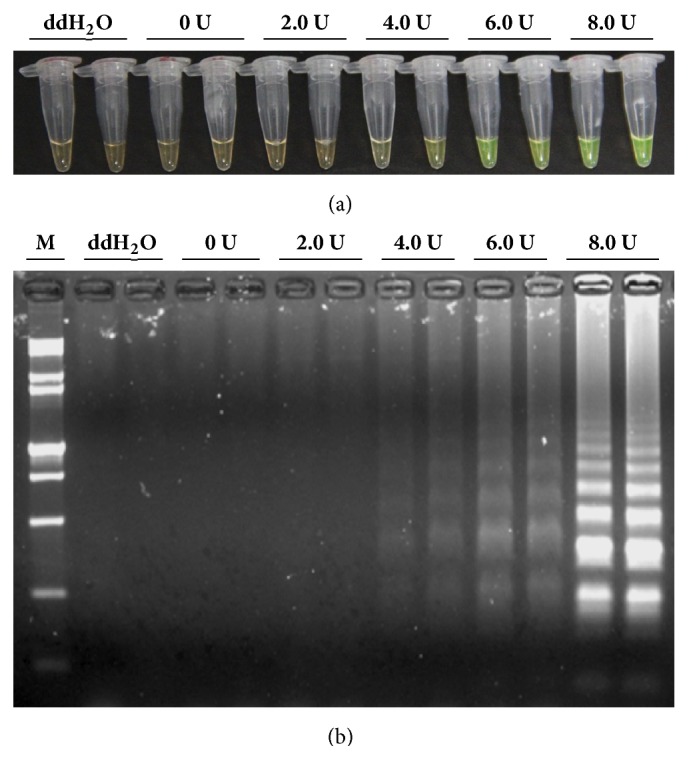
Effects of different concentrations of Bst DNA polymerase on the LAMP reaction. (a) Detection of Lxx-LAMP products based on color change; (b) agarose gel electropherograms of Lxx-LAMP products. Lane M: 15,000 + 2,000 bp molecular marker; Lanes 0 U to 8.0 U: LAMP reaction mixture containing 20 ng/μL Lxx-positive plasmid plus 0 U, 2.0 U, 4.0 U, 6.0 U, and 8.0 U Bst DNA polymerase, respectively.
As shown in Figure 8(a), faint green fluorescence was observed at 4.0 U of the Bst DNA polymerase, while more intense green fluorescence was observed when 6.0 U or 8.0 U of the Bst DNA polymerase was used. The fluorescence intensity was readily detectable by the naked eye when 6.0 U of Bst DNA polymerase was included in the Lxx-LAMP reaction.
4. Discussion
RT-qPCR is a highly sensitive nucleic acid quantification technique based on PCR. This method is advantageous over conventional PCR because it offers higher accuracy, has tremendous sensitivity [28, 29], can be highly sequence-specific [46, 47], and requires simple yet rapid experimental procedures, little to no postamplification processing, and no agarose gel electrophoresis, making it less labor-intensive [48]. As a result, RT-qPCR has had increasing applications in the diagnosis and quantification of plant pathogens [30–32, 49]. Gao et al. utilized RT-qPCR for the diagnosis and quantification of Fusarium solani f. sp. glycines in soybean sudden death syndrome (SDS), which was the first report of using the comparative threshold cycle (Ct) method to quantify the DNA of a plant pathogen relative to its host DNA [31]. Liu et al. (2014) used multiplex PCR and SYBR Green real-time PCR to facilitate the simultaneous detection of three rice pathogens, Xanthomonas oryzae pv. oryzae, Xanthomonas oryzae pv. oryzicola, and Burkholderia glumae [49]. Kokkinos et al. developed an RT-qPCR protocol for diagnosis and quantification of the sweet potato feathery mottle virus (SPFMV), sweet potato virus G (SPVG), Ipomoea vein mosaic virus (IVMV), Crinivirus sweet potato chlorotic stunt virus (SPCSV), and the Begomovirus sweet potato leaf curl virus (SPLCV) directly from infected sweet potato plants. They found lower titers of SPFMV, IVMV, and SPVG in singly infected sweet potato plants than singly infected Ipomoea setosa Ker. and I. nil cv. Scarlet O'Hara plants. Kokkinos concluded that RT-qPCR was a more efficient method for detection of SPLCV than conventional PCR assay [30]. Sayler et al. successfully developed an RT-qPCR method that is effective for detecting and identifying the bacterium Burkholderia glumae in rice seed lots [32]. Grisham et al. developed an RT-qPCR assay to quantitatively detect the RSD causal pathogen Leifsonia xyli subsp. xyli (Lxx) from the sugarcane leaf tissue [33]. Because of its quantitative nature, RT-qPCR was used to rank cultivars for susceptibility to Lxx infection. In Grisham et al.'s study, two pairs of primers (Lxx202F/Lxx331R and Lxx82F/Lxx22R) that were suitable for Lxx detection were compared. Since Lxx82F/Lxx22R amplified nonspecific products, the Lxx202F/Lxx331R primer pair was considered optimal for Lxx amplification. In our study, however, Lxx82F/Lxx22R did not amplify nonspecific products and also showed higher amplification efficiency than Lxx202F/Lxx331R. We also demonstrated that the sensitivity of RT-qPCR was 10-fold higher than that of the conventional PCR method.
Currently, PCR and ELISA are widely used techniques for RSD diagnosis. However, PCR requires more expensive instruments and takes more than 2 h [4, 50]. ELISA also has some limitations, such as the requirement of a higher titer of Lxx pathogen in infected juice for detection [33]. Since the LAMP method was first described by Notomi et al. in 2000 [34], it has been widely applied in the diagnosis of bacterial and viral infection, as well as transgenic plant detection [37–45, 51]. Li et al. designed 5 primers targeting the hlyA gene of Listeria monocytogenes (CMCC54001) and established a LAMP method for its detection [44]. Fukuta et al. developed immunocapture reverse transcription loop-mediated isothermal amplification (IC/RT-LAMP) for the detection of tomato spotted wilt virus (TSWV) [39]. This method enabled sensitive, reproducible, and specific detection of TSWV from chrysanthemum plants and was 100 times more sensitive than IC/RT-PCR. Liu et al. designed four specific primers targeting the cp gene of tomato aspermy virus (TAV), optimized reaction conditions, and established a LAMP method for TAV detection [41]. Their results demonstrated that the LAMP method was 1,000 times more sensitive than RT-PCR. Wang et al. investigated application of the LAMP method for detection of genetically modified crops. In their study, the Cauliflower mosaic virus 35S (CaMV35S) promoter gene, a widespread genetic element, was specifically amplified by the LAMP method [38]. Their results indicated that the LAMP method could detect a specific promoter containing CaMV35S and was 10 times more sensitive than conventional PCR.
Based on an Lxx-LAMP method developed by Liu et al. in 2013 [43], we added two additional loop primers to the Lxx-LAMP reaction system and reduced the total reaction time to 20–30 min. In addition, the presence or absence of target LAMP amplification products can be detected based on color change visible by the naked eye after SYBR Green I fluorescent dye staining. Moreover, when Lxx-infected juice DNA was used as a template, the Lxx-LAMP method was 10-fold and 100-fold more sensitive than RT-qPCR and conventional PCR, respectively. The Lxx-LAMP was 10-fold more sensitive than conventional PCR when Lxx-pMD18-T plasmid was used as a template. These results were consistent with those reported by Liu et al. [43], who found that the Lxx-LAMP method was 10-fold more sensitive than conventional PCR when Lxx+ juice DNA was used as a template.
In this study, three techniques, conventional PCR, Lxx-LAMP, and RT-qPCR, were used to detect the RSD casual pathogen Lxx. RSD is specifically caused by Lxx infection, which was first confirmed using the two nonpathogenic model bacteria of Leifsonia subsp. When Lxx-infected juice DNA was used as a template, the lowest DNA concentration that could be detected by conventional PCR, Lxx-LAMP, or RT-qPCR was 0.4 ng/μL, 0.004 ng/μL, and 0.04 ng/μL, respectively. The results from the three different techniques conducted on different dates were consistent, demonstrating that all three techniques provided satisfactory reproducibility. The Lxx-LAMP method offered the highest sensitivity, being 10- and 100-folder higher than RT-qPCR and conventional PCR, respectively. When Lxx-pMD18-T was used as a template, the lowest DNA concentration that could be detected by conventional PCR was 10−7 ng/μL, while for Lxx-LAMP and RT-qPCR they were both 10−8 ng/μL and the results were reproducible in experiments conducted on different dates. These results demonstrated that the sensitivity of Lxx-LAMP and that of RT-qPCR are comparable, but that both are 10-fold higher than conventional PCR.
5. Conclusions
Conventional PCR, Lxx-LAMP, and RT-qPCR all provide reproducible results for its detection, with Lxx-LAMP being the most sensitive technique to detect Lxx. In addition, when two additional loop primers were added to the Lxx-LAMP reaction mixture, the reaction was accelerated and the reaction time reduced. Moreover, the optimal amount of Bst DNA polymerase large fragment was found to be 6.0 U when taking the cost and the feasibility of detecting the color change by the naked eye into account.
Acknowledgments
This work was supported by the earmarked fund for the Modern Agriculture Technology of China (CARS-17), the 948 Program on the Introduction of International Advanced Agricultural Science and Technique of Department of Agriculture (2014-S18), and the Program on Agricultural Technology Experiment and Demonstration (K4215003A).
Contributor Information
Liping Xu, Email: xlpmail@126.com.
Youxiong Que, Email: queyouxiong@hotmail.com.
Data Availability
The data supporting the conclusions of this article are all within the paper.
Disclosure
This work was presented at the American Phytopathological Society (APS) Annual Meeting, August 5-9, San Antonio, Texas. The Abstract was published in Phytopathology 107 (12S): S5.3.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
Qibin Wu, Liping Xu, and Youxiong Que conceived and designed the experiments. Qibin Wu, Dinggang Zhou, and Shiwu Gao performed the experiments. Qibin Wu, Jinlong Guo, and Yachun Su analyzed the data. Qibin Wu, Yong-Bao Pan, Michael P. Grisham, Liping Xu, and Youxiong Que wrote the paper. All authors read and approved the final version of the paper.
References
- 1.Martin J. P., Abbott E. V., Hughes E. G. Sugarcane Diseases of the World. Vol. 1. Beijing, China: China Agriculture Press; 1982. Translated by Chen Q. L. [Google Scholar]
- 2.Davis M. J., Gillaspie A. G., Jr., Vidaver A. K., Harris R. W. Clavibacter: a new genus containing some phytopathogenic coryneform bacteria, including Clavibacter xyli subsp. xyli sp. nov., subsp. nov. and Clavibacter xyli subsp. cynodontis subsp. nov., pathogens that cause ratoon stunting disease of sugarcane and Bermudagrass stunting disease. International Journal of Systematic Bacteriology. 1984;34(2):107–117. doi: 10.1099/00207713-34-2-107. [DOI] [Google Scholar]
- 3.Evtushenko L. I., Dorofeeva L. V., Subbotin S. A., Cole J. R., Tiedje J. M. Leifsonia poae gen. nov., sp. nov., isolated from nematode galls on Poa annua, and reclassification of ‘Corynebacterium aquaticum’ Leifson 1962 as Leifsonia aquatica (ex Leifson 1962) gen. nov., nom. rev., comb. nov. and Clavibacter xyli Davis et al. 1984 with two subspecies as Leifsonia xyli (Davis et al. 1984) gen. nov., comb. nov. International Journal of Systematic and Evolutionary Microbiology. 2000;50(1):371–380. doi: 10.1099/00207713-50-1-371. [DOI] [PubMed] [Google Scholar]
- 4.Xu J. S., Xu L. P., Que Y. X., Gao S. J., Chen R. K. Advances in the ratoon stunting disease of sugarcane. Journal of Tropical and Subtropical Botany. 2008;16:184–188. [Google Scholar]
- 5.James G. A review of ratoon stunting disease. International Sugar Journal. 1996;98(1174):532–541. [Google Scholar]
- 6.Bailey R. A., Bechet G. R. Further evidence of the effects of ratoon stunting disease on production under irrigated and rainfed conditions. Proceedings of the South African Sugar Technologists Association; 1997; pp. 97–101. [Google Scholar]
- 7.Que Y. X., JS X. u., LP X. u., Gao S. J., Chen R. K. PCR detection for Leifsonia xyli subsp. xyli, pathogen of the sugarcane ratoon stunting disease. Fujian Journal of Agricultural Sciences. 2008;23:364–367. [Google Scholar]
- 8.Luo L. F., Wei C. Z., Tang J. H. Progress on detection technologies of sugarcane ratoon stunting disease. Agriculture Research and Application. 2011;6:25–27. [Google Scholar]
- 9.Damann K. E. Jr. Alkaline-induced metaxylem autofluorescence: a diagnostic symptom of ratoon stunting disease of sugarcane. Journal of Phytopathology. 1988;78(2):p. 233. doi: 10.1094/Phyto-78-233. [DOI] [Google Scholar]
- 10.Roach B. T. Sampling and diagnostic procedures for testing sugarcane resistance to ratoon stunting disease by phase contrast microscopy. Proceedings of the 1990 conference of the Australian Society of Sugar Cane Technologists; 1988; Townsville, Queensland, Australia. pp. 111–119. [Google Scholar]
- 11.Hoy J. W., Grisham M. P., Damann K. E. Spread and increase of ratoon stunting disease of sugarcane and comparison of disease detection methods. Plant Disease. 1999;83(12):1170–1175. doi: 10.1094/PDIS.1999.83.12.1170. [DOI] [PubMed] [Google Scholar]
- 12.Harrison N. A., Davis M. J. Comparison of serological techniques for diagnosis of ratoon stunting disease. Sugar Cane (Spring Supplement) 1990:5–9. [Google Scholar]
- 13.Croft B. J., Greet A. D., Leaman T. M., Teakle D. S. RSD diagnosis and varietal resistance screening in sugarcane using the EB-EIA technique. Australian Society of Sugar Cane Technologists (ASSCT) 1994;16:143–151. [Google Scholar]
- 14.Davis M. J., Dean J. L., Miller J. D., Shine J. M. A method to screen for resistance to ratoon stunting disease of sugarcane. Sugar Cane. 1994;6:9–16. [Google Scholar]
- 15.Davis M. J., Gillaspie A. G., Jr., Harris R. W., Lawson R. H. Ratoon stunting disease of sugarcane: isolation of the causal bacterium. Science. 1980;210(4476):1365–1367. doi: 10.1126/science.210.4476.1365. [DOI] [PubMed] [Google Scholar]
- 16.Matthews A. New Method Makes Identification of RSD More Efficient. Vol. 44. BSES Bulletin; 1993. [Google Scholar]
- 17.Shen W. K., Deng H. H., Zhou G. H. Comparison of diagnostic methods for sugarcane ratoon stunting disease. Acta Agariculture Universitatis Jiangxiensis. 2007;29:561–565. [Google Scholar]
- 18.Shen W. K., Deng H. H., Chen Z. H., Chen Z. J. Survey of sugarcane ratoon stunting disease in Wengyuan sugarcane regions of northern Guangdong. Guangdong Agricultural Sciences. 2009;1:60–62. [Google Scholar]
- 19.Li W. F., Huang Y. K., Wang X. Y., CW W. u., Luo Z. M., Chen X. K., et al. Rapid detection of sugarcane ratoon stunting disease by TBIA. Journal of Yunnan Agricultural University. 2010:25–132. [Google Scholar]
- 20.Pan Y.-B., Grisham M. P., Burner D. M., Damann K. E., Jr., Wei Q. A polymerase chain reaction protocol for the detection of Clavibacter xyli subsp. xyli, the causal bacterium of sugarcane ratoon stunting disease. Plant Disease. 1998;82(3):285–290. doi: 10.1094/pdis.1998.82.3.285. [DOI] [PubMed] [Google Scholar]
- 21.Fegan M., Croft B. J., Teakle D. S., Hayward A. C., Smith G. R. Sensitive and specific detection of Clavibacter xyli subsp. xyli, causal agent of ratoon stunting disease of sugarcane, with a polymerase chain reaction-based assay. Plant Pathology. 1998;47(4):495–504. doi: 10.1046/j.1365-3059.1998.00255.x. [DOI] [Google Scholar]
- 22.Deng Z. Y., Wang B. H., Liu H. B., Zhu Q. Z., Li M., Wang W. Z., et al. Occurrence and pathogen detection of sugarcane ratoon stunting disease in Guangxi province. Sugar Crops of China. 2004:35–38. [Google Scholar]
- 23.Deng Z. Y., Liu H. B., Li M., Wang B. H., Zhu Q. Z., Wang W. Z., et al. PCR detection of sugarcane ratoon stunting disease pathogen in Guangxi. Southwest China Journal of Agricultural Sciences. 2004;17:324–327. [Google Scholar]
- 24.Shen W. K., Zhou G. H., Deng H. H., Zhou L. Y. Detection of sugarcane ratoon stunting disease pathogen with polymerase chain reaction (PCR) and nucleotide sequence analysis. Chinese Agricultural Science Bulletin. 2006;22:413–416. [Google Scholar]
- 25.Dan M., Li S., Yu K. X., Liu L. M., Liu H. J., Dai Y. M. Detection of sugarcane ratoon stunting disease in virus-free seed cane of Saccharum officinarum by PCR. Agricultural Science and Technology. 2010;11:111–113. [Google Scholar]
- 26.Zhou L. Y., Zhou G. H. PCR techniques for detection of Leifsonia xyli subsp. xyli infecting sugarcane. Journal of Guangxi Agricultural and Biological Science. 2006;2:172–174. [Google Scholar]
- 27.Kazeem S. A., Ikotun B., Awosusi O. O., Wintola A. P. O., Wada A. C. Status of ratoon stunting disease of sugarcane (Leifsonia xyli subsp. xyli) in Nigeria. Tropical Plant Pathology. 2015;40(5):350–354. doi: 10.1007/s40858-015-0049-1. [DOI] [Google Scholar]
- 28.Balaji B., Bucholtz D. B., Anderson J. M. Barley yellow dwarf virus and cereal yellow dwarf virus quantification by real-time polymerase chain reaction in resistant and susceptible plants. Journal of Phytopathology. 2003;93(11):1386–1392. doi: 10.1094/PHYTO.2003.93.11.1386. [DOI] [PubMed] [Google Scholar]
- 29.Deprez R. H. L., Fijnvandraat A. C., Ruijter J. M., Moorman A. F. Sensitivity and accuracy of quantitative real-time polymerase chain reaction using SYBR green I depends on cDNA synthesis conditions. Analytical Biochemistry. 2002;307(1):63–69. doi: 10.1016/S0003-2697(02)00021-0. [DOI] [PubMed] [Google Scholar]
- 30.Kokkinos C. D., Clark C. A. Real-time PCR assays for detection and quantification of sweetpotato viruses. Plant Disease. 2006;90(6):783–788. doi: 10.1094/PD-90-0783. [DOI] [PubMed] [Google Scholar]
- 31.Gao X., Jackson T. A., Lambert K. N., Li S., Hartman G. L., Niblack T. L. Detection and quantification of Fusarium solani f. sp. glycines in soybean roots with real-time quantitative polymerase chain reaction. Plant Disease. 2004;88(12):1372–1380. doi: 10.1094/PDIS.2004.88.12.1372. [DOI] [PubMed] [Google Scholar]
- 32.Sayler R. J., Cartwright R. D., Yang Y. Genetic Characterization and Real-Time PCR Detection of Burkholderia glumae, a newly emerging bacterial pathogen of rice in the United States. Plant Disease. 2006;90(5):603–610. doi: 10.1094/PD-90-0603. [DOI] [PubMed] [Google Scholar]
- 33.Grisham M. P., Pan Y.-B., Richard E. P., Jr. Early detection of Leifsonia xyli subsp. xyli in sugarcane leaves by real-time polymerase chain reaction. Plant Disease. 2007;91(4):430–434. doi: 10.1094/pdis-91-4-0430. [DOI] [PubMed] [Google Scholar]
- 34.Notomi T., Okayama H., Masubuchi H., et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Research. 2000;28(12) doi: 10.1093/nar/28.12.e63.E63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li F., Yan W., Long L., Qi X., Li C., Zhang S. Development and application of loop-mediated isothermal amplification assays for rapid visual detection of cry2Ab and cry3A genes in genetically-modified crops. International Journal of Molecular Sciences. 2014;15(9):15109–15121. doi: 10.3390/ijms150915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou D., Guo J., Xu L., et al. Establishment and application of a loop-mediated isothermal amplification (LAMP) system for detection of cry1Ac transgenic sugarcane. Scientific Reports. 2015;4(1) doi: 10.1038/srep04912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan X., Guo J., Shen P., Yang L., Zhang D. Visual and rapid detection of two genetically modified soybean events using loop-mediated isothermal amplification method. Food Analytical Methods. 2010;3(4):313–320. doi: 10.1007/s12161-010-9132-x. [DOI] [Google Scholar]
- 38.Wang Y., Lan Q. K., Zhao X., Zhu Z., Cheng Y. Development and application of loop-mediated isothermal amplification for detection of genetically modified crops. Scientia Agriculture Sinica. 2009;42:1473–1477. [Google Scholar]
- 39.Fukuta S., Ohishi K., Yoshida K., Mizukami Y., Ishida A., Kanbe M. Development of immunocapture reverse transcription loop-mediated isothermal amplification for the detection of tomato spotted wilt virus from chrysanthemum. Journal of Virological Methods. 2004;121(1):49–55. doi: 10.1016/j.jviromet.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 40.Nie X. Reverse transcription loop-mediated isothermal amplification of DNA for detection of potato virus Y. Plant Disease. 2005;89(6):605–610. doi: 10.1094/PD-89-0605. [DOI] [PubMed] [Google Scholar]
- 41.Liu J., Huang C. L., Wu Z. Y., Zhang X. H., Wang Y. Q. Detection of tomato aspermy virus infecting chrysanthemums by LAMP. Scientia Agriculture Sinica. 2010;43:1288–1294. [Google Scholar]
- 42.Su Y., Yang Y., Peng Q., et al. Development and application of a rapid and visual loop-mediated isothermal amplification for the detection of Sporisorium scitamineum in sugarcane. Scientific Reports. 2016;6(1) doi: 10.1038/srep23994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J., Xu L., Guo J., Chen R., Grisham M. P., Que Y. Development of loop-mediated isothermal amplification for detection of Leifsonia xyli subsp. xyli in sugarcane. BioMed Research International. 2013;2013:8. doi: 10.1155/2013/357692.357692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X. M., Wang Y., Zhang X. Z., Wang X. L., Ma X. Y., Zhang H. Y., et al. Research in LAMP for detection of Listeria monocytogenes. Journal of Anhui Agricultural Science. 2010;38:13122–13134. [Google Scholar]
- 45.Nemoto J., Sugawara C., Akahane K., et al. Rapid and specific detection of the thermostable direct hemolysin gene in vibrio parahaemolyticus by loop-mediated isothermal amplification. Journal of Food Protection. 2009;72(4):748–754. doi: 10.4315/0362-028X-72.4.748. [DOI] [PubMed] [Google Scholar]
- 46.Pan B Y., Grisham M. P., Wei Q. J. PCR diagnosis of sugarcane leaf scald and ratoon stunting disease. Proceedings of the International Society of Sugar Cane; 2001; pp. 607–608. [Google Scholar]
- 47.Wong M. L., Medrano J. F. Real-time PCR for mRNA quantitation. BioTechniques. 2005;39(1):75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
- 48.Ririe K. M., Rasmussen R. P., Wittwer C. T. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Analytical Biochemistry. 1997;245(2):154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 49.Lu W., Pan L., Zhao H., et al. Molecular detection of Xanthomonas oryzae pv. oryzae, Xanthomonas oryzae pv. oryzicola, and Burkholderia glumae in infected rice seeds and leaves. The Crop Journal. 2014;2(6):398–406. doi: 10.1016/j.cj.2014.06.005. [DOI] [Google Scholar]
- 50.Rott P., Davis M. J., Baudin P. Serological variability in Xanthomonas albilineans, causal agent of leaf scald disease of sugarcane. Plant Pathology. 1994;43(2):344–349. doi: 10.1111/j.1365-3059.1994.tb02694.x. [DOI] [Google Scholar]
- 51.Chen S., Ge B. Development of a toxR-based loop-mediated isothermal amplification assay for detecting Vibrio parahaemolyticus. BMC Microbiology. 2010;10 doi: 10.1186/1471-2180-10-41.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the conclusions of this article are all within the paper.