

Abstract
NAFLD is a clinically progressive disease with steatosis, inflammation, endothelial dysfunction and fibrosis being the stages where clinical intervention becomes necessary. Lack of early biomarkers and absence of a FDA approved drug obstructs efforts for effective treatment. NAFLD progression is strongly linked to a balance between liver injury, tissue regeneration and the functioning of endogenous defense mechanisms. The failure of the defense pathways to resist the tissue damage arising from redox stress, one of the “multiple hits” in disease progression, give rise to heightened inflammation and occasional fibrosis. We introduce an endogenous defense mechanism in the liver that is mediated by TRPV4, a transient receptor potential calcium-permeable ion channel that responds to the cytotoxic liver environment and negatively regulates CYP2E1, a cytochrome p450 enzyme. Using Trpv4−/− mice and cultured primary cells, we show that TRPV4 is activated both by damage associated molecular pattern HMGB1 and collagen in diseased Kupffer cells that in turn activate the endothelial NOS (NOS3) to release nitric oxide (NO). The diffusible NO acts in a paracrine fashion in neighboring hepatocytes to deactivate the redox toxicity induced by CYP2E1. We also find that CYP2E1-mediated TRPV4 repression in late stages causes an unrestricted progression of disease. Thus, TRPV4 functions as a sensor of cell stress in the diseased fatty liver and constitutes an endogenous defense molecule, a novel concept with potential for therapeutic approaches against NAFLD, perhaps also against hepatic drug toxicity in general.
Keywords: Oxidative stress, Lipid peroxidation, Hydroxynonenal, Liver, Cation channel mechanosensor
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is progressive in nature and often arises from an underlying condition of obesity [1–4]. Features include insulin resistance, increased levels of circulating leptin, increased hepatic CYP2E1-induced redox stress and inflammation, hepatic sinusoidal endothelial dysfunction, inflammation and in more progressive cases, fibrosis [1,5–9]. NAFLD encompasses dysregulation of metabolic function and represents a hepatic manifestation of metabolic syndrome [10]. The initiation and trigger of the progressive nature of NAFLD from a condition of benign steatosis has been a focus of research interest in the last several decades. “Two hit theory”/“multiple hit theories” to account for chronic progression of NAFLD have guided the field as constructive working hypotheses, but the molecular identity of endogenous defense mechanisms that counteract disease progression has remained unclear and, perhaps, under-appreciated [11,12].
We recorded increased methylation of the promoter region of the Trpv4 gene among many other proteins in a methylomics-screen aiming to elucidate epigenetic dysregulation that underlies NAFLD. We decided to zero in on this remarkable finding because (i) TRPV4 is a Ca2+ permeable ion channel that has been found expressed in hepatic cells, and (ii) Ca2+ dynamics in liver cells can be an important switch for detoxifying redox systems such as the CYP2E1 cytochrome P450 system [7,13–16]. Interestingly detoxifying redox systems that involve CYP2E1 have been shown to play an important role in the pro-inflammatory processes in NAFLD [7,17,18]. Though CYP2E1’s role is crucial for NAFLD progression the molecular underpinnings of CYP2E1 regulation has remained elusive.
TRPV4 is a prototypic osmo-mechano TRP, a multimodally-activated TRP ion channel, activated by physical and chemical cues, and involved in multiple physiologies and pathophysiologies [19]. Its expression in many organs and cells, including liver has been demonstrated previously [19–21], and evidence suggestive of its role as a hepatic osmosensor was provided [14]. TRPV4 was first identified to be activated by hypotonicity-induced cell swelling [22,23]. The gene for this Ca2+ permeable ion channel is localized on chromosome 12q23-q24.1 in humans. TRPV4 channel proteins show a high degree of sequence homology amongst vertebrates, and it has been found to function orthologously in C. elegans lacking the primordial OSM-9 osmo-mechano-sensory TRPV ion channel [19–22].
Recent reports focus on the roles of TRPV4 in TGF-β-induced hepatic stellate cell (HSC) proliferation, an important marker of tissue regeneration and a pathophysiological event in progressive NAFLD [13]. Studies revealed that there was a significant increase in TRPV4 expression in liver fibrotic tissues. Moreover, blocking TRPV4 via incubation of HSCs with TRPV4 antagonists significantly inhibited HSC proliferation [13]. In addition, TRPV4 was found to increase autophagy and suppress HSC apoptosis [24].
Although these recent studies were interesting as they uncovered regulated expression of hepatic TRPV4 associated with HSC proliferation, they do not define a role for TRPV4 in hepato-protection or injury via the essential CYP450 pathway. We hypothesized TRPV4 to function in a hepato-protective manner during NAFLD progression by blocking CYP2E1 function via activating Kupffer cell NOS3 and releasing NO in a paracrine fashion. The TRPV4 activation and release of NO precedes its activation by damage associated molecular patterns and collagen from injured hepatocytes. Epigenetically-mediated repression of Trpv4 gene expression during NAFLD pathogenesis then accelerates disease progression resulting in a vicious cycle of an early resistance to disease progression followed by a lame surrender to the higher assault by tissue injury mediators [25–28]. Results as uncovered in our current experiments – using both in-vitro and in-vivo studies - support this novel concept and provide a mechanism, namely TRPV4-mediated regulation of CYP2E1 via NOS3. In a disease where an FDA-approved drug is not available, these novel insights have the potential to ferment new therapeutic avenues [29].
2. Results
2.1. Complex TRPV4 expression regulated by CYP2E1 suggest co-contributory role for TRPV4 in NAFLD
Increased CYP2E1 activity in progressive NAFLD has been reported. Here we show that increased CYP2E1 activity is closely associated with change of TRPV4 protein expressions across cell types in the diseased liver. TRPV4 protein (red) showed increased presence in the hepatocytes (albumin positive cells, green) of human and mouse NAFLD control tissues (Fig. S1A and B) as shown by arrows. Progressive stage of the disease as characterized by increased inflammation, lobular necrosis that correlated with increased CYP2E1 activity showed a rapid shift in TRPV4 protein expression from hepatocytes to CD68-positive macrophages that are essentially Kupffer cells (Fig. S1B). CD68 positive macrophages showed increased TRPV4 expression in both human and mice livers (Fig. S1B) (Albumin-green, upper panel; CD68-green, lower panel). CYP2E1 protein activation in the NAFLD mouse liver was associated with strong DNA methylation in the TRPV4 promoter region (p=0.002) causing significantly decreased protein expression and was CYP2E1 protein dependent since CYP2E1 knockout (CYP2E1 KO) mice showed no such change in protein expression at 4 weeks (4w) following CYP2E1 activation by a specific substrate BDCM (Fig. S2A, D–G). TRPV4 DNA promoter methylation might be ascribed to increased activity of DNA methyl transferase I (Dnmt1) in mouse livers with increased CYP2E1 activity (Fig. S2B and C) when compared to controls (those that had only steatosis and no significant rise in CYP2E1 activity). However, Dnmt1 expression is significantly decreased in CYP2E1 KO mice and Resveratrol (known natural antioxidant compound found in red grapes) treated mice co-exposed with pyrazole (Fig. S2C) when compared with NAFLD mice treated with pyrazole. This data clearly suggest that Dnmt1 expression and activation depends on CYP2E1 mediated oxidative stress. Interestingly TRPV4 protein was increased at 1 week (1w) following introduction of the CYP2E1 substrate in mouse NAFLD liver while it was unchanged in CYP2E1 KO mice (Fig. S2D–G). All the above results were significant at p < 0.05 levels (individual p values are provided in the figures). The results suggested that CYP2E1 activity regulated TRPV4 protein expression in NAFLD mouse livers with an initial increase in its expression followed by a rapid decrease as the disease progressed probably because of Trpv4 promoter methylation initiated by CYP2E1-mediated oxidative stress that is unrestricted at this stage of disease progression. In parallel to the above observations, increased CYP2E1 activity led to significantly higher lipid peroxidation (4-hydroxynoeanal (4-HNE), an indirect measure of the reductive metabolism by CYP2E1), tyrosyl radical adducts (3-NTR)(a free radical process where tyrosyl radical formation takes place following oxidative stress), increased CYP2E1 and HMGB1 mRNA (measure of cell damage)(Fig. S3A–F), increased mRNA expression of MCP-1, IL-Iβ, and IFN-γ, all known to be hallmarks of NAFLD-induced inflammation (Fig. S4A) (data were significant at p < 0.05). Further, CYP2E1 activity led to increased circulating insulin, higher than the levels seen in obesity, increased tissue expression of MMP-1A, MMP-9, TIMP1, as well as markers of increased extracellular matrix deposition in the form of collagen, and Collagen-1-α-1 mRNA. These findings strongly suggest an important role of CYP2E1 activity in the NAFLD disease progression, which is also characterized by increased extracellular matrix formation and tissue stiffness [30]. In turn, this is not only an interesting regulation, but also possibly relevant in the context of our present study because changes in tissue stiffness feedback to mechanically-sensitive TRPV4.
2.2. Trpv4 loss of function increases liver injury and inflammation in progressive NAFLD
In order to better understand the role of TRPV4 in NAFLD, encouraged by the above findings, we studied hepatic injury in an NAFLD-model in mice, using Trpv4−/− (Trpv4 KO) mice. Trpv4 loss of function in mice led to a worsening of NAFLD at 1 week following CYP2E1 activation (Fig. 1). Liver histopathology revealed markedly higher hepatocellular necrosis, ballooning, formation of Mallory-Denk bodies and increased bipolar nuclei in Trpv4 KO-NAFLD mice treated with CYP2E1 substrate pyrazole (PYR) when compared to either NAFLD (WT-NAFLD or Trpv4 KO-NAFLD) control alone, or WT-NAFLD+PYR (Fig. 1A and Table S1). Trpv4 KO-NAFLD+PYR mice showed significantly increased Kupffer cell activation (CD68) and IL-1β levels when compared to NAFLD control (WT-NAFLD or Trpv4 KO-NAFLD) alone or WT-NAFLD+PYR groups (Fig. 1B–E)(p < 0.05). mRNA expressions of F4/80 (macrophage marker), IL-1β and MCP-1 significantly increased in Trpv4 KO-NAFLD+PYR mice when compared to respective NAFLD control (WT-NAFLD or Trpv4 KO-NAFLD) alone or WT-NAFLD+PYR groups (Fig. 1H). Trpv4 gene deletion also showed increased mRNA and protein expression of damage associated molecular pattern HMGB1 when compared to respective NAFLD control (WT-NAFLD or Trpv4 KO-NAFLD) alone or WT-NAFLD+PYR treated groups (Fig. F,G, J). Finally the expression of MMP-1A, MMP-9 and TIMP1, proteolytic regulators of extracellular matrix and substrates of tissue rigidity, increased significantly in Trpv4 KO-NAFLD+PYR mice when compared to either respective NAFLD control (WT-NAFLD or Trpv4 KO-NAFLD) alone or NAFLD-WT+PYR treated groups (Fig. 1I). The marked increase in TIMP-1A in TRPV4 KO-NAFLD+PYR group was not significant (p=0.284) when compared to NAFLD-WT+PYR group (Fig. 1I). Similarly, we also have observed that in human NAFLD, CYP2E1 activity (lipid peroxidation) marker 4-HNE, damage associated molecular pattern HMGB1, Inflammatory response cytokine IL-1β and tissue stiffness marker Collagen-1 were significantly higher as compared to human control groups (Fig. S5A–D). The results above suggest strongly that TRPV4 functions in controlling oxidative stress and inflammation while, release of damage-associated molecules and stiffness of extracellular matrix might not be controlled by TRPV4 though all of them are known pathophysiological accelerator mechanisms in disease progression of NAFLD. Interestingly, higher collagen that is linked to liver tissue stiffness might act as an activator for TRPV4.
Fig. 1. Loss of function of Trpv4space increases liver injury in progressive NAFLD.
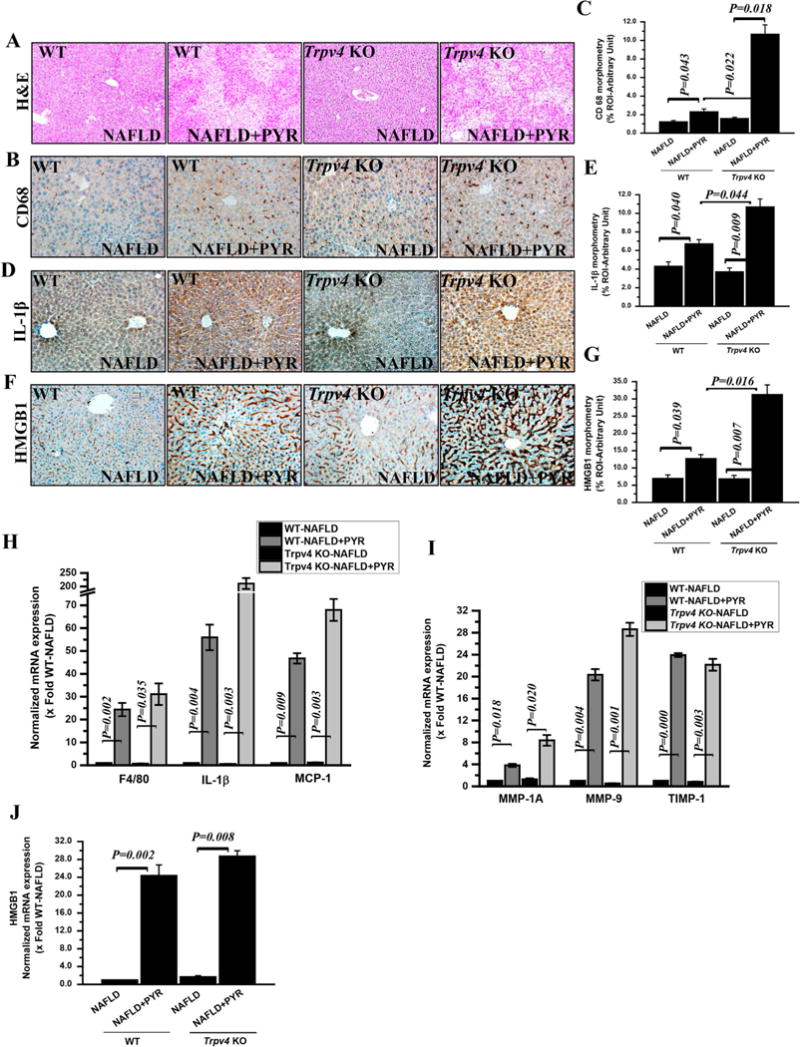
(A) Representative Hematoxylin and Eosin stained (H & E) images of liver sections showed NAFLD pathophysiology of wild type NAFLD (WT-NAFLD, n=4) as a control, wild type NAFLD treated with CYP2E1 substrate pyrazole (WT-NAFLD+PYR, n=3) as a wild type progressive NAFLD, Trpv4 KO-NAFLD (n=3) as a knockout control and Trpv4 Knockout (Trpv4 KO) treated with pyrazole (Trpv4KO-NAFLD+PYR, n=4) as a progressive NAFLD in absence of Trpv4 functional gene. Images were taken at 10X magnification. (B, D and F) Representative immunohistochemistry images depicting immunoreactivity of Kupffer cell activation marker CD68 (B), Interleukin-1β cytokine (IL-1β) (D) and HMGB1 (F) in the liver section of WT-NAFLD, WT-NAFLD+PYR, Trpv4 KO-NAFLD and Trpv4 KO-NAFLD+PYR mice. (C, E and G) Morphometric analysis of CD68 (C), IL-1β (E) and HMGB1 (G) immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (analysis done on images from three separate microscopic fields). All microscopic images were taken at 20X magnification. (H, I and J) mRNA expression of pan macrophage marker F4/80, IL-1β and MCP-1 (H), MMP-1A, MMP-9, TIMP-1 (I) and HMGB1 (J) genes in the liver of WT-NAFLD (n=4), WT-NAFLD+PYR (n=3), Trpv4 KO-NAFLD (n=3) and Trpv4 KO-NAFLD+PYR (n=4) groups of mice. Normalized mRNA expression is represented as fold change of NAFLD on Y-axis. Data points represented with means ± SEM. P value was calculated using unpaired t-test.
2.3. Trpv4 loss of function causes increase in CYP2E1 protein levels and function in NAFLD
The above results argued for a significant role of TRPV4 in NAFLD pathophysiology. We did not fail to notice the profile of the hepatic injury as a result of Trpv4 KO-NAFLD+PYR group resembling well-established consequences of increased CYP2E1 function in NAFLD [16]. In order to address whether hepatic CYP2E1 protein levels depended on Trpv4, we found that CYP2E1 protein levels were significantly higher in Trpv4 KO (Trpv4 KO+NAFLD+PYR) mice when compared to NAFLD mice alone either WT or Trpv4 KO groups (Fig. 2A and B)(p < 0.05). Levels of 4-HNE, a marker for lipid peroxidation and 3-nitrotyrosine, a marker for tyrosine radical adducts that were previously shown by us as co-markers of CYP2E1 function in NAFLD were significantly higher in Trpv4 KO mice exposed to pyrazole when compared to NAFLD groups (WT-NAFLD or Trpv4 KO-NAFLD) or WT-NAFLD+PYR groups respectively (Fig. 2C–F)(p < 0.05). These results indicate TRPV4 to be functional in down-regulating CYP2E1 expression and function in NAFLD progression.
Fig. 2. Loss of function of Trpv4 increases CYP2E1 expression and activity in progressive NAFLD.
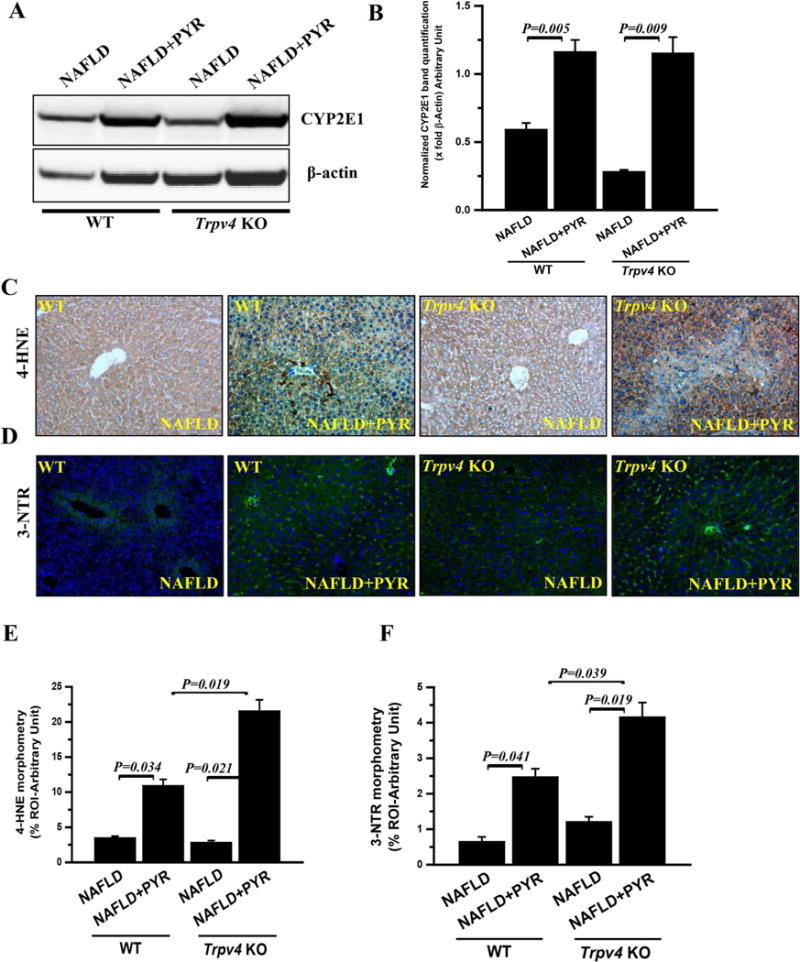
(A and B) Immunoblot of CYP2E1 (A) and morphometric analysis (B) in WT-NAFLD (n=4), WT-NAFLD+PYR (n=3), Trpv4 KO-NAFLD (n=3) and Trpv4 KO-NAFLD+PYR (n=4) liver homogenate. (C and D) Representative immunohistochemistry images depicting 4-HNE immunoreactivity (C) and Immunofluorescence images depicting 3-Nitrotyrosine (D) immunoreactivity (green) and counterstained with DAPI (blue) in WT-NAFLD (n=4), WT-NAFLD+PYR (n=3), Trpv4 KO-NAFLD (n=3) and Trpv4 KO-NAFLD+PYR (n=4) group of mice. Images were taken at 20X magnification. (E and F) Bar graph showed morphometric analysis of 4-HNE (E) and 3-Nitrotyrosine (F) immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=3) Data points represented with means ± SEM, unpaired t-test. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.4. CYP2E1-inhibition in Trpv4 KO mice with NAFLD decreased oxidative stress, and attenuated the NAFLD pathology
In view of the above finding, given the known pro-injurious role of CYP2E1 in NAFLD progression, we treated Trpv4 KO-NAFLD+PYR mice with specific CYP2E1 inhibitor diallyl sulfide (DAS). As a result, these mice had significantly decreased lipid peroxidation than vehicle-treated (no DAS) Trpv4 KO-NAFLD+PYR mice (Fig. 3A and C). Administration of DAS to Trpv4 KO-NAFLD+PYR mice also significantly decreased activated Kupffer cells as shown by significantly decreased protein levels of CD68 (Fig. 3B and D)(p < 0.05) vs vehicle-treated Trpv4 KO-NAFLD+PYR mice. In keeping, histopathology of DAS-treated Trpv4 KO-NAFLD+PYR mice showed significantly decreased hepatocellular necrosis, ballooning, Mallory-Denk bodies and lobular inflammation (Fig. 3E and Table S2). These observations establish the concept that blocking CYP2E1 function in Trpv4 KO mice that had NAFLD and were primed for higher CYP2E1 activity attenuated the severity of the disease. This was corroborated and extended by our findings that DAS treatment in Trpv4 KO-NAFLD+PYR mice significantly decreased tyrosyl adducts (Fig. S6A and B), mRNA expressions of MCP-1, IL-1β, IFN-γ, F4/80 and Kupffer cell activation marker CD68 (Fig. S6C) when compared to WT-NAFLD+PYR and Trpv4 KO-NAFLD+PYR groups, as was expression of extracellular matrix-remodeling and pro-inflammatory metalloproteases MMP-1A, MMP-9 and TIMP1 (Fig. S6D) (all data sets were significant below the p=0.05 levels). Thus, we provide substantial evidence for our new concept, namely that inhibition of CYP2E1 function attenuates excess hepatic damage in a mouse NAFLD-model, as a consequence of genetically-encoded absence of Trpv4. We believe that this is important because inhibition of CYP2E1 appears beneficial to slow down NAFLD progression. Interestingly, CYP2E1 inhibition in Trpv4 KO-NAFLD+PYR mice also resulted in significant decrease in the expression of primary DNA methyltransferase I (Dnmt1) showing that Trpv4 promoter methylation, as we observed, had a direct bearing on the increased activity of CYP2E1 found due to lack of an opposing action from TRPV4 (Fig. S7A and B). This set of findings begets the question how TRPV4 function in NAFLD can be hepato-protective.
Fig. 3. Inhibition of CYP2E1 activity showed decreased oxidative stress and inflammation in progressive NAFLD.
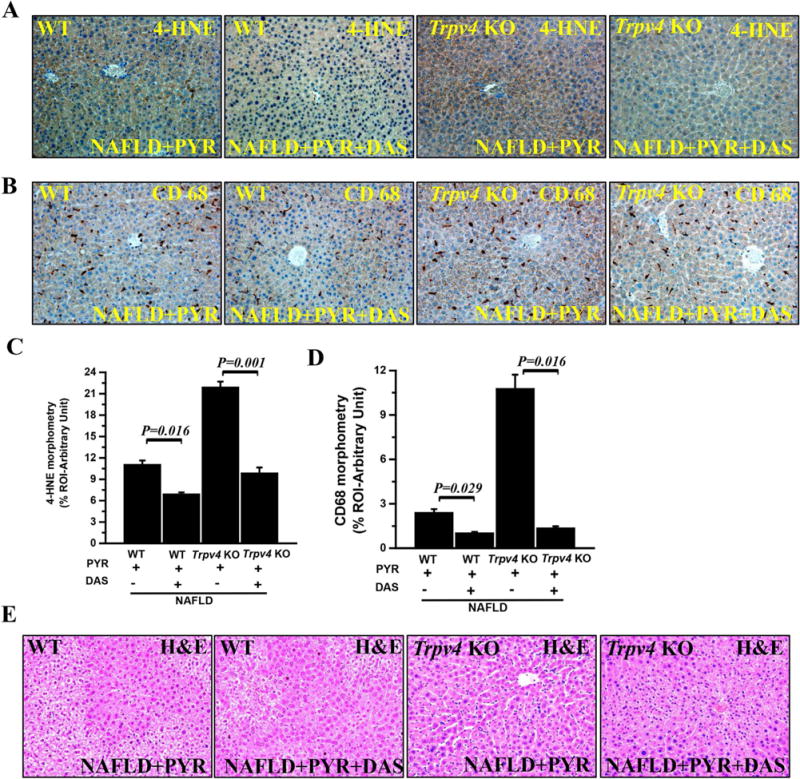
To study the role of CYP2E1 in progressive NAFLD, both wild type and Trpv4 KO mice were co-exposed with pyrazole and diallyl sulfide (DAS) known inhibitor of CYP2E1. (A and B) Representative immunohistochemistry images depicting lipid peroxidation marker (4-HNE) immunoreactivity (A) and Kupffer cell activation marker (CD68) immunoreactivity (B) in WT-NAFLD+PYR (n=3), WT-NAFLD+PYR+DAS (n=3), Trpv4 KO-NAFLD+PYR (n=4) and Trpv4 KO-NAFLD+PYR+DAS (n=3) groups of mice liver. Images were taken at 20X magnification. (C and D) Bar graph showed morphometric analysis (% ROI) of 4-HNE (C) and CD68 (D) immunoreactivity (n=3). (E) Representative Hematoxylin and Eosin staining of liver sections showed pathophysiology of WT-NAFLD+PYR (n=3), WT-NAFLD+PYR+DAS (n=3), Trpv4 KO-NAFLD+PYR (n=4) and Trpv4 KO-NAFLD+PYR+DAS (n=3) liver. The morphometric data point was represented as means ± SEM. P value was calculated with unpaired t-test.
2.5. Trpv4 loss of function decreases hepato-protective nitric oxide (NO) bioavailability
We and others have shown previously that in the liver, NO bioavailability plays a crucial role in attenuating CYP2E1-mediated oxidative stress, inflammation and NAFLD pathology [7,31]. We identified the NOS3-dependent NO pathway as a candidate mechanism to function in a TRPV4-dependent manner, based on previous findings in endothelial cells [32]. Results showed that CYP2E1 activation did not significantly triggered NO release in WT-NAFLD+PYR mice when compared to WT-NAFLD mice but was severely attenuated in Trpv4 KO-NAFLD+PYR mice (Fig. 4A). We identified decreased hepatic NOS3 phosphorylation in these animals as likely cause. We found that Trpv4 KO-LEAN, Trpv4 KO-NAFLD and Trpv4 KO-NAFLD+PYR groups showed significantly decreased eNOS (NOS3) activation when compared to either WT-LEAN, WT-NAFLD or WT-NAFLD+PYR respectively (Fig. 4B and C). Interestingly, ratio of hepatic pNOS3/NOS3 was highest in unchallenged WT mice, decreasing in NAFLD mice, and further going down in NAFLD mice treated with pyrazole. In striking contrast, the ratio remained low and unchanged in Trpv4 KO mice, clearly suggesting the critical role of the Trpv4 gene, and also corroborating our concept that NAFLD as it progresses is associated with Trpv4 loss-of-function that becomes more profound with NAFLD-progression. To determine the role of NO in enhanced NAFLD pathogenesis in Trpv4 KO mice, NO donor was administered to both WT-NAFLD+PYR and Trpv4 KO-NAFLD+PYR groups that had increased CYP2E1 activation (Fig. 4). This led to decreased lipid peroxidation (4-HNE) and CD68 immunoreactivity in the liver, indicating improved outcomes as compared to vehicle-treated Trpv4 KO-NAFLD+PYR mice (Fig. 4D,E,G,H). Trpv4 KO-NAFLD+PYR mice administered with NO donor showed decreased tyrosyl radical formation (Fig. S8A and B), decreased inflammatory indicators MCP-1, IL-1β, IFN-γ,F4/80 and HMGB1 expression when compared to vehicle-treated Trpv4 KO-NAFLD+PYR mice (Fig. S8C-D). Hematoxylin and Eosin stained liver sections from Trpv4 KO-NAFLD+PYR mice treated with NO donor vs vehicle-treated Trpv4 KO-NAFLD+PYR had decreased hepatocellular necrosis, decreased ballooning, decreased lobular inflammation and decreased Mallory-Denk bodies indicative of attenuated NAFLD (Fig. 4F and Table S3). Thus, increased NO bioavailability in the liver can rescue hepato-injurious loss-of-function of Trpv4. Interestingly, increased NO bioavailability through NO donor administration also significantly decreased Dnmt1 expression in WT-NAFLD+PYR+NO or WT-NAFLD +BDCM(1w)+NO groups as compared to vehicle treated WT-NAFLD +PYR or WT-NAFLD+BDCM (1w) groups suggesting that TRPV4-mediated NO release might be the mechanism of resisting CYP2E1-induced promoter methylation of Trpv4 in late stages of NAFLD (Fig. S9A and B)(p < 0.05).
Fig. 4. Nitric oxide (NO) release and its role in attenuating CYP2E1 activity and resulting in decreased inflammation in progressive NAFLD.
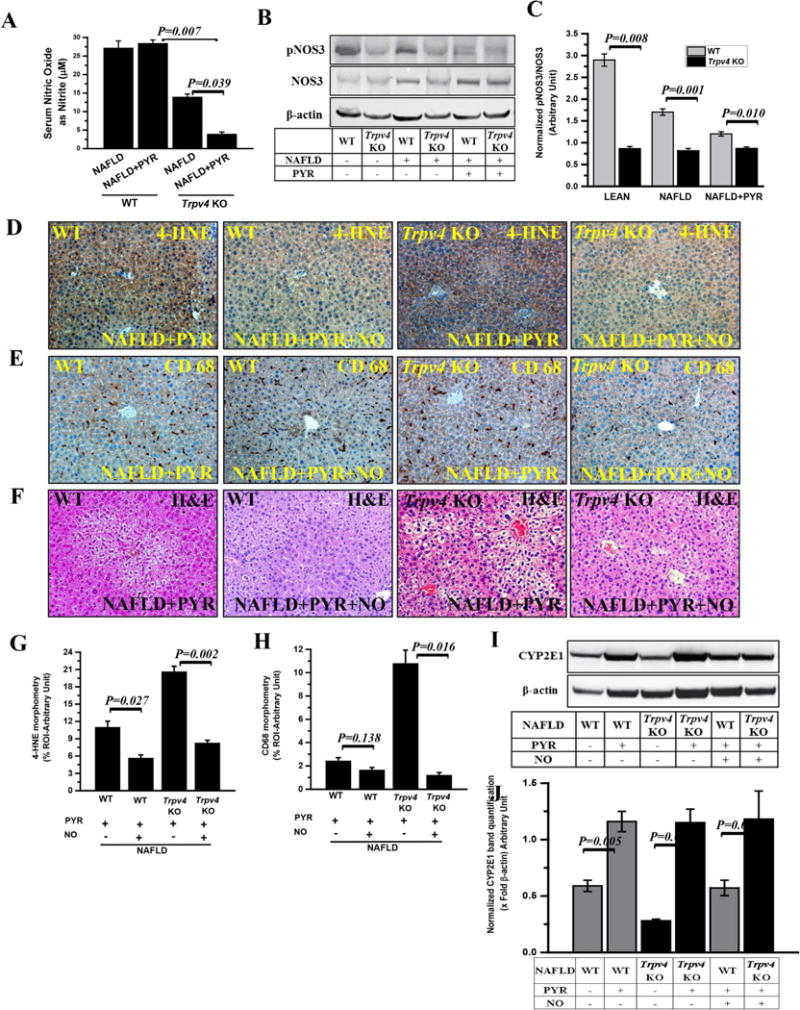
(A) Nitric oxide release measured as total nitrite (μM) in mouse serum of WT-NAFLD, WT-NAFLD+PYR, Trpv4 KO-NAFLD and Trpv4 KO-NAFLD+PYR mice. (B) Immunoblot of phosphorylated NOS3- Ser 1177 (pNOS3) and NOS3 protein in liver homogenate of both wild-type (WT) and Trpv4 KO mice fed with chow diet (LEAN, n=3), high-fat- diet fed (NAFLD, n=4) and high-fat-diet fed mice exposed with pyrazole (NAFLD+PYR, n=3). (C) Bar graphs represents protein band quantification analysis of western blot and β-actin normalized pNOS3/NOS3 ratio in respective groups of mice was plotted on y-axis. (D and E) To study the role of nitric oxide, both WT and Trpv4 KO mice groups were co-exposed with pyrazole and nitric oxide donor (NO, DETANONOate). Representative immunohistochemistry images depicting 4-HNE immunoreactivity (D) and CD68 immunoreactivity (E) in WT-NAFLD+PYR (n=3), WT-NAFLD+PYR+NO (n=3), Trpv4 KO-NAFLD+PYR (n=4) and Trpv4 KO-NAFLD+PYR+NO (n=3) liver. Images were taken at 20X magnification. (G and H) Bar graph showed morphometric analysis (% ROI) of 4-HNE (G) and CD68 (H) immunoreactivity (n=3). (F) Representative Hematoxylin and Eosin stained images of liver sections showed pathophysiology of WT-NAFLD+PYR (n=3), WT-NAFLD +PYR+NO (n=3), Trpv4 KO-NAFLD+PYR (n=4) and Trpv4 KO-NAFLD+PYR+NO (n=3). Images were taken at 20X magnification. (I and J) Immunoblot of CYP2E1 (I) and morphometric analysis (J) in WT-NAFLD (n=4), WT-NAFLD+PYR (n=3), Trpv4 KO-NAFLD (n=3), Trpv4 KO-NAFLD+PYR (n=4), WT-NAFLD+PYR+NO (n=3) and Trpv4 KO-NAFLD+PYR+NO (n=3) liver homogenate. Data points were represented with means ± SEM, unpaired t-test.
2.6. TRPV4-induced NO from Kupffer cells attenuates CYP2E1 function in hepatocytes: a new mechanistic Kupffer cell-hepatocyte feedback loop involving Trpv4 in NAFLD
TRPV4 has been shown to activate endothelial nitric oxide synthase (NOS3) and generate nitric oxide in human brain microvascular endothelial cells [32]. Further it has been shown to regulate mouse colonic motility by activating nitric oxide (NO)-dependent enteric neurotransmission, renal function and endothelium dependent relaxation in pulmonary arteries [33–36]. Based on this background, we performed experiments in hepatocytes and Kupffer cells by studying the TRPV4-mediated NO generation in these cells. Human hepatocytes (HepaRG) showed a significant increase in TRPV4 protein levels following stimulation by a selective Trpv4 activator (GSK1016790A in cells that were stimulated with CYP2E1 substrate pyrazole (P < 0.05) but failed to generate significant levels of NO as measured in the cell supernatant of both HepaRG and primary human hepatocytes (Fig. S10A–D). NO levels in the Rat Kupffer cells were significantly higher when primed with HMGB1 when compared to unprimed cells (Fig. 5A). Co-stimulation with Trpv4 activator had significant elevation of NO levels when compared to HMGB1 alone or co-stimulated with Trpv4 antagonist (P < 0.05) with a slight decrease in the cells that were co-stimulated with a Trpv4 antagonist (Fig. 5A). Normalized ratio of p-NOS3/NOS3 as indicator of eNOS activation was significantly elevated in HMGB1 primed cells (p < 0.05) while co-stimulation with a Trpv4 antagonist significantly decreased the eNOS activation in these cells (p < 0.05)(Fig. 5C and E). Co-stimulation with a Trpv4 agonist restored the eNOS activation and was significantly higher than that of the antagonist treatment group (Fig. 5C and E)(p < 0.05). Similar result patterns were obtained when the cells were primed with collagen, an important constituent of extracellular matrix that is shown to be increased when there is a rise in tissue stiffness and progressive stages of NAFLD or is released by damaged hepatocytes. Collagen priming of Kupffer cells significantly increased NO production and eNOS activation while co-stimulation with a TRPV4 antagonist significantly decreased NO generation through a NOS3-dependent process (Fig. 5B, D and F)(P < 0.05). Co-stimulation with a Trpv4 activator restored NO generation, which was significantly higher than the antagonist treated group (Fig. 5B, D, and F)(P < 0.05). These results suggest that Kupffer cells but not hepatocytes are the seats of NO generation by the TRPV4-eNOS pathway and might play a vital role in the liver microenvironment in regulating the increased CYP2E1-mediated redox toxicity that characterizes progressive stages of NAFLD.
Fig. 5. Trpv4 activation and nitric oxide release in primed rat Kupffer cells.
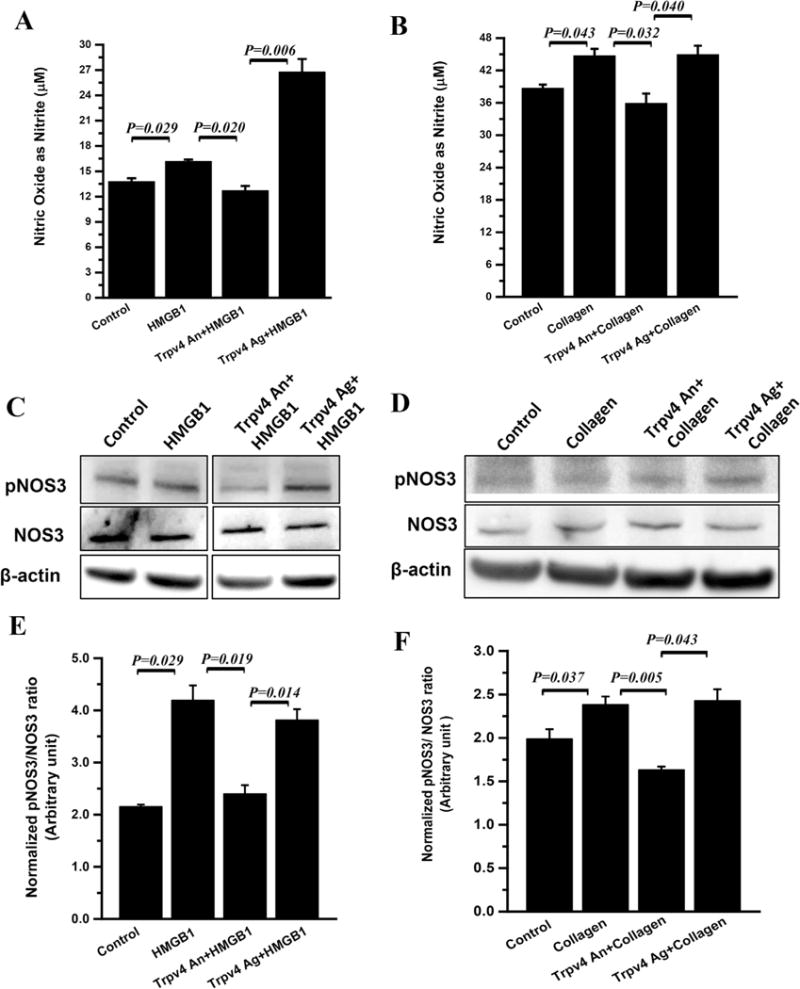
To study the role of Kupffer cell in production of extracellular nitric oxide, the rat Kupffer cells were primed with HMGB1 or collagen in the presence of Trpv4 antagonist or agonist. (A and B) Nitric oxide release measured as total nitrite (μM) in culture supernatant of control, HMGB1, Trpv4 An+ HMGB1, and Trpv4 Ag+ HMGB1 groups (HMGB1 primed groups, A) and control, collagen, Trpv4 An+ Collagen, and Trpv4 Ag+ Collagen (collagen primed groups, B). (C and D) Immunoblot of pNOS3, NOS3 protein of the total Kupffer cell lysate of HMGB1 primed groups (C) and collagen primed groups (D). (E and F) Bar graphs represent protein band quantification analysis of HMGB1 primed groups (E) and collagen primed groups (F) and y-axis represent β-actin normalized pNOS3/NOS3 ratio in respective groups. Data points were represented with means ± SEM and p value was calculated using unpaired t-test.
2.7. Kupffer cell-Hepatocyte crosstalk for regulation of CYP2E1-dependent redox toxicity and HMGB1 release is regulated by TRPV4-mediated NO release
Failure of the hepatocyte to generate significant NO in spite of higher TRPV4 protein levels following stimulation with CYP2E1 substrate pyrazole led us to hypothesize that TRPV4-mediated NO in Kupffer cells might act in a paracrine fashion to regulate hepatocyte function. Our previously published results of NO-mediated regulation of CYP2E1-redox toxicity in NAFLD supported the rationale for experiments involving hepatocytes primed with Kupffer cell conditioned medium. Results showed that rat primary hepatocytes stimulated with pyrazole had a significant increase in CYP2E1 protein as seen by immunofluorescence imaging and morphometry (Fig. 6A panel i–viii, and B)(P < 0.05) while administration of CYP2E1 inhibitor DAS significantly decreased the above levels. Co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen (CM1) did not show any change in CYP2E1 protein levels. Co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen and Trpv4 antagonist (CM2) showed a significant increase in CYP2E1 protein levels while treatment with an agonist (CM3) significantly decreased CYP2E1 protein levels when compared to pyrazole-stimulated hepatocyte group (Fig. 6Aand B )(P < 0.05). To ensure that TRPV4 mediated its effect through endothelial NOS (NOS3) the Kupffer cell supernatants were generated using Kupffer cells primed with selective eNOS inhibitor L-NAME (CM4 and CM5). Co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen and L-NAME (CM4) showed a significant increase in CYP2E1 protein levels when compared to similar agonist (CM5) group (P < 0.05). These results suggested that CYP2E1 protein levels in hepatocytes were dependent on TRPV4 mediated NO release in Kupffer cells.
Fig. 6. NO released from Kupffer cells attenuate CYP2E1 function in Hepatocytes.
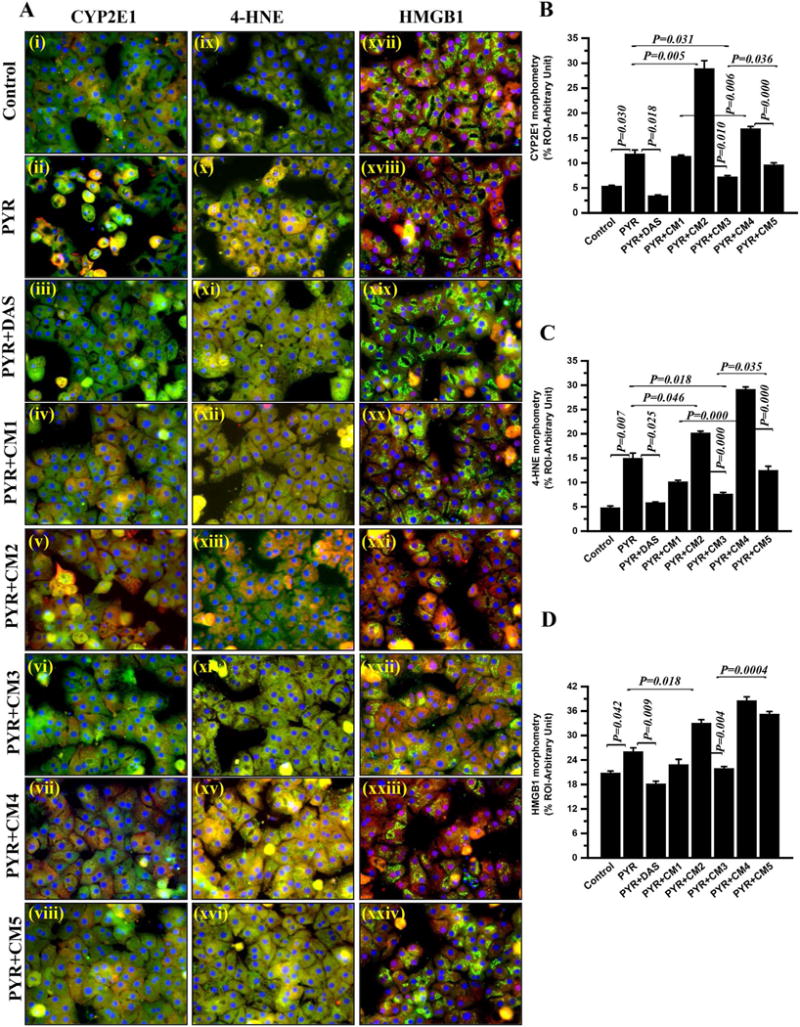
Primary hepatocytes from wild type Rat liver plated on coverslips were co-exposed with conditioned media (Kupffer cell supernatant, CM) and/or pyrazole. The hepatocytes exposed with vehicle control Kupffer cell supernatant (CM0) (control); with Pyrazole and vehicle control Kupffer cell supernatant (CM0) (PYR); with pyrazole and diallyl sulfide in vehicle control Kupffer cell supernatant (CM0) (PYR+DAS); with pyrazole and Kupffer cell supernatant treated with collagen (CM1) (PYR+CM1); with pyrazole and Kupffer cell supernatant treated with collagen+Trpv4 antagonist (CM2) (PYR+CM2); with pyrazole and Kupffer cell supernatant treated with collagen+Trpv4 agonist (CM3) (PYR+CM3); with pyrazole and Kupffer cell supernatant treated with collagen+ Nitric oxide synthase inhibitor L-NAME (CM4) (PYR+CM4); with pyrazole and Kupffer cell supernatant treated with collagen+Trpv4 agonist+L-NAME (CM5) (PYR+CM5). (A) Representative merged immunofluorescence images of CYP2E1 (red) and α-smooth muscle actin (green) (i–viii); 4-HNE (red) and α-smooth muscle actin (green) (ix–xvi); and HMGB1 (red) and β-actin (green) (xvii–xxiv) showed CYP2E1, 4-HNE and HMGB1 expression and localization in respective groups of exposed primary rat hepatocytes. All cells were counter stained with DAPI (blue) and minimum 3 images were taken from different microscopic field at 40X magnification. The morphometric analysis (calculated as % ROI of red immunofluorescence image (n=3)) of images were plotted as bar graph as CYP2E1 (B), 4-HNE (C) and HMGB1 (D). Data points were represented with means ± SEM (n=3). One-way analysis of variance (ANOVA) and p value was calculated using unpaired t-test. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Since CYP2E1 has been shown to contribute to redox toxicity in hepatocytes we studied the stable lipid peroxidation marker 4-HNE and damage associated molecular pattern HMGB1 [16,17,37]. Co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen (CM1) showed a marked decrease in the levels of both 4-HNE and HMGB1 when compared to pyrazole-stimulated hepatocyte group (Fig. 6A, C and D). Co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen and Trpv4 antagonist (CM2) showed a significant increase in both 4-HNE and HMGB1 levels in hepatocytes when compared to Pyrazole treated hepatocyte group (Fig. 6A, C and D)(P < 0.05). On the other hand co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen and Trpv4 agonist (CM3) significantly decreased the levels of 4HNE and HMGB1 respectively in the hepatocytes when compared to Pyrazole only treatment (P < 0.05). Interestingly co-incubation of pyrazole stimulated hepatocytes with Kupffer cell conditioned medium that were primed with collagen, and eNOS inhibitor L-NAME (CM4) significantly increased lipid peroxidation marker 4-HNE and HMGB1 when compared to hepatocytes primed with Pyrazole and treated with Kupffer cell conditioned medium that were co-incubated with TRPV4 agonist and L-NAME (CM5) (Fig. 6A, C and D)(P < 0.05). Thus, significant cross talk likely exists between hepatocytes and Kupffer cells in the damaged liver lobule. Moreover, a mechanistic role for TRPV4 in resisting tissue injury in NAFLD arises (Fig. 7). Our previous data as presented in earlier figures show that in liver injury associated with NAFLD, there is an initial activation of CYP2E1-driven redox toxicity and release of damage associated molecular pattern HMGB1 and extracellular collagen. The release of HMGB1 and collagen increases TRPV4 protein levels in both these cells but the activation of TRPV4 and subsequent release of NO through an eNOS-dependent pathway takes place in the Kupffer cells only. Following the release of NO, it diffuses into the hepatocytes to block CYP2E1mediated redox toxicity and resists tissue damage (Fig. 7). However, the increase in Dnmt1 levels following CYP2E1 activation explains the promoter methylation of Trpv4 and subsequent drop in TRPV4 levels at later stages of NAFLD where TRPV4 function declines. This in turn evokes unrelenting CYP2E1-mediated redox toxicity, resulting in tissue damage and unrestricted inflammation, hallmark features of advanced steatohepatitis.
Fig. 7. Model of CYP2E1 and Trpv4 concerted interaction in progressive NAFLD.
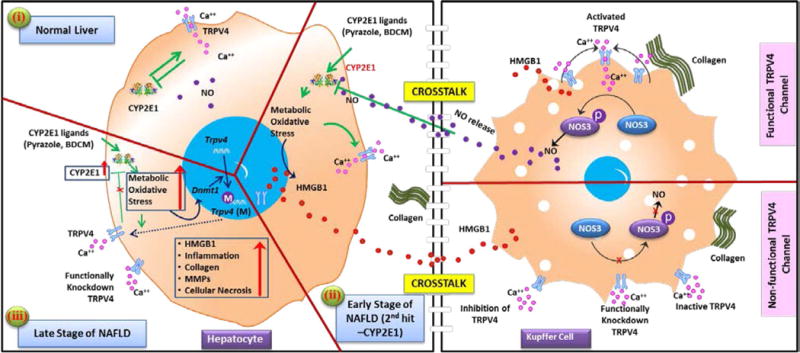
Graphical representation of molecular mechanism of the CYP2E1 activity attenuation by Trpv4 and cross talk between hepatocytes and Kupffer cells in microenvironment of the liver at normal stage (panel-i), early stage (panel-ii) and late stage (panel-iii) of progressive NAFLD.
3. Discussion
In this report we describe a novel role of the Ca2+-permeable ion channel TRPV4 in attenuating hepatic oxidative stress and inflammation, hallmarks of progressive NAFLD, by suppressing the activity of CYP2E1, a CYP450 enzyme key to the development of NAFLD. Our results show that in progressive NAFLD, in hepatic genomic DNA, the Trpv4 gene is methylated in its promoter regions, likely repressing Trpv4 gene expression so that protein levels of the channel became decreased with NAFLD progression. To elucidate Trpv4-dependent NAFLD mechanisms, we recapitulated an NAFLD model in Trpv4 KO mice. CYP2E1 activity was induced by either administering Pyrazole or bromodichloromethane (BDCM) [7,17]. In the genetically-encoded absence of Trpv4, we observed a worsened outcome of NAFLD caused by CYP2E1-mediated oxidative stress and inflammation. Highlighting an exciting new Trpv4-dependent mechanism in-vivo, the poor outcome in these mice could be attenuated by blocking CYP2E1-activity or administration of NO donor. We were intrigued by these two rescue pathways depending on Trpv4 gene function. Our results suggest that NO derived from hepatic Kupffer cells attenuated CYP2E1 function in hepatocytes, and that the NAFLD-attenuating NO appears produced by Kupffer cells in a Trpv4-dependent manner. Thus, TRPV4 functions in a hepato-protective manner in NAFLD. We therefore harbor hope that our findings will open a new chapter of improved understanding of NAFLD pathogenesis, by defining the role of a powerful new hepato-protective player, TRPV4, at increased resolution. Our results hint at a hitherto unknown translational-medical approach toward prevention and treatment of NAFLD, perhaps other liver diseases that rely on oxidative stress and inflammation, by boosting hepatic Trpv4 gene expression and channel function.
The results of this study show the important role that boosting of TRPV4 function could assume in attenuating CYP2E1-induced oxidative stress and inflammation. This represents a finding with strong translational impact, yet at the same time our findings bring along the challenge that potent TRPV4 activating molecules have proven lethal in mammalian species because of disintegration of the lung alveolar barrier as a direct result of TRPV4 activation after systemic application [38]. For the sake of improved treatment perspectives in NAFLD, this challenge will have to be overcome. At the level of mechanistic insight, another challenge will be to precisely define the role of TRPV4 in specific liver cells by generating cell-specific and inducible knockout mouse lines so that Trpv4 is knocked down only in hepatocytes, Kupffer cells, and so that developmental compensation of Trpv4 pan-knockout and non-conditional knockout can be obviated. These objectives will be worthy subjects of future studies, as will be the studies involving other cell types in the liver such as hepatic stellate cells and liver sinusoidal endothelial cells.
Thus in summary, the study establishes a novel mechanistic definition of TRPV4-CYP2E1 crosstalk in the NAFLD liver, especially as NAFLD progresses. The pathways of TRPV4-mediated production of hepato-protective NO by Kupffer cells, which affects hepato-injurious CYP2E1-function appear as rewarding future therapeutic targets in NAFLD and forms the basis of novel discoveries in an ever growing disease that does not have an FDA approved drug for its successful management.
4. Materials and methods
4.1. Materials
All other chemicals were of analytical grade and were purchased from Sigma Chemical Company unless otherwise specified in Supplementary material.
5. Mouse model
5.1. Diet-induced NAFLD model
C57BL/6J (WT), CYP2E1 gene deleted mice 29/Sv-Cyp2e1tm1Gonz/ J (CYP2E1KO) and mice that contained the deleted exon 12 (codes for the pore-loop and adjacent transmembrane domain) of the trpv4 gene (Trpv4 KO) were fed with high-fat-diet (60% kCal) from 8 weeks to 12 weeks for diet-induced NAFLD model. All mice had ad libitum access to food and water and were housed in a temperature-controlled room at 23–24 °C with a 12-h light/dark cycle. All animals were treated in strict accordance with the NIH Guide for the Humane Care and Use of Laboratory Animals and local IACUC standards. The study was approved by the institutional review board both at Duke University and the University of South Carolina at Columbia.
5.2. Induction of liver injury: administration of CYP2E1 ligand (BDCM, Pyrazole)
Wild-type and gene specific knockout mice, at 12 weeks were administered with vehicle (corn oil or normal saline, NAFLD), Bromodichloromethane (1 mmole/kg, BDCM, two dosages per week), Pyrazole (250 mg/kg, PYR, three dosages per week), and/or diallyl sulfide (50 mg/kg, DAS), and/or nitric oxide donor DETANONOate (1 mg/kg, NO) through the intraperitoneal route for one week. DAS or NO was administered 1 h prior to each BDCM or PYR exposure. Mice were euthanized at the completion of dosage and serum and liver tissue was collected for further processing.
5.3. Human liver tissue and primary human hepatocytes
Formalin-fixed, paraffin-embedded Human liver tissue blocks from both normal (control) and progressive NAFLD individuals and freshly isolated primary human hepatocytes were obtained from Liver Tissue Cell Distribution System, University of Minnesota, Minneapolis, MN.
5.4. Kupffer cell culture and treatments
Immortalized rat Kupffer cell line (SV40) was grown and maintained in complete DMEM media containing high glucose, L-glutamine and 15% FBS at 37 °C in a humidified atmosphere of 5% CO2. After overnight serum starvation (1.5% FBS) the cells were then treated with vehicle (control, CM0), recombinant Rat HMGB1 (50 ng/mL), collagen-1 (40 μg/mL, CM1), Trpv4 antagonist (GSK2193874, 100 nM) and Trpv4 agonist (GSK1016790A,100 nM), L-NAME (200 μM) separately or in combinations for 24 h. Kupffer cells conditioned media (CM) was generated for collagen-1+Trpv4 antagonist (Trpv4 An+collagen, CM2), collagen-1+Trpv4 agonist (Trpv4 Ag+collagen, CM3), collagen-1+ L-NAME (collagen+L-NAME, CM4) and collagen-1+Trpv4 agonist+L-NAME (Trpv4 Ag+collagen+L-NAME, CM5) groups. Upon completion of treatment cells and supernatant were processed for future experiments.
5.5. Hepatocytes culture and treatments
Frozen NoSpin HepaRG cells and freshly isolated human and rat hepatocytes plated on cover glass maintained in maintenance media as recommended by vendor until treated. Cells were treated with vehicle (control), pyrazole (PYR, 5 mM), diallyl sulfide (DAS, 2 mM), Trpv4 antagonist (100 nM) and Trpv4 agonist (100 nM), Kupffer cells conditioned media (CM0-CM5) separately or in combination as (i) HepaRG groups: Control, pyrazole (PYR), pyrazole+Trpv4 antagonist (Trpv4 An +PYR) and pyrazole+Trpv4 agonist (Trpv4 Ag+PYR) and (ii) Fresh Rat primary hepatocytes groups: control+CM0 (control), pyrazole +CM0 (PYR), pyrazole+CM0+DAS (PYR+DAS), pyrazole+CM1 (PYR+CM1), pyrazole+CM2 (PYR+CM2), pyrazole+CM3 (PYR +CM3), pyrazole+CM4 (PYR+CM4) and pyrazole+CM5 (PYR +CM5) for 24 h. All HepaRG cell were processed for western blot analysis. However, primary Rat hepatocytes were used for immunofluorescence imaging.
6. Laboratory analysis
6.1. Quantitative Real-Time Polymerase Chain Reaction (qRTPCR)
mRNA expression of the gene in tissue samples was measured by two step qRTPCR. Threshold Cycle (Ct) value for the selected genes was normalized against 18 s (internal expression control) value in the same sample. Each reaction was carried out in triplicates for each gene and for each tissue sample. Relative fold change was calculated using NAFLD as a control.
6.2. Western blot
30 μg of denatured protein (RIPA buffer extract) from each liver sample was loaded per well of novex 4–12% bis-tris gradient gel and subjected for standard SDS-PAGE. Separated protein bands were transferred to nitrocellulose membrane using Trans – Blot Turbo transfer system. Further, blots were blocked with 5% non-fat milk solution for 1 h and then incubated with primary antibodies for 2 h at RT or overnight at 4 °C. Species-specific anti-IgG secondary antibody conjugated with HRP were used to tag primary antibody. ECL western blotting substrate was used to develop the blot. Finally, the blot was imaged using G:Box Chemi XX6 and subjected to densitometry analysis using Image J software.
6.3. Detection of 4HNE-histidine adduct and serum insulin (ELISA)
Immunoreactivity of 4HNE histidine adduct was detected in liver homogenate using OxiSelect HNE-His adduct ELISA kit (Cell Biolabs, Inc. San Diego, CA) and serum insulin was quantified using Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, IL) following manufacturer’s protocol.
6.4. Colorimetric detection of Nitric Oxide (NO)
The nitric oxide in mouse serum or in the culture supernatant was quantified using an indirect method of NO determination. The total NO2 was measured using Griess Reagent System (Promega, Madison, WI).
6.5. Histopathology and Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded, 5 μm thick liver sections were used for histopathology by standard hematoxylin and eosin (H & E) staining method and IHC staining. Deparaffinization of the sections followed by heat based epitope retrieval and both endogenous peroxidase blocking (3% H2O2, 5 min) and serum blocking (5% normal serum, 1 h) were carried out. Further, sections were incubated with primary antibody (1:500) for 2 h at RT and then species-specific anti-IgG secondary antibodies and conjugation with HRP was performed using Vectastain Elite ABC kit following manufacturer’s protocols. Finally, 3,3′ Diaminobenzidine (DAB) were used as a chromogen substrate and counter stained with Mayer’s hematoxylin. Morphometric analysis was done using CellSens Software from Olympus America.
6.6. Immuno-fluorescence microscopy
The deparaffinized liver tissue section from all the mice groups was subjected to epitope retrieval and permeabilization (0.01% triton X-100) followed by blocking with 5% normal serum. Fixed Rat primary hepatocytes on the cover glass were subjected to permeabilization (0.01% triton X-100) followed by blocking with 10% normal serum. Further, both tissue sections and cells were incubated with primary antibody (1:250) overnight at 4 °C. Species-specific anti-IgG secondary antibody conjugated with Alexa fluor 633 (red) and Alexa fluor 488 (green) was used to observe the antigen-specific immunoreactivity. Sections were mounted in ProLong gold antifade reagent with DAPI (counter stain). Morphometric analysis was done using CellSens Software from Olympus America.
6.7. Statistical analyses
All in vivo experiments were repeated three times with at least 3 mice per group (N=3; data from each group of three mice were pooled). All in vivo experiments laboratory analysis experiments were repeated three times. The statistical analysis was carried out by unpaired t-test and analysis of variance (ANOVA). For all analysis P < 0.05 was considered statistically significant.
Supplementary Material
One sentence summary.
TRPV4-mediated nitric oxide pathway stands guard against progression of nonalcoholic fatty liver disease.
Acknowledgments
The authors gratefully acknowledge the technical services of Benny Davidson at the IRF, University of South Carolina School of Medicine and AML Labs (Baltimore MD). We also thank the Instrumentation resource facility (IRF) at the University of South Carolina for equipment usage and consulting services.
Funding
This work has been supported by NIH Pathway to Independence Award, R00ES019875 and P01AT003961 to Saurabh Chatterjee, US Department of Defense (W81XWH-13-1-0299) and a Harrington Discovery Institute (Cleveland OH) Scholar-Innovator Award to Wolfgang Liedtke. R01DK053792 to Anna Mae Diehl, P01AT003961, P20GM103641, R01AT006888, R01ES019313, R01MH094755 and VA Merit Award BX001357 to Mitzi Nagarkatti and Prakash S. Nagarkatti.
Appendix A. Supplementary material
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.freeradbiomed.2016.11.047.
Footnotes
Author contributions
R.S designed and performed experiments, maintained the Trpv4 colony and analyzed data. S.D., DD, V.C, F.A performed experiments AMD and GM provided materials, edited manuscript and analyzed data. PDB provided materials. WL designed experiments, analyzed data, edited manuscript. S.C. designed, performed experiments, wrote manuscript, edited manuscript, analyzed and interpreted data.
Competing interests
The authors declare that there is no conflict of interest.
Data and materials availability
All the data are available with corresponding author, if required data will be provided.
References
- 1.Abdelmalek MF, Diehl AM. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med Clin N Am. 2007;91(6):1125–1149. doi: 10.1016/j.mcna.2007.06.001. http://dx.doi.org/10.1016/j.mcna.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Diehl AM. Hepatic complications of obesity. Gastroenterol Clin N Am. 2010;39(1):57–68. doi: 10.1016/j.gtc.2009.12.001. http://dx.doi.org/10.1016/j.gtc.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl. 1) doi: 10.1002/hep.20973. http://dx.doi.org/10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 4.Yilmaz Y, Younossi ZM. Obesity-associated nonalcoholic fatty liver disease. Clin Liver Dis. 2014;18(1):19–31. doi: 10.1016/j.cld.2013.09.018. http://dx.doi.org/10.1016/j.cld.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 5.Bugianesi E, Zannoni C, Vanni E, Marzocchi R, Marchesini G. Non-alcoholic fatty liver and insulin resistance: a cause-effect relationship? Dig Liver Dis: Off J Ital Soc Gastroenterol Ital Assoc Study Liver. 2004;36(3):165–173. doi: 10.1016/j.dld.2003.12.008. http://dx.doi.org/10.1016/j.dld.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Ganini D, Tokar EJ, Kumar A, Das S, Corbett J, et al. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental nonalcoholic steatohepatitis. J Hepatol. 2012 doi: 10.1016/j.jhep.2012.11.035. http://dx.doi.org/10.1016/j.jhep.2012.11.035. [DOI] [PMC free article] [PubMed]
- 7.Seth RK, Das S, Pourhoseini S, Dattaroy D, Igwe S, Ray JB, et al. M1 polarization bias and subsequent nonalcoholic steatohepatitis progression is attenuated by nitric oxide donor DETA NONOate via inhibition of CYP2E1-induced oxidative stress in obese mice. J Pharmacol Exp Ther. 2015;352(1):77–89. doi: 10.1124/jpet.114.218131. http://dx.doi.org/10.1124/jpet.114.218131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasarin M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One. 2012;7(4):e32785. doi: 10.1371/journal.pone.0032785. http://dx.doi.org/10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pourhoseini S, Seth RK, Das S, Dattaroy D, Kadiiska MB, Xie G, et al. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS One. 2015;10(2):e0116780. doi: 10.1371/journal.pone.0116780. http://dx.doi.org/10.1371/journal.pone.0116780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim CH, Younossi ZM. Nonalcoholic fatty liver disease: a manifestation of the metabolic syndrome. Clevel Clin J Med. 2008;75(10):721–728. doi: 10.3949/ccjm.75.10.721. Epub 2008/10/23. [DOI] [PubMed] [Google Scholar]
- 11.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. Epub 1998/04/18. [DOI] [PubMed] [Google Scholar]
- 12.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi: 10.1002/hep.24001. http://dx.doi.org/10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 13.Song Y, Zhan L, Yu M, Huang C, Meng X, Ma T, et al. TRPV4 channel inhibits TGF-beta1-induced proliferation of hepatic stellate cells. PLoS One. 2014;9(7):e101179. doi: 10.1371/journal.pone.0101179. http://dx.doi.org/10.1371/journal.pone.0101179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carreno FR, Ji LL, Cunningham JT. Altered central TRPV4 expression and lipid raft association related to inappropriate vasopressin secretion in cirrhotic rats. Am J Physiol Regul Integr Comp Physiol. 2009;296(2):R454–R466. doi: 10.1152/ajpregu.90460.2008. http://dx.doi.org/10.1152/ajpregu.90460.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, et al. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139(1) doi: 10.1053/j.gastro.2010.04.010. http://dx.doi.org/10.1053/j.gastro.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced nonalcoholic steatohepatitis. J Hepatol. 2012;57(4):860–866. doi: 10.1016/j.jhep.2012.05.019. http://dx.doi.org/10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, et al. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol Sci: Off J Soc Toxicol. 2013 doi: 10.1093/toxsci/kft104. http://dx.doi.org/10.1093/toxsci/kft104. [DOI] [PMC free article] [PubMed]
- 18.Seth RK, Das S, Kumar A, Chanda A, Kadiiska MB, Michelotti G, et al. CYP2E1-dependent and leptin-mediated hepatic CD57 expression on CD8+ T cells aid progression of environment-linked nonalcoholic steatohepatitis. Toxicol Appl Pharmacol. 2014;274(1):42–54. doi: 10.1016/j.taap.2013.10.029. http://dx.doi.org/10.1016/j.taap.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol. 2010;103(1):2–17. doi: 10.1016/j.pbiomolbio.2009.10.002. http://dx.doi.org/10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Birder L, Kullmann FA, Lee H, Barrick S, de Groat W, Kanai A, et al. Activation of urothelial transient receptor potential vanilloid 4 by 4alpha-phorbol 12,13-didecanoate contributes to altered bladder reflexes in the rat. J Pharmacol Exp Ther. 2007;323(1):227–235. doi: 10.1124/jpet.107.125435. http://dx.doi.org/10.1124/jpet.107.125435. [DOI] [PubMed] [Google Scholar]
- 21.Gevaert T, Vriens J, Segal A, Everaerts W, Roskams T, Talavera K, et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Investig. 2007;117(11):3453–3462. doi: 10.1172/JCI31766. http://dx.doi.org/10.1172/jci31766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA. 2003;100(23):13698–13703. doi: 10.1073/pnas.1735416100. http://dx.doi.org/10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62(24):2985–3001. doi: 10.1007/s00018-005-5181-5. http://dx.doi.org/10.1007/s00018-005-5181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan L, Yang Y, Ma TT, Huang C, Meng XM, Zhang L, et al. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol Cell Biochem. 2015;402(1–2):9–22. doi: 10.1007/s11010-014-2298-6. http://dx.doi.org/10.1007/s11010-014-2298-6. [DOI] [PubMed] [Google Scholar]
- 25.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–194. doi: 10.1038/nri3623. http://dx.doi.org/10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 26.Xie G, Diehl AM. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am J Physiol Gastrointest liver Physiol. 2013;305(12):G881–G890. doi: 10.1152/ajpgi.00289.2013. http://dx.doi.org/10.1152/ajpgi.00289.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maciejewska D, Ossowski P, Drozd A, Ryterska K, Jamiol-Milc D, Banaszczak M, et al. Metabolites of arachidonic acid and linoleic acid in early stages of nonalcoholic fatty liver disease—a pilot study. Prostaglandins Other Lipid Mediat. 2015;121(Pt B):184–189. doi: 10.1016/j.prostaglandins.2015.09.003. http://dx.doi.org/10.1016/j.prostaglandins.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Schuck RN, Zha W, Edin ML, Gruzdev A, Vendrov KC, Miller TM, et al. The cytochrome P450 epoxygenase pathway regulates the hepatic inflammatory response in fatty liver disease. PLoS One. 2014;9(10):e110162. doi: 10.1371/journal.pone.0110162. http://dx.doi.org/10.1371/journal.pone.0110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung O, Sanyal AJ. Recent advances in nonalcoholic fatty liver disease. Curr Opin Gastroenterol. 2009;25(3):230–237. doi: 10.1097/mog.0b013e3283294a18. Epub 2009/04/28. [DOI] [PubMed] [Google Scholar]
- 30.Palmeri ML, Wang MH, Rouze NC, Abdelmalek MF, Guy CD, Moser B, et al. Noninvasive evaluation of hepatic fibrosis using acoustic radiation force-based shear stiffness in patients with nonalcoholic fatty liver disease. J Hepatol. 2011;55(3):666–672. doi: 10.1016/j.jhep.2010.12.019. http://dx.doi.org/10.1016/j.jhep.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu D, Cederbaum A. Nitric oxide donors prevent while the nitric oxide synthase inhibitor L-name increases arachidonic acid plus CYP2E1-dependent toxicity. Toxicol Appl Pharmacol. 2006;216(2):282–292. doi: 10.1016/j.taap.2006.05.019. http://dx.doi.org/10.1016/j.taap.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 32.Pu J, Wang Z, Zhou H, Zhong A, Ruan L, Jin K. Role of TRPV4 channels in regulation of eNOS expression in brain microvascular endothelial cells under the condition of mechanical stretch, Zhong nan da xue xue bao Yi xue Ban. J Cent South Univ Med Sci. 2015;40(9):960–966. doi: 10.11817/j.issn.1672-7347.2015.09.003. http://dx.doi.org/10.11817/j.issn.1672-7347.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Fichna J, Poole DP, Veldhuis N, MacEachern SJ, Saur D, Zakrzewski PK, et al. Transient receptor potential vanilloid 4 inhibits mouse colonic motility by activating NO-dependent enteric neurotransmission. J Mol Med (Berlin, Germany) 2015;93(2015):1297–1309. doi: 10.1007/s00109-015-1336-5. http://dx.doi.org/10.1007/s00109-015-1336-5. [DOI] [PubMed] [Google Scholar]
- 34.Cabral PD, Garvin JL. TRPV4 activation mediates flow-induced nitric oxide production in the rat thick ascending limb. Am J Physiol Ren Physiol. 2014;307(6) doi: 10.1152/ajprenal.00619.2013. http://dx.doi.org/10.1152/ajprenal.00619.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishijima Y, Zheng X, Lund H, Suzuki M, Mattson DL, Zhang DX. Characterization of blood pressure and endothelial function in TRPV4-deficient mice with l-NAME- and angiotensin II-induced hypertension. Physiol Rep. 2014;2(1):e00199. doi: 10.1002/phy2.199. http://dx.doi.org/10.1002/phy2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sukumaran SV, Singh TU, Parida S, Reddy ChE Narasimha, Thangamalai R, Kandasamy K, et al. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res: Off J Ital Pharmacol Soc. 78(2013):18–27. doi: 10.1016/j.phrs.2013.09.005. http://dx.doi.org/10.1016/j.phrs.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Lieber CS. CYP2E1: from ASH to NASH. Hepatol Res: Off J Jpn Soc Hepatol. 2004;28(1):1–11. doi: 10.1016/j.hepres.2003.08.001. Epub 2004/01/22. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99(9):988–995. doi: 10.1161/01.RES.0000247065.11756.19. http://dx.doi.org/10.1161/01.res.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.