

Abstract
Background
MicroRNAs (miRNAs) regulate gene expression and influence cancer. Primary transcripts of miRNAs (pri-miRNAs) are poorly annotated and little is known about the role of germline variation in miRNA genes and breast cancer (BC). We sought to identify germline miRNA variants associated with BC risk and tumor subtype among African-American (AA) women.
Methods
Under the African American Breast Cancer Epidemiology and Risk (AMBER) Consortium, genotyping and imputed data from four studies on BC in AA women were combined into a final dataset containing 224,188 miRNA gene single nucleotide polymorphisms (SNPs) for 8350 women: 3663 cases and 4687 controls. The primary miRNA sequence was identified for 566 miRNA genes expressed in Encyclopedia of DNA Elements (ENCODE) Tier 1 cell types and human pancreatic islets. Association analysis was conducted using logistic regression for BC status overall and by tumor subtype.
Results
A novel BC signal was localized to an 8.6-kb region of 17q25.3 by four SNPs (rs9913477, rs1428882938, rs28585511, and rs7502931) and remained statistically significant after multiple test correction (odds ratio (OR) = 1.44, 95% confidence interval (CI) = 1.26–1.65; p = 3.15 × 10−7; false discovery rate (FDR) = 0.03). These SNPs reside in a genomic location that includes both the predicted primary transcript of the noncoding miRNA gene MIR3065 and the first intron of the gene for brain-specific angiogenesis inhibitor 1-associated protein 2 (BAIAP2). Furthermore, miRNA-associated SNPs on chromosomes 1p32.3, 5q32, and 3p25.1 were the strongest signals for hormone receptor, luminal versus basal-like, and HER2 enrichment status, respectively. A second phase of genotyping (1397 BC cases, 2418 controls) that included two SNPs in the 8.6-kb region was used for validation and meta-analysis. While neither rs4969239 nor rs9913477 was validated, when meta-analyzed with the original dataset their association with BC remained directionally consistent (OR = 1.29, 95% CI = 1.16–1.44 (p = 4.18 × 10–6) and OR = 1.33, 95% CI = 1.17–1.51 (p = 1.6 × 10–5), respectively).
Conclusion
Germline genetic variation indicates that MIR3065 may play an important role in BC development and heterogeneity among AA women. Further investigation to determine the potential functional effects of these SNPs is warranted. This study contributes to our understanding of BC risk in AA women and highlights the complexity in evaluating variation in gene-dense regions of the human genome.
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0964-4) contains supplementary material, which is available to authorized users.
Keywords: microRNA, miRNA, SNP, Breast cancer, African American, Case-control
Background
MicroRNAs (miRNAs) are small noncoding RNAs that were formally recognized in 2001 [1] as one of the largest classes of gene regulators in eukaryotes [2]. miRNAs undergo a complex, multistep process of biogenesis summarized by Lin and Gregory in 2015 [3]. Briefly, within the nucleus, a primary miRNA transcript (pri-miRNA)—usually several hundred nucleotides (nt) to greater than 1 megabase (Mb) in length—is cleaved to create a precursor miRNA (pre-miRNA) approximately 70 nt in length which folds to form a stem-loop intermediate. This intermediate is exported from the nucleus and further processed to a miRNA duplex, approximately 22 nt in length. One strand of the miRNA duplex is loaded onto the RNA-induced silencing complex (RISC) to form a functional mature miRNA. Cleavage and processing of the pri- and pre-miRNA require sequence and secondary structure recognition by several RNA-binding proteins and their partners. Approximately 30% of mature miRNAs are processed from introns or exons of coding genes, while the remaining miRNAs are intergenic and expressed from independent transcription units. Mature miRNAs bind to the 3’ untranslated region (UTR) of target genes to silence them by either translational repression or messenger RNA (mRNA) degradation [4]. There are over 2500 identified human miRNAs [5] and each may bind to hundreds or even thousands of different target genes, coordinating expression of a large number of mRNAs; this makes them key players in gene regulatory networks [3].
miRNAs have been shown to influence numerous molecular pathways and pathological conditions, including cancer [3, 6–10], and can function as both oncogenes and tumor suppressors depending on the context. Furthermore, oncoproteins such as MYC bind to the promoters of key miRNAs, activating oncogenic miRNAs (oncomiRs) and downregulating tumor suppressor miRNAs [11–13]. In breast cancer (OMIM #114480), miRNAs have been implicated in the regulation of genes involved in pathways critically relevant to disease etiology and severity including apoptosis, cell cycle checkpoints, cell migration, invasion, and metastasis [14–17]. To a large extent, the miRNA repertoire that is present in normal and paired tumor tissue from the same organ is quite similar; however, specific miRNAs are often aberrantly elevated or suppressed in the tumor [18]. In 2011, Persson et al. performed one of the first comprehensive characterizations via next-generation sequencing (NGS) of miRNAs in paired normal and tumor breast tissue and identified 361 new miRNAs [18]. While the functionality of some of the miRNAs identified by deep sequencing remains unknown, about two-thirds of these newly identified miRNAs were expressed in other tissues, and nearly half were associated with components of the RISC and were found in estrogen receptor-positive, invasive breast ductal carcinoma cells. Germline single nucleotide polymorphisms (SNPs) in critical regions of miRNA genes including the promoter and primary transcripts may contribute to the dysregulation in miRNA biogenesis and expression differences common in breast cancer.
Over the last decade there has been tremendous progress made in the field of miRNAs and cancer, particularly centered on miRNA expression patterns that are emerging as promising diagnostic tools and predictive markers because of their correlation with cancer progression and patient survival [3]. However, little is known about the role of germline variation in miRNAs and susceptibility to cancer.
Currently, known germline genetic variation primarily from studies of European women explains only 50% of the familial aggregation of breast cancer (BC), suggesting that numerous other susceptibility gene variants have yet to be uncovered [19]. Several molecular epidemiologic studies have assessed the association of common germline miRNA gene variation in mature and precursor miRNA sequences with disease risk, including BC [20–29]. Few epidemiologic studies have evaluated the association between a large number of germline genetic variants in the promoter and primary sequences of miRNAs and BC risk, particularly among African-American (AA) women. We sought to identify large numbers of germline miRNA gene variants associated with BC risk and subtype among women participating in a large AA BC consortium.
Methods
Study population
This research was conducted using data from the African American Breast Cancer Epidemiology and Risk (AMBER) Consortium, a collaboration of two case-control studies of BC in AA women (the Carolina Breast Cancer Study (CBCS) [30] and the Women’s Circle of Health Study (WCHS) [31, 32]) and two cohort studies (the Black Women’s Health Study (BWHS) [33] and the Multiethnic Cohort (MEC) [34]). AMBER has been described previously [35]. All study participants provided written informed consent and all studies obtained Institutional Review Board approval.
This analysis utilizes data from 3663 cases and 4687 controls in AMBER who provided either blood or saliva for DNA analysis. For the case-control studies, controls were identified either through Division of Motor Vehicles lists (age < 65 years) and Health Care Financing Administration lists (age ≥ 65) (CBCS), or random digit dialing and community controls (WCHS). For BWHS and MEC, controls were chosen from among women without BC, and were frequency matched to cases on geographical region, sex, race, and 5-year age group. Eligible cases were AA women with incident invasive BC or ductal carcinoma in situ (DCIS). Estrogen receptor (ER), progesterone receptor (PR), epidermal growth factor receptor 2 (HER2) receptor, and invasive status for cases was determined using pathology data from hospital or cancer registry records.
Genotyping and quality control (QC)
Genotyping of DNA from participants in the BWHS, CBCS, and WCHS was performed by the Center for Inherited Disease Research (CIDR) using the Illumina Human Exome BeadChip v1.1. This array includes > 200,000 coding variants, as well as tag SNPs for genome-wide association study (GWAS) hits, a grid of common variants, and ancestry informative markers (AIMs). A description of the exome chip design is available from http://genome.sph.umich.edu/wiki/Exome_Chip_Design. In addition to the standard BeadChip, the chip included approximately 159,000 SNPs of custom content focused on BC pathways (e.g., steroid hormone metabolism, insulin and insulin-like growth factors, inflammatory and immune factors, and vitamin D).
A total of 405,555 SNPs were genotyped, and 300,008 SNPs remained after excluding variants that failed technical filters imposed by CIDR, or QC filters recommended by the University of Washington. Briefly, genotypes with a GenCall score < 0.15 were classified as missing, and SNPs were removed if they were monomorphic, had poor cluster properties (ex. cluster separation < 0.2 or < 0.3 depending on allele frequency), call rates < 0.98, Hardy-Weinberg Equilibrium p < 1 × 10−4, > 1 Mendelian error in trios from HapMap, or > 2 discordant calls in duplicate samples. Mitochondrial and Y chromosome SNPs were also removed. Genotypes were attempted for 6936 participants from the BWHS, CBCS, and WCHS, and were completed with a call rate > 98% for 6828 participants, which included 3130 cases (963 ER negative, 1674 ER positive, 493 ER unknown) and 3698 controls. Imputation was performed by the University of Washington using the IMPUTE2 software [36] and the 1000 Genomes Phase I reference panel (5/21/2011 1000 Genomes data, December 2013 haplotype release).
Genetic data from 533 cases (135 ER negative, 309 ER positive, and 89 ER unknown) and 989 controls in the MEC were available from a previous GWAS on the Illumina Human 1 M-Duochip [37]. SNPs from MEC were imputed to the same release of 1000 Genomes and combined with the genotype data from the Illumina Human Exome BeadChip v1.1. Additional exclusion criteria applied to the four-study merged dataset were: variants with mismatching alleles or allele frequencies that were different by more than 0.15 in MEC when compared with the other three studies; variants with allele frequencies < 0.5%; and variants with imputation score INFO < 0.5 in either MEC or any of the other three studies. The final merged dataset included genotypes from 8350 women, 3663 cases (1983 ER positive, 1098 ER negative, 582 unknown), and 4687 controls.
miRNA annotation, SNP selection and QC
Among the genotyped and imputed SNPs, miRNA variants were defined as those within promoter, pri-miRNA, pre-miRNA, mature, or downstream regions of a known human miRNA. Mature and pre-miRNA sequence locations were identified from the miRNA database, miRBase release 21 [5, 38] . Pri-miRNAs were identified by integrative analysis of chromatin immunoprecipitation and massively parallel DNA sequencing (ChIP-seq) data from the Encyclopedia of DNA Elements (ENCODE) project using an algorithm described previously [39]. Five hundred and sixty six miRNA genes with pri-miRNA sequence expressed in six cell lines and tissue types (all ENCODE Tier 1 cell types plus human pancreatic islets) were the focus of this analysis. We extended the pri-miRNA 5 kilobases (kb) upstream of the 5′-end (putative promoter) and 1 kb downstream of the 3′-end (additional putative regulatory region). Variants that could be defined as having multiple miRNA locations were defined by their most unique location with the following priority: mature > precursor > primary > promoter > downstream. For example, a variant in the mature miRNA sequence is also by default in the pri-miRNA; however, according to our prioritization it would be defined as a mature miRNA sequence variant. SNPs were restricted to those variants with minor allele frequencies (MAF) ≥ 1%. Annotation defined a total of 224,188 miRNA gene SNPs, with MAF ≥ 1%, from the following miRNA gene regions: 10,435 promoter, 182,593 primary, 272 precursor, 158 mature, and 2150 downstream variants. The impact of genotype platform was evaluated by quantile-quantile plots both with and without MEC genotypes, both yielding a lambda = 0.991.
Association analysis
Single variant analyses were conducted using logistic regression as implemented in PLINK version 1.07. Models were adjusted for age group (by ~ 10-year intervals), study site, geographic group of residence, DNA source, and ancestry by including principal components 5, 6, and 8 in the model given their association with BC at p < 0.1 [40]. Models were run for all cases versus all controls and for all hormone receptor subtyped (ER, PR, and HER2) cases versus controls, respectively. Additional models were run for case-only subtype analyses (n = 3663, eligible cases with biomarker and covariate information) using ER, PR, and HER2 receptor marker status. Specifically, the following three case-only subtype analyses were performed: 1) hormone receptor positive (ER positive or PR positive, n = 2081) versus hormone receptor negative (ER negative and PR negative, n = 997); 2) luminal (ER positive or PR positive, n = 1613) versus basal-like (ER negative, PR negative, and HER2 negative, n = 405) [41]; and 3) HER2 enriched (n = 1356) versus HER2 negative (n = 344). P values were corrected within subtype analyses for multiple comparisons using the false discovery rate (FDR) at 5% [42]. In all analyses, both invasive and in situ cases were combined.
Validation and meta-analysis
A second phase of genotyping (1397 BC cases, 2418 controls) conducted in three of the four studies within AMBER (CBCS, WCHS, and BWHS) on the Illumina’s Infinium Multi-Ethnic Genotyping Array (MEGA) Chip that included study-specific content and SNPs rs4969239 and rs9913477 was used for validation and meta-analysis. Similar to the association analysis, logistic regression implemented in PLINK version 1.07 was used and models were adjusted for age group (by ~ 10-year intervals), study site, DNA source, and ancestry by including principal component 1 in the model given its association with BC at p < 0.1. Validation for each variant was evaluated for directional consistency and tested at the p < 0.05 level. In the meta-analysis, both the original and the second phase of genotyping were combined and the p value corrected for multiple comparisons using an FDR at 5% [42].
Power was calculated for detecting an odds ratio (OR) of 1.44 and an OR of 1.30 (a 10% reduction in effect estimate assuming the original OR is an overestimate of the actual effect) using a two-sided, p = 0.05 significance, log-additive mode of inheritance, allele frequency of 0.06 (the same as rs9913477 MAF in the study population), control to case ratio of 1.7 with 1397 cases, and prevalence of disease of 10%.
Results
Table 1 provides a distribution of key characteristics of the study population by case or control status and includes age at diagnosis, study site, DNA source, as well as clinical parameters (tumor stage and receptor status). The study population originates from a broad geographical region of the United States with most cases from the Northeast and South. Overall, the vast majority of the cases have known ER or PR receptor status; however, over half do not have known HER2 receptor status. Among cases with known receptor status for all three markers, approximately 20% are triple negative.
Table 1.
Characteristics of the study population
| Controls (n = 4687) | Cases (n = 3663) | |||
|---|---|---|---|---|
| Frequency | Mean (SD) or % | Frequency | Mean (SD) or % | |
| Age at enrollment (years) | 4687 | 55.62 (12.01) | 3663 | 54.94 (11.74) |
| Age at enrollment (years) | ||||
| 18–29 | 24 | 0.51 | 30 | 0.82 |
| 30–39 | 396 | 8.45 | 306 | 8.35 |
| 40–49 | 1107 | 23.62 | 945 | 25.8 |
| 50–59 | 1461 | 31.17 | 1087 | 29.68 |
| 60–69 | 986 | 21.04 | 819 | 22.36 |
| 70–79 | 609 | 12.99 | 433 | 11.82 |
| 80+ | 104 | 2.22 | 43 | 1.17 |
| DNA source | ||||
| Blood | 1817 | 38.77 | 1961 | 53.54 |
| Mouthwash | 2243 | 47.86 | 853 | 23.29 |
| Saliva | 627 | 13.38 | 849 | 23.18 |
| Study | ||||
| BWHS | 2249 | 48.98 | 901 | 24.6 |
| WCHS | 834 | 17.79 | 821 | 22.41 |
| CBCS | 615 | 13.12 | 1408 | 38.44 |
| MEC | 989 | 21.1 | 533 | 14.55 |
| Location | ||||
| New Jersey (NJ) | 573 | 12.23 | 613 | 16.73 |
| Northeast (except NJ) | 1245 | 26.56 | 441 | 12.04 |
| South | 1476 | 31.49 | 1720 | 46.96 |
| Midwest | 238 | 5.08 | 200 | 5.46 |
| West | 1155 | 24.64 | 689 | 18.81 |
| Stage | ||||
| In situ | NA | 376 | 10.26 | |
| Invasive | NA | 2528 | 69.01 | |
| Unknown | NA | 759 | 20.72 | |
| Tumor receptor status | ||||
| ER | ||||
| Positive | NA | 1983 | 54.14 | |
| Negative | NA | 1098 | 29.98 | |
| Unknown | NA | 582 | 15.89 | |
| PR | ||||
| Positive | NA | 1580 | 43.13 | |
| Negative | NA | 1343 | 36.66 | |
| Unknown | NA | 740 | 20.2 | |
| HER2 | ||||
| Positive | NA | 344 | 9.39 | |
| Negative | NA | 1356 | 37.02 | |
| Unknown | NA | 1963 | 53.59 | |
| Triple negative | ||||
| Yes | NA | 405 | 11.06 | |
| No | NA | 1613 | 44.03 | |
| Unknown | NA | 1645 | 44.91 | |
BWHS, Black Women’s Health Study; CBCS, Carolina Breast Cancer Study; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; MEC, Multiethnic Cohort; NA, not applicable; PR, progesterone receptor; SD, standard deviation; WCHS, Women’s Circle of Health Study
Genomic location of novel miRNA SNPs associated with BC in African-American women: case-control analysis
The main case-control association analysis identified seven SNPs (five imputed and two genotyped) in a 16.5-kb region on chromosome 17q25.3 (Fig. 1 and Table 2), with imputed rs9913477 (INFO r2 = 0.99; MAF = 0.06; OR = 1.44, 95% confidence interval (CI) = 1.26–1.65; p = 3.15 × 10−7; FDR = 0.03) emerging as the top hit. Following FDR correction, four of the seven remained significantly associated with BC risk, spanning an 8.6-kb region (Table 2). All four SNPs reside in a genomic region that includes the first intron of the brain-specific angiogenesis inhibitor 1-associated protein 2 (BAIAP2), as well as the predicted primary transcript for MIR3065. Linkage disequilibrium (LD) between the top hit (rs9913477) and the other three statistically significant SNPs was high (r2 = 0.94) for two (rs1428882938 and rs28585511) and perfect (r2 = 1.0) for the third (rs7502931), suggesting they are all tagging the same signal in this population. Subsequently, ER/PR subtype analysis was conducted for all seven SNPs with p < 5 × 10–6 in the full analysis (Additional file 1: Table S1) and identified that the signal and pattern of association in this region was statistically significant based on FDR in ER+ versus controls, most likely because it had the largest sample size. While the other subtype analyses versus controls were not significant, the magnitude of the odds ratio was similar to that observed in ER+ versus controls. However, when we look at the case-only subtype analyses (e.g., ER+ versus ER–, PR+ versus PR–) we see a reduction in the magnitude of the odds ratio suggesting that this region is more likely to be associated generally with the development of breast cancer rather than a particular subtype. Additionally, in a subanalysis of ER positive cases (n = 1983) versus controls (n = 4687) and PR positive cases (n = 1580) versus controls (n = 4687) the same 17q25.3 locus top hit (rs9913477) emerged, but was statistically significantly associated with BC after FDR correction only for the largest subgroup of ER positive cases (INFO r2 = 0.99, MAF = 0.58; OR = 1.53, 95% CI = 1.30–1.81; p = 4.29 × 10–7; FDR = 0.027). The variant rs9913477 was also the second most significant SNP in the ER positive plus PR-positive case group versus control analysis but did not reach statistical significance after FDR correction (data not shown). In the ER negative, PR-negative, and ER negative plus PR-negative cases versus control analysis, rs80339298 located in the primary sequence of MIR761 on chromosome 1 emerged as the top SNP but did not reach statistical significance after FDR correction (data not shown).
Fig. 1.
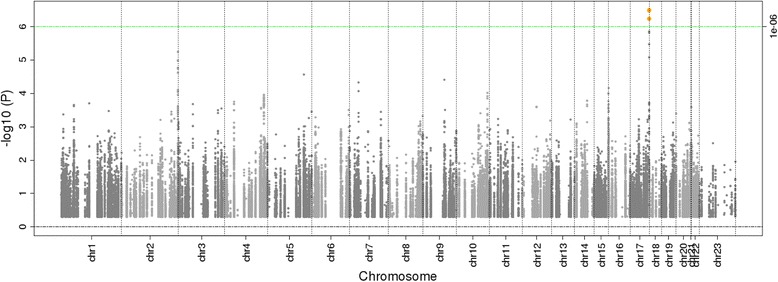
Manhattan plot of miRNA SNP and breast cancer risk in the four-site AMBER Consortium (n = 8350, with 3663 cases and 4687 controls). The green line represents a significant p value threshold of 1 × 10−6 at a false discovery rate (FDR) of 5%
Table 2.
Association of the top seven miRNA SNPs with p < 5 × 10−6 and breast cancer risk
| SNP | Chromosome:positiona | Effect/other allele | EAF | OR (95% CI) | p b | FDR pc | INFO (r2) |
|---|---|---|---|---|---|---|---|
| rs142882938 | 17:79010031 | C/CT | 0.06 | 1.45 (1.24–1.70) | 5.9 × 10–7 | 0.03 | 0.97 |
| rs4969239d | 17:79010544 | G/A | 0.08 | 1.35 (1.20–1.52) | 1.4 × 10–6 | 0.06 | – |
| rs28585511 | 17:79010609 | T/A | 0.06 | 1.45 (1.26–1.66) | 5.8 × 10–7 | 0.03 | 0.98 |
| rs4969351 | 17:79011141 | A/G | 0.08 | 1.35 (1.20–1.52) | 3.4 × 10–6 | 0.11 | 0.99 |
| rs9913477 | 17:79015698 | G/A | 0.06 | 1.44 (1.26–1.65) | 3.2 × 10–7 | 0.03 | 0.99 |
| rs7502931 | 17:79018677 | G/A | 0.06 | 1.44 (1.26–1.65) | 3.4 × 10–7 | 0.03 | 0.99 |
| rs4969366d | 17:79026572 | G/A | 0.05 | 1.45 (1.24–1.70) | 1.5 × 10–6 | 0.06 | – |
The seven single nucleotide polymorphisms (SNPs) are intronic to BAIAP2 and located in the primary transcript of miR-3065
Significant FDR results are shown in bold
CI, confidence interval; EAF, effect allele frequency; FDR, false discovery rate; INFO, imputation quality score; OR, odds ratio
aHuman Genome GRCh37/hg19 assembly, NT_010783.15
bAdditive genetic models were adjusted for age group (by ~ 10-year intervals), study site, geographic region of residence, DNA source, and ancestry (PCs 5, 6, and 8 associated with cancer trait, p < 0.1). Sample size: 3663 cases and 4687 controls
cAdjustment for multiple comparisons using the FDR
dGenotyped SNPs, with the other SNPs having been imputed to 1000 Genome Project data
Case-only subtype analysis
Top SNPs identified in each of the three subtype analyses are provided in Table 3. These top SNPs were located on chromosomes 1p32.3 (rs80339298, OSBPL9 intron 11, NT_032977.10), 5q32 (rs147821319, PPARGC1B intron 7, NM_001172698), and 3p25.1 (rs116367195, intergenic between BTD and ANKRD2, NM_001195099) from the GRCh38.p2 assembly for hormone receptor, luminal versus basal-like, and HER2 enrichment status, respectively. All three SNPs were low frequency (MAF < 5%) and none were statistically significant after FDR correction.
Table 3.
Top SNP hits for breast cancer subtype analyses
| Breast Cancer Subtype | Hormone Receptor +/− | Luminal / Basal-like | HER2 +/− |
|---|---|---|---|
| Sample size | 2081/997 | 1613/405 | 1356/344 |
| SNP ID | rs80339298 | rs147821319 | rs116367195 |
| Chromosome:Position* | 1:52244019 | 5:149217038 | 3:15693446 |
| Effect/Other | A/G | A/G | G/A |
| Reference Sequence | NT_032977.10 | NM_001172698 | NM_001195099 |
| OR (95%CI) | 2.11 (1.54, 2.89) | 2.20 (1.52, 3.19) | 2.70 (1.72, 4.24) |
| EAF | 0.02 | 0.04 | 0.97 |
| p-value** | 2.90 × 10−6 | 2.34 × 10−5 | 1.59 × 10−5 |
| FDR p-value*** | 0.16 | 0.37 | 0.84 |
Abbreviations: OR: odds ratio; 95% CI of the OR; EAF: effect allele frequency; FDR: false discovery rate
* Chromosome: position from GRCh37/hg19 Assembly
** Additive genetic model was adjusted for age group (by ~ 10 year intervals), study site, geographic region of residence, DNA source, and ancestry (PCs 5, 6 and 8 - associated with cancer trait, p-value< 0.1)
***Adjustment for multiple comparisons using the False Discovery Rate (FDR) within each subtype analysis
Validation and meta-analysis
A stage 2 analysis of rs4969239 (OR = 1.07, 95% CI = 0.83–1.39; p = 5.78 × 10–1 and rs9913477 (OR = 0.86, 95% CI = 0.62–1.18; p = 3.56 × 10–1) failed to validate their association with BC at a nominal p value. However, when meta-analyzed with the original dataset, the association of rs4969239 (OR = 1.29, 95% CI = 1.16–1.44); p = 4.18 × 10–6) and rs9913477 (OR = 1.33, 95% CI = 1.17–1.51; p = 1.60 × 10–5) with BC remained directionally consistent (Table 4).
Table 4.
Stages 1 and 2 and meta-analysis of rs9913477 and rs4969239 located in the primary transcript of miR-3065
| SNP | Chromosome:Positiona | Effect/other allele | Stageb | Sample size |
EAF | OR (95% CI) | p |
|---|---|---|---|---|---|---|---|
| rs4969239 | 17:79010544 | G/A | Stage 1 | 8350 | 0.08 | 1.35 (1.2–1.52) | 1.40 × 10–6 |
| Stage 2 | 3814 | 0.08 | 1.07 (0.83–1.39) | 5.78 × 10–1 | |||
| Meta-analysis | 12,164 | 0.08 | 1.29 (1.16–1.44) | 4.18 × 10–6 | |||
| rs9913477 | 17:79015698 | G/A | Stage 1 | 8350 | 0.06 | 1.44 (1.30–1.58) | 3.15 × 10–7 |
| Stage 2 | 3815 | 0.06 | 0.86 (0.62–1.18) | 3.56 × 10–1 | |||
| Meta-analysis | 12,165 | 0.06 | 1.33 (1.17–1.51) | 1.60 × 10–5 |
CI, confidence interval; EAF, effect allele frequency; OR, odds ratio; SNP, single nucleotide polymorphism
aHuman Genome GRCh37/hg19 assembly, NT_010783.15
bStage 1 model: Additive genetic models were adjusted for age group (by ~ 10-year intervals), WCHS study site, geographic region of residence, DNA source, and ancestry (PCs 5, 6 and 8 associated with cancer trait, p < 0.1); Stage 2 model: Additive genetic models were adjusted for age group (by ~ 10-year intervals), DNA source, and ancestry (PCs 1); Meta-analysis was performed in METAL [67]; heterogeneity I2 = 88.1 for rs9913477 and 58.1 for rs4969239
Power calculations for the detection of a SNP associated with BC at ORs of 1.44 and 1.30 at a significance of p ≤ 0.05 indicated that validation among the study set undergoing the second phase of genotyping was 97% and 77%, respectively.
Discussion
In a combined analysis of four large studies of BC in AA women, we identified and annotated a novel genomic region on chromosome 17q25.3 significantly associated with BC and extended its functional interpretation with a comprehensive evaluation of miRNA gene sequence. Specifically, we have localized the BC association signal to an 8.6-kb region on chromosome 17 marked by four tightly linked, significantly associated SNPs, with rs9913477 demonstrating the strongest association. Using a second phase of genotyping we were unable to validate the association of either rs4969239 or rs9913477 with BC; however, in a meta-analysis these SNPs remained directionally consistent (OR = 1.29 and 1.33, respectively). Power calculations indicate that the validation analysis was well powered (97%) at an OR of 1.44 (our original finding and likely an overestimate of effect size) and slightly underpowered (77%) at an OR of 1.3, which represents an effect estimate 10% less that the original OR. No statistically significant miRNA SNP associations were identified in the subanalyses of hormone receptor-negative tumors. Additionally, a case-only analysis that encompassed comparisons of hormone receptor status, luminal versus basal-like subtypes, and HER2 enrichment status did not identify any statistically significant associations with BC after FDR correction. The most strongly associated miRNA-associated SNPs for each subtype analysis identified regions on chromosomes 1p32.3, 5q32, and 3p25.1, respectively. None of these regions or SNPs have been previously implicated in BC GWAS according to the GWAS Catalog (release dated 12 June 2016) [43].
Given their genomic location within the intron of BAIAP2 and pri-MIR3065 sequence, these four SNPs have the potential to impact BAIAP2 expression, BAIAP2 gene intron 1 binding proteins, and/or MIR3065 biogenesis. Mature MIR3065 resides in a gene adjacent to BAIAP2 known as apoptosis-associated tyrosine kinase (AATK) where it is located in the seventh intron and is transcribed in the opposite direction from its host gene. In this gene (AATK) and miRNA-rich region, mature MIR3065 and mature MIR338 share the same genomic location but are transcribed from opposite DNA strands (Fig. 2) [44]. This critical miRNA sequence region is highly conserved across species [44].
Fig. 2.
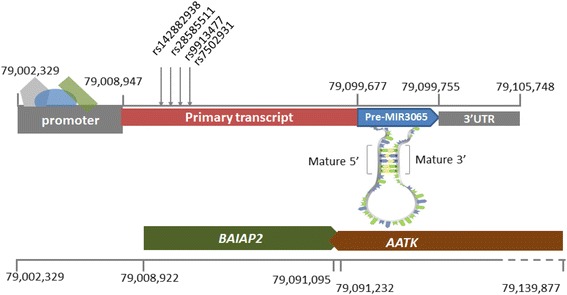
Chromosome 17 position GRCh37/hg19: 79,008,947–79,105,748, encompassing breast cancer-associated SNPs, defined promoter, primary, precursor, mature and 3’-UTR, miR-3065 gene regions and other overlapping coding genes. Note: BAIAP2 and AATK are transcribed in opposite directions. Additionally, AATK contains three more miRNAs in close proximity to miR-3065: miR-657, miR-338 and miR-1250
To better understand the implications of inherited susceptibility to BC that may involve BAIAP2, we examined expression of this gene in human tissues using data from the Genotype-Tissue Expression (GTEx) project portal version 6 [45]. Human brain-specific angiogenesis inhibitor 1-associated protein 2 (BAIAP2) demonstrates a range of expression across various human tissues including brain and breast [45] (Additional file 2: Figure S1). Using data from the Human Protein Atlas project that includes immunochemistry results on 83 different normal cell types from 44 tissue types, we note that moderate BAIAP2 protein expression is observed in human breast tissue when compared with other normal tissue types [46, 47]. Furthermore, when examining RNA sequencing gene expression of BAIAP2 in 47 invasive breast carcinoma cell lines from the Cancer Cell Line Encyclopedia [48], we note differential expression with the highest levels (10-fold or more) of BAIAP2 occurring in four cell lines: EFM-192A, HCC1937, HCC202, and ZR-75-30. Of these four cell lines, two are derived from metastatic sites, with one of these from an African-American woman, the other Caucasian. Of the remaining two cell lines (HCC1937, HCC202), both are from Caucasian women, from primary ductal carcinoma, are ER and PR negative, p53 mutation negative, and positive for EFP2 and CK19 expression; however, they differed in BRCA1 mutation and HER2 status.
The 17q25.3 region containing the top four BC-associated SNPs is extensively marked in the human mammary epithelial cell (HMEC) line by regulatory chromatin states from DNase and histones H3K27ac and H3K4me1, reflecting a number of active promoter and enhancer sequences in the region [44, 49–54]. Furthermore, a number of regulatory sequence motifs (e.g., sequence-specific binding sites for transcription factors) located within intron 1 of BAIAP2 are altered by these SNPs. Specifically rs9913477 alters regulatory motifs for CDP1 and SOX3 binding while rs7502931 alters a regulatory motif for ZNF143 [55]. No expression quantitative trait loci (eQTL) were identified in GTEx for any of the four top SNPs [45].
Several epidemiological studies, including both admixture mapping and association analysis of the insulin-related pathway, have examined the 17q region for association with BC in AMBER. A recently published genome-wide case-only admixture scan using 2624 AIMs in the AMBER consortium identified a novel region of excess African ancestry associated with BC risk at 17q25.1 (confirmed in a case-control admixture analysis in the same consortium) [56]. In this admixture scan, AIM rs496948172 provided the largest Z score and marked a wide 17q25.1 region of approximately 4.6 Mb where Z scores remained above 3.7 indicating excess African ancestry associated with BC. Elevated Z scores of 2.4 for this case analysis extend into the 17q25.3 region, where the top hit for the current association analysis (rs9913477) is approximately 1 Mb from the top admixture hit (rs4969481), although LD between these variants is limited (D’ = 0.19, r2 = 0.0009) based on AFR 1000 Genomes reference panel [57]. Thus, the region of excess African ancestry associated with BC marked by an AIM at 17q25.1 could include the more distal 17q25.3 region as well, providing evidence that variation in this genomic region associated with BC may contribute to disparity in risk. A second AMBER gene-based analysis of 184 genes in the insulin/insulin-like growth factor, leptin, and growth hormone pathways identified BAIP2 and CALM2, and AIAP2 and CSNK2A1 as the most significant gene associations (gene-based p ≤ 0.01) with both overall and ER positive BC, respectively [58]. Thus, both admixture and insulin pathway-specific association analyses in AMBER provide suggestive evidence of an association with overall and ER-positive BC in the 17q21–25 region. However, due to the relatively less dense SNP set used to evaluate this genomic region in the admixture scan and the less granular nature of the gene-based insulin pathway analysis, neither identified the specific set of four miRNA-associated SNPs localizing a statistically significant association with BC reported here. Additionally, each study strategy for multiple test correction varied in accordance with its statistical methods, with the admixture analysis using a more conservative Bonferroni correction, the gene-based method utilizing a gene-level correction factor, and our current study using a false discovery rate.
A recently published epidemiological study examining miRNA genes and BC among women of African ancestry found fourteen miRNA SNPs associated with overall BC risk at the significance level of 0.05 [29]. Included among these SNPs was rs73410309 within the precursor sequence of MIR4739 located on chromosome 17q25.3 (OR = 1.1; p = 0.039), which is approximately 1.5 Mb from our top hit in MIR3065 (rs9913477) and not in high LD with this SNP (D’ = 0.007 and R2 = 0.0) based on AFR 1000 Genomes reference panel [57]. This study was restricted to SNPs within miRNA precursor and mature sequences and thus would not have included the SNP in the primary miRNA sequence identified in our study, but highlights the potential role for miRNA SNPs in BC risk among women of African ancestry in this genomic region.
While no GWAS hits have been reported in the 17q25.3 region, this region has been implicated in several studies of BC tissues where recurrent gain of this genomic region is associated with subtype and recurrence [59, 60]. Gene expression studies of 17q25.3 have identified significant overexpression of 17q25.3 genes in BRCA1 mutated triple-negative breast cancer (TNBC) as compared with BRCA1 nonmutated TNBC [59], highlighting the important role that overexpressed sets of genes in this region may play in BC. Given the major role that miRNAs play in global gene regulation it is possible that, even in the absence of a copy number gain, abnormal expression of miRNA genes intended to suppress expression across multiple oncogenes could lead to similar upregulation of sets of genes in this region with similar BC effect. Studies of higher-order chromatin organization have identified regional epigenetic regulation (RER) in breast tumors and BC cell lines that are independent of copy number [61], where 26 regions of coordinate expression were identified between breast tumors and BC cell lines with nine RER showing upregulated gene expression relative to normal breast tissue. Included among these upregulated regions was a 0.9-Mb 17q25.1 region with correlated expression of KCTD2, GGA3, MRPS7, and GRB2, and a 0.58-Mb 17q25.3 region (approximately 900 kb from the four associated SNPs reported here) with correlated expression of STRA13, RFNG, CSNK1D, and SECTM1.
Perhaps the most compelling support implicating MIR3065 and BC comes from a recent study by Perrson et al. in which NGS expression analysis in paired normal and breast tumor tissue demonstrates a strikingly disparate expression pattern for MIR3065 [18]. Among the 361 newly NGS identified miRNA precursors, tumor identity was defined by differences in expression level of a large and common set of miRNAs rather than tissue specific expression [18]. While tissue MIR3065 expression was highest in breast tumors in a panel of nine human tissues, both lung and placenta demonstrated the next highest expression levels. Similar to previous studies, through BAC array comparative genomic hybridization (CGH), MIR3065 was also identified as a gene encoded in a region with high-level genomic amplification in luminal B, ERBB2/HER2-positive, ER positive, and ER negative subtypes. Among TargetScan’s (release 6.2, June 2012) MIR3065 gene top 15 predicted gene targets are the top hit AT-rich interaction domain 4B, ARID4B (alias BRCAA1, breast cancer antigen epitope-1) and RAB22A, a member of the RAS oncogene family of small GTPases involved in signal transduction [62–65]. Immunohistochemically, ARID4B/BRCAA1 was expressed in 65% of BC specimens but not in noncancerous tissues. and expression was closely associated with ER- and PR-positive status [66]. BC patients also had significantly higher titers of this epitope than healthy donors (p < 0.001). Given that two of the top predicted targets are likely oncogenes, it is possible that the role of MIR3065 is to suppress expression of these oncogenes. It is also possible that the pri-MIR3065 SNP associated with BC (or a SNP in LD) impairs MIR3065 processing, leading to lower levels of mature MIR3065 and reduced inhibition of these oncogenes. Of course, specific gene targets of this new miRNA are not yet fully known and new information may emerge as target prediction algorithms improve and functional data become available. For example, in addition to supporting the potential role of MIR3065 in BC, Perrson et al. also uncovered a new miRNA in a very well-studied region within the intron of ERBB2/HER2, a major predictive marker in BC [18]. This discovery highlights the importance of evaluating genomic regulation beyond the protein coding gene level to examine the major role that noncoding genes, such as miRNAs, may play in cancer development and heterogeneity. These insights will prove invaluable in our understanding of disease development, identifying at-risk populations and providing targets for cancer treatment.
Although this study was limited in scope to miRNAs with SNPs represented or imputed from the Illumina Human Exome BeadChip v1.1 and AMBER custom content (and thus only surveys one-third of all SNPS in the miRBase), as well as by miRNAs with predicted primary sequence from six cell lines, this study is one of the largest evaluations of miRNAs for association with BC. Moreover, it is the largest investigation among African-American women with BC annotated for subtype. Furthermore, predicted boundaries of primary transcripts at both 5′ and 3′ ends were extended from the start of the H3K4me3 peak at the 5′ end (which is often upstream from the actual transcription start site (TSS)) and through the end of the H3K79me2 or H3K36me3 signal (which may or may not be downstream of the transcription termination site). Thus, SNPs defined as retained within the primary transcript may reside just upstream in the miRNA promoter region or may reside just downstream beyond the 3′ end of the primary transcript, thus potentially altering our interpretation of function. Specifically, we predicted that the BC-associated SNPs identified may affect miRNA processing; however, if in fact they reside in the promoter regions, they may influence miRNA expression through other mechanisms. More experimental validation of discrete TSS and end sites is needed for the majority of known miRNAs. Additionally, while this is one of the largest populations of African-Americans with BC examined for miRNA gene association, the subtype analyses remain underpowered for the genetic effect sizes anticipated.
Despite these limitations, this study provides valuable new information about the relationship between numerous miRNA genes and BC in an understudied population, African-American women. It emphasizes the complexity of SNP association analyses and interpretation of function in gene-dense regions, and also the complex interplay of evidence from studies of coding genes, copy number variation, epigenetic regulation, and admixture mapping in an important chromosomal region associated with BC. Functional assessment of the BC-associated SNPs in BAIAP2 and MIR3065 are needed to identify the potential molecular mechanism behind their association with BC risk, in particular the risk of ER positive BC, the most common subtype. Larger studies of African-American women are needed to address subtype-specific biology and genetics, including those related to miRNAs.
Conclusions
This study reports a novel BC signal within an 8.6-kb locus on chromosome 17q25.3, where germline genetic variation is associated with overall and ER positive BC risk among African-American women. This complex and gene-dense region contains BAIAP2, a protein-coding gene, and MIR3065, an important nonprotein coding regulatory gene, which may play key roles in BC development and heterogeneity among AA women. An understanding of the potentially functional implications of variation in these genes is necessary and may uncover important genetic risk factors and mechanisms for BC in general and, more specifically, for ER positive BC, the most common subtype. Understanding risk factors and mechanisms for BC may lead to improved screening, risk stratification, and novel treatments.
Additional files
Table S1. Association of the top seven miRNA SNPs from the full analyses with p < 5 × 10−6 in case versus control and case-only subtype analyses. (XLSX 72 kb)
Figure S1. BAIAP2 gene expression (from Gene-Tissue Expression project, GTEx) in human tissues [46]. (DOCX 193 kb)
Acknowledgements
We thank the participants and staff of the contributing studies. We wish also to acknowledge the late Robert Millikan, DVM, MPH, PhD, who was instrumental in the creation of this consortium. Pathology data were obtained from numerous state cancer registries (Arizona, California, Colorado, Connecticut, Delaware, District of Columbia, Florida, Georgia, Hawaii, Illinois, Indiana, Kentucky, Louisiana, Maryland, Massachusetts, Michigan, New Jersey, New York, North Carolina, Oklahoma, Pennsylvania, South Carolina, Tennessee, Texas, and Virginia). The results reported do not necessarily represent their views or the views of the National Institutes of Health.
Funding
This research was funded by the National Institutes of Health: P01 CA151135 (CBA, JRP, and AFO), R01 CA058420 (LR), UM1 CA164974 (JRP and LR), R01 CA098663 (JRP), R01 CA100598 (CBA), UM1 CA164973 (CAH), R01 CA54281 (CAH), P50 CA58223 (MAT and AO), U01 CA179715 (MAT and AO), KL2TR001109 (KLY), R01CA185623 (EVB and CCH), R25 5R25GM089569 (KC), the Komen for the Cure Foundation, the Breast Cancer Research Foundation (CBA), and the University Cancer Research Fund of North Carolina (JTB, AFO, and MAT).
Availability of data and materials
The datasets generated and/or analyzed during the current study are publicly available in dbGAP (https://www.ncbi.nlm.nih.gov/gap).
Abbreviations
- AA
African-American
- AIM
Ancestry informative marker
- AMBER
African American Breast Cancer Epidemiology and Risk
- BC
Breast cancer
- BWHS
Black Women’s Health Study
- CBCS
Carolina Breast Cancer Study
- CGH
Comparative genomic hybridization
- ChIP-seq
Chromatin immunoprecipitation and massively parallel DNA sequencing
- CI
Confidence interval
- CIDR
Center for Inherited Disease Research
- DCIS
Ductal carcinoma in situ
- ENCODE
Encyclopedia of DNA Elements
- eQTL
Expression quantitative trait loci
- ER
Estrogen receptor
- FDR
False discovery rate
- GTEx
Genotype-Tissue Expression
- GWAS
Genome-wide association study
- HER2
Human epidermal growth factor receptor 2
- HMEC
Human mammary epithelial cell
- kb
Kilobase
- LD
Linkage disequilibrium
- MAF
minor allele frequencies
- Mb
Megabase
- MEC
Multiethnic Cohort
- MEGA
Multi-Ethnic Genotyping Array
- miRNA
MicroRNA
- mRNA
Messenger RNA
- NGS
Next-generation sequencing
- nt
Nucleotide
- oncomiR
Oncogenic miRNA
- OR
Odds ratio
- PR
Progesterone receptor
- pre-miRNA
Precursor microRNA
- pri-miRNA
Primary microRNA transcript
- QC
Quality control
- RER
Regional epigenetic regulation
- RISC
RNA-induced silencing complex
- SNPs
Single nucleotide polymorphisms
- TNBC
Triple-negative breast cancer
- TSS
Transcription start site
- UTR
untranslated region
- WCHS
Women’s Circle of Health Study
Authors’ contributions
JTB played a central role in the study design and data interpretation and made a major contribution to the manuscript as the primary author. MG made a major contribution to the analysis and interpretation of genetic association data and provided supportive details from public genetic and expression databases. KLY analyzed and interpreted genetic association data. PS provided miRNA promoter annotation using six cell lines and tissue types (all ENCODE Tier 1 cell types plus human pancreatic islets) and integrative analysis of ChIP-seq data from the NIH ENCODE project and algorithm described previously. Provided input on functional interpretation of miRNA SNPs associated with breast cancer. JP provided extensive bioinformatics support for gene annotation and SNP identification and selection. CVP provided clinical input and functional interpretation of miRNA SNPs associated with breast cancer. KC provided miRNA promoter annotation using six cell lines and tissue types (all ENCODE Tier 1 cell types plus human pancreatic islets) and integrative analysis of ChIP-seq data from the NIH ENCODE project and algorithm described previously. CAH facilitated access to MEC biospecimens and data and provided input regarding genetic data interpretation in AMBER. SAH, ERA-N, C-CH, LES-C, QZ, SL, SY, EVB, and LR provided input regarding genetic data interpretation in AMBER. KLL made a major contribution to the evaluation, quality control, coordination and management, and interpretation of the genetic data for AMBER and linkage with other study-specific clinical and demographic variables. CBA facilitated access to WCHS biospecimens and data and provided input regarding genetic data interpretation in AMBER. JRP facilitated access to BWHS biospecimens and data and provided input regarding genetic data interpretation in AMBER. MAT and AFO facilitated access to CBCS biospecimens and data and provided input regarding genetic data interpretation in AMBER. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All study subjects provided informed consent and all study protocols were institutionally reviewed and approved by local IRBs and were titled: Epidemiology of Breast Cancer Subtypes in African-American Women: a Consortium:
University of North Carolina at Chapel Hill (CBCS) IRB #11-1277
Boston University (BWHS) IRB # H-31079
Roswell Park Cancer Institute (WCHS) IRB # I-177810
University of Hawaii Cancer Center (MEC) IRB CHS #19251
University of Southern California (MEC) IRB# HS-11-00427
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0964-4) contains supplementary material, which is available to authorized users.
Contributor Information
Jeannette T. Bensen, Phone: 919-843-1017, Email: jbensen@med.unc.edu
Mariaelisa Graff, migraff@email.unc.edu.
Kristin L. Young, Email: kristin.young@unc.edu
Praveen Sethupathy, Email: praveens@cornell.edu.
Joel Parker, parkerjs@email.unc.edu.
Chad V. Pecot, pecot@email.unc.edu
Kevin Currin, kwcurrin@email.unc.edu.
Stephen A. Haddad, Email: shaddad7@msn.com
Edward A. Ruiz-Narváez, Email: eruiznar@umich.edu
Christopher A. Haiman, Email: Christopher.Haiman@med.usc.edu
Chi-Chen Hong, Email: Chi-Chen.Hong@RoswellPark.org.
Lara E. Sucheston-Campbell, Email: sucheston-campbell.1@osu.edu
Qianqian Zhu, Email: Qianqian.Zhu@RoswellPark.org.
Song Liu, Email: Song.Liu@RoswellPark.org.
Song Yao, Email: Song.Yao@RoswellPark.org.
Elisa V. Bandera, Email: banderel@cinj.rutgers.edu
Lynn Rosenberg, Email: lrosenbe@bu.edu.
Kathryn L. Lunetta, Email: klunetta@bu.edu
Christine B. Ambrosone, Email: Christine.Ambrosone@RoswellPark.org
Julie R. Palmer, Email: jpalmer@bu.edu
Melissa A. Troester, Email: troester@unc.edu
Andrew F. Olshan, Email: andy_olshan@unc.edu
References
- 1.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294(5543):862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 2.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37(7):766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 3.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15(6):321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions—beyond repression of gene expression. Nat Rev Genet. 2014;15(9):599–612. doi: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, Hatzigeorgiou AG, Antonarakis SE. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am J Hum Genet. 2007;81(2):405–413. doi: 10.1086/519979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra PJ, Humeniuk R, Longo-Sorbello GS, Banerjee D, Bertino JR. A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc Natl Acad Sci U S A. 2007;104(33):13513–13518. doi: 10.1073/pnas.0706217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landi D, Gemignani F, Naccarati A, Pardini B, Vodicka P, Vodickova L, Novotny J, Försti A, Hemminki K, Canzian F, et al. Polymorphisms within micro-RNA-binding sites and risk of sporadic colorectal cancer. Carcinogenesis. 2008;29(3):579–584. doi: 10.1093/carcin/bgm304. [DOI] [PubMed] [Google Scholar]
- 9.Brendle A, Lei H, Brandt A, Johansson R, Enquist K, Henriksson R, Hemminki K, Lenner P, Försti A. Polymorphisms in predicted microRNA-binding sites in integrin genes and breast cancer: ITGB4 as prognostic marker. Carcinogenesis. 2008;29(7):1394–1399. doi: 10.1093/carcin/bgn126. [DOI] [PubMed] [Google Scholar]
- 10.Larrea E, Sole C, Manterola L, Goicoechea I, Armesto M, Arestin M, Caffarel MM, Araujo AM, Araiz M, Fernandez-Mercado M, et al. New concepts in cancer biomarkers: circulating miRNAs in liquid biopsies. Int J Mol Sci. 2016;17(5)627. doi:10.3390/ijms17050627. [DOI] [PMC free article] [PubMed]
- 11.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 12.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heneghan HM, Miller N, Lowery AJ, Sweeney KJ, Kerin MJ. MicroRNAs as novel biomarkers for breast cancer. J Oncol. 2009;2009:950201. doi: 10.1155/2010/950201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12(2):201. doi: 10.1186/bcr2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Z, Baserga R, Chen L, Wang C, Lisanti MP, Pestell RG. microRNA, cell cycle, and human breast cancer. Am J Pathol. 2010;176(3):1058–1064. doi: 10.2353/ajpath.2010.090664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang J, Ahmad A, Sarkar FH. The role of microRNAs in breast cancer migration, invasion and metastasis. Int J Mol Sci. 2012;13(10):13414–13437. doi: 10.3390/ijms131013414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, Loman N, Jonsson G, Naya H, Hoglund M, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res. 2011;71(1):78–86. doi: 10.1158/0008-5472.CAN-10-1869. [DOI] [PubMed] [Google Scholar]
- 19.Aloraifi F, Boland MR, Green AJ, Geraghty JG. Gene analysis techniques and susceptibility gene discovery in non-BRCA1/BRCA2 familial breast cancer. Surg Oncol. 2015;24(2):100–109. doi: 10.1016/j.suronc.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Duan S, Mi S, Zhang W, Dolan ME. Comprehensive analysis of the impact of SNPs and CNVs on human microRNAs and their regulatory genes. RNA Biol. 2009;6(4):412–425. doi: 10.4161/rna.6.4.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman AE, Zheng T, Yi C, Leaderer D, Weidhaas J, Slack F, Zhang Y, Paranjape T, Zhu Y. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Cancer Res. 2009;69(14):5970–5977. doi: 10.1158/0008-5472.CAN-09-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu Z, Liang J, Wang Z, Tian T, Zhou X, Chen J, Miao R, Wang Y, Wang X, Shen H. Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Hum Mutat. 2009;30(1):79–84. doi: 10.1002/humu.20837. [DOI] [PubMed] [Google Scholar]
- 23.Kontorovich T, Levy A, Korostishevsky M, Nir U, Friedman E. Single nucleotide polymorphisms in miRNA binding sites and miRNA genes as breast/ovarian cancer risk modifiers in Jewish high-risk women. Int J Cancer. 2010;127(3):589–597. doi: 10.1002/ijc.25065. [DOI] [PubMed] [Google Scholar]
- 24.Akkız H, Bayram S, Bekar A, Akgöllü E, Ulger Y. A functional polymorphism in pre-microRNA-196a-2 contributes to the susceptibility of hepatocellular carcinoma in a Turkish population: a case-control study. J Viral Hepat. 2011;18(7):e399–e407. doi: 10.1111/j.1365-2893.2010.01414.x. [DOI] [PubMed] [Google Scholar]
- 25.Mittal RD, Gangwar R, George GP, Mittal T, Kapoor R. Investigative role of pre-microRNAs in bladder cancer patients: a case-control study in North India. DNA Cell Biol. 2011;30(6):401–406. doi: 10.1089/dna.2010.1159. [DOI] [PubMed] [Google Scholar]
- 26.Okubo M, Tahara T, Shibata T, Yamashita H, Nakamura M, Yoshioka D, Yonemura J, Ishizuka T, Arisawa T, Hirata I. Association between common genetic variants in pre-microRNAs and gastric cancer risk in Japanese population. Helicobacter. 2010;15(6):524–531. doi: 10.1111/j.1523-5378.2010.00806.x. [DOI] [PubMed] [Google Scholar]
- 27.Jedlinski DJ, Gabrovska PN, Weinstein SR, Smith RA, Griffiths LR. Single nucleotide polymorphism in hsa-mir-196a-2 and breast cancer risk: a case control study. Twin Res Hum Genet. 2011;14(5):417–421. doi: 10.1375/twin.14.5.417. [DOI] [PubMed] [Google Scholar]
- 28.Bensen JT, Tse CK, Nyante SJ, Barnholtz-Sloan JS, Cole SR, Millikan RC. Association of germline microRNA SNPs in pre-miRNA flanking region and breast cancer risk and survival: the Carolina Breast Cancer Study. Cancer Causes Control. 2013;24(6):1099–1109. doi: 10.1007/s10552-013-0187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian F, Feng Y, Zheng Y, Ogundiran TO, Ojengbede O, Zheng W, Blot W, Ambrosone CB, John EM, Bernstein L, et al. Genetic variants in microRNA and microRNA biogenesis pathway genes and breast cancer risk among women of African ancestry. Hum Genet. 2016;135(10):1145–1159. doi: 10.1007/s00439-016-1707-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman B, Moorman PG, Millikan R, Qaqish BF, Geradts J, Aldrich TE, Liu ET. The Carolina Breast Cancer Study: integrating population-based epidemiology and molecular biology. Breast Cancer Res Treat. 1995;35(1):51–60. doi: 10.1007/BF00694745. [DOI] [PubMed] [Google Scholar]
- 31.Ambrosone CB, Ciupak GL, Bandera EV, Jandorf L, Bovbjerg DH, Zirpoli G, Pawlish K, Godbold J, Furberg H, Fatone A, et al. Conducting molecular epidemiological research in the age of HIPAA: a multi-institutional case-control study of breast cancer in African-American and European-American women. J Oncol. 2009;2009:871250. doi: 10.1155/2009/871250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bandera EV, Chandran U, Zirpoli G, McCann SE, Ciupak G, Ambrosone CB. Rethinking sources of representative controls for the conduct of case-control studies in minority populations. BMC Med Res Methodol. 2013;13:71. doi: 10.1186/1471-2288-13-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg L, Adams-Campbell L, Palmer JR. The Black Women's Health Study: a follow-up study for causes and preventions of illness. J Am Med Womens Assoc. 1995;50(2):56–58. [PubMed] [Google Scholar]
- 34.Kolonel LN, Henderson BE, Hankin JH, Nomura AM, Wilkens LR, Pike MC, Stram DO, Monroe KR, Earle ME, Nagamine FS. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am J Epidemiol. 2000;151(4):346–357. doi: 10.1093/oxfordjournals.aje.a010213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer JR, Ambrosone CB, Olshan AF. A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes Control. 2014;25(3):309–319. doi: 10.1007/s10552-013-0332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen F, Chen GK, Stram DO, Millikan RC, Ambrosone CB, John EM, Bernstein L, Zheng W, Palmer JR, Hu JJ, et al. A genome-wide association study of breast cancer in women of African ancestry. Hum Genet. 2013;132(1):39–48. doi: 10.1007/s00439-012-1214-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sethupathy P. Illuminating microRNA transcription from the epigenome. Curr Genomics. 2013;14(1):68–77. doi: 10.2174/138920213804999183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haddad SA, Ruiz-Narváez EA, Haiman CA, Sucheston-Campbell LE, Bensen JT, Zhu Q, Liu S, Yao S, Bandera EV, Rosenberg L, et al. An exome-wide analysis of low frequency and rare variants in relation to risk of breast cancer in African American Women: the AMBER Consortium. Carcinogenesis. 2016;37(9):870-877. doi: 10.1093/carcin/bgw067. Epub 2016 Jun 7. [DOI] [PMC free article] [PubMed]
- 41.Allott EH, Cohen SM, Geradts J, Sun X, Khoury T, Bshara W, Zirpoli GR, Miller CR, Hwang H, Thorne LB, et al. Performance of three-biomarker immunohistochemistry for intrinsic breast cancer subtyping in the AMBER consortium. Cancer Epidemiol Biomark Prev. 2016;25(3):470–478. doi: 10.1158/1055-9965.EPI-15-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. [Google Scholar]
- 43.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The GTEx Consortium. Lonsdale JTJ, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Kyung H, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis T, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Little AR, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, Huber W, Jupp S, Keays M, Kryvych N, et al. Expression Atlas update—a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42(Database issue):D926–D932. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapushesky M, Adamusiak T, Burdett T, Culhane A, Farne A, Filippov A, Holloway E, Klebanov A, Kryvych N, Kurbatova N, et al. Gene Expression Atlas update—a value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012;40(Database issue):D1077–D1081. doi: 10.1093/nar/gkr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26(17):2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, Nguyen N, Paten B, Zweig AS, Karolchik D, et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014;30(7):1003–1005. doi: 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kent WJ, Hsu F, Karolchik D, Kuhn RM, Clawson H, Trumbower H, Haussler D. Exploring relationships and mining data with the UCSC Gene Sorter. Genome Res. 2005;15(5):737–741. doi: 10.1101/gr.3694705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23(5):800–811. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruiz-Narváez EA, Sucheston-Campbell L, Bensen JT, Yao S, Haddad S, Haiman CA, Bandera EV, John EM, Bernstein L, Hu JJ, et al. Admixture mapping of African-American women in the AMBER consortium identifies new loci for breast cancer and estrogen-receptor subtypes. Front Genet. 2016;7:170. doi: 10.3389/fgene.2016.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruiz-Narváez EA, Lunetta KL, Hong CC, Haddad S, Yao S, Cheng TD, Bensen JT, Bandera EV, Haiman CA, Troester MA, et al. Genetic variation in the insulin, insulin-like growth factor, growth hormone, and leptin pathways in relation to breast cancer in African-American women: the AMBER consortium. NPJ Breast Cancer. 2016;2:16034. [DOI] [PMC free article] [PubMed]
- 59.Toffoli S, Bar I, Abdel-Sater F, Delrée P, Hilbert P, Cavallin F, Moreau F, Van Criekinge W, Lacroix-Triki M, Campone M, et al. Identification by array comparative genomic hybridization of a new amplicon on chromosome 17q highly recurrent in BRCA1 mutated triple negative breast cancer. Breast Cancer Res. 2014;16(6):466. doi: 10.1186/s13058-014-0466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hwang KT, Han W, Cho J, Lee JW, Ko E, Kim EK, Jung SY, Jeong EM, Bae JY, Kang JJ, et al. Genomic copy number alterations as predictive markers of systemic recurrence in breast cancer. Int J Cancer. 2008;123(8):1807–1815. doi: 10.1002/ijc.23672. [DOI] [PubMed] [Google Scholar]
- 61.Rafique S, Thomas JS, Sproul D, Bickmore WA. Estrogen-induced chromatin decondensation and nuclear re-organization linked to regional epigenetic regulation in breast cancer. Genome Biol. 2015;16:145. doi: 10.1186/s13059-015-0719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 63.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18(10):1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui D, Jin G, Gao T, Sun T, Tian F, Estrada GG, Gao H, Sarai A. Characterization of BRCAA1 and its novel antigen epitope identification. Cancer Epidemiol Biomark Prev. 2004;13(7):1136–1145. [PubMed] [Google Scholar]
- 67.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genome-wide association scans. Bioinformatics. 2010;26(17):2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Association of the top seven miRNA SNPs from the full analyses with p < 5 × 10−6 in case versus control and case-only subtype analyses. (XLSX 72 kb)
Figure S1. BAIAP2 gene expression (from Gene-Tissue Expression project, GTEx) in human tissues [46]. (DOCX 193 kb)
Data Availability Statement
The datasets generated and/or analyzed during the current study are publicly available in dbGAP (https://www.ncbi.nlm.nih.gov/gap).