

Abstract
In all living organisms, coenzyme A (CoA) is an essential cofactor with a unique design allowing it to function as an acyl group carrier and a carbonyl-activating group in diverse biochemical reactions. It is synthesized in a highly conserved process in prokaryotes and eukaryotes that requires pantothenic acid (vitamin B5), cysteine and ATP. CoA and its thioester derivatives are involved in major metabolic pathways, allosteric interactions and the regulation of gene expression. A novel unconventional function of CoA in redox regulation has been recently discovered in mammalian cells and termed protein CoAlation. Here, we report for the first time that protein CoAlation occurs at a background level in exponentially growing bacteria and is strongly induced in response to oxidizing agents and metabolic stress. Over 12% of Staphylococcus aureus gene products were shown to be CoAlated in response to diamide-induced stress. In vitro CoAlation of S. aureus glyceraldehyde-3-phosphate dehydrogenase was found to inhibit its enzymatic activity and to protect the catalytic cysteine 151 from overoxidation by hydrogen peroxide. These findings suggest that in exponentially growing bacteria, CoA functions to generate metabolically active thioesters, while it also has the potential to act as a low-molecular-weight antioxidant in response to oxidative and metabolic stress.
Keywords: coenzyme A, Gram-negative and -positive bacteria, post-translational modification, redox signaling
Introduction
Coenzyme A (CoA) is a ubiquitous and essential cofactor in all living cells, where it functions as a carbonyl-activating group and a carrier for activated acyl groups in numerous metabolic and catabolic processes. The biosynthesis of CoA in prokaryotes and eukaryotes is a conserved process that requires pantothenic acid (vitamin B5), cysteine and ATP. The presence of a thiol group in the CoA structure is at the core of its biochemical behavior. In cells, CoA forms a diverse range of thioester derivatives, such as Acetyl CoA, malonyl CoA and 3-hydroxy-3-methylglutaryl (HMG) CoA, which play central roles in many biochemical reactions in protein, carbohydrate and lipid metabolism [1–4]. These include the synthesis and oxidation of fatty acids, isoprenoid and cholesterol biosynthesis, amino acid metabolism, the Krebs cycle and the synthesis of peptidoglycans. In addition, CoA derivatives function as substrates for protein acylation (e.g. lysine acetylation succinylation, malonylation, propionylation and butyrylation), which has emerged as an important mechanism in the regulation of transcription, chromatin maintenance and cellular metabolism [5–7].
One aspect of CoA biochemistry that has not been well investigated is the role of this central metabolic coenzyme in thiol-disulfide exchange reactions and redox regulation. It has been reported that CoA undergoes copper-catalyzed air oxidation at a rate which is 4-fold slower than GSH (glutathione) and 720-fold less rapidly than cysteine, making it an appropriate protective thiol in all living cells [8]. As a thiol-containing molecule, CoA has been found to form CoA disulfides (CoASSCoA) and mixed disulfides with other low-molecular-weight (LMW) thiols (e.g, CoA-cysteine and CoA-glutathione, CoASSG) or cysteine residues in specific proteins. The CoASSG heterodimer has been isolated from bacteria, yeast, human myocardial tissue and parathyroid glands [9–11]. Potent vasoconstrictive and proliferative effects of CoASSG were observed in cultured vascular smooth muscle cells [12]. CoASSG was also shown to inhibit the activity of bacterial RNA polymerase [13].
Exposed protein thiols are the predominant targets of redox-linked regulation mediated by post-translational modifications, including oxidation, S-acylation, S-nitrosation, persulfhydration and S-thiolation [14,15]. When the cysteine thiol group is oxidized by reactive oxygen species (ROS) to an unstable sulfenic acid intermediate, it can react with nearby thiols leading to the formation of intra- and intermolecular disulfides or mixed disulfides with LMW thiols, such as cysteine, glutathione, bacillithiol and CoA. Formed disulfides are reversible regulatory events and function to protect unstable sulfenic acids against overoxidation to sulfinic and sulfonic acids which may alter irreversibly the structure, function and subcellular localization of modified proteins [16].
The formation of mixed disulfides between CoA and cysteines of specific proteins has been reported in several biochemical and crystallographic studies [17–21]. The CoA-modified forms of acetyl-CoA acetyltransferase and glutamate dehydrogenase were detected immunohistochemically in rat liver mitochondria [17,18]. In these studies, the activity and half-life of acetyl-CoA acetyltransferase were shown to be modified by covalent attachment of CoA. Furthermore, covalent modification of phenol sulfotransferase by CoA was shown to inhibit its activity in a dose- and time-dependent manner [19]. In Klebsiella pneumoniae, CoA binding to flavodoxin NifF was found to halt the N2 fixation by blocking electron transfer from pyruvate-flavodoxin oxidoreductase NifJ to nitrogenase NifH [20]. Covalent binding of CoA to the Bacillus subtilis organic peroxide sensor OhrR was reported, but the consequence of this modification on the transcription repressor activity of OhrR in oxidative stress response has not been examined [21]. Despite the existence of these sporadic studies, investigation into the extent of covalent protein modification by CoA and the mechanism of regulation in eukaryotes and prokaryotes has long been overdue. Recent studies from our laboratory revealed extensive covalent modification of cellular proteins by CoA in mammalian cells and tissues, which we termed protein CoAlation [22]. We showed that protein CoAlation is a reversible post-translational modification induced in mammalian cells by oxidizing agents and metabolic stress. To uncover protein CoAlation as a common post-translational modification and to reveal its role in redox regulation, we have developed a range of new research tools and methodologies, including: (a) unique anti-CoA mAbs which work efficiently in various immunological assays; (b) in vitro protein CoAlation and assay and (c) a reliable strategy for the identification of CoAlated proteins by LC–MS/MS.
In the present paper, we provide evidence that protein CoAlation occurs at a low level in Gram-negative and Gram-positive bacteria under normal growth conditions, but is strongly induced in response to oxidizing agents and metabolic stress. Approximately 12% of the predicted Staphylococcus aureus proteome was found to be CoAlated in diamide-treated bacteria. SaGAPDH (S. aureus glyceraldehyde-3-phosphate dehydrogenase), a key enzyme in glycolysis, was found to be readily CoAlated in Escherichia coli treated with hydrogen peroxide (H2O2), diamide and sodium hypochlorite (NaOCl). Furthermore, in vitro CoAlation of recombinant SaGAPDH prevented overoxidation and irreversible loss of its activity in the presence of exogenous H2O2. Altogether, our findings suggest that in bacteria, protein CoAlation is a widespread redox-regulated post-translational modification with a potential to protect critical reactive cysteines against irreversible overoxidation.
Experimental
Reagents and chemicals
All common chemicals were obtained from Sigma–Aldrich unless otherwise stated. The generation and characterization of the anti-CoA antibody (1F10) was described recently [23]. For Western blotting, anti-CoA antibody was diluted in Odyssey blocking buffer (0.17 µg/ml) containing 0.01% Tween 20. Secondary antibodies [Alexa Fluor 680 goat antimouse IgG H&L (Life Technologies)] were diluted in Odyssey blocking buffer (1 : 10 000) containing 0.02% sodium dodecyl sulfate (SDS).
Bacterial species, growth conditions and treatments
Following bacterial species were used in the present study: E. coli SG13009 and DH5alpha, Bacillus megaterium NCTC10342 and S. aureus DSM11729. B. megaterium cells were cultured overnight in Nutrient Broth 3 (NB3) medium, while E. coli and S. aureus cells were grown in Luria Bertani (LB) medium. The overnight cultures were diluted 1 : 100 in the same media and incubated until the optical density of 0.7 at 600 nm (OD600). The samples of cells were then treated with or without oxidizing agents for 30 min at 37°C: hydrogen peroxide (10 and 100 mM), diamide (2 mM) and NaOCl (150 µM). To induce metabolic stress, bacterial cultures at OD600 of 0.7 were harvested by centrifugation and resuspended in M9 minimal medium supplemented with or without glucose as a source of carbohydrate.
Cell lysis and protein extraction
Protein extracts from harvested bacteria were prepared in the following ways: (a) the pellet of harvested E. coli, B. megaterium and S. aureus was resuspended in buffer containing 100 mM Tris–HCl, pH 7.5, 100 mM NaCl, 100 mM NEM and a cocktail of protease inhibitors (Roche). SDS was added (1% final), and the homogenate was sonicated to reduce viscosity before centrifuging at 21 000 g for 10 min at RT. The supernatant was collected and analyzed by western blotting. (b) The pellet of harvested S. aureus was resuspended in buffer containing 100 mM Tris–HCl, pH 7.5, 100 mM NaCl, 100 mM NEM and a cocktail of protease inhibitors (Roche). To solubilize cell wall proteins, lysostaphin (22 U/ml) was added and the lysate was incubated at 37°C for 30 min. After the addition of SDS (1% final), the homogenate was sonicated to reduce viscosity before centrifuging at 21 000 g for 10 min at RT. The supernatant was collected for further analysis.
Western blot analysis
Samples of bacterial extracts containing ∼30–40 µg of proteins were heated for 5 min at 99°C in SDS loading buffer with or without dithiothreitol (DTT, 100 mM final) and separated by SDS–polyacrylamide gel electrophoresis (PAGE) on 4–20% Mini-PROTEAN TGX Precast Gels (Bio-Rad Laboratories). Separated proteins were transferred from the gel to a low-fluorescence polyvinylidene fluoride membrane (Bio-Rad Laboratories), which was then blocked with Odyssey blocking buffer (LI-COR Biosciences). Primary anti-CoA antibody was diluted in Odyssey blocking buffer (0.17 µg/ml) and incubated with the membrane for 2 h at RT or overnight at 4°C. Immunoreactive protein bands were visualized using infrared dye-conjugated secondary antibodies and the Odyssey infrared imaging system (Odyssey Scanner CLx and Image Studio Lite software, LI-COR Biosciences).
Expression and affinity purification of SaGAPDH
The full coding sequence of SaGAPDH was cloned into the pQE3/SaGAPDH expression plasmid with the N-terminal His-tag sequences as previously described [24]. Expression of His-tagged SaGAPDH was carried out in exponentially growing SG13009 cells in the presence of 1 mM isopropyl- β-d-thiogalactopyranoside (IPTG). After 3 h induction at 37°C, the cells were harvested and stored at −80°C. Affinity purification of His-tagged SaGAPDH was performed using Ni-NTA chromatography. Eluted preparations were examined by SDS–PAGE and stored at −80°C.
GAPDH enzymatic assay
Recombinant SaGAPDH activity was determined by measuring the absorbance change at 340 nm and 25°C resulting from the production of NADH. The reaction was carried out in a 150 µl assay mixture containing 20 mM Tris–HCl (pH 8.7), 0.36 µM SaGAPDH, 1.25 mM NAD+, 1.25 mM ethylenediaminetetraacetic acid and 15 mM sodium arsenate. The reaction was started by the addition of 0.25 mM glyceraldehyde 3-phosphate. Initial reaction rates were calculated as described recently [25], by determining the slope in the linear part of the curve during the first 80 s of the reaction (GraphPad, linear regression function). The percentage of SaGAPDH activity was calculated as: Rate of inactivated/Rate of untreated × 100%. The results are presented as mean ± SEM from at least three separate experiments.
For the inactivation experiments, SaGAPDH was preincubated with 1 µM, 10 µM, 100 µM and 10 mM H2O2 for 10 min or with 10 mM CoASSCoA for 30 min. About 2 µl of the mixture was then added to the assay mixture and the remaining activity was measured as described. To reduce it, the enzyme was incubated with 10 mM DTT for 15 min. After treatments, excess H2O2, CoASSCoA and DTT were removed using Micro Biospin 6 columns (Bio-Rad).
Purification and activity assay of Nudix 7 hydrolase
Recombinant His-Nudix 7 hydrolase was expressed in bacteria and purified by Ni-NTA affinity chromatography. His-Nudix 7 (1.7 µg) was incubated in a total volume of 100 µl containing 50 mM (NH4)HCO and 0.2 mM CoASSG at 37°C for 20 min with or without 5 mM MgCl2. Reaction products and substrates were analyzed by HPLC as described, except that elution was monitored at 205 nm [26].
Preparation and enrichment of CoAlated peptides from diamide-treated S. aureus for MS analysis
The pellet of diamide-treated S. aureus (2 mM for 30 min) was resuspended in buffer containing 100 mM Tris–HCl, pH 7.5, 100 mM NaCl, 100 mM NEM and a cocktail of protease inhibitors (Roche). The lysate was incubated with lysostaphin (20 U/ml) at 37°C for 30 min to solubilize cell wall proteins. SDS was then added (1% final), and the homogenate was sonicated to reduce viscosity before centrifuging at 21 000 g for 10 min at RT. Proteins in the supernatant were precipitated with 90% methanol. The protein pellet was resuspended in 50 mM (NH4)HCO3 (pH 7.8) supplemented with 6.4 mM iodoacetamide (IAM) and digested with endoproteinases Lys C and trypsin (sequencing grade, Promega). After heat inactivation (99°C, 10 min) of digestive enzymes, CoAlated peptides were immunoprecipitated with anti-CoA antibody cross-linked to Protein G Sepharose. Trypsin digested and immunoprecipitated peptide mixtures were dried down completely in a SpeedVac and resolubilized in 20 µl of 50 mM ammonium bicarbonate (Ambic). After mixing for 2 min, 2.3 µl of 50 mM MgCl2 was added followed by 1 µl of Nudix 7 phosphatase. The solution was incubated at 37°C for 20 min then acidified, desalted with a C18 Stage tip that contained 1.5 µl of Poros R3 resin and partially dried in a SpeedVac. Modified peptides were further enriched using Phos-Select IMAC resin (Sigma). Desalted peptides were resuspended in 100 µl of 30% MeCN, 0.25 M acetic acid (loading solution) and 30 µl of IMAC beads, previously equilibrated with the loading solution was added. After 45 min incubation at room temperature, beads were washed four times with loading solution and CoAlated peptides were eluted twice with 500 mM imidazole (pH 7.6) and once with 30% MeCN/500 mM imidazole (pH 7.6). CoAlated peptides were acidified, dried and desalted with a C18 Stage tip that contained 1.5 µl of Poros R3 resin. This solution was then partially dried down using a SpeedVac and was ready for mass spectrometry analysis.
In vitro CoAlation of SaGAPDH
About 100 µg of affinity-purified SaGAPDH was CoAlated in buffer containing 50 mM Tris-HCl, pH 7.5 and 250 µM CoASSCoA for 30 min. Excess of unbound CoASSCoA was removed using Micro Biospin 6 columns (Bio-Rad).
Preparation and enrichment of CoAlated peptides from in vitro CoAlated SaGAPDH
Solution samples of in vitro CoAlated SaGAPDH (2 µg) in 50 mM ammonium bicarbonate (NH4CO3) and 8 mM iodoacetamide were digested with endoproteinases Lys C and elastase (Promega, U.K.). To the peptide mixture, 3.1 µl of 50 mM MgCl2 was added followed by 1.35 µl of Nudix-7 phosphatase and the mixture was incubated at 37°C for 20 min. The peptide mixture was then acidified, desalted on a C18 Stage tip (3M Empore) containing 0.7 µl of Poros R3 resin (Applied Biosystems, U.K.) and partially dried in a SpeedVac.
CoAlated peptides were enriched using Phos-Select IMAC resin (Sigma, U.K.). Desalted peptides were resuspended in 100 µl of 30% (v/v) acetonitrile (MeCN), 0.25 M acetic acid (loading solution) and 10 µl of IMAC resin, previously equilibrated with the loading solution was added. After 45 min incubation at room temperature, the resin was washed four times with loading solution and the CoAlated peptides were eluted with 500 mM imidazole (pH 7.6) followed by 30% (v/v) MeCN/500 mM imidazole (pH 7.6). CoAlated peptides were acidified, dried and desalted with a C18 Stage tip containing 0.5 µl of Poros R3 resin (Applied Biosystems, U.K.). The solution was then partially dried down using a SpeedVac prior to analysis by mass spectrometry.
Mass spectrometry and data acquisition
Mass spectrometry data acquisition liquid chromatography was performed on a fully automated Ultimate U3000 Nano LC System (Dionex) fitted with a 100 µm × 2 cm PepMap100 C18 Nano-Trap column and a 75 μm × 25 cm reverse-phase PepMap100 C18 Nano-Trap column (Dionex). Peptides were separated using an acetonitrile gradient and sprayed directly via a nano-flow electrospray ionization source into the mass spectrometer (Orbitrap Velos, Thermo Scientific). The mass spectrometer was operated in a standard data-dependent mode, performed survey full scan (m/z = 350–1600) in the Orbitrap analyzer, with a resolution of 60 000 at m/z = 400, followed by MS/MS acquisitions of the 20 most intense ions in the LTQ ion trap. Maximum FTMS scan accumulation times were set at 250 ms and maximum ion trap MSn scan accumulation times were set at 200 ms. The Orbitrap measurements were internally calibrated using the lock mass of polydimethylcyclosiloxane at m/z 445.120025. Dynamic exclusion was set for 30 s with exclusion list of 500.
Data processing
LC–MS/MS raw data files were processed as standard samples using MaxQuant version 1.5.2.8, which incorporates the Andromeda search [27]. MaxQuant processed data were searched against a Uniprot — S. aureus (November 2015) database. Carbamidomethyl cysteine, acetyl N-terminal, N-ethylmaleimide cysteine, oxidation of methionines, CoAlation of cysteine with λ mass 338, 356 and 765 were set as variable modifications. For all data sets, the default parameters in MaxQuant were used, except MS/MS tolerance which was set at 0.6 Da and the second peptide ID was unselected.
Using MQ viewer, CoA_356 peptides were first visually checked. Those matched MS/MS spectra that did not have continuous 4 y or b ion series were checked manually.
Functional characterization of identified proteins
Gene ontology (GO) [28] terms describing the function(s) of the identified proteins were either extracted from UniProtKB (UniProt Release June 2017) or predicted using a protein domain-based function prediction pipeline [29,30]. The functions of the proteins were then classified into major functional categories and protein classes were based on the inferred GO terms.
Molecular dynamics simulations
A high-resolution (1.7 Å) X-ray crystal structure of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) complexed with NAD+, from S. aureus, was obtained from the Protein DataBank (PDBID: 3LVF). Missing residues were modeled with MODELER [31]. The molecular dynamics (MD) simulations were performed using the code GROMACS 4 [32]. To enhance the sampling, we used the Metadynamics algorithm as implemented the PLUMED plug-in [33,34]. The protein was described by the Amber99SB*-ILDN force field which includes the dihedral corrections of Best and Hummer, while CoA was parametrized with the general Amber force field (GAFF) and RESP charges derived from ab initio calculations at the Hartree–Fock level of theory [35,36]. The system was solvated with ∼19 000 tip3p water molecules and enclosed in a dodecahedron box with periodic boundary conditions for a total of more than 60 000 atoms. The van der Waals interactions were smoothly shifted to zero between 0.8 and 1.0 nm; the long-range electrostatic interactions were calculated by the particle mesh Ewald algorithm, with mesh spaced 0.12 nm, combined with a switch function for the direct space between 0.8 and 1.0 nm for better energy conservation [37,38]. Following an initial conjugate gradient optimization to relax the structure and remove possible atomic clashes, a brief NPT equilibration was run with a Berendsen thermostat and target pressure of 1 bar. The system evolved in the canonical ensemble with a time step of 2 fs and was coupled with a velocity-rescale thermostat to maintain the temperature at 300 K.
Statistical analysis
Where appropriate, values are given as means ± SEM. Graphs were produced and statistics were calculated using GraphPad Prism (version 6.07 for Windows, GraphPad Software, La Jolla, CA, U.S.A.; www.graphpad.com).
Results
Oxidizing agents induce strong protein CoAlation in bacteria
Bacteria employ a diverse range of molecular mechanisms to cope with ROS/reactive nitrogen species and to repair the resulting damage [39]. These include the production of antioxidant enzymes (superoxide dismutase, catalases and peroxiredoxins) and LMW thiols (glutathione, bacillithiol and mycothiol). While significant progress has been made to understand the antioxidant function of glutathione and to some extent bacillithiol and mycothiol, the role of the small thiol CoA in redox regulation in bacteria remains to be elucidated. This was mainly due to the lack of specific antibodies which can recognize CoA in various immunological assays and a reliable mass spectrometry-based protocol for identifying CoA-modified peptides in CoAlated proteins. The developed research tools and methodologies and the identification of extensive protein CoAlation in mammalian cells induced by oxidizing agents and metabolic stress prompted us to investigate the magnitude and relevance of this post-translational modification in bacteria [22]. To do so, we used both Gram-negative (E. coli) and Gram-positive (S. aureus and B. megaterium) bacteria, which have different expression profiles of LMW thiols. In Gram-negative bacteria and eukaryotes, glutathione is the major LMW thiol and antioxidant; however, it is absent in most Gram-positive bacteria. Instead, the differential expression of bacillithiol and mycothiol has been reported in different species of Gram-positive bacteria, where they function as a thiol redox buffer in the detoxification of ROS, toxins and antibiotics [40]. In contrast, CoA is a ubiquitous and highly expressed LMW thiol in all living cells, whose function has been mainly associated with the regulation of cellular metabolism and gene expression.
Initially, we examined the effect of H2O2 on protein CoAlation in E. coli, B. megaterium and S. aureus. Here, bacteria were grown to mid-log phase (OD600 = 0.7) in rich medium (LB medium for E. coli and S. aureus; NB3 medium for B. megaterium) at 37°C and then treated with and without 10 or 100 mM H2O2. After 30 min, cells were collected and bacterial protein extracts were prepared as described in the Experimental procedures. Separation of protein extracts under non-reducing conditions and Western blot analysis with anti-CoA antibody 1F10 revealed a weak immunoreactive signal in control samples and cells treated with 10 mM H2O2 (Figure 1A). However, exposure of cells to 100 mM H2O2 induced readily detectable protein CoAlation in all three bacterial species. These data indicate that bacteria can cope efficiently with oxidative stress induced by 10 mM H2O2, without engaging CoA in the antioxidant response. We also observed extensive protein CoAlation in cells treated with the disulfide stress inducer diamide at a concentration of 2 mM. Notably, the pattern of CoA-modified proteins in cells treated with H2O2 and diamide was similar except for several differentially CoAlated proteins (Figure 1A). Hypochlorous acid (HOCl) is produced by neutrophils to kill engulfed bacteria and is commonly used as an antimicrobial disinfectant [41]. The bactericidal effect of HOCl is associated with the production of various ROS. LMW thiols, such as GSH, protect bacteria from HOCl by direct interaction and the formation of less harmful substances. The treatment of bacteria with NaOCl was shown to induce the formation of mixed disulfides between LMW thiols and proteins, mediated by the disulfide exchange mechanism [25,42]. The treatment of exponentially growing bacteria with 100 µM NaOCl showed the strongest induction of protein CoAlation, when compared with control, H2O2 or diamide. The pattern of protein CoAlation differed significantly from that of H2O2- or diamide-treated cells. Ponceau staining of protein blots revealed that the treatment of cells with 100 µM NaOCl caused a significant change in the pattern of separated proteins, when compared with control and H2O2- or diamide-treated cells (Figure 1A).
Figure 1. Induction of protein CoAlation in bacteria by a panel of oxidizing agents.

Protein CoAlation in Gram-positive and Gram-negative bacteria is induced by different oxidizing agents (A) in a DTT-sensitive manner (B). E. coli, B. megaterium and S. aureus were grown to mid-log phase in rich medium at 37°C and then treated for 30 min with and without 10 mM or 100 mM H2O2, 2 mM diamide and 100 µM NaOCl. Cells were lysed as described in Experimental procedures and protein CoAlation examined by anti-CoA immunoblot. DTT (200 mM final) was added to protein extracts before SDS–PAGE analysis to demonstrate that the protein-CoA binding involves a reversible disulfide bond formation.
To demonstrate that the protein-CoA binding involves a reversible disulfide bond formation, the disulfide reducing agent DTT was added to protein extracts before SDS–PAGE analysis. As shown in Figure 1B, the presence of 200 mM DTT in the sample buffer efficiently abrogated immunoreactive signal in H2O2- and diamide-treated cells. In case of hypochlorite stress, the reduction of protein-CoA disulfide bonds was not complete and possibly required a higher DTT concentration in the sample buffer.
To find out whether protein CoAlation is a reversible post-translational modification, exponentially growing E. coli, B. megaterium and S. aureus were treated with 2 mM diamide for 30 min. Bacteria were harvested by centrifugation and then incubated in fresh LB or NB3 media for various periods of time to recover from the oxidative stress. As shown in Figure 2, diamide-induced protein CoAlation in E. coli was reversed to the level of untreated cells in a time-dependent manner. The reversibility of protein CoAlation in B. megaterium and S. aureus also occurred in a time-dependent manner, but did not reach baseline levels within 60 min.
Figure 2. Diamide-induced protein CoAlation in E. coli, B. megaterium and S. aureus is a reversible post-translational modification.
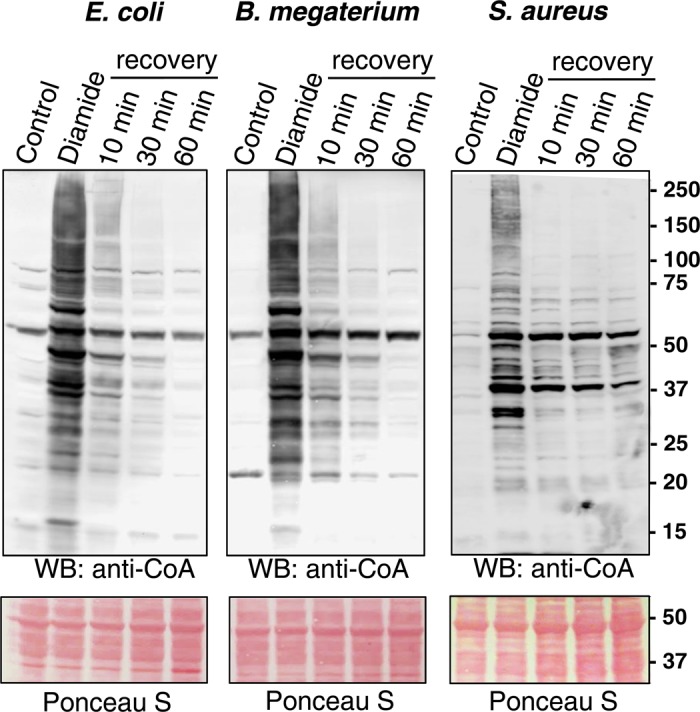
Exponentially growing bacteria were treated with 2 mM diamide for 30 min. The medium was then replaced with fresh media without the oxidant and cells were incubated for the indicated times. Protein CoAlation was examined by anti-CoA immunoblot.
Induction of protein CoAlation by glucose deprivation
Bacteria have evolved elaborate strategies that enable them to adapt to challenging growth environments. When the nutrient supply becomes limiting, bacteria employ general starvation-response mechanisms, such as the stringent response and carbon catabolite repression, which are associated with ROS production and oxidative stress [43]. We were interested to examine whether under nutrient deprivation CoA is also used for S-thiolation of redox-sensitive cysteine residues, resulting in the formation of mixed disulfides with proteins. Taking into account that the examined bacterial species use carbon catabolism for energy generation, we employed the model of glucose starvation.
In the present study, all three types of bacteria were cultured in nutrient-rich medium until OD600 = 0.7 and then transferred in the medium lacking glucose or any other source of carbohydrates. As shown in Figure 3, protein CoAlation was at a very low level in bacteria cultured in nutrient-rich medium, but strongly induced under the condition of glucose starvation for 60 and 120 min. The pattern of CoAlated proteins in glucose-starved E. coli and B. megaterium was similar, but differed significantly from that in S. aureus. Comparing the pattern of CoA-modified proteins induced by glucose starvation and the treatment with oxidizing agents revealed little similarity in all three types of examined bacteria, indicating the involvement of different redox-sensing and responding strategies.
Figure 3. Induction of protein CoAlation by metabolic stress.
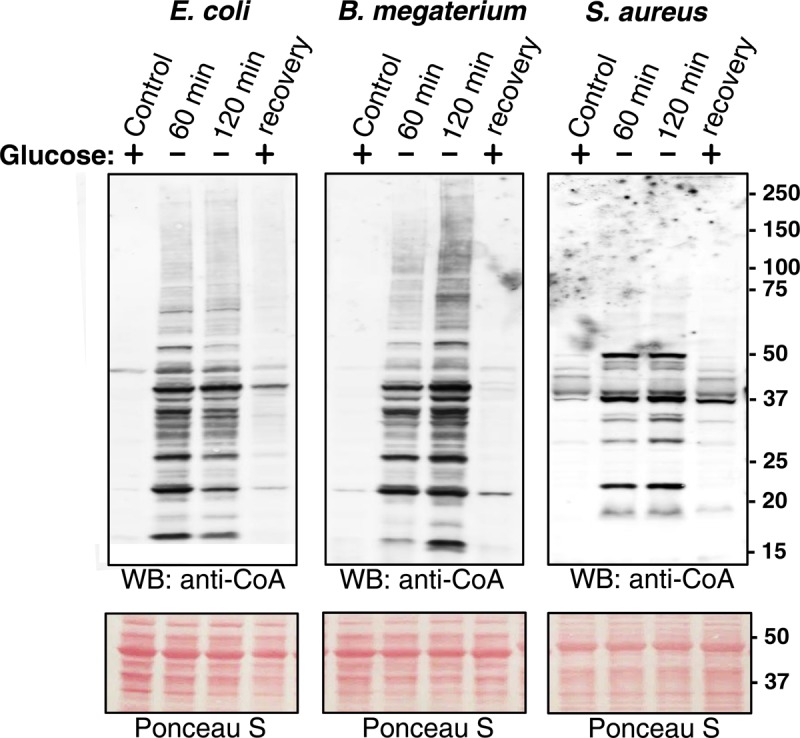
Glucose depravation induces protein CoAlation in E. coli, B. megaterium and S. aureus, which is reversed by the re-addition of glucose. E. coli, B. megaterium and S. aureus were grown to mid-log phase in rich medium at 37°C and then transferred and cultured in the medium lacking glucose or any other source of carbohydrates for the indicated times. The cultures of glucose-starved bacteria were then supplemented with 20 mM glucose and incubated at 37°C for 30 min. Protein CoAlation in total protein extracts was examined by anti-CoA immunoblot.
We then examined whether protein CoAlation induced by glucose starvation can be reversed with the re-addition of glucose to starved bacterial cultures. The results presented in Figure 3 clearly indicate that supplementing cultures of glucose-starved bacteria with glucose for 30 min resulted in near complete deCoAlation of CoA-modified proteins.
Mass spectrometry-based identification of CoAlated proteins in diamide-treated S. aureus
Extensive protein CoAlation which we observed in Gram-negative and Gram-positive bacteria in response to oxidative and metabolic stress encouraged us to identify CoA-modified proteins using the developed methodology [22]. Our efforts were focused on determining the identity of CoAlated proteins in S. aureus under diamide-induced disulfide stress. Exponentially growing S. aureus were treated with 2 mM diamide for 30 min and protein extracts were prepared as described in Experimental procedures. In brief, to prevent in vitro modification of protein thiols by free CoA, 25 mM NEM was added to the lysis buffer. Extracted proteins were digested with Lys C/trypsin in the presence of IAM and CoAlated peptides were immunoprecipitated with anti-CoA antibody. Then, immune complexes were incubated with Nudix 7 hydrolase to remove the ADP moiety of CoA and to produce a distinctive MS/MS fragmentation signature of Cys + 356, corresponding to covalently attached 4PP (4′-phosphopantetheine). Representative MS/MS spectrum of a cysteine-containing peptide from SaGAPDH is shown in Figure 4A. In total, the LC–MS/MS analysis revealed the identity of 440 CoAlated cysteine-containing peptides which correspond to 356 proteins in the S. aureus proteome (Table 1). Bioinformatic pathway analysis revealed that a large number of CoAlated proteins are involved in major metabolic pathways, regulation of transcription, protein synthesis and stress response (Figure 4B). Among identified proteins, we found those which use CoA as the covalent intermediate in catalytic reactions or function as CoA-regulated proteins. These include succinate-CoA ligase, acyl-CoA ligase, HMG-CoA synthase, an acetyl-CoA carboxylase and acyl-CoA dehydrogenase.
Figure 4. Development of methodology and the identification of CoAlated proteins.

(A) Strategy for the identification of CoA-modified proteins S. aureus in response to diamide. (B) Pie chart showing the major functional categories of the proteins that were identified to be CoAlated in diamide-treated S. aureus.
Table 1. Proteomic identification of CoAlated proteins in S. aureus treated with diamide. CoAlated peptides identified by MS/MS analysis and corresponding proteins are shown. Perspective CoA-modified cysteine residues within the identified peptides are marked by asterisks.
| Gene name | Protein name | MW (kDa) | Sequence | Score |
|---|---|---|---|---|
| HMPREF0769_11633 | Resolvase, N-terminal domain protein | 21.845 | LNAYGC*EK | 72.531 |
| MW2460 | MW2460 protein | 63.757 | LC*EDVAVYNHQIEK | 97.291 |
| ybaK | Cys-tRNA(Pro)/Cys-tRNA(Cys) deacylase | 17.89 | GGC*SPVGMK | 41.689 |
| pyrE | Orotate phosphoribosyltransferase | 22.057 | SPIYC*DNR | 105.25 |
| binL | BinL protein | 22.491 | LNTHGC*EK | 98.156 |
| SAOUHSC_02733 | Membrane protein, putative | 67.572 | NVNVC*TIPFK | 116.98 |
| SAUSA300_1090 | Pseudouridine synthase | 34.634 | DYTLVEC*QLETGR | 88.803 |
| SAKOR_01553 | Uncharacterized protein | 50.22 | LIEESPC*AALTEER | 106.87 |
| pyrG | Cytidine 5′-triphosphate synthase | 59.991 | ESVIEC*R | 131.12 |
| pyrG | Cytidine 5′-triphosphate synthase | 59.991 | IALFC*DINK | 153.34 |
| pyrG | Cytidine 5′-triphosphate synthase | 59.991 | LGLYPC*SIK | 86.114 |
| yisK | Fumarylacetoacetate hydrolase family protein | 33.113 | SLTGGC*PMGPYIVTK | 108.45 |
| deoC2 | Deoxyribose-phosphate aldolase 2 | 23.341 | SVC*VNPTHVK | 99.941 |
| polC | DNA polymerase III PolC-type | 162.69 | NC*GFDIDK | 94.767 |
| AYM28_04315 | Phage protein | 10.633 | ENYFC*DR | 58.132 |
| V070_02571 | Uncharacterized protein | 13.806 | SC*VEVAR | 83.647 |
| pflB | Formate acetyltransferase | 84.861 | AAC*EAYGYELDEETEK | 166.57 |
| pflB | Formate acetyltransferase | 84.861 | IPYDC*C*K | 101.46 |
| rpmF | 50S ribosomal protein L32 | 6.48 | NC*GSYNGEEVAAK | 159.4 |
| purB | Adenylosuccinate lyase | 49.603 | EELDEC*FDPK | 109.15 |
| pheT | Phenylalanine–tRNA ligase β subunit | 88.901 | AC*YLLQTYANGK | 110.22 |
| SAZ172_1072 | Uncharacterized protein | 8.7688 | NAGKFEETPC*EFVDGSKGVR | 236.03 |
| ddl | d-alanine–d-alanine ligase | 40.23 | ATDC*SGLVR | 136.81 |
| ddl | d-alanine–d-alanine ligase | 40.23 | C*NNEAELK | 100.09 |
| HMPREF0769_11996 | Response regulator receiver domain protein | 27.021 | KDGIDVC*K | 104.94 |
| gapA2 | Glyceraldehyde-3-phosphate dehydrogenase 2 | 36.979 | SC*NESIIPTSTGAAK | 122.83 |
| N/A | Truncated catalase-like protein | 38.88 | GVGIENIC*PFSR | 173.19 |
| mvaD | Mevalonate diphosphate decarboxylase | 36.831 | EAGYPC*YFTMDAGPNVK | 116.79 |
| rpoC | DNA-directed RNA polymerase subunit β′ | 135.41 | C*GVEVTK | 114.89 |
| rpoC | DNA-directed RNA polymerase subunit β′ | 135.41 | DGLFC*ER | 87.806 |
| rpoC | DNA-directed RNA polymerase subunit β′ | 135.41 | DWEC*.SC*.GK | 121.61 |
| rpoC | DNA-directed RNA polymerase subunit β′ | 135.41 | MYQC*GLPK | 123.86 |
| gluD | NAD-specific glutamate dehydrogenase | 45.76 | C*GIVNLPYGGGK | 135.81 |
| gluD | NAD-specific glutamate dehydrogenase | 45.76 | GGIVC*DPR | 110.53 |
| SAKOR_01005 | Phosphoenolpyruvate–protein phosphotransferase | 63.276 | LC*LAQQDIFR | 135.02 |
| SAKOR_02109 | Uncharacterized protein | 12.497 | TAETNYFWLNC*GYNR | 141.34 |
| HMPREF0776_2410 | ABC transporter, ATP-binding protein | 13.659 | FTEGNC*YGLIGANGAGK | 128.85 |
| SAOUHSC_00756 | Uncharacterized protein | 41.797 | IAELC*HK | 96.034 |
| yloU | General stress protein, Gls24 family | 13.345 | AVEC*YGIVGMASR | 161.48 |
| rap | 50S ribosomal protein L2 | 30.026 | MILSTC*R | 128.6 |
| cmk | Cytidylate kinase | 24.595 | GQC*VILDNEDVTDFLR | 91.622 |
| sarS | HTH-type transcriptional regulator SarS | 29.889 | KIVSDLC*YK | 205.53 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | DWFDGKPC*ER | 108.32 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | VVTYC*TGGIR | 139.78 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | YINCANPEC*NK | 111.07 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | YLGAC*SYDCAK | 92.151 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | YLGACSYDC*AK | 104.7 |
| SAR2771 | UPF0176 protein SAR2771 | 36.938 | YTTIDDPEQFAQDHLAFC*K | 97.823 |
| HMPREF0769_10405 | Metallo-β-lactamase domain protein | 29.57 | EVLLC*DTDK | 190.66 |
| ctsR | Transcriptional regulator CtsR | 17.841 | FDC*VPSQLNYVIK | 189.52 |
| rplN | 50S ribosomal protein L14 | 13.135 | FDENAC*VIIR | 96.464 |
| rplN | 50S ribosomal protein L14 | 13.135 | TANIGDVIVC*TVK | 196.01 |
| RK97_03585 | Nitrogen fixation protein NifU | 16.631 | C*ATLAWK | 95.81 |
| RK97_03585 | Nitrogen fixation protein NifU | 16.631 | GVLDNGSMTVDMNNPTC*GDR | 109.28 |
| proC | Pyrroline-5-carboxylate reductase | 29.825 | QQLEC*QNPVAR | 108.23 |
| aspS | Aspartate–tRNA ligase | 55.836 | C*FRDEDLR | 94.767 |
| infB | Translation initiation factor IF-2 | 31.699 | VGTIAGC*YVTEGK | 132.15 |
| pyrB | Aspartate carbamoyltransferase | 33.257 | GESLYDTC*K | 130.69 |
| tsaD | tRNA N6-adenosine threonylcarbamoyltransferase | 37.069 | QSLADQC*K | 72.289 |
| mshB | Bacillithiol biosynthesis deacetylase BshB2 | 24.894 | ERELEEAC*K | 158.76 |
| SAOUHSC_00547 | Uncharacterized protein | 58.418 | QC*QDISQYIENK | 94.309 |
| murI | Glutamate racemase | 29.702 | C*PYGPRPGEQVK | 103.13 |
| clpC | ATP-dependent Clp protease | 91.141 | GELQC*IGATTLDEYRK | 190.1 |
| SAV0414 | Uncharacterized protein | 83.287 | C*AIVTDLDEQAIPSEHR | 77.668 |
| SAV0414 | Uncharacterized protein | 83.287 | IVEFEAC*R | 149.94 |
| SAV0414 | Uncharacterized protein | 83.287 | SNLNFC*INENYDK | 231.38 |
| murE | UDP-N-acetylmuramoyl-l-alanyl-d-glutamate–l-lysine ligase | 54.104 | FC*QNVADQGCK | 82.515 |
| SAOUHSC_02158 | Uncharacterized protein | 48.119 | IAFSC*VEK | 131.42 |
| ribH | 6,7-Dimethyl-8-ribityllumazine synthase | 16.41 | GATSHYDYVC*NEVAK | 54.094 |
| nadE | NH(3)-dependent NAD(+) synthetase | 30.682 | EEGIDC*TFIAVK | 139.77 |
| glyA | Serine hydroxymethyltransferase | 45.172 | EAEETLDSVGITC*NK | 114.55 |
| glyA | Serine hydroxymethyltransferase | 45.172 | GGMILC*K | 85.212 |
| AYM28_08455 | Integral membrane protein | 36.972 | YSYIC*EK | 125.75 |
| ble | Bleomycin resistance protein | 14.922 | SIGFYC*DK | 117.25 |
| SAOUHSC_01732 | Uncharacterized protein | 12.768 | KEGQGC*ISLK | 92.247 |
| typA | GTP-binding protein TypA | 37.865 | EIDGVMC*EPFER | 191.89 |
| secA | Protein translocase subunit SecA | 11.664 | NDDC*.PC*.GSGK | 70.68 |
| vraX | Protein VraX | 6.5003 | C*DDSFSDTEIFK | 128.06 |
| AYM28_07645 | YqiW-like protein | 16.014 | DAFDENC*K | 129 |
| AYM28_05325 | UPF0738 protein AYM22_05325 | 13.529 | IKDDILYC*YTEDSIK | 108.28 |
| dltA | d-alanine–poly(phosphoribitol) ligase subunit 1 | 54.67 | AGC*GYVPVDTSIPEDR | 167.93 |
| SAOUHSC_02613 | Uncharacterized protein | 25.225 | AVC*GFSK | 90.601 |
| SAOUHSC_01744 | Uncharacterized protein | 85.61 | VLNDQC*PTSVK | 153.41 |
| AYM28_02495 | GIY-YIG catalytic domain protein | 9.1542 | C*SDGSLYTGYAK | 114.31 |
| tagF_3 | CDP-glycerol–poly(glycerophosphate) glycerophosphotransferase | 66.074 | IDGNQFVC*R | 111.5 |
| mqo1 | Probable malate : quinone oxidoreductase 1 | 54.785 | C*TNQEVIDR | 135.04 |
| mqo1 | Probable malate : quinone oxidoreductase 1 | 54.785 | DGTVDC*SK | 147.3 |
| N/A | Putative uncharacterized protein | 7.9711 | GEC*DDKWEGLYSK | 104.76 |
| gnd | 6-Phosphogluconate dehydrogenase | 51.802 | DGASC*VTYIGPNGAGHYVK | 98.76 |
| gnd | 6-Phosphogluconate dehydrogenase | 51.802 | IC*SYAQGFAQMR | 155.33 |
| alr1 | Alanine racemase 1 | 42.823 | VC*MDQTIVK | 114.72 |
| SAOUHSC_00696 | Uncharacterized protein | 34.777 | C*GIILPSK | 110.41 |
| ychF | Ribosome-binding ATPase YchF | 13.516 | EVDAIC*QVVR | 196.02 |
| tsf | Elongation factor Ts | 32.511 | TGAGMMDC*K | 78.324 |
| xpt | Xanthine phosphoribosyltransferase | 20.884 | LEEAGLTVSSLC*K | 79.332 |
| mqo2 | Probable malate : quinone oxidoreductase 2 | 55.998 | EGC*MNHLR | 122.94 |
| gmk | Guanylate kinase | 24.037 | IQC*IVEAEHLK | 100.11 |
| HMPREF0776_1272 | Mu transposase domain protein | 32.351 | NDTNWPVC*GIPEK | 69.314 |
| MW1126 | Ribosome biogenesis GTPase A | 33.403 | IGNYC*FDIFK | 65.809 |
| fabG | β-Ketoacyl-ACP reductase | 24.616 | GVFNC*IQK | 121.73 |
| SAKOR_02572 | Transcriptional regulator, TetR family protein | 22.356 | ALLQC*IEAGNNK | 89.484 |
| SAKOR_02572 | Transcriptional regulator, TetR family protein | 22.356 | SDLC*YYVIQR | 129.82 |
| nth | Endonuclease III | 25.668 | LC*SVIPR | 78.264 |
| SA0511 | Uncharacterized epimerase/dehydratase | 36.052 | QGIANSWPDSIDTSC*SR | 155.06 |
| SA0511 | Uncharacterized epimerase/dehydratase | 36.052 | VAGELLC*QYYFK | 119 |
| tagF_2 | Putative teichoic acid biosynthesis protein | 45.954 | IC*QTLFK | 66.056 |
| mnmG | tRNA uridine 5-carboxymethylaminomethyl modification enzyme MnmG | 69.507 | YC*PSIEDK | 131.09 |
| rpoA | DNA-directed RNA polymerase subunit alpha | 35.011 | SYNC*LKR | 61.593 |
| tagB | Teichoic acid biosynthesis protein B | 18.212 | C*LPGYTLINK | 116.74 |
| SAOUHSC_01812 | Uncharacterized protein | 35.096 | C*IEDNDTIIIHR | 141.34 |
| SAV1710 | Putative universal stress protein SAV1710 | 18.38 | HAPC*DVLVVR | 131.83 |
| srtA | LPXTG-specific sortase A | 23.541 | QLTLITC*DDYNEK | 152.32 |
| ST398NM01_1322 | Uncharacterized protein | 28.118 | SELEELC*K | 118.23 |
| recG | ATP-dependent DNA helicase RecG | 78.343 | C*IFFNQPYLK | 125.72 |
| SAKOR_00906 | Oligopeptide transport ATP-binding protein oppF | 16.074 | GETLGLVGESGC*GK | 102.03 |
| murD | MURD | 29.82 | NQTEEDYLIC*NYHQR | 92.151 |
| SA0315 | Uncharacterized protein | 20.923 | C*DAPMEVNK | 73.435 |
| HMPREF0776_0655 | 3-Demethylubiquinone-9 3-methyltransferase domain protein | 16.768 | VLC*SDSFGR | 132.03 |
| SAOUHSC_02324 | Uncharacterized protein | 24.634 | QFFTEWNC*HD | 141.58 |
| rnc | Ribonuclease III | 27.922 | ATIVC*EPSLVIFANK | 95.273 |
| topA | DNA topoisomerase I | 79.07 | C*NDGDVVER | 92.151 |
| topA | DNA topoisomerase I | 79.07 | C*NQYLVENK | 126.12 |
| rpmJ | 50S ribosomal protein L36 | 4.3 | VMVIC*ENPK | 183.99 |
| SAZ172_2022 | Uncharacterized protein | 24.952 | TVIIDC*VTK | 109.44 |
| N/A | Uncharacterized protein | 13.573 | GIDLNMGC*PVANVAK | 167.93 |
| polC_2 | DNA polymerase III PolC-type | 4.1918 | TSIC*SVGMVK | 67.952 |
| ackA | Acetate kinase | 44.056 | IISC*HIGNGASIAAIDGGK | 50.404 |
| SAOUHSC_02264 | Accessory gene regulator protein C | 13.754 | C*ADDIPR | 101.46 |
| rnpA | Ribonuclease P protein component | 11.093 | QFVVYTC*NNK | 166.48 |
| rpmG | 50S ribosomal protein L33 | 5.87 | VNVTLAC*TEC*GDR | 134.08 |
| rpmG | 50S ribosomal protein L33 | 5.87 | VNVTLAC*TEC*GDR | 100.01 |
| N/A | mRNA interferase PemK | 20.91 | FLC*DSLK | 100.25 |
| graR | Response regulator protein GraR | 26.066 | YDGFYWC*R | 123.79 |
| SAKOR_02101 | Uncharacterized protein | 42.899 | VVVLGC*PAEEGGENGSAK | 95.755 |
| SAKOR_02241 | Molybdopterin biosynthesis MoeB protein | 37.894 | YATLC*GR | 87.806 |
| cshB | DEAD-box ATP-dependent RNA helicase CshB | 51.064 | C*NAQPQLIIGTPTR | 86.016 |
| SACOL0939 | NifU domain protein | 7.9651 | DGGDC*SLIDVEDGIVK | 163.45 |
| SAKOR_00091 | Ornithine cyclodeaminase family protein | 37.776 | VVVDDWSQC*NR | 91.592 |
| SA2277 | Uncharacterized protein | 50.847 | TILC*ALDVR | 107.34 |
| glnA | Glutamine synthetase | 50.854 | GFTAVC*NPLVNSYK | 229.96 |
| glnA | Glutamine synthetase | 50.854 | LIC*DVYK | 111.61 |
| glnA | Glutamine synthetase | 50.854 | LVPGYEAPC*YIAWSGK | 134.32 |
| glnA | Glutamine synthetase | 50.854 | YADAVTAC*DNIQTFK | 154.34 |
| whiA | Putative sporulation transcription regulator WhiA | 35.868 | LVNC*ETANLNK | 154.72 |
| whiA | Putative sporulation transcription regulator WhiA | 35.868 | NNIYIC*R | 157.91 |
| SAKOR_00683 | Transcriptional regulator, MarR family protein | 17.089 | EQLC*FSLYNAQR | 207.66 |
| menB | 1,4-Dihydroxy-2-naphthoyl-CoA synthase | 30.411 | EIWYLC*R | 154.12 |
| menB | 1,4-Dihydroxy-2-naphthoyl-CoA synthase | 30.411 | VEDETVQWC*K | 185.57 |
| perR | Peroxide-responsive repressor PerR | 17.183 | MEIYGVC*K | 120.45 |
| SAOUHSC_00462 | Uncharacterized protein | 29.281 | GLSYEEVC*EQTTK | 170 |
| tpx | Probable thiol peroxidase | 18.005 | LISVVPSIDTGVC*DQQTR | 81.656 |
| SAOUHSC_00960 | Uncharacterized protein | 57.927 | VTMTDYC*YR | 87.216 |
| ybhF_3 | ABC transporter ATP-binding protein | 32.952 | LEDIELIC*DR | 121.42 |
| BN1321_100031 | MutT/NUDIX family protein | 15.067 | C*VCLVEETADK | 193.28 |
| SAKOR_00705 | Uncharacterized protein | 14.205 | YDEVTIYC*K | 153.74 |
| SAOUHSC_01677 | Uncharacterized protein | 25.002 | TAQTVTDLRPAGIIFC*ENER | 111.77 |
| SAKOR_00374 | Phosphoglycerate mutase family protein | 22.777 | SADDLC*DYFK | 121.56 |
| sufB | Fe–S cluster assembly protein SufB | 52.531 | YPNC*VLLGEGAK | 121.45 |
| ccpA | Catabolite control protein A | 12.161 | NGLQLGDTLNC*SGAESYK | 140.07 |
| SAV0485 | Signal peptidase II-like protein | 23.935 | DAENALILC*K | 85.672 |
| SAV0485 | Signal peptidase II-like protein | 23.935 | LVNC*EYTLDK | 97.223 |
| sucD | Succinate-CoA ligase [ADP-forming] subunit alpha | 31.542 | LVGPNC*PGVITADEC*K | 249.56 |
| sucD | Succinate-CoA ligase [ADP-forming] subunit alpha | 31.542 | LVGPNC*PGVITADEC*K | 179.85 |
| sucD | Succinate-CoA ligase [ADP-forming] subunit alpha | 31.542 | TLNSC*GVK | 91.469 |
| SAKOR_01205 | Transcriptional regulator, GntR family protein | 26.976 | EQSNHNIC*YADTEIEAVNYEPR | 97.621 |
| SAKOR_01205 | Transcriptional regulator, GntR family protein | 26.976 | TADGEPVVYC*LDK | 129.88 |
| SAOUHSC_02755 | Uncharacterized protein | 39.192 | YGC*ALAIEVLK | 79.283 |
| ugtP | Processive diacylglycerol β-glucosyltransferase | 44.547 | SANAQVVMIC*GK | 213.99 |
| ugtP | Processive diacylglycerol β-glucosyltransferase | 44.547 | YATQTIC*R | 122.18 |
| MW0660 | MW0660 protein | 28.342 | TGC*SASTIR | 104.07 |
| MW0924 | Uncharacterized protein | 18.511 | SC*VDATYR | 89.171 |
| MW0924 | Uncharacterized protein | 18.511 | VAGC*IISYSGENELK | 145.17 |
| MW0924 | Uncharacterized protein | 18.511 | WSLNC*DINNEAALK | 126.04 |
| SAOUHSC_01973 | Uncharacterized protein | 35.778 | EAEILC*YIDNIDAR | 77.505 |
| SAOUHSC_01973 | Uncharacterized protein | 35.778 | SIC*DIYPLLNK | 112.44 |
| upp | Uracil phosphoribosyltransferase | 23.05 | FMC*LIAAPEGVEK | 131.96 |
| SAOUHSC_02727 | Uncharacterized protein | 22.28 | C*IYVQPHSYTIENQQQNK | 119.37 |
| MW0535 | MW0535 protein | 29.857 | AGEVYEASNAQYFVVDPVMVC*K | 55.621 |
| HMPREF0776_0347 | CHAP domain protein | 12.753 | NLYTSGQC*TYYVFDR | 78.615 |
| SAOUHSC_02574 | Uncharacterized protein | 40.742 | SCLNDC*YDK | 139.74 |
| SAV2378 | Uncharacterized protein | 13 | C*ANEEER | 63.624 |
| tagH_1 | Teichoic acids export ATP-binding protein TagH | 29.762 | MLC*MGFK | 101.28 |
| CH52_06005 | 2-Dehydropantoate 2-reductase | 32.358 | QLLLDGC*R | 82.279 |
| glyS | Glycine–tRNA ligase | 53.62 | IIDDEGIVC*PVSK | 195.45 |
| glyS | Glycine–tRNA ligase | 53.62 | YIPYC*IEPSLGADR | 107.98 |
| tpiA | Triosephosphate isomerase (TIM) (TPI) | 27.261 | HGMTPIIC*VGETDEER | 135.95 |
| tpiA | Triosephosphate isomerase (TIM) (TPI) | 27.261 | SSTSEDANEMC*AFVR | 142.93 |
| SAOUHSC_01716 | Uncharacterized protein | 47.655 | C*TLSNHMTAR | 60.901 |
| SAOUHSC_01716 | Uncharacterized protein | 47.655 | TGATGIIVADPLIIETC*K | 92.8 |
| vraS_1 | Histidine kinase (nitrate/nitrite sensor protein) (EC 2.7.3.-) (two-component sensor protein) | 41.88 | ALQEC*INNVK | 101.61 |
| vicR | DNA-binding response regulator (PhoP family transcriptional regulator) | 27.192 | DGMEVC*R | 117.81 |
| SAOUHSC_02218 | Conserved hypothetical phage protein | 11.1 | EISNGHC*NYWK | 144.51 |
| SAOUHSC_01696 | Uncharacterized protein | 22.463 | TIDC*LNYYNYSDER | 154.72 |
| hsdS_2 | Restriction endonuclease subunit S | 23.781 | IPC*LTEQDK | 102.87 |
| sufA | Chaperone involved in Fe–S cluster assembly | 12.485 | VAGNPENC* | 106.16 |
| SAKOR_00641 | Ferrichrome transport ATP-binding protein fhuC | 29.496 | TGKPLLVTYDLC*R | 90.755 |
| SAKOR_00641 | Ferrichrome transport ATP-binding protein fhuC | 29.496 | VTSIIGPNGC*GK | 164.66 |
| HMPREF0769_12132 | ROK family protein | 35.077 | IILAADVGGTTC*K | 103.13 |
| nadK | NAD kinase | 30.769 | GDGLC*VSTPSGSTAYNK | 102.24 |
| mraZ | Transcriptional regulator MraZ | 17.237 | EC*TVIGVSNR | 109.16 |
| yutD | Uncharacterized protein conserved in bacteria | 15.401 | EC*FNEEQFIAR15.401 | 149.35 |
| SAZ172_0295 | Uncharacterized protein | 15.751 | C*FEEEDFER | 164.48 |
| SAZ172_0295 | Uncharacterized protein | 15.751 | YIDC*LEVGPTLSTK | 170 |
| gatA | Glutamyl-tRNA(Gln) amidotransferase subunit A | 52.82 | DNIITNGLETTC*ASK | 128.66 |
| MW0675 | MW0675 protein | 22.322 | YHSLIADGATFPNC*LK | 80.905 |
| rpsR | 30S ribosomal protein S18 | 9.3098 | VC*YFTANGITHIDYK | 100.69 |
| fusA | Elongation factor G | 64.009 | DTGTGDTLC*GEK | 188 |
| fusA | Elongation factor G | 64.009 | KC*DPVILEPMMK | 134.61 |
| fusA | Elongation factor G | 64.009 | KEFNVEC*NVGAPMVSYR | 171.39 |
| fusA | Elongation factor G | 64.009 | QATTNVEFYPVLC*GTAFK | 81.594 |
| rocD2 | Ornithine aminotransferase 2 | 43.417 | EEGLLC*K | 133.86 |
| hutU | Urocanate hydratase | 60.632 | GLSIEC*K | 80.231 |
| narH | NarH protein | 59.446 | RDEDGIVLVDQDAC*R | 95.988 |
| SAOUHSC_00882 | Uncharacterized protein | 15.517 | LTIIDPHETFC*QR | 93.839 |
| SAOUHSC_02811 | Uncharacterized protein | 27.165 | EC*ATEITEVEDK | 115.68 |
| NWMN_2186 | Acyl-CoA dehydrogenase-related protein | 34.413 | METLLLC*AR | 163.9 |
| fabF | 3-Oxoacyl-[acyl-carrier protein] synthase 2 | 42.433 | ALSTNDDIETAC*R | 94.61 |
| fabF | 3-Oxoacyl-[acyl-carrier protein] synthase 2 | 42.433 | GPNGATVTAC*ATGTNSIGEAFK | 70.501 |
| N/A | Putative uncharacterized protein | 24.022 | VDMIAC*EDTR | 92.295 |
| ppaC | Probable manganese-dependent inorganic pyrophosphatase | 34.068 | AEPVGC*TATILYK | 136.26 |
| ppaC | Probable manganese-dependent inorganic pyrophosphatase | 34.068 | IANFETAGPLC*YR | 229.02 |
| ppaC | Probable manganese-dependent inorganic pyrophosphatase | 34.068 | SPTC*TQQDVK | 163.33 |
| AYM28_13750 | Nicotianamine synthase | 31.096 | SLQYITAQC*VK | 120.21 |
| HMPREF0769_12162 | Transketolase, pyridine binding domain protein | 36.033 | SNNDWQC*PLTIR | 110.57 |
| nos | Nitric oxide synthase oxygenase | 41.71 | EC*HYETQIINK | 55.899 |
| nos | Nitric oxide synthase oxygenase | 41.71 | YAGYDNC*GDPAEKEVTR | 147.84 |
| SAKOR_00998 | Hydroxymethylpyrimidine transport ATP-binding protein | 53.302 | VLLLGPSGC*GK | 54.898 |
| SAOUHSC_01872 | Uncharacterized protein | 46.176 | C*SQFVYK | 68.657 |
| SAV2122 | Putative aldehyde dehydrogenase SAV2122 | 51.968 | VVNNTGQVC*TAGTR | 225.69 |
| SAOUHSC_02064 | Phi ETA orf 25-like protein | 15.401 | DVNLTWIC*K | 79.089 |
| HUNSC491_pPR9_p11 | ATP-binding protein p271 (ATP-binding protein, putative) | 7.4555 | YQYIGIC*YGQPGVGK | 91.065 |
| AYM28_02415 | Acetyltransferase (GNAT) family protein | 19.908 | AQEYSTVVVDHC*FDYFEK | 87.963 |
| accD | Acetyl-coenzyme A carboxylase carboxyl transferase subunit β | 31.52 | IIDYC*TENR | 143.42 |
| ldh2 | l-lactate dehydrogenase 2 (l-LDH 2) | 34.42 | AGEYEDC*KDADLVVITAGAPQKPGETR | 129.82 |
| gltX | Glutamate–tRNA ligase | 18.695 | C*YMTEEELEAER | 204.18 |
| srpF | Alpha-helical coiled-coil protein | 19.257 | TYVC*EDMSK | 193.22 |
| mnmA | tRNA-specific 2-thiouridylase MnmA | 42.15 | DSTGIC*FIGEK | 84.173 |
| mnmA | tRNA-specific 2-thiouridylase MnmA | 42.15 | TPNPDVMC*NK | 145.52 |
| V070_01284 | Uncharacterized protein | 48.871 | LPYTLC*YISR | 145.61 |
| tmk | Uncharacterized protein | 51.081 | AQLIEC*LEK | 79.886 |
| trxA_1 | Thiol reductase thioredoxin | 11.454 | IDLNFYPQFC*K | 83.204 |
| pdxT | Pyridoxal 5′-phosphate synthase subunit PdxT | 20.63 | VGQGVDILC*K | 126.71 |
| mcsB | Protein-arginine kinase | 38.61 | SLGILQNC*R | 63.694 |
| ydaG | General stress protein 26 | 15.886 | EDPELC*VLR | 152.7 |
| HMPREF0769_12370 | SWIM zinc finger domain protein | 15.906 | GFNYYQSEC*VINLK | 157.73 |
| HMPREF0769_10247 | Oxidoreductase, FAD-binding protein | 42.831 | AFLANKPEIYIC*GGTK | 107.15 |
| tarI1 | Ribitol-5-phosphate cytidylyltransferase 1 | 26.656 | SILSDAC*K | 122.69 |
| SACOL2177 | Zinc-type alcohol dehydrogenase-like protein | 32.773 | QETTEWC*EK | 218.02 |
| SAOUHSC_02146 | Uncharacterized protein | 40.354 | ESGC*TVFQGK | 93.345 |
| SAOUHSC_02146 | Uncharacterized protein | 40.354 | LILENC*R | 125.68 |
| SACOL2396 | Uroporphyrinogen III methylase SirB, putative | 36.29 | INDC*IVEAAR | 113.37 |
| glpK | Glycerol kinase | 55.625 | ATLESLC*YQTR | 158.91 |
| glpK | Glycerol kinase | 55.625 | QTQSIC*SELKQQGYEQTFR | 125.84 |
| HMPREF3211_00337 | Methyltransferase domain protein | 21.763 | ALDIGC*GSGLLVEK | 55.031 |
| tarJ | Ribulose-5-phosphate reductase 1 | 38.451 | IPEGLTFDHAFEC*VGGR | 60.968 |
| MW2550 | MW2550 protein | 29.096 | LLIMC*GK | 113.41 |
| gpmI | 2,3-Bisphosphoglycerate-independent phosphoglycerate mutase | 56.423 | AIEAVDEC*LGEVVDK | 144.75 |
| SAR1875 | Putative membrane protein insertion efficiency factor | 8.9865 | FYPTC*SEYTR | 109.95 |
| serS | Serine–tRNA ligase | 48.639 | EISSC*.SNC*.TDFQAR | 122.29 |
| serS | Serine–tRNA ligase | 48.639 | FTGQSAC*FR | 123.26 |
| serS | Serine–tRNA ligase | 48.639 | MTGILC*R | 123.21 |
| serS | Serine–tRNA ligase | 48.639 | VILC*TGDIGFSASK | 124.45 |
| MW2545 | MW2545 protein | 25.27 | GC*TLILDEAK | 83.204 |
| femB | Aminoacyltransferase FemB | 49.675 | YLQQHQC*LYVK | 83.53 |
| mfd | Transcription-repair-coupling factor | 134.3 | LLC*GDVGYGK | 84.605 |
| N/A | Kanamycin nucleotidyl transferase protein | 27 | IC*YTTSASVLTEAVK | 119.62 |
| sdhA | SdhA protein | 65.502 | EIFDVC*INQK | 265.75 |
| sdhA | SdhA protein | 65.502 | GLFAAGEC*DFSQHGGNR | 110.77 |
| HMPREF0769_12639 | PHP domain protein | 8.9913 | ASLQVAC*ENGK | 121.95 |
| SAZ172_1861 | Ribosomal large subunit pseudouridine synthase D-like protein | 31.387 | C*VSPTGQR | 88.056 |
| NWMN_0748 | Uncharacterized protein | 28.19 | GIVTMC*APMGGK | 138.55 |
| SAZ172_0851 | Pathogenicity island protein | 15.839 | IIC*DFSTEREEK | 134.38 |
| SAKOR_01965 | RecT protein | 16.895 | NQC*YFIPYGNK | 86.772 |
| gtf1 | Glycosyltransferase Gtf1 | 58.273 | SSFVTC*YLQNEQK | 187.56 |
| fbp | Fructose-1,6-bisphosphatase class 3 | 76.213 | VC*LANLLR | 91.087 |
| glmU | Bifunctional protein GlmU [includes: UDP-N-acetylglucosamine pyrophosphorylase] | 48.532 | EGTTIVVC*GDTPLITK | 109.28 |
| glmU | Bifunctional protein GlmU [includes: UDP-N-acetylglucosamine pyrophosphorylase] | 48.532 | TNIGC*GTITVNYDGENK | 155.26 |
| SAOUHSC_02464 | Uncharacterized protein | 32.803 | NIEAC*TSLK | 62.162 |
| trxA_2 | Thiol reductase thioredoxin | 12.141 | FEAGWCPDC*R | 119.87 |
| aroC | Chorismate synthase | 43.059 | VAVGALC*K | 123.35 |
| MW0527 | MW0527 protein | 23.895 | AC*GLTEPSSK | 168.85 |
| MW0527 | MW0527 protein | 23.895 | C*GEVATQSAFK | 97.273 |
| lolD_1 | ABC transporter ATP-binding protein | 24.698 | AC*IIVTHDER | 73.435 |
| pckA | Phosphoenolpyruvate carboxykinase | 59.377 | NGVFNIEGGC*YAK | 207.21 |
| glmS | Glutamine–fructose-6-phosphate aminotransferase | 65.835 | C*GIVGYIGYDNAK | 159.41 |
| ST398NM01_0974 | Uncharacterized protein | 81.447 | GELHC*IGATTLNEYR | 166.21 |
| SAV0406 | Uncharacterized protein | 29.041 | GWNTLC*TYLK | 147.39 |
| SAOUHSC_02980 | Uncharacterized protein | 20.729 | SC*DIESVESWK | 130.19 |
| lysA_1 | Diaminopimelate decarboxylase | 9.757 | AFTC*IQMVK | 106.26 |
| HMPREF0776_0362 | HTH domain protein | 26.531 | QC*LSLPQTR | 63.966 |
| SAKOR_02509 | Transcriptional regulator, MarR family protein | 16.544 | VYMAC*LTEK | 137.01 |
| SAOUHSC_00118 | Capsular polysaccharide biosynthesis protein Cap5E, putative | 38.591 | SEQTLIC*GTR | 184.51 |
| SAOUHSC_00118 | Capsular polysaccharide biosynthesis protein Cap5E, putative | 38.591 | VIC*LSTDK | 157.66 |
| SAOUHSC_02364 | Uncharacterized protein | 12.686 | MEVC*PYLEETFK | 158.89 |
| SAOUHSC_02584 | Uncharacterized protein | 30.385 | AC*HETVLK | 114.97 |
| SAOUHSC_02584 | Uncharacterized protein | 30.385 | GEGAFC*NGIK | 114.72 |
| SAOUHSC_02584 | Uncharacterized protein | 30.385 | LIC*SWLK | 118.33 |
| map | Methionine aminopeptidase | 27.358 | EIGYIC*AK | 128.86 |
| QU38_16080 | Acyl-CoA ligase | 59.748 | LGVAIIPC*SEMLR | 111.07 |
| AYM28_10805 | 6-Phosphogluconolactonase | 38.546 | AGTGC*YVSISEDKR | 105.63 |
| AYM28_10805 | 6-Phosphogluconolactonase | 38.546 | EGEQC*GVASLK | 153 |
| AYM28_10805 | 6-Phosphogluconolactonase | 38.546 | ITLC*DNTR | 151.13 |
| SAOUHSC_02891 | Uncharacterized protein | 21.555 | SCELNSEAFC*NK | 123.72 |
| SA2075 | Sulfur carrier protein FdhD | 19.75 | LYGFC*IQR | 106.68 |
| pth | Peptidyl-tRNA hydrolase | 21.703 | C*IVGLGNIGK | 84.31 |
| dnaK | Chaperone protein DnaK | 66.361 | IIGIDLGTTNSC*VTVLEGDEPK | 89.507 |
| SAOUHSC_01064 | Pyruvate carboxylase | 18.812 | C*AEEGIK18.812 | 77.73 |
| sarR | HTH-type transcriptional regulator SarR | 13.669 | C*SEFKPYYLTK | 98.421 |
| lepA | Elongation factor 4 | 28.674 | C*YGGDISR | 128.35 |
| asnS | Asparagine–tRNA ligase | 49.157 | SVLENC*KLELK | 125.97 |
| pfkA | ATP-dependent 6-phosphofructokinase | 34.839 | C*PEFKEQEVR | 108.97 |
| SAKOR_01872 | Uncharacterized protein | 12.527 | FILSTSDDSDYIC*K | 91.589 |
| lipA_2 | Lipoyl synthase | 34.885 | HC*QAGPLVR | 83.08 |
| lipA_2 | Lipoyl synthase | 34.885 | NLNTVC*EEAK | 222.56 |
| V070_00687 | Uncharacterized protein | 27.971 | LINPDC*K | 128.35 |
| SAOUHSC_00532 | Uncharacterized protein | 42.89 | NDAILSDELNHASIIDGC*R | 87.411 |
| rnj2 | Ribonuclease J 2 (RNase J2) | 62.603 | LIVSC*YASNFIR | 118.71 |
| MW1645 | MW1645 protein | 44.233 | C*FEIEER | 74.165 |
| miaB_1 | MiaB family protein, possibly involved in tRNA or rRNA modification | 50.955 | STVAFHTLGC*K | 79.614 |
| ung | Uracil-DNA glycosylase (UDG) | 24.967 | ELADDIGC*VR | 138.24 |
| glpD | Aerobic glycerol-3-phosphate dehydrogenase | 62.387 | KDYGLTFSPC*NTK | 250.46 |
| SAV0941 | NADH dehydrogenase-like protein | 44.104 | IATPIVAC*NEK | 236.05 |
| SAV0941 | NADH dehydrogenase-like protein | 44.104 | IPELC*SK | 154.01 |
| MW2452 | MW2452 protein | 24.558 | LDC*KDEFIK | 89.189 |
| SA1530 | Uncharacterized peptidase | 39.606 | QVLFC*PK | 132.76 |
| pheT_2 | Phenylalanine–tRNA ligase β subunit | 12.153 | GVASSGMIC*SMK | 86.772 |
| SAKOR_02579 | Putative cytosolic protein | 11.547 | YMFDYSAC*K | 90.614 |
| AYM28_07495 | DNA-binding protein | 12.72 | HYQQLINQC*K | 121.29 |
| gcvPB | Probable glycine dehydrogenase (decarboxylating) subunit 2 | 22.485 | NFGVDNGFYPLGSC*TMK | 102.15 |
| NWMN_0123 | Uncharacterized protein | 151.91 | SLLEC*VK | 99.013 |
| adh | Alcohol dehydrogenase (ADH) | 36.061 | LDPAAASSITC*AGVTTYK | 210.33 |
| dnaJ | DnaJ | 29.458 | TEQVC*PK | 88.495 |
| guaB | Inosine-5′-monophosphate dehydrogenase | 52.85 | VGIGPGSIC*TTR | 119.46 |
| N/A | UPF0413 protein | 25.089 | C*QAQSTSNFDNIALAYK | 125.39 |
| SAKOR_00478 | VEG protein | 9.9982 | NSIDC*HVGNR | 76.868 |
| mutS | MutS protein | 48.889 | SEYQDC*LLFFR | 79.885 |
| mutS | MutS protein | 48.889 | VAIC*EQMEDPK | 125.36 |
| yibN | Putative sulfur transferase | 14.803 | KDQPVYLC*DANGIASYR | 78.191 |
| ileS | Isoleucine–tRNA ligase | 104.74 | C*KEFALEQIELQK | 116.61 |
| SAOUHSC_01907 | Uncharacterized protein | 31.471 | VENDENC*MESVK | 159.04 |
| nagB | Glucosamine-6-phosphate deaminase | 28.467 | QASFYVAC*ELYK | 106.82 |
| SAKOR_02240 | Molybdenum cofactor biosynthesis protein B | 18.5 | DFDTDKGGQC*VR | 93.716 |
| nirB | Nitrite reductase [NAD(P)H], large subunit | 46.979 | SC*VESGVK | 65.627 |
| tetM | Tetracycline resistance protein TetM | 70.346 | GPSELC*GNVFK | 69.261 |
| thyA | Translation initiation factor IF-3 | 36.825 | LSC*QLYQR | 55.721 |
| SAOUHSC_01781 | Uncharacterized protein | 36.431 | FANC*TQELTIEK | 110.6 |
| miaB | Uncharacterized protein | 58.916 | AWVNIMYGC*DK | 135.91 |
| miaB | RNA methyltransferase TrmA family protein | 58.916 | YEGQTVTVLC*EGSSK | 188.32 |
| pgcA | Fructose-1,6-bisphosphate aldolase | 62.376 | C*PNFDDVAQK | 151.3 |
| SAKOR_02003 | Ribosomal protein-serine acetyltransferase | 27.906 | C*HNSFVVNR | 98.407 |
| SAKOR_02003 | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase TrmFO | 27.906 | IFIC*EDDPK | 142.19 |
| SAKOR_02003 | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase TrmFO | 27.906 | IIDC*LETAHTR | 170.95 |
| SAV1153 | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase TrmFO | 19.255 | GNC*DFYPEFENEAVAK | 76.341 |
| ftsH | Peptide methionine sulfoxide reductase MsrB | 77.812 | IC*GLLGGR | 124.08 |
| HMPREF0769_10485 | Putative peptide methionine | 18.28 | LDSPYDGYAEC*VK | 115.26 |
| sepF | Cell division protein SepF | 20.686 | MC*LFEPR | 120.15 |
| SAOUHSC_02898 | Uncharacterized protein | 24.931 | VNSLAYC*SSK | 128.85 |
| sucC | Succinate-CoA ligase [ADP-forming] subunit β | 42.056 | C*DVIAEGIVEAVK | 189.04 |
| sucC | Succinate-CoA ligase [ADP-forming] subunit β | 42.056 | RLYIEEGC*AIQK | 231.07 |
| SA2102 | Putative formate dehydrogenase | 111.29 | FAEEC*AK | 160.82 |
| SA2102 | Putative formate dehydrogenase | 111.29 | GHNNVQGC*SDMGSMPDK | 88.108 |
| SA2102 | Putative formate dehydrogenase | 111.29 | QVIGTNNVDNC*SR | 175.19 |
| SA2102 | Putative formate dehydrogenase | 111.29 | YC*QAPATK | 117.25 |
| SAKOR_00737 | Ferric anguibactin transport ATP-binding protein | 28.62 | STLLSAIC*R | 103.43 |
| SAOUHSC_01323 | Uncharacterized protein | 29.821 | QDFDEIVDYC*R | 127.05 |
| SAOUHSC_02248 | Uncharacterized protein | 17.196 | IIGLSGMC*K | 68.132 |
| taqD | Glycerol 3-phosphate cytidyltransferase | 15.789 | C*EVIYLK | 51.528 |
| AYM28_05950 | Uncharacterized protein | 15.185 | FQMINDC*AEK | 64.52 |
| queA | S-adenosylmethionine:tRNA ribosyltransferase-isomerase | 38.97 | IIAEC*IK | 138.08 |
| SAOUHSC_00086 | 3-Ketoacyl-acyl-carrier protein reductase, putative | 27.215 | IINATSQAGVEGNPGLSLYC*STK | 70.572 |
| glcT | Protein GlcT | 32.822 | NHYPIC*YNTAYK | 118.52 |
| AYM28_01135 | AraC family transcriptional regulator | 29.599 | VVIC*DDER | 129.82 |
| AYM28_01135 | AraC family transcriptional regulator | 29.599 | YLQMSPSDYC*K | 115.09 |
| AYM28_13045 | Putative 3-methyladenine DNA glycosylase | 22.771 | AIDGATLNDC*R | 116.58 |
| tyrC | Arogenate dehydrogenase | 40.395 | C*LNYSEAIK | 68.676 |
| SAOUHSC_02899 | Uncharacterized protein | 38.194 | AIELC*QK | 143.37 |
| SAR2150 | Protein SprT-like | 17.186 | ANYEYYC*TK | 166.62 |
| SAR2150 | Protein SprT-like | 17.186 | FC*NSIESYQQR | 97.797 |
| SA0314 | Uncharacterized protein | 20.027 | LDC*AEIIR | 73.841 |
| SA1974 | Probable uridylyltransferase | 44.865 | LVNVDC*K | 82.417 |
| hemH | Ferrochelatase | 35.056 | VVC*DDIGANYYRPK | 112.24 |
| hpt | Hypoxanthine-guanine phosphoribosyltransferase | 20.154 | EVLLTEEDIQNIC*K | 103.39 |
| SAOUHSC_00548 | Uncharacterized protein | 58.418 | GFLSC*SR | 94.309 |
| SAOUHSC_00531 | Uncharacterized protein | 43.657 | VRPGAFFLTGC*GNESK | 74.296 |
| tnp | Putative transposase | 8.5839 | GIEC*IYALYK | 90.614 |
| cap5G | Capsular polysaccharide biosynthesis protein Cap5G | 42.851 | C*FDQNVPEEINR | 186.96 |
| SAOUHSC_00973 | Uncharacterized protein | 27.727 | VC*YQVFYDEK | 138.02 |
| ykaA | Phosphate transport regulator | 22.598 | EFETNC*DGILR | 133.48 |
| hprK | HPr kinase/phosphorylase | 34.481 | LC*RPETPAIIVTR | 98.508 |
| murA1 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase 1 | 44.995 | LGHAIVALPGGC*AIGSR | 67.846 |
| rocA | 1-Pyrroline-5-carboxylate dehydrogenase | 56.867 | GC*TSAVVGYHPFGGFK | 103.67 |
| HMPREF0769_10271 | Oxidoreductase, NAD-binding domain protein | 39.204 | AAC*AAEAYGTDNAK | 78.917 |
| ahpC | Alkyl hydroperoxide reductase | 20.976 | KNPGEVC*PAKWEEGAK | 127.78 |
| mvaS | HMG-CoA synthase | 43.217 | EAC*YAATPAIQLAK | 226.7 |
| SAKOR_00677 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase | 20.889 | ALAPLC*DTK | 137.4 |
| SAKOR_00677 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase | 20.889 | IAVYC*GASK | 122.33 |
| pcrA | ATP-dependent DNA helicase PcrA | 84.073 | IC*YVAITR | 109.83 |
| N/A | Putative uncharacterized protein | 15.429 | EQGSDIDAAC*GQLR | 137.52 |
| rimP | Ribosome maturation factor RimP | 17.627 | EGGVDLNDC*TLASEK | 197.6 |
| asp1 | Accessory Sec system protein Asp1 | 53.78 | EC*ITSVNEEYQAK | 188 |
| SAUSA300_2158 | Uncharacterized protein | 14.457 | DDNILC*EEFSYK | 79.393 |
| SAKOR_00594 | Trp repressor-binding protein | 20.243 | VILVGGDC*PK | 186.76 |
| SAOUHSC_02447 | Uncharacterized protein | 36.266 | VPVC*GAISSYNHPEADIGPR | 126.62 |
| SAOUHSC_00497 | Uncharacterized protein | 53.954 | NGLTLQEC*LDR | 114.64 |
| SAOUHSC_00497 | Uncharacterized protein | 53.954 | SIFPSC*R | 68.557 |
| pyk | Pyruvate kinase | 63.102 | C*DILNSGELK | 126.71 |
| pyk | Pyruvate kinase | 63.102 | IVC*TIGPASESEEMIEK | 96.561 |
| pyk | Pyruvate kinase | 63.102 | QC*SIVWGVQPVVK | 172.11 |
| rpoB | DNA-directed RNA polymerase subunit β | 133.22 | FMDDEVVC*R | 92.247 |
| yigZ | ABC transporter | 23.868 | EAVPC*IVTLNYDQTGK | 120.21 |
| yigZ | ABC transporter | 23.868 | LDVHNAC*VVVTR | 98.182 |
| N/A | Uncharacterized protein | 32.909 | NESLC*ELKK | 140.63 |
| SAOUHSC_01365 | Uncharacterized protein | 37.855 | SHLVNLC*K | 99.788 |
| SA2162 | Ferredoxin–NADP reductase | 38.164 | C*NTLLSETSSK | 186.08 |
| SA2162 | Ferredoxin–NADP reductase | 38.164 | LDMHDDC*R | 64.224 |
| infC | Translation initiation factor IF-3 | 20.244 | YADEC*KDIATVEQKPK | 88.311 |
| SAOUHSC_02827 | Uncharacterized protein | 10.548 | IIASC*SFAK | 135.1 |
| SAOUHSC_02579 | Uncharacterized protein | 41.89 | VLYQGYTC*FR | 109.99 |
| NWMN_1835 | RNA methyltransferase TrmA family protein | 51.682 | IVYISC*NPATQQR | 216.34 |
| fba | Fructose-bisphosphate aldolase | 30.836 | EC*QELVEK | 97.472 |
| SAKOR_00338 | Ribosomal protein-serine acetyltransferase | 20.294 | YC*FEELDLNR | 77.644 |
| trmFO | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase | 48.371 | FAELVC*SNSLR | 144.4 |
| trmFO | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase | 48.371 | YDKGEAAYLNC*PMTEDEFNR | 90.259 |
| trmFO | Methylenetetrahydrofolate–tRNA-(uracil-5-)-methyltransferase | 48.371 | YFEGC*MPFEVMAER | 85.937 |
| msrB | Peptide methionine sulfoxide reductase MsrB | 16.277 | FHSEC*GWPSFSK | 125.33 |
| msrB | Peptide methionine sulfoxide reductase MsrB | 16.277 | YC*INSAAIQFIPYEK | 144.3 |
| ytqA | Fe–S oxidoreductase | 36.053 | VALDGGFDC*PNR | 116.87 |
| yjlD | NADH dehydrogenase | 39.399 | IYNC*DEPK | 160.72 |
| SAZ172_2586 | Mutator mutT protein | 11.465 | C*DLIVGDK | 63.073 |
| AYM28_03635 | Protein of uncharacterized function | 14.312 | IMYC*FNK | 132.08 |
| SAKOR_01397 | ATP-dependent helicase, DinG family protein | 104.19 | C*LVLFTSYK | 112.13 |
| SAOUHSC_02393 | Uncharacterized protein | 25.34 | C*LANNDVQIMNSIK | 78.674 |
| panD | Aspartate 1-decarboxylase | 14.05 | IC*LNGAASR | 112.64 |
| purA | Adenylosuccinate synthetase | 47.551 | IC*TAYELDGK | 104.59 |
| fhs | Formate–tetrahydrofolate ligase | 59.871 | QFKENGWDNYPVC*MAK | 200 |
| SAZ172_2084 | 5-Amino-6-(5-phosphoribosylamino)uracil reductase | 15.666 | AFQILHEQYGC*K | 51.727 |
| gatB_2 | Aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase subunit B | 53.656 | C*DANISLRPYGQEK | 76.574 |
| SAZ172_2659 | d-specific d-2-hydroxyacid dehydrogenase-like protein | 37.264 | DAVFVNC*AR | 73.435 |
| folP | Dihydropteroate synthase | 29.532 | SEVAEAC*LK | 115.33 |
| AYM28_03210 | Deoxyguanosinetriphosphate triphosphohydrolase | 50.595 | GGEVLLNNC*LK | 164.22 |
Furthermore, the prevalence of hydrophobic and positively charged amino acids flanking modified cysteines was observed when linear amino acid sequences and available 3D structures of CoAlated proteins were examined using computational methods (manuscript in preparation).
To sense and overcome the oxidative stress, S. aureus employs oxidation-sensing transcriptional regulators, such as MgrA, SarZ and SarA, and the quorum-sensing Agr system which controls global gene expression via the redox-active Cys residues [44,45]. It was interesting to find redox-sensing transcriptional regulators detected among CoAlated proteins. In total, 16 CoA-modified peptides from 12 transcriptional regulators, including SarR, CtsR, AgrA, PerR and SarS, were found in diamide-treated S. aureus. Induction of the CtsR and PerR regulons in NaOCl-treated B. subtilis was found to be indicative of the disulfide stress response [46]. PerR exists in Gram-positive bacteria as a functional homolog of OxyR and functions as a sensor of oxidative stress. PerR possesses four cysteine residues, which are involved in Zn coordination, dimerization and DNA binding. One of these cysteines, Cys142, was found to be CoAlated in diamide-treated S. aureus. The effect of PerR CoAlation on its DNA-binding and transcriptional activities in response to oxidative and metabolic stress remains to be investigated.
The quorum-sensing transcriptional regulator AgrA was CoAlated on Cys6 and Cys199 in diamide-treated cells. The oxidation-sensing role of Cys199 in AgrA was revealed in mutational, biochemical and mass spectrometric analyses [47]. The formation of the intracellular disulfide bond between Cys199 and Cys228 was shown to cause the dissociation of AgrA from DNA. It would be interesting to examine the pattern of AgrA CoAlation in S. aureus exposed to different stresses, including nitrogen and carbohydrate deprivation, exposure to heat, UV and oxidizing chemicals. Furthermore, in vitro CoAlation of recombinant AgrA at Cys199 has the potential to modify its DNA-binding and transcriptional activities.
Other targets of protein CoAlation are antioxidant proteins, including thioredoxin (Trx), alkyl hydroperoxide reductase C (AhpC), thiol peroxidase (Tpx), malate : quinone oxidoreductases 1 and 2 (Mqo1/2), FAD-binding protein oxidoreductase (HMPREF0769_10247) and Fe–S oxidoreductase (YtqA). In Tpx, diamide-induced CoAlation occurs on the active site Cys60 and the relevance of this modification is yet to be fully understood. Interestingly, Tpx peroxidase was shown to be S-mycothiolated at Cys60 in Corynebacterium glutamicum and its activity inhibited by in vitro S-mycothiolation [42].
In addition, many ribosomal proteins were found to be CoAlated, including L12, S12, L14, S18, L32, L33 and L36. Recently, ribosomal proteins L33 (RpmG3) and L36 (RpmJ) were found to be S-bacillithiolated within the CXXC motifs at conserved Zn-binding sites in S. aureus treated with NaOCl [25]. Oxidation of cysteines in ribosomal proteins, especially in the CXXC motif, was reported in various studies, and it has been suggested that disulfide stress could lead to a stalling of the ribosomes and thus, to the release of the alarmone ppGpp [48,49]. Furthermore, glutathionylation of ribosomal protein S12 was found in oxidatively stressed human T lymphocytes, but the importance of this modification has not been investigated [50]. The identification of many ribosomal proteins as targets for CoAlation may suggest the inhibitory effect of this modification on protein biosynthesis under oxidative stress.
The largest functional group of CoAlated proteins includes metabolic enzymes that function in diverse anabolic and catabolic pathways for carbohydrates, amino acids, nucleotides, fatty acids, coenzymes and antioxidants (Figure 4B and Table 1). Among CoAlated proteins, we found key players of the citric acid cycle, glycolysis, gluconeogenesis, glycerol catabolism and the glyoxylate shunt. These included glyceraldehyde-3-phosphate dehydrogenase 2 (GapA2), pyruvate kinase (Pyk), ATP-dependent 6-phosphofructokinase (PfkA), acetate kinase (AckA), alcohol dehydrogenase (Adh), aldehyde dehydrogenase 1 (AldA1), triose phosphate isomerase (TpiA), manganese-dependent inorganic pyrophosphatase (PpaC), fructose-1,6-bisphosphate aldolase (Fbp), glycerol kinase (GlpK), inosine-5′-monophosphate dehydrogenase (GuaB), malate dehydrogenase (Mdh) and others. In the list of CoAlated metabolic enzymes, we found several enzymes, which are known to be modified by other LMW thiols, including glutathione, bacillithiol and mycothiol. For example, GAPDH, GuaB and AldA were shown to be S-bacillithiolated in S. aureus under NaOCl stress [25]. Mycothiolation of GuaB and Fbp in C. glutamicum treated with NaOCl was previously reported [34]. In Gram-negative bacteria, GAPDH, TpiA and PpaC were shown to be glutathionylated in oxidative stress response [51,52]. Extensive CoAlation of metabolic enzymes in response to oxidative stress may reflect the potential involvement of CoA in regulatory and/or feedback mechanisms which control metabolic pathways by balancing the redox state.
In vivo CoAlation of SaGAPDH in response to various oxidizing agents
GAPDH homologs in eukaryotic and prokaryotic cells have been found in numerous studies as targets for oxidation and subjects for redox-controlled post-translational modifications, including S-glutathionylation, bacillithiolation and mycothiolation. These modifications were mapped to a strictly conserved catalytic site cysteine and shown to inhibit the activity of GAPDH, and to protect the catalytic cysteine from irreversible overoxidation. Two homologs of GAPDH have been identified in S. aureus, termed GAPDH1 and GapA2. In the present study, diamide-induced CoAlation was shown to modify the GapA2 isoform at a non-catalytic Cys202. Protein sequence analysis revealed that trypsin/Lys C digestion of SaGAPDH and GapA2 would produce very long peptides, containing catalytic active cysteines 151 and 153, respectively, making their analysis by LC–MS/MS less feasible. To date, detailed structure–function analysis has been carried out mainly for the SaGAPDH isoform. Therefore, we investigated SaGAPDH CoAlation in vitro and in vivo, and examined the effect of this modification on its activity. Initially, our efforts were focused on validating in vivo CoAlation of SaGAPDH in response to diamide and testing the effect of other oxidizing agents. To do so, E. coli transformed with the pQE3/SaGAPDH plasmid were grown at 37°C in LB medium to mid-log phase (OD600 = 0.7) and induced with 0.1 mM IPTG for 3 h at 30°C. Bacterial cultures were then treated for 30 min at 37°C with 2 mM diamide, 10 mM H2O2 and 100µM NaOCl. Ni-NTA Sepharose was used to capture His-SaGAPDH from lysed cells and the pulled-down proteins were analyzed by SDS–PAGE and immunoblotting with anti-CoA antibody. As shown in Figure 5A (right panel), the level of pulled-down His-SaGAPDH was nearly the same in all examined samples. No CoAlation of His-SaGAPDH was detected in control cells, while the treatment of cells with 2 mM diamide induced strong CoAlation of His-SaGAPDH. The strongest CoAlation of His-SaGAPDH was observed in response to NaOCl and a weak immunoreactive signal was detected in H2O2-treated cells.
Figure 5. In vitro and in vivo CoAlation of SaGAPDH.

(A) CoAlation of SaGAPDH overexpressed in E. coli is strongly induced by oxidizing agents. The expression of His-tagged SaGAPDH in E. coli transformed with the pQE3/SaGAPDH plasmid was induced with 0.1 mM IPTG for 3 h at 30°C. Bacterial cultures were then treated with 2 mM diamide, 10 mM H2O2 and 100 µM NaOCl for 30 min. Ni-NTA Sepharose was used to pull-down His-SaGAPDH and protein CoAlation analyzed by immunoblotting with anti-CoA antibody. (B) In vitro CoAlation of recombinant SaGAPDH. Recombinant preparations of His-SaGAPDH were incubated with 2 mM CoA dimer (CoASSCoA). NEM (25 mM) was added and samples were heated in loading buffer with or without DTT. CoAlation of enzymes was examined by anti-CoA immunoblot. (C) LC–MS/MS spectrum of a CoAlated peptide derived from in vitro CoAlated SaGAPDH. The spectrum shows a peptide from SaGAPDH (LDGSETVVSGASC*TTNSLAPVAK), containing CoAlated catalytic cysteine 151.
Next, we investigated the effect of in vitro CoAlation on the activity of SaGAPDH. In the present study, recombinant His-SaGAPDH was purified by Ni-NTA Sepharose chromatography from E. coli transformed with the pQE3/His-SaGAPDH plasmid. To produce CoAlated SaGAPDH, an in vitro CoAlation assay was performed in the presence of recombinant SaGAPDH and CoASSCoA. Immunoblotting of reaction mixtures with the 1F10 antibody revealed a strong immunoreactive signal corresponding to the SaGAPDH sample incubated with CoA disulfide (Figure 5B). No immunoreactivity was detected in the sample separated under reducing conditions (100 mM DTT). The LC–MS/MS analysis of in vitro CoAlated SaGAPDH showed that catalytic Cys151 is CoAlated in the LDGSETVVSGASC*TTNSLAPVAK peptide (Figure 5C).
In vitro CoAlation of SaGAPDH prevents irreversible inhibition of its enzymatic activity by H2O2
GAPDH homologs in eukaryotic and prokaryotic cells have been shown to be readily inhibited by a variety of ROS and the inhibitory effect is mediated by direct oxidation of the catalytic active cysteine located in a highly conserved CTTNC motif. Studies from several laboratories revealed that SaGAPDH is very sensitive to irreversible oxidation to sulfonic acid when S. aureus is treated with 100 mM H2O2 [25,53].
We initially examined a dose-dependent inhibition of recombinant SaGAPDH with H2O2. Purified His-SaGAPDH was incubated in the absence or presence of 1 μM, 10 μM, 100 μM, 1 mM or 10 mM H2O2 for 10 min before the activity was measured spectrophotometrically, as described in Experimental procedures. As shown in Figure 6A, exposure of SaGAPDH to 10 μM H2O2 results in a ∼50% decrease of its catalytic activity. The presence of 1 mM H2O2 in the reaction mixture resulted in 95% inhibition, while 10 mM H2O2 completely blocked SaGAPDH activity. These data indicate that SaGAPDH is efficiently inhibited by ROS-mediated direct oxidation of its catalytic active cysteine. Further analysis revealed that the inactivation of SaGAPDH activity by 10 mM H2O2 was only partially reversible, as only 40% of SaGAPDH activity could be recovered with 10 mM DTT (Figure 6B). The addition of DTT to the untreated sample of SaGAPDH increased its activity by ∼50%, indicating partial and reversible oxidation of recombinant preparations of His-SaGAPDH during the storage.
Figure 6. Testing the effect of CoAlation on SaGAPDH activity in vitro.

In vitro CoAlation prevents H2O2-induced overoxidation of recombinant SaGAPDH dose-dependent inhibition of SaGAPDH activity by H2O2 in vitro. (B) In vitro CoAlation inhibits SaGAPDH activity. (C) H2O2-induced inhibition of SaGAPDH activity is only partially reversed by DTT. (D) The inhibition of SaGAPDH activity by in vitro CoAlation is fully reversed by DTT.
Next, we investigated the effect of CoAlation on SaGAPDH catalytic activity. As shown in Figure 6C, in vitro CoAlation of SaGAPDH resulted in 90% inhibition of its activity. The inhibitory effect of CoAlation on SaGAPDH activity was completely reversed by the addition of 10 mM DTT to the reaction mixture. In line with these results, we have recently reported significant inhibition (∼80%) of mammalian GAPDH by in vitro CoAlation, which was fully reversed by DTT [22]. These findings prompted us to examine whether CoAlation can protect SaGAPDH catalytic activity against irreversible overoxidation by H2O2. In the agreement with the above findings, SaGAPDH was fully inactivated with 10 mM H2O2 and the addition of 10 mM DTT recovered only 40% of the enzymatic activity (Figure 6D). We subsequently found that in vitro CoAlation of SaGAPDH before exposure to 10 mM H2O2 resulted in nearly 100% recovery of its activity with DTT. These results indicate that SaGAPDH CoAlation can prevent irreversible overoxidation of the catalytic Cys151 under the oxidative stress.
Analysis of GAPDH/CoA interaction by MD simulation
In the glycolysis pathway, GAPDH functions to convert glyceraldehyde 3-phosphate to the energy-rich intermediate 1,3-bisphosphoglycerate and uses inorganic phosphate to harness the energy into NADH [54]. Biochemical, mutational and crystallographic studies of GAPDH from prokaryotic and eukaryotic cells have revealed that the active site cysteine is essential for the catalytic activity of the enzyme. GAPDH consists of four identical non-covalently connected subunits, which possess the NAD+-binding domain and the catalytic domain. Each subunit contains a strictly conserved catalytic cysteine, which is susceptible to various thiol modifications in cellular response to oxidative stress, including sulfenylation, glutathionylation, nitrosylation, bacillithiolation and mycothiolation [55]. These modifications inhibit GAPDH activity and glucose metabolism, and divert a metabolic flux through the pentose phosphate cycle to increase NADPH production.
Recently, a crystal structure of overoxidized SaGAPDH revealed an apo form of the enzyme lacking NAD+ in the binding pocket [25]. Taking this into account and the fact that SaGAPDH is CoAlated in response to H2O2 and NaOCl, we hypothesized that the ADP moiety of CoA may also be involved in mediating the interaction with SaGAPDH by occupying the vacant nucleotide-binding pocket. To investigate the binding mode between CoA and SaGAPDH, we performed MD simulations. In the present study, the structure of the CoA : GAPDH complex was modeled onto the GAPDH structure with NAD+ (PDBID: 3LVF). The adenine moieties of both cofactors were aligned to start from a structure with CoA bound in the NAD adenine-binding site. CoA remains firmly bound to the protein during a long 400 ns MD run in explicit solvent, suggesting that CoA can indeed bind to the NAD+ binding site. As (un)binding events can occur on a wide range of timescales, from μs to hours, they cannot be captured by the limited timescales accessible to standard MD simulations. For this reason, we employed metadynamics to enhance the sampling and accelerate the occurrence of these rare events. The free energy landscape reconstructed after a 400 ns metadynamics run is shown in Figure 7A. In this map, the most stable conformation (labeled MD-A) has CoA-folded onto itself and occupies the deep cavity in proximity of the nicotinamide-binding site. In this conformation, the CoA thiol group is quite close to catalytic Cys151 (at ∼7–8 Å, highlighted as a red sphere in the figure). A nearby secondary minimum (MD-B) is also present, structurally similar to A, but for which the CoA thiol group and Cys151 are within bond distance (SH–SH distance of 4 Å). This conformation is only 1.8 kcal/mol higher in energy, suggesting that interactions between the CoA thiol and Cys151 are not only possible, but might lead to covalent binding. An alternative metastable basin (MD-C) is also observed in the free energy map. In this conformation, CoA is bound to the NAD+ adenine-binding site, as in the starting X-ray structure. As in the case of MD-B, this minimum is very close in energy to lowest basin MD-A, being only 1.3 kcal/mol less stable. Interestingly, CoA can approach Cys151 also from this conformation, by assuming a bridge conformation that connects the adenine- and nicotinamide-binding sites (labeled MD-D in Figure 7A).
Figure 7. The mode of interaction between SaGAPDH and CoA.

(A) Free energy landscape of the CoA : GAPDH complex as a function of the distance between Cys151 (shown as a red ball in the exemplary structures) and the CoA SH tail (x-axis) and the PO3 group attached to the sugar moiety (y-axis). The contour lines are drawn every 2 kcal/mol. CoA and, for reference, NAD are shown in blue and magenta sticks, respectively. The computed free energy landscape reveals how CoA can assume different conformations, folded onto itself (labeled as A in the map) near the nicotinamide-binding site (as shown in the corresponding figure on the lower left corner), or binding the adenine-binding site (MD-B). The CoA tail can approach Cys151 from both these minima or by assuming the conformations found in basin MD-D (top left corner) when starting from the extended conformation observed in basin MD-C (top right corner). The latter basins are slightly higher energy when compared with the absolute minimum, but still accessible. (B) A proposed model of CoA binding to SaGAPDH, where CoA binds to the nicotinamide-binding site and its tail approaches Cys151 forming a covalent bond.
In the holoenzyme structure, the NAD+ occupies the nucleotide-binding pocket and prevents the formation of bonds with the ADP moiety of CoA. The absence of NAD+ in the apoenzyme structure allows this ADP moiety to occupy the nucleotide-binding pocket.
Taken together the above findings and published studies in the literature, we propose that the interaction between CoA and the oxidized form of GAPDH is facilitated by the ADP moiety of CoA which can occupy the vacant nucleotide-binding pocket, while permitting the pantetheine tail of CoA to form a covalent disulfide bond with Cys151.
Discussion
The presence of a highly reactive thiol group and the ADP moiety in the CoA structure offer diverse functions in biochemical processes. Using a thiol group, CoA reacts with carboxylic acids to form diverse thioesters, thus functioning in cellular metabolic processes as a master acyl group carrier. It also functions as a carbonyl-activating group in numerous anabolic and catabolic processes, including the citric acid cycle and fatty acid metabolism. In addition, CoA provides the 4-phosphopantetheine prosthetic group to proteins that play key roles in fatty acid, polyketide and non-ribosomal peptide biosynthesis. A novel unconventional function of CoA in redox regulation, involving oxidative S-thiolation of cellular proteins (termed protein CoAlation), has recently emerged as a new research field [22]. In this original study, extensive protein CoAlation was observed in mammalian cells and tissues in response to oxidative and metabolic stress. Developed research tools and a mass spectrometry methodology allowed the identification of 587 CoAlated proteins under various experimental conditions in cell-based and animal models ([22] and unpublished data). Bioinformatic pathway analysis of CoAlated proteins showed that they are involved in diverse cellular processes, including metabolism, protein synthesis and stress response. Furthermore, catalytic activities of several metabolic enzymes, including creatine kinase, isocitrate dehydrogenase 2, pyruvate dehydrogenase kinase 2 and GAPDH, were shown to be inhibited by in vitro CoAlation. Here, we demonstrate for the first time that protein CoAlation also occurs in prokaryotic cells and is associated with redox regulation. Evidence is provided that exponentially growing Gram-negative and Gram-positive bacteria exhibit a basal level of protein CoAlation, while exposure to oxidizing agents and glucose deprivation induce strong covalent protein modification by CoA in a DTT-sensitive manner.
The intracellular concentration of CoA and its derivatives in bacteria varies from 0.4 mM in E. coli to low millimolar level in S. aureus [1,56,57]. The level of CoA and the ratio between CoA and its thioesters fluctuate depending on the growth conditions and are regulated by the availability of nutrients, intracellular metabolites and the exposure to stress. In exponentially growing E. coli, four CoA species (CoASH, acetyl CoA, succinyl CoA and malonyl CoA) compose the bulk of the CoA pool, where acetyl CoA is the dominant component (79.8%) and the level of CoASH is significantly lower (13.8%) [57]. When glucose in the medium is depleted, CoASH becomes the major component (82%) of the CoA pool at the expense of the acetyl CoA derivatives. The same effect was also observed in cells treated with the oxidizing agent or cultured in the glucose-deprived medium [57]. The production of metabolically active CoA thioesters has been associated with cell growth, while the increase in the level of CoASH under adverse growth conditions may allow bacteria to sense, respond and adapt to excessive ROS accumulation via thiol-mediated protein CoAlation.
The redox proteome analysis of S. aureus enabled us to identify 356 CoAlated proteins which belong to diverse functional classes. The main targets of CoAlation are proteins involved in cellular metabolism, translation and antioxidant response, and this pattern correlates with that in mammalian cells and tissues [22]. In contrast with mammalian CoAlome, many redox-dependent transcription factors, whose DNA-binding activity is modulated in response to cysteine oxidation, have been identified in diamide-treated S. aureus. These include transcriptional regulators SarR, CtsR, AgrA, PerR and SarS, which control the expression of genes involved in oxidative stress response, antibiotic resistance, virulence or catabolism of aromatic compounds.
To examine the effect of CoAlation on the activity of modified proteins, our efforts were focused on SaGAPDH, a major target of S-thiolation in prokaryotic and eukaryotic cells in response to oxidative and metabolic stress. We provide evidence that CoAlation protects the catalytic cysteine in SaGAPDH against overoxidation under H2O2 stress in vitro and offers a reversible mode of regeneration of this essential glycolytic enzyme during the recovery from oxidative stress. Using MD simulations to examine the binding mode between CoA and SaGAPDH, we found that in the most stable conformation, the ADP moiety of CoA occupies the deep cavity where the nicotinamide-binding pocket is located, and the CoA thiol group and Cys151 are within the distance permitting covalent disulfide bond formation. Based on these findings and a recently reported crystal structure of overoxidized SaGAPDH which lacks NAD+ in the binding pocket, we propose a double anchor model for GAPDH CoAlation in response to oxidative or metabolic stress. In this model, the ADP moiety of CoA anchors to the nucleotide-binding pocket in the oxidized form of GAPDH and positions the CoA thiol group in the flexible pantetheine tail in close vicinity for covalent bond formation with catalytic Cys151 (Figure 7B).
Acknowledgements
We thank the members of Cell Regulation Laboratory at the Department of Structural and Molecular Biology (UCL) for their valuable inputs throughout the present study. We are grateful to A.K. Das for providing the pQE3/SaGAPDH expression plasmid; A. Edwards and S. Cutting for critical reading of the manuscript and UCL Darwin Research Facility analytical biochemistry support.
Abbreviations
- 4PP
4′-phosphopantetheine
- AhpC
alkyl hydroperoxide reductase C
- CoA
coenzyme A
- CoASSCoA
CoA disulfide
- CoASSG
CoA-cysteine and CoA-glutathione
- DTT
dithiothreitol
- Fbp
fructose-1,6-bisphosphate aldolase
- GapA2
glyceraldehyde-3-phosphate dehydrogenase 2
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GO
Gene Ontology
- GSH
glutathione
- GuaB
inosine-5′-monophosphate dehydrogenase
- H2O2
hydrogen peroxide
- HMG
3-hydroxy-3-methylglutaryl
- HOCl
hypochlorous acid
- IAM
iodoacetamide
- IPTG
isopropyl-β-d-thiogalactopyranoside
- LB
Luria Bertani
- LMW
low-molecular-weight
- MD
molecular dynamics
- Mqo1/2
malate : quinone oxidoreductases 1 and 2
- NaOCl
sodium hypochlorite
- NB3
Nutrient Broth 3
- OD600
optical density at 600 nm
- PAGE
polyacrylamide gel electrophoresis
- PpaC
manganese-dependent inorganic pyrophosphatase
- ROS
reactive oxygen species
- SaGAPDH
S. aureus glyceraldehyde-3-phosphate dehydrogenase
- SDS
sodium dodecyl sulfate
- TpiA
triose phosphate isomerase
- Tpx
thiol peroxidase
Author Contribution
The present study was conceived by I.G. and Y.T. I.G., A.Z., N.T. and J.W. performed cell-based experiments. J.B. and C.N. carried out enzymatic assays. A.Z. and J.B. purified recombinant His-SaGAPDH and carried out in vitro CoAlation studies. V.F. developed and characterized anti-CoA Mabs. G.S., F.C. and F.L.G. designed and performed MD simulation. S.D. and C.O. carried out bioinformatics analysis. Y.T., S.Y.P.-C. and M.S. prepared the samples and analyzed protein CoAation by LC–MC/MS. I.G. wrote the manuscript with the assistance and approval of all authors.
Funding
This work was supported by University College London Business [13-014 and 11-018] and the Biotechnology and Biological Sciences Research Council [BB/L010410/1] to I.G.; National Academy of Sciences of Ukraine [0110U000692] to V.F. F.L.G., G.S. and F.C. acknowledge EPSRC [EP/M013898/1, EP/P022138/1 and EP/P011306/1] for financial support. HecBioSim [EPSRC grant EP/L000253/1] and PRACE are acknowledged for computer time.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Leonardi R., Zhang Y.-M., Rock C.O. and Jackowski S. (2005) Coenzyme A: back in action. Prog. Lipid Res. 44, 125–153 10.1016/j.plipres.2005.04.001 [DOI] [PubMed] [Google Scholar]
- 2.Davaapil H., Tsuchiya Y. and Gout I. (2014) Signalling functions of coenzyme A and its derivatives in mammalian cells. Biochem. Soc. Trans. 42, 1056–1062 10.1042/BST20140146 [DOI] [PubMed] [Google Scholar]
- 3.Srinivasan B. and Sibon O.C.M. (2014) Coenzyme A, more than ‘just’ a metabolic cofactor. Biochem. Soc. Trans. 42, 1075–1079 10.1042/BST20140125 [DOI] [PubMed] [Google Scholar]
- 4.Theodoulou F.L., Sibon O.C.M., Jackowski S. and Gout I. (2014) Coenzyme A and its derivatives: renaissance of a textbook classic. Biochem. Soc. Trans. 42, 1025–1032 10.1042/BST20140176 [DOI] [PubMed] [Google Scholar]
- 5.Choudhary C., Weinert B.T., Nishida Y., Verdin E. and Mann M. (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 10.1038/nrm3841 [DOI] [PubMed] [Google Scholar]
- 6.Chen Y., Sprung R., Tang Y., Ball, H., Sangras, B., Kim S.C. et al. (2007) Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 6, 812–819 10.1074/mcp.M700021-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin H., Su X. and He B. (2012) Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 7, 947–960 10.1021/cb3001793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.delCardayré S.B., Stock K.P., Newton G.L., Fahey R.C. and Davies J.E. (1998) Coenzyme A disulfide reductase, the primary low molecular weight disulfide reductase from Staphylococcus aureus. J. Biol. Chem. 273, 5744–5751 10.1074/jbc.273.10.5744 [DOI] [PubMed] [Google Scholar]
- 9.Loewen P.C. (1978) Levels of coenzyme A — glutathione mixed disulfide in Escherichia coli. Can. J. Biochem. 56, 753–759 10.1139/o78-113 [DOI] [PubMed] [Google Scholar]
- 10.Jankowski J., Schröter A., Tepel M., van der Giet M., Stephan N., Luo J. et al. (2000) Isolation and characterization of Coenzyme A glutathione disulfide as a parathyroid-derived vasoconstrictive factor. Circulation 102, 2548–2552 10.1161/01.CIR.102.20.2548 [DOI] [PubMed] [Google Scholar]
- 11.Luo J., Jankowski V., Henning L., Schlüter H., Zidek W. and Jankowski J. (2006) Endogenous coenzyme A glutathione disulfide in human myocardial tissue. J. Endocrinol. Invest. 29, 688–693 10.1007/BF03344177 [DOI] [PubMed] [Google Scholar]
- 12.Schlüter H., Meissner M., van der Giet M., Tepel M., Bachmann J., Gro, I. et al. (1995) Coenzyme A glutathione disulfide: a potent vasoconstrictor derived from the adrenal gland. Circ. Res. 76, 675–680 10.1161/01.RES.76.4.675 [DOI] [PubMed] [Google Scholar]
- 13.Bees W.C.H. and Loewen P.C. (1979) Partial characterization of the mode of inhibition of Escherichia coli RNA polymerase by the mixed disulfide, CoASSG. Can. J. Biochem. 57, 336–345 10.1139/o79-043 [DOI] [PubMed] [Google Scholar]
- 14.Van Laer K., Hamilton C.J. and Messens J. (2013) Low-molecular-weight thiols in thiol–disulfide exchange. Antioxid. Redox Signal. 18, 1642–1653 10.1089/ars.2012.4964 [DOI] [PubMed] [Google Scholar]
- 15.Jacob C., Battaglia E., Burkholz T., Peng D., Bagrel D. and Montenarh M. (2012) Control of oxidative posttranslational cysteine modifications: from intricate chemistry to widespread biological and medical applications. Chem. Res. Toxicol. 25, 588–604 10.1021/tx200342b [DOI] [PubMed] [Google Scholar]
- 16.Wouters M.A., Iismaa S., Fan S.W. and Haworth N.L. (2011) Thiol-based redox signalling: rust never sleeps. Int. J. Biochem. Cell Biol. 43, 1079–1085 10.1016/j.biocel.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 17.Huth W., Pauli, C. and Möller, U. (1996) Immunochemical detection of CoA-modified mitochondrial matrix proteins. Biochem. J. 320, 451–457 10.1042/bj3200451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwerdt G., Möller U. and Huth W. (1991) Identification of the CoA-modified forms of mitochondrial acetyl-CoA acetyltransferase and of glutamate dehydrogenase as nearest-neighbour proteins. Biochem. J. 280, 353–357 10.1042/bj2800353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chodavarapu S., Hertema H., Huynh T., Odette J., Miller R., Fullerton A. et al. (2007) Reversible covalent inhibition of a phenol sulfotransferase by coenzyme A. Arch. Biochem. Biophys. 457, 197–204 10.1016/j.abb.2006.10.016 [DOI] [PubMed] [Google Scholar]
- 20.Thorneley R.N.F., Abell C., Ashby G.A., Drummond M.H., Eady R.R., Huff S. et al. (1992) Posttranslational modification of Klebsiella pneumoniae flavodoxin by covalent attachment of coenzyme A, shown by phosphorus-31 NMR and electrospray mass spectrometry, prevents electron transfer from the nifJ protein to nitrogenase. A possible new regulatory mechanism for biological nitrogen fixation. Biochemistry 31, 1216–1224 10.1021/bi00119a035 [DOI] [PubMed] [Google Scholar]
- 21.Lee J.-W., Soonsanga S. and Helmann J.D. (2007) A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl Acad. Sci. U.S.A. 104, 8743–8748 10.1073/pnas.0702081104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuchiya Y., Peak-Chew S.Y., Newell C., Miller-Aidoo S., Mangal S., Zhyvoloup A. et al. (2017) Protein CoAlation: a redox-regulated protein modification by coenzyme A in mammalian cells. Biochem. J. 474, 2489–2508 10.1042/BCJ20170129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malanchuk O.M., Panasyuk G.G., Serbyn N.M., Gout I.T. and Filonenko V.V. (2015) Generation and characterization of monoclonal antibodies specific to Coenzyme A. Biopolym. Cell 31, 187–192 10.7124/bc.0008DF [DOI] [Google Scholar]
- 24.Mukherjee S., Dutta D., Saha B. and Das A.K. (2010) Crystal structure of glyceraldehyde-3-phosphate dehydrogenase 1 from methicillin-resistant Staphylococcus aureus MRSA252 provides novel insights into substrate binding and catalytic mechanism. J. Mol. Biol. 401, 949–968 10.1016/j.jmb.2010.07.002 [DOI] [PubMed] [Google Scholar]
- 25.Imber M., Huyen N.T.T., Pietrzyk-Brzezinska A.J., Loi V.V., Hillion M., Bernhardt J. et al. (2018) Protein S-Bacillithiolation functions in thiol protection and redox regulation of the glyceraldehyde-3-phosphate dehydrogenase gap in Staphylococcus aureus under hypochlorite stress. Antioxid. Redox Signal. 28, 410–430 10.1089/ars.2016.6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuchiya Y., Pham U., Hu W., Ohnuma S.-i. and Gout I. (2014) Changes in acetyl CoA levels during the early embryonic development of Xenopus laevis. PLoS ONE 9, e97693 10.1371/journal.pone.0097693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox J. and Mann M. (2008) Maxquant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 28.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S., Lee D., Sillitoe I., Dawson N.L., Lees J.G. and Orengo C.A. (2015) Functional classification of CATH superfamilies: a domain-based approach for protein function annotation. Bioinformatics 31, 3460–3467 10.1093/bioinformatics/btv398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S., Sillitoe I., Lee D., Lees J.G., Dawson N.L., Ward J. et al. (2015) CATH FunFHMMer web server: protein functional annotations using functional family assignments. Nucleic Acids Res. 43, W148–W153 10.1093/nar/gkv488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Šali A. and Blundell T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 32.Hess B., Kutzner C., van der Spoel D. and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 33.Cavalli A., Spitaleri A., Saladino G. and Gervasio F.L. (2015) Investigating drug–target association and dissociation mechanisms using metadynamics-based algorithms. Acc. Chem. Res. 48, 277–285 10.1021/ar500356n [DOI] [PubMed] [Google Scholar]
- 34.Bonomi M., Branduardi D., Bussi G., Camilloni C., Provasi D., Raiteri P. et al. (2009) PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 180, 1961–1972 10.1016/j.cpc.2009.05.011 [DOI] [Google Scholar]
- 35.Lindorff-Larsen K., Piana, S., Palmo, K., Maragakis, P., Klepeis, J.L., Dror, R.O. et al. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 10.1002/prot.22711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Best R.B. and Hummer G. (2009) Optimized molecular dynamics force fields applied to the helix–coil transition of polypeptides. J. Phys. Chem. B 113, 9004–9015 10.1021/jp901540t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W. and Klein M.L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 38.Essmann U., Perera L. and Berkowitz M.L. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 39.Ezraty B., Gennaris A., Barras F. and Collet J.-F. (2017) Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 15, 385–396 10.1038/nrmicro.2017.26 [DOI] [PubMed] [Google Scholar]
- 40.Loi V.V., Rossius M. and Antelmann H. (2016) Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 6, 187 10.3389/fmicb.2015.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winterbourn C.C. and Kettle A.J. (2013) Redox reactions and microbial killing in the neutrophil phagosome . Antioxid. Redox Signal. 18, 642–660 10.1089/ars.2012.4827 [DOI] [PubMed] [Google Scholar]
- 42.Chi B.K., Busche T., Van Laer K., Bäsell K., Becher D., Clermont L. et al. (2014) Protein S-mycothiolation functions as redox-switch and thiol protection mechanism in Corynebacterium glutamicum under hypochlorite stress. Antioxid. Redox Signal. 20, 589–605 10.1089/ars.2013.5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimizu K. (2016) Metabolic regulation and coordination of the metabolism in bacteria in response to a variety of growth conditions. Adv. Biochem. Eng. Biotechnol. 155, 1–54 10.1007/10_2015_320 [DOI] [PubMed] [Google Scholar]
- 44.Hillion M. and Antelmann H. (2015) Thiol-based redox switches in prokaryotes. Biol. Chem. 396, 415–444 10.1515/hsz-2015-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Painter K.L., Krishna A., Wigneshweraraj S. and Edwards A.M. (2014) What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol. 22, 676–685 10.1016/j.tim.2014.09.002 [DOI] [PubMed] [Google Scholar]
- 46.Chi B.K., Gronau K., Mäder U., Hessling B., Becher D. and Antelmann H. (2011) S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell Proteomics 10, M111.009506 10.1074/mcp.M111.009506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun F., Liang H., Kong X., Xie S., Cho H., Deng X. et al. (2012) Quorum-sensing agr mediates bacterial oxidation response via an intramolecular disulfide redox switch in the response regulator AgrA. Proc. Natl Acad. Sci. U.S.A. 109, 9095–9100 10.1073/pnas.1200603109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eymann C., Homuth G., Scharf C. and Hecker, M. (2002) Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J. Bacteriol. 184, 2500–2520 10.1128/JB.184.9.2500-2520.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leichert L.I., Gehrke F., Gudiseva H.V., Blackwell T., Ilbert M., Walker A.K. et al. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl Acad. Sci. U.S.A. 105, 8197–8202 10.1073/pnas.0707723105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fratelli M., Demol H., Puype M., Casagrande S., Eberini I., Salmona M. et al. (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl Acad. Sci. U.S.A. 99, 3505–3510 10.1073/pnas.052592699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chardonnet S., Sakr S., Cassier-Chauvat C., LeMarechal P., Chauvat F., Lemaire S.D. et al. (2015) First proteomic study of S-glutathionylation in cyanobacteria. J. Proteome Res. 14, 59–71 10.1021/pr500625a [DOI] [PubMed] [Google Scholar]
- 52.Lillig C.H., Potamitou A., Schwenn J.-D., Vlamis-Gardikas A. and Holmgren A. (2003) Redox regulation of 3′-phospho adenylyl sulfatereductase from Escherichia coli by glutathione and glutaredoxins. J. Biol. Chem. 278, 22325–22330 10.1074/jbc.M302304200 [DOI] [PubMed] [Google Scholar]
- 53.Weber H., Engelmann S., Becher D. and Hecker M. (2004) Oxidative stress triggers thiol oxidation in the glyceraldehyde-3-phosphate dehydrogenase of Staphylococcus aureus. Mol. Microbiol. 52, 133–140 10.1111/j.1365-2958.2004.03971.x [DOI] [PubMed] [Google Scholar]
- 54.Nicholls C., Li H. and Liu J.-P. (2012) GAPDH: a common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 39, 674–679 10.1111/j.1440-1681.2011.05599.x [DOI] [PubMed] [Google Scholar]
- 55.Hildebrandt T., Knuesting J., Berndt C., Morgan B. and Scheibe R. (2015) Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 396, 523–537 10.1515/hsz-2014-0295 [DOI] [PubMed] [Google Scholar]
- 56.Jackowski S. and Rock C.O. (1981) Regulation of coenzyme A biosynthesis. J. Bacteriol. 148, 926–932 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jackowski S. and Rock C.O. (1986) Consequences of reduced intracellular coenzyme A content in Escherichia coli. J. Bacteriol. 166, 866–871 10.1128/jb.166.3.866-871.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]