

SUMMARY
Translation of mRNAs is tightly regulated and constantly surveyed for errors. Aberrant translation can trigger co-translational protein and RNA quality control processes, impairments of which cause neurodegeneration by still poorly understood mechanism(s). Here we show that quality control of translation of mitochondrial outer membrane (MOM)-localized mRNA intersects with the turnover of damaged mitochondria, both orchestrated by the mitochondrial kinase PINK1. Mitochondrial damage causes stalled translation of complex-I 30 kD subunit (C-I30) mRNA on MOM, triggering the recruitment of co-translational quality control factors Pelo, ABCE1, and NOT4 to the ribosome/mRNA-ribonucleoprotein complex. Damage-induced ubiquitination of ABCE1 by NOT4 generates poly-Ubiquitin signals that attract autophagy receptors to MOM to initiate mitophagy. In the Drosophila PINK1 model, these factors act synergistically to restore mitophagy and neuromuscular tissue integrity. Thus ribosome-associated co-translational quality control generates an early signal to trigger mitophagy. Our results have broad therapeutic implications for the understanding and treatment of neurodegenerative diseases.
Keywords: mitophagy, mitochondrial quality control, PINK1, ABCE1, NOT4, co-translational quality control, ribosome stalling, ribosome/mRNP remodeling, ubiquitination, autophagy receptor
eTOC Blurb
Removal of damaged mitochondrial is essential for maintaining cellular vitality but the earliest signal that initiates the mitophagy process is not well defined. Wu et al. show that mitochondrial damage causes stalled translation of OXPHOS-related mRNAs on the mitochondrial surface. Co-translational quality control of stalled ribosomes generates ubiquitin-containing signals that trigger mitophagy.
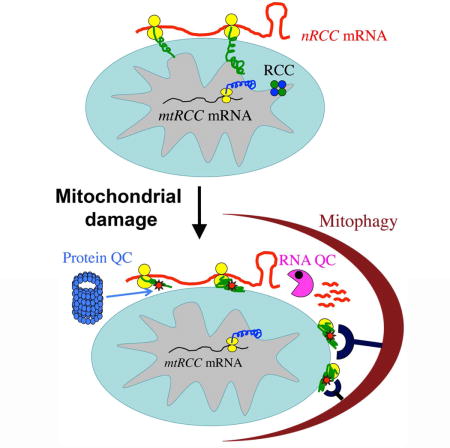
INTRODUCTION
Mitochondria are dynamic and complex organelles with essential roles in many aspects of cellular physiology, ranging from energy production and intermediary metabolism to calcium and redox homeostasis and cell death. These broad functions also position mitochondria as a central player in human health. Thus, mitochondrial dysfunction can cause a wide range of diseases, including cancer, diabetes, and neurodegenerative diseases, and has been implicated in aging and age-related conditions (Chan, 2006; Schon and Przedborski, 2011; Wallace, 2005). Autophagy, an evolutionarily conserved process that helps cells survive stress conditions by degrading self-constituents, has also been linked to the human disease and aging conditions associated with mitochondrial dysfunction (Green and Levine, 2014; Levine and Kroemer, 2008). It thus follows that mitochondrial function and autophagy are intimately linked in health and disease.
Elaborate quality control systems are deployed to maintain mitochondrial integrity and functionality (Narendra and Youle, 2011; Rugarli and Langer, 2012). Mitophagy, the selective removal of unwanted or damaged mitochondria by autophagy, is a key mechanism by which eukaryotic cells exercise mitochondrial quality control (Bingol and Sheng, 2016; Durcan and Fon, 2015; Pickrell and Youle, 2015). Defects in this process can lead to the accumulation of dysfunctional or damaged mitochondria, releasing damaging signals such as reactive oxygen species and cytochrome C and leading to inflammation, genotoxic stress, tumorigenesis, neurodegeneration, and possibly premature aging. PINK1 (Valente et al., 2004) and Parkin (Kitada et al., 1998), two proteins associated with familial Parkinson’s disease (PD), have been identified as central players in a cellular pathway that directs mitochondrial quality control. Studies in mammalian cells showed that Parkin, a normally cytosolic protein, is recruited in a PINK1-dependent manner to dysfunctional mitochondria exhibiting reduced membrane potential (MMP) (Narendra et al., 2008; Narendra et al., 2010). Subsequent work showed that Parkin induces broad ubiquitination and degradation of mitochondrial outer membrane (MOM) proteins through the ubiquitin proteasome system (Chan et al., 2011b; Sarraf et al., 2013). The prevailing view is that ubiquitinated MOM proteins generated by Parkin serve as a ‘eat me’ signal on damaged mitochondria, which is recognized by ubiquitin-binding autophagy receptor proteins such as Optineurin, NDP52, p62, and their regulator TBK1, resulting in the recruitment of autophagy machinery to clear the damaged organelles (Nguyen et al., 2016). Importantly, while the central role of PINK1 in the mitophagy process is indisputable, the role of Parkin appears to be facilitating and amplifying, as autophagy receptor recruitment and mitophagy can occur in the absence of Parkin activity (Lazarou et al., 2015; Richter et al., 2016). The Parkin-independent events downstream of PINK1 in the mitophagy process, for example the key substrate(s) and the E3 ligase(s) responsible for generating the mitophagy-inducing poly-Ub signals, however are largely unknown.
In addition to its role in mitophagy, PINK1 has been implicated in a number of other mitochondrial processes in Drosophila, including complex-I function (Liu et al., 2011; Vilain et al., 2012), fission/fusion dynamics (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008), transport (Liu et al., 2012; Wang et al., 2011), and localized translation of nuclear encoded respiratory chain component (nRCC) mRNAs such as C-I30 mRNA encoding the 30 kD subunit of complex-I (Gehrke et al., 2015). Regulated mRNA translation on MOM by PINK1 allows co-translational protein import, offering an elegant way to coordinate the assembly of the electron transport chain complexes, whose components are dually encoded by the nuclear and mitochondrial genomes. We wondered whether the role of PINK1 in the control of MOM-associated nRCC mRNA translation might be mechanistically linked to its other role in mitophagy induction, as nascent peptide chain synthesis on translating ribosomes are subjected to dynamic ubiquitination and quality control (Brandman and Hegde, 2016; Lykke-Andersen and Bennett, 2014), and defects in ribosomes-associated quality control processes can lead to neurodegeneration through still poorly understood mechanism (Chu et al., 2009; Ishimura et al., 2014).
By characterizing the composition and modification of C-I30 mRNA-ribonucleoprotein (mRNP) complex during the course of mitophagy induction, we tested the hypothesis that mitochondrial damage-induced remodeling of the nRCC mRNP on MOM may mediate the function of PINK1 in maintaining normal mitochondrial function and in removing damaged mitochondria. Our results support this model and further identify biochemical interaction between ribosome-associated quality control factors NOT4 and ABCE-1 as a key event in generating poly-ubiquitinated ABCE1, which serves as a molecular signal to recruit autophagy receptors and initiate mitophagy. These findings expand our view of PINK1-directed mitochondrial quality control into a continuum of quality control process encompassing co-translational RNA and protein quality control and mitophagy, with important implications for our understanding of the molecular mechanisms of mitophagy and the pathogenesis of neurodegeneration associated with defective ribosome-associated translational surveillance.
RESULTS
Recruitment of Autophagy Receptors to MOM-Associated mRNPs in Response to Mitochondrial Damage
Through biochemical analysis of purified mitochondria, we found that the autophagy receptors OPTN, NDP52, p62, and their regulator TBK1 (Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016; Wong and Holzbaur, 2014), showed increased recruitment to mitochondria in HeLa cells in response to treatment of cells with 20 μM CCCP (Figure 1A), a condition commonly used to induce mitophagy (Narendra et al., 2008). In contrast, PINK1 knockout (PINK1 −/−) HeLa cells did not show such response, although there is still basal recruitment of autophagy receptors to mitochondria (Figure S1A). As Parkin expression is silenced in HeLa cells (Narendra et al., 2008), these results are consistent with the notion of PINK1-dependent, but Parkin-independent, mitophagy induction (Lazarou et al., 2015; Richter et al., 2016). The purity of the Percoll-gradient purified mitochondria was verified by the absence of marker proteins from other organelles, including the ER (Figure S1B; data not shown). In GFP-Parkin stably transfected HeLa cells, the mitochondrial recruitment of autophagy receptors was enhanced compared to HeLa cells (Figure 1A), suggesting that although not required, Parkin can facilitate this process. Intriguingly, treatment with RNase A released autophagy receptors, poly-ubiquitinated proteins, and LC3B-II-the autophagy-associated form of LC3B (Kabeya et al., 2000) from mitochondria, with more release seen from mitochondria experiencing shorter CCCP treatment (0.5hr vs. 3hr, 20 μM CCCP; Figure 1B, S1C). Mitochondrial proteins, including the MOM proteins Tom20 and Tom40, were not released, suggesting that mitochondrial integrity was not compromised (Figure 1B, S1C). This result pointed to the presence of an RNase-sensitive component that recruits autophagy receptors to MOM. EDTA treatment, which is commonly used to dissociate ribosome subunits (Utsunomiya and Roth, 1966), resulted in more efficient autophagy receptors release (Figure 1B, S1C). These data together support a role of ribosome/mRNP in anchoring autophagy receptors to MOM. Mitochondrial poly-Ub signals were more readily releasable from HeLa cells than HeLa/GFP-Parkin cells, indicating that the Parkin-independent poly-Ub signals in HeLa cells were mostly contributed by MOM-associated mRNPs, whereas Parkin-dependent poly-Ub signals, e.g. those added to MOM proteins Miro and Mfn (Chan et al., 2011a), may reside on mitochondria independently of ribosome/mRNPs.
Figure 1. Recruitment of Autophagy Receptors to Mitochondria-Localized mRNPs in PINK1-Activated Mitophagy.
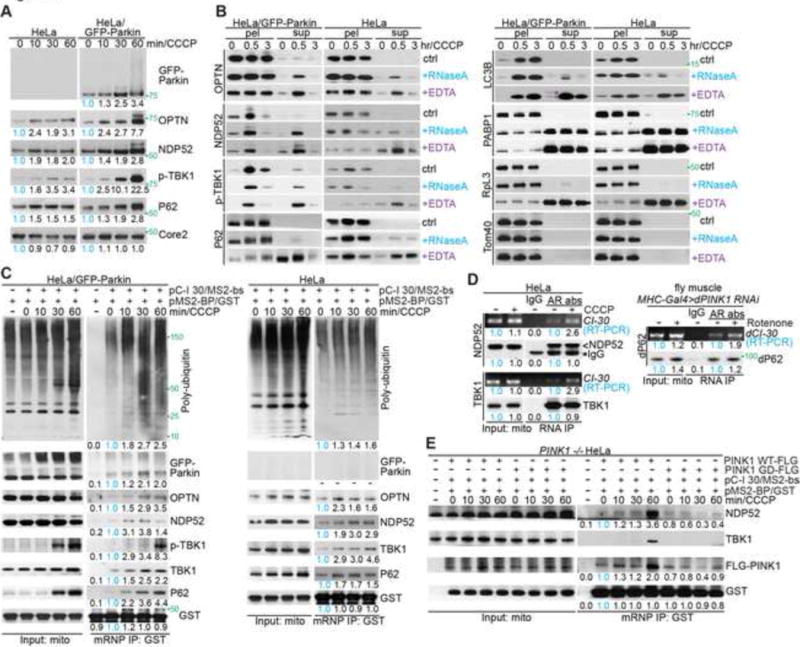
(A) Immunoblots of mitochondrial fractions from HeLa or HeLa/GFP-Parkin cells treated with 20 μM CCCP. Ubiquinol-cytochrome c reductase complex core protein 2 (Core 2) serves as mitochondrial loading control. Values below the blots represent quantification of normalized protein levels.
(B) RNase A and EDTA treatments of mitochondrial fractions from HeLa or HeLa/GFP-Parkin cells. Immunoblots of mitochondrial pellets (pel) and released fractions (sup) are shown. * and < mark LC3B-I and LC3B-II, respectively.
(C) Immunoblots of C-I30 mRNP purified from mitochondria of HeLa or HeLa/GFP-Parkin cells co-transfected with plasmids expressing MS2-bs-tagged C-I30 mRNA and GST-tagged MS2 protein and treated with 20 μM CCCP.
(D) RNA-IP showing mitochondrial association of C-I30 mRNA with TBK1 and NDP52 in HeLa cells treated with 20 μM CCCP, or with p62 in muscle tissue from flies treated with rotenone (200 μM supplied in fly food). AR: autophagy receptor.
(E) Immunoblots of C-I30 mRNP purified from mitochondria of PINK1(−/−) HeLa cells transfected with PINK1-WT or pathogenic PINK1-G309D.
See also Figure S1.
As CCCP is a strong depolarizer of mitochondria, and it can depolarize other organelles and membranes, we next performed a number of control experiments to assess the specificity of the CCCP effect. First, we showed that the effects of CCCP on mitochondrial membrane potential and ATP production were reversible (Figure S1D, S1E), and that there is no cell death during the period of drug treatment when cells were analyzed (Figure S1F, S1G). Second, in the period (0-1hr) when we observed autophagy receptor recruitment to CCCP-damaged mitochondria, there was no drop of ATP production (Figure S1H) or global protein synthesis inhibition (actin, Figure S1I; also see Figure S5M later for C-I75, Tom20, and Tubulin). Third, the effect of mitochondrial damage on autophagy receptor recruitment was also observed when cells were treated with electron transport chain inhibitors Antimycin A (10 μM) and Oligomycin (10 μM), suggesting that it was not unique to CCCP (Figure S1J, input: mito panel).
PINK1 was recently shown to recruit several nuclear-encoded respiratory chain component (nRCC) mRNAs to MOM where it controls their localized translation. One such nRCC mRNA is C-I30 (Gehrke et al., 2015). Using a MS2 binding site (MS2-bs)-tagging strategy that allows one-step affinity purification of C-I30 mRNPs (Gehrke et al., 2015), we found that autophagy receptors are recruited to mitochondrial C-I30 mRNPs in HeLa and HeLa/GFP-Parkin cells (Figure 1C), but not PINK1−/− HeLa cells (Figure S2A), indicating that they are recruited to the mRNPs in a PINK1-dependent, but Parkin-independent manner. RNA immunoprecipitation (RNA-IP) showed that endogenous C-I30 mRNA also associated with autophagy receptors in response to mitochondrial damage in HeLa cells and fly tissues (Figure 1D), and RT-PCR showed that near physiological level of MS2-bs-C-I30 mRNA expression (Figure S2B), suggesting that the mRNP/autophagy receptor association detected above was not specific to MS2-bs-tagged mRNAs. Compared to PINK1-WT, disease-associated PINK1-G309D mutant (Valente et al., 2004) was defective in recruiting autophagy receptors to C-I30 mRNP when introduced into PINK1(−/−) mutant cells (Figure 1E), and interfered with the activity of endogenous PINK1 in this respect (Figure S2C), supporting the disease relevance of this PINK1 function.
We investigated the mechanism by which autophagy receptors are recruited to C-I30 mRNPs. By immunofluorescence detection of MS2-bs-tagged C-I30 mRNP in situ in cells, we found that within minutes of mitochondrial damage, there was increased C-I30 mRNP recruitment to mitochondria (Figure 2A, B). Endogenous C-I30 mRNA also exhibited enhanced PINK1-dependent mitochondrial recruitment in response to mitochondrial damage by CCCP in mammalian cells (Figure S2D) or by rotenone in Drosophila muscle (Figure S2E). Oxidative stress with H2O2, or mitochondrial unfolded protein stress had no obvious effect (Figure S2F, G), suggesting that specific types of mitochondrial damage, presumably those that affect the electron transport chain or cause dramatic MMP loss, are capable of inducing mRNP recruitment to MOM. Increased recruitment of PINK1 mRNA in response to mitochondrial damage was also observed in fly muscle (Figure S2E), the significance of which awaits further investigation.
Figure 2. Association of Co-Translational Quality Control Factors with Stalled Ribosomes on Damaged Mitochondria.
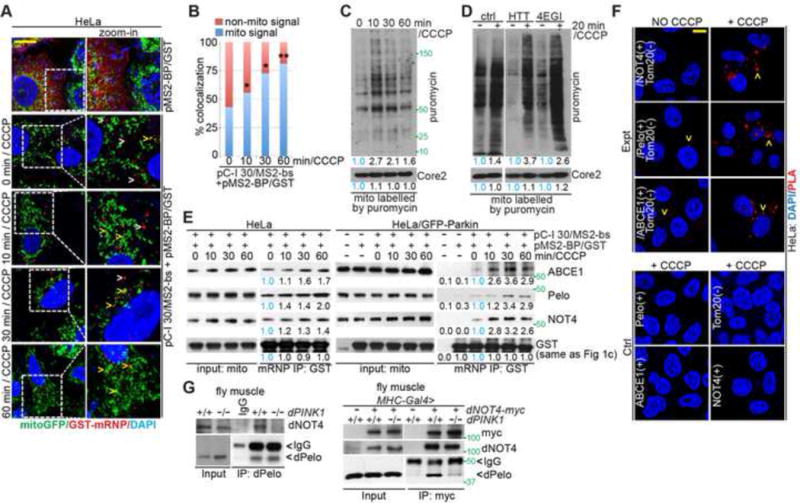
(A) Imaging of C-I 30 mRNA localization to MOM in HeLa cells using the MS2-bs/MS2-GST system. Arrowheads: white, free mRNPs; yellow: mitochondrially localized mRNPs.
(B) Qualification of data shown in A, with % of C-I 30 mRNA signals colocalizing with mitochondria indicated. *, p<0.05; **, p<0.01; Chi-squared test. More than 150 foci from 15~20 cells in each group were counted.
(C) Puromycin labeling of nascent peptide chains (NPCs) on MOM in CCCP treated cells. Cells untreated (time 0) or treated with CCCP were labeled with puromycin/emetin 5 mins before the end of the indicated CCCP treatment times. Mitochondrial fractions were purified and analyzed by western blot analysis with the indicated antibodies.
(D) Puromycin labeling of NPCs on MOM in cells pretreated with homoharringtonine (HTT) or 4EGI. HTT and 4EGI were added 10 min before the addition of puromycin/emetin to inhibit new translation initiation and allow active ribosomes to finish translation. Inhibition of new translation by HTT or 4EGI was evident by comparing the no-CCCP lanes with that of control (no drug treatment) samples. Mitochondrial fractions were analyzed as in C.
(E) Immunoblots of C-I30 mRNP purified from mitochondria of HeLa cells.
(F) Close proximity between co-translational quality control factors and MOM marker Tom20 as detected by PLA. Lower panels are negative controls.
(G) Reciprocal co-IP showing PINK1-dependent interaction between dPelo and dNOT4 in fly muscle.
See also Figure S2.
Stalled Translation of MOM-Associated C-I30 mRNA on Damaged Mitochondria
To assess the translational status of MOM-associated ribosome/mRNPs, we treated purified mitochondria with puromycin, which can be incorporated into elongating as well as stalled nascent peptide chains (NPCs) (Graber et al., 2013). We observed increased synthesis of NPCs on MOM minutes after CCCP treatment (Figure 2C). Control experiments showed that puromycin was incorporated mainly into MOM-associated cytosolic mRNP/ribosomes, not the mitochondrial ones in the matrix (Figure S2H). Remarkably, pre-treatment of cells with the translation inhibitor 4EGI (Moerke et al., 2007), or homoharringtonine (HTT) (Huang, 1975), to inhibit new translation initiation while allowing active/elongating ribosomes to run off, reduced basal puromycin incorporation but not CCCP-induced incorporation (Figure 2D), suggesting that CCCP causes stalling of MOM-associated NPC synthesis. Translational stalling activates co-translational quality control mechanisms that target aberrant or incompletely synthesized/folded NPCs and the template mRNAs for degradation. We observed increased ubiquitination, primarily K48-linked ubiquitination, of MOM-associated NPCs immediately upon CCCP treatment (Figure S2I). Moreover, MOM-associated C-I30 mRNA level was reduced upon longer CCCP treatment (60 min) in HeLa/GFP-Parkin cells (Figure S2D), suggesting the activation of a RNA quality control mechanism.
Recruitment of No-Go Decay-Related Co-translational Quality Control Factors to MOM-Associated mRNPs That Involves PINK1
We next sought to determine what mechanism is activated for the surveillance of MOM-associated translation of C-I30 mRNAs. We analyzed C-I30 mRNPs purified from the mitochondrial fractions of control cells and cells treated with 20 μM CCCP at different time points within an hour. Pelo and ABCE1, critical components of the No-Go Decay (NGD) pathway that form a complex during co-translational quality control of stalled ribosomes to dissociate ribosomes from mRNAs (Doma and Parker, 2006; Pisareva et al., 2011; Shao and Hegde, 2014; Shoemaker et al., 2010), were recruited to C-I30 mRNP upon CCCP treatment in HeLa or HeLa/GFP-Parkin cells (Figure 2E). NOT4, originally identified as a component of the CCR4-NOT complex involved in transcription and RNA degradation (Collart and Struhl, 1994) but could also act as a ribosome-associated E3 ligase that targets NPCs on stalled ribosomes for ubiquitination and degradation (Dimitrova et al., 2009), was also recruited to C-I30 mRNP on MOM (Figure 2E). The recruitment of NGD factors to MOM-associated C-I30 mRNP was observed in cells treated with high (20 μM, Figure 2E) and low (5 μM, Figure S3A) concentrations of CCCP, or in cells treated with Antimycin A/Oligomycin (Figure S1J). Pelo, ABCE1, and NOT4 association with endogenous C-I30 mRNA, and enhancement of ABCE1 association with mRNA by CCCP were also observed (Figure S3B). Using proximity ligation assay, we confirmed CCCP-induced colocalization of Pelo, ABCE1 and NOT4 with the MOM marker Tom20 (Figure 2F), and ABCE1 with C-I30 mRNP in HeLa cells (Figure S3C). Recruitment of NGD-related factors to C-I30 mRNP, and evidence of ribosomal subunit splitting, as shown by the time-dependent decrease of 40S subunit protein RpS6 but increase of 60S subunit protein RpL7a on mitochondrial surface (Figure S3D), further supported the notion that CCCP causes translational stalling. The resulting 60S:peptidyl-tRNA complex may be the substrate of the ribosome-associated quality control complex (RQC) as shown in yeast (Brandman and Hegde, 2016). Mitochondrial depolarization by CCCP presumably arrest the co-translational import of C-I30 and other NPCs through the TOM/TIM complex, which depends on MMP for function (Neupert, 1997), leading to stalled translation. Supporting in vivo relevance, we observed recruitment of ABCE1 and Pelo to damaged mitochondria in PINK1 RNAi animals (Figure S3E, F).
Consistent with Pelo/ABCE1 and NOT4 being recruited to MOM through mRNP/ribosomes, the association of these factors with mitochondria was EDTA-and RNase A-sensitive (Figure S3G). In HEK293 cells expressing PINK1-G309D, Pelo and ABCE1 recruitment to C-I30 mRNP was increased, whereas NOT4 recruitment was reduced (Figure S3H). Thus, Pelo/ABCE1 is recruited to C-I30 mRNP on MOM in response to mitochondrial damage by CCCP or pathogenic PINK1, but the recruitment of NOT4 to C-I30 mRNP appears to depend on PINK1. Although Pelo, ABCE1, and NOT4 all act as co-translational quality control factors, they seem to be differentially regulated by PINK1.
We further investigated the biochemical relationships between the co-translational QC factors and PINK1. Pelo and ABCE1 both exhibited co-IP with NOT4 and PINK1 (Figure S3I, J); however, the interaction between Pelo and NOT4 was abolished in PINK1 mutant (Figure 2G). Notably, NOT4 overexpression in flies significantly increased the steady-state levels of Pelo (Figure S3K) and ABCE1 protein (see Figure 5A later), suggesting the existence of feedback regulatory mechanisms that reinforce the connection between the co-translational quality control factors in vivo. Similar regulation of ABCE1 and Pelo levels by NOT4 were also observed in HeLa cells (Figure S3L).
Figure 5. Direct Ubiquitination of ABCE1 by NOT4 Generates a Poly-Ub Signal for Autophagy Receptor Recruitment and Mitophagy Induction.

(A) Immunoblots of denaturing HA IP from Mhc>PINK1 RNAi fly muscle expressing dABCE1-HA and with dNOT4 overexpressed or knocked down. Arrowheads mark unmodified dABCE1-HA.
(B) Immunoblot of Ub showing in vitro ubiquitination of ABCE1 by NOT4. Diagram depicts the effect of K20R mutations in ABCE1 on its ubiquitination by NOT4.
(C) Immunoblots of C-I30 mRNP purified from mitochondria of HeLa cells transfected with NOT4 cDNA, with or without ABCE1 knockdown, and with or without 20 μM CCCP treatment.
(D) Immunoblots of Flag IP from HeLa cells transfected with Flag-ABCE1 or Flag-ABCE1-K20R.
(E) Immunoblots showing the effect of WT vs. K20R mutant ABCE1 transfection on the recruitment of autophagy receptors to C-I30 mRNP in HeLa cells treated with 20 μM CCCP.
(F) Immunostaining showing the rescue of ABCE1 siRNA-induced mitochondrial clearance defect by the WT but not K20R form of ABCE1. Blue fluorescence represents expression of Flag tag carried by the rs ABCE1 cDNA constructs. Mitochondria are stained with anti-Tom20. Note the aggregated mitochondrial morphology caused by 24 hr CCCP treatment.
(G) Quantification of data shown in F. *, #, p<0.05 in Chi-squared test. For the siRNA knockdown groups, more than 200 cells were counted; while for the rsABCE1 rescuing groups, more than 60 positive cells were found and counted.
See also Figure S5.
NGD-Related Quality Control Factors Interact with Autophagy Receptors and Are Required for Their Recruitment in PINK1-Activated Mitophagy
We next tested the role of the co-translational quality control factors in the recruitment of autophagy receptors to C-I30 mRNP on MOM. Pelo, ABCE1, or NOT4 knockdown by siRNA attenuated damage-induced early recruitment of autophagy receptors to mitochondria or MOM-associated C-I30 mRNP in HeLa (Figure S4C) or HeLa/GFP-Parkin (Figure 3A) cells. The RNAi effects were verified for specificity and rescued by the co-transfection of siRNA-resistant (rs) cDNAs (Figure S4A–G). Pelo, ABCE1, or NOT4 knockdown significantly reduced the levels of poly-Ub associated with purified mitochondria or C-I30 mRNP (Figure 3A), impaired Parkin recruitment (Figure 3B, S4H) and removal of damaged mitochondria in HeLa/GFP-Parkin cells treated with 20 μM CCCP for 24 hrs (Figure 3C, D; S4F, G), demonstrating the critical involvement of co-translational quality control factors in PINK1-directed mitophagy.
Figure 3. Co-Translational Quality Control Factors Are Required for Autophagy Receptor Recruitment and Mitophagy Induction.
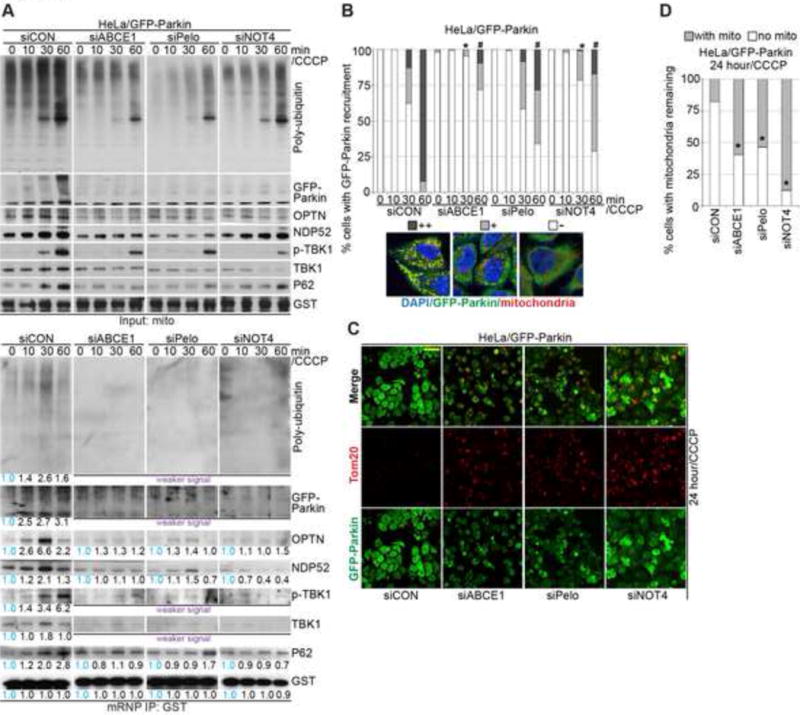
(A) Immunoblots of C-I30 mRNP purified from mitochondrial fractions from HeLa/GFP-Parkin cells transfected with control siRNA (siCON) or siRNAs against ABCE1, Pelo, or NOT4. In this figure and some following figures, some panels with weaker signals than control or with negligible signals are indicated, but due to the low signals they are not quantified.
(B) Effect of ABCE1, Pelo, or NOT4 knockdown on Parkin recruitment to damaged mitochondria in HeLa/GFP-Parkin cells. The images show representative categories of Parkin-GFP recruitment phenotypes: ++, strong; +, weak; −, negligible. *, p<0.05 vs. siCON in 30 min 20 μM CCCP treatment group; #, p<0.05 vs. siCON in 60 min 20 μM CCCP treatment group; Chi-squared test. More than 200 cells were counted in each group.
(C) Effect of ABCE1, Pelo, or NOT4 knockdown on mitochondrial clearance in HeLa/GFP-Parkin cells treated with 20 μM CCCP for 24 hrs.
(D) Quantification of data shown in C. *, p<0.05 vs. siCON in Chi-squared test.
See also Figure S3. Around 100 remaining cells were counted in each group.
Ubiquitination of ABCE1 by NOT4 in Response to Mitochondrial Damage
Interestingly, we found that ABCE1 was present in a poly-ubiquitinated form (poly-Ub-ABCE1) in both 20 μM CCCP-treated HeLa cells (Figure 4A) and PINK1-RNAi flies (Figure 4B), with K48-linked ubiquitination being more prominent than K63-or K11-linked one (Figure S5A). These results raised the interesting possibility that poly-Ub-ABCE1 in the stalled mRNPs on MOM might serve as an autophagy receptor-recruiting signal. Indeed, we found that ABCE1 (Figure 4A, Figure S5B) but not Pelo (Figure S5B, C) physically interact with the autophagy receptors. Moreover, ABCE1 and NOT4 promoted the recruitment of autophagy receptors to MOM-associated mRNPs and to mitochondria in HeLa cells (Figure 4C), HeLa/GFP-Parkin cells (Figure S5D), and fly tissue (Figure S5E), and the formation of autophagosome-like structures on HeLa cell mitochondria (Figure 4D). Notably, the level of poly-Ub-ABCE1 in response to mitochondrial damage was positively regulated by NOT4 (Figure 5A), and similar to Pelo (Figure S3K), ABCE1 protein accumulated under NOT4 overexpression condition in flies (Figure 5A, input panel). Promotion of ABCE1 poly-ubiquitination and its stabilization instead of degradation by NOT4 are consistent with poly-Ub-ABCE1 serving some signaling function.
Figure 4. Interaction between ABCE1 and Autophagy Receptors.
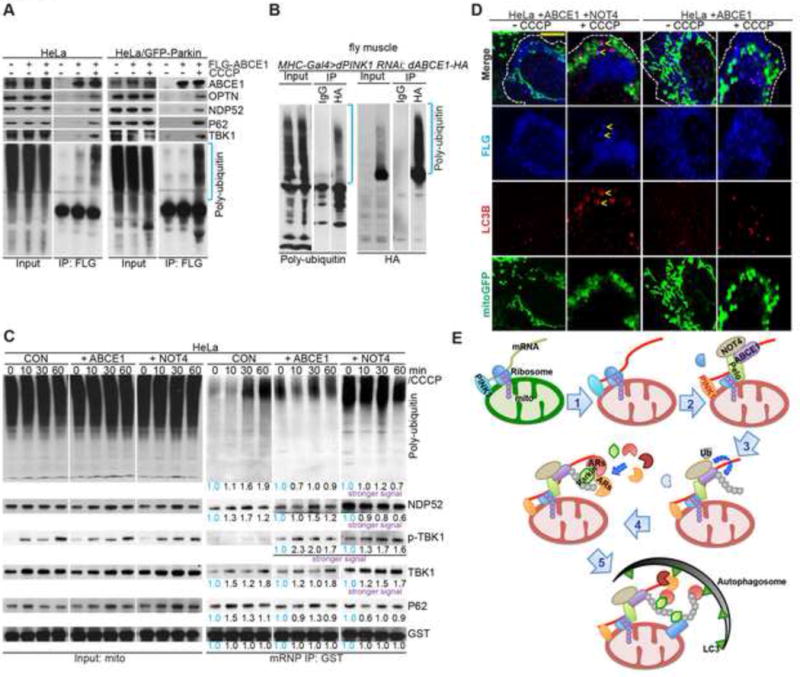
(A) Immunoblots of Flag IP from HeLa or HeLa/GFP-Parkin cells transfected with Flag-ABCE1.
(B) Immunoblots of denaturing HA IP from Mhc>PINK1 RNAi fly muscle co-expressing dABCE1-HA.
(C) Immunoblots of C-I30 mRNP purified from mitochondria of HeLa cells transfected with ABCE1 or NOT4 cDNA and treated with 20 μM CCCP at different time points.
(D) Immunostaining of HeLa cells transfected with ABCE1 or co-transfected with ABCE1 and NOT4. Arrows mark the induction of LC3-and ABCE1-positive structures on mitochondria in ABCE1/NOT4 co-transfected cells treated with 20 μM CCCP.
(E) Diagram illustrating the sequence of events from co-translational quality control of MOM-associated nRCC mRNA, mRNP remodeling, to autophagy machinery recruitment and mitophagy.
See also Figure S4.
We tested whether NOT4 might directly ubiquitinate ABCE. To this end, we performed in vitro ubiquitination assays using affinity-purified NOT4 and ABCE1 as the E3 ligase and substrate, respectively. ABCE1 from PINK1−/− HeLa cells, which is present largely as a non-ubiquitinated form (Figure S5F), was used for this purpose. NOT4 was able to directly ubiquitinate ABCE1 in vitro (Figure 5B). Moreover, using mutant forms of ubiquitin, we found that consistent with the ubiquitination pattern observed in vivo, ubiquitination of ABCE1 by NOT4 in vitro occurs primarily via K48-linked modification, as a K48-only form of Ub, but not a K48R mutant, was as efficiently incorporated into ABCE1 by NOT4 as wild type Ub (Figure S5G). The specificity of ABCE1 ubiquitination by NOT4 was confirmed by comparing the effect of NOT4 with a few other E3-ubiquitin ligases in both fly muscle tissues (Figure S5H) and human cells (Figure S5I).
Ubiquitination of ABCE1 is Critically Involved in Mitophagy Induction
We further examined the importance of NOT4-mediated ubiquitination of ABCE1 in mitophagy. While NOT4 overexpression stimulated the generation of poly-Ub signals and recruitment of autophagy receptors to MOM in HeLa cells, ABCE1 RNAi attenuated this effect (Figure 5C). Moreover, we generated a mutant form of ABCE1 in which all predicted potential ubiquitination sites are mutated (ABCE1-K20R). Compared to wild type ABCE1, ABCE1-K20R exhibited abolished ubiquitination by NOT4 in vitro (Figure 5B) and in cells (Figure 5D), and diminished abilities to recruit autophagy receptors to damaged mitochondria and to C-I30 mRNPs (Figure 5E), or to rescue the mitophagy defects caused by ABCE1-RNAi (Figure 5F, G). Thus, functional interplays between the co-translational quality control factors generate a key molecular signal (poly-Ub-ABCE1) on MOM that promotes mitophagy.
Remodeling of Protein-Protein Interactions Within MOM-Associated mRNPs May Dictate the Outcome of PINK1-Directed Mitochondrial Quality Control
PINK1-dependent increase of nRCC mRNA recruitment to MOM immediately upon mitochondrial stress, and mitophagy induction after prolonged mitochondrial damage, raised the intriguing possibility that PINK1 is deployed to promote repair of mildly damaged mitochondria by stimulating translation of nRCC mRNAs on one hand, and clearance of severely damaged mitochondria on the other hand. We first tested the hypothesis that PINK1-dependent recruitment of nRCC mRNA to MOM provides a protective mechanism against mild mitochondrial damage. We found that within the first 0.5hr treatment of HeLa cells with low (5 μM) concentration of CCCP, there was a boost of C-I30 protein expression (Figure S1I). This correlated well with the increased C-I30 mRNA recruitment during this period (Figure S2D). C-I30 protein level declined after 2hrs of CCCP treatment, likely due to stalled translation and induction of co-translational RNA quality control. This boost of C-I30 protein expression was not observed on PINK1(−/−) mutant HeLa cells treated with CCCP (Figure S5J). Measurement of ATP levels showed that there was a gradual decline of ATP production starting from 5 min of CCCP treatment in PINK1(−/−) mutant HeLa cells (Figure S5K), whereas wild type HeLa cells maintained ATP level well within the first hour of CCCP treatment and started to decline at 2 hrs of treatment when C-I30 level also started to decline (Figure S1I). These results support the notion that PINK1-dependent recruitment of nRCC mRNAs reflects a cellular repair mechanism to protect against mitochondrial damage.
We next tested whether remodeling of the mRNPs on MOM may mediate this shift of PINK1 function. In HeLa cells treated with 20 μM CCCP, Pelo/ABCE1 interaction, known to be critical for ribosome recycling and translation efficiency (Mancera-Martinez et al., 2017), was enhanced starting from the first 5 min of treatment but decreased after longer treatment, while the NOT4/ABCE1 interaction and ABCE1 poly-ubiquitination increased steadily during the treatment (Figure S5J). These molecular changes were not observed in PINK1 −/− HeLa cells (Figure S5L). Interestingly, while we observed reduced translation of C-I30 upon CCCP treatment of HeLa cells, for another complex-I subunit (C-I75), both the precursor and mature forms actually accumulated upon CCCP treatment (Figure S5M), presumably due to arrested import of the precursor protein translated in the cytosol caused by MMP loss. Cytosolic translation apparently is not stalled by CCCP treatment. Mitochondrial damage thus causes translational stalling specifically for those mRNAs locally translated on the MOM, such as C-I30 mRNA (Gehrke et al., 2015), and remodeling of MOM-associate mRNPs induced by mitochondrial damage may dictate the switch of PINK1 function from stimulation of translation to activation of mitophagy.
NGD-Related Quality Control Factors Mediate PINK1 Function in Drosophila
We next sought to examine the in vivo effects of the co-translational quality control factors on PINK1 regulated mitochondrial function and neuromuscular tissue maintenance. PINK1 flies exhibit defective mitochondrial morphology, function, and indirect flight muscle integrity (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). We found that ectopic expression of Pelo, ABCE1, or NOT4 restored mitophagy as indicated by the removal of aggregated mitochondria (Figure 6A, B), mitochondrial function as measured by ATP production (Figure 6C), and flight muscle integrity as indicated by wing posture (Figure 6D). Pelo, ABCE1, or mild NOT4 overexpression in wild type background had no obvious effect on muscle mitochondrial morphology or wing posture (Figure S6A, B), and DA neuron mitochondrial morphology or neuronal number (Figure S6D, E), although they increased ATP production to various degrees (Figure S6C), supporting their importance to mitochondrial function in vivo. In disease-relevant DA neurons, ectopic expression of Pelo, ABCE1, and NOT4 effectively rescued the mitochondrial aggregation and neuronal loss in PINK1 mutant (Figure 6E, F). Ectopic expression of Pelo, ABCE1, and especially NOT4 rescued C-I30 deficit in PINK1 mutant, and induced the formation of LC3-II (Figure S6F), suggesting that they exerted rescuing effects through autophagy induction. Consistently, inhibition of the key autophagy regulator ATG1 attenuated the abilities of these factors to rescue PINK1 mutant effects on C-I30 translation (Figure S6G), mitochondrial morphology in muscle (Figure 6G, H) and DA neurons (Figure 6I, J), and DA neuron survival (Figure S6H). In PINK1(−/−) mutant HeLa cells, we found that co-expression of ABCE1 and NOT4 also promoted the formation of LC3-positive autophagic rings encircling mitochondria in a portion of PINK1(−/−) mutant HeLa cells, which were never observed in non-transfected or ABCE1 singly transfected mutant cells (Figure S6I, J), suggesting that the function of co-translational quality control factors in mediating the induction of mitophagy downstream of PINK1 may be conserved.
Figure 6. In Vivo Genetic Data Demonstrating the Involvement of Co-Translational Quality Control Factors in PINK1-Directed Mitophagy.
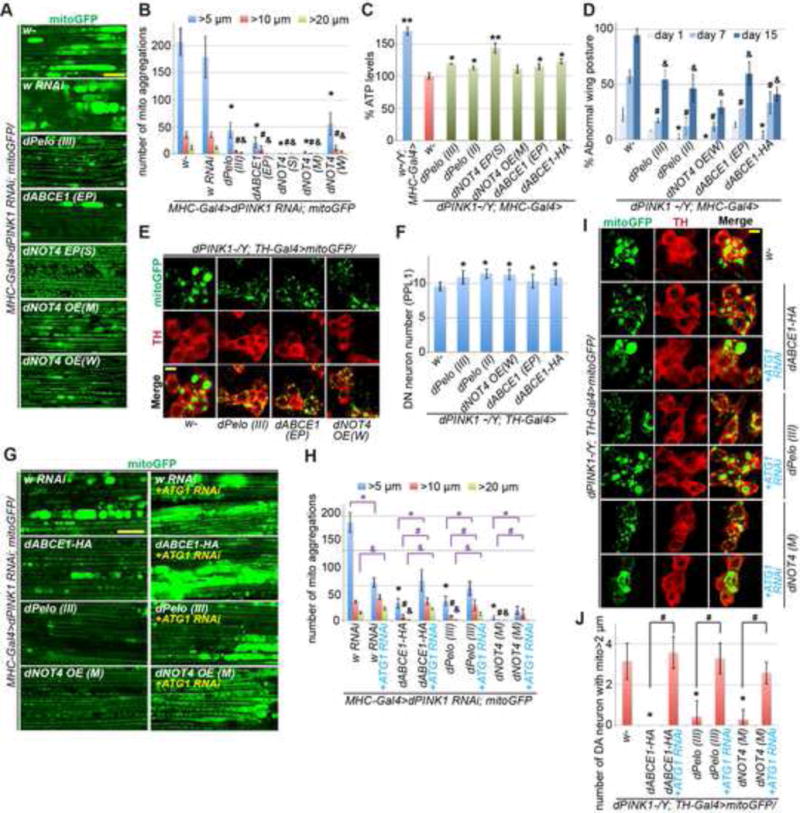
(A) Removal of aggregated mitochondria by overexpression of co-translational quality control factors in muscle tissue of dPINK1 RNAi flies. Scale bar, 25μm. The fly lines used are: w-, white eyed wild type control; w RNAi, control white RNAi line; dPelo(III), a UAS-pelo transgene on the 3rd chromosome; dABCE1 (EP), an enhancer P element induced dABCE1 overexpression line; dNOT4 EP(S), (M), (W): strong (S), moderate (M), or weak (W) EP overexpression lines of dNOT4.
(B) Quantification of data shown in A. *, #, or &, p<0.05 in Student’s t-test vs. w-control group for aggregates >5 μm, >10μm, or >20μm, respectively.
(C) Restoration of ATP level by co-translational quality control factors in dPINK1 mutant flies. Error bars: s.d. from 3 independent measurements normalized and compared to dPINK1 mutant (red bar). *, p<0.05; **, p<0.01; Student’s t-test.
(D) Rescue of wing posture defect by co-translational quality control factors in dPINK1 mutant condition. *, #, or &, p<0.05 in Student’s t-test vs. dPINK1 mutant group at day1, 7, or 15, respectively.
(E, F) Rescue of DA neuron mitochondrial morphology (E) and neuronal number (F) by co-translational quality control factors in dPINK1 mutant condition. F shows quantification of data shown in E. *, p<0.05 in Student’s t-test.
(G-J) Blockage by ATG1 RNAi of the rescuing effect of co-translational quality control factors on mitochondrial morphology in muscle tissue (G, H) and DA neurons (I, J) of dPINK1 mutant flies. H and J show quantification of data shown in G or I, respectively. *, #, or &, p<0.05 in Student’s t-test.
See also Figure S6.
To further test the in vivo role of co-translational quality control factors in mitophagy induction, we examined their effects on p62, the sole autophagy receptor in Drosophila (Nezis et al., 2008). Prominent p62 puncta were present in the muscle cytoplasm of PINK1 RNAi flies, but few were localized to mitochondria (Figure 7A, B). Recruitment of p62 to mitochondria was enhanced by ectopic ABCE1 or Pelo expression, but reduced when these factors were inhibited, especially when they were simultaneously inhibited (Figure 7C, D). NOT4 exerted intriguing dosage-sensitive effects. While its weak (W) overexpression promoted p62 recruitment to mitochondria, moderate (M) to strong (S) overexpression (Figure S5E) promoted mitophagy, resulting in removal of mitochondrial aggregates (Figure 7C, D). Interestingly, whereas NOT4 (M) overexpression frequently led to p62-positive membrane structures with internal mitochondria removed, concomitant knockdown of ATG1 caused the accumulation of enlarged mitochondria encircled by p62 signals (Figure 7E), suggesting that activation of NOT4 is sufficient to induce the recruitment of autophagy receptors and initiate mitophagy in vivo. Fly p62 was present in MOM-associated mRNPs prepared by sucrose gradient ultra-centrifugation, where the level of p62 is positively regulated by Pelo/ABCE1 and dose-dependently regulated by NOT4 (Figure S5E). Correlating with the biochemical interaction between NOT4 and ABCE1, when ABCE1 and NOT4 were co-expressed, near complete removal of mitochondria was observed in wild type or PINK1 RNAi condition, indicating excessive mitophagy, whereas knockdown of ABCE1 blocked the mitophagy-promoting effect of NOT4 (Figure 7F, G).
Figure 7. In vivo Genetic Interaction Studies and Analysis of PD Patient Brain Samples Supporting the Roles of ABCE1 in Mitophagy Induction and Disease Pathogenesis.
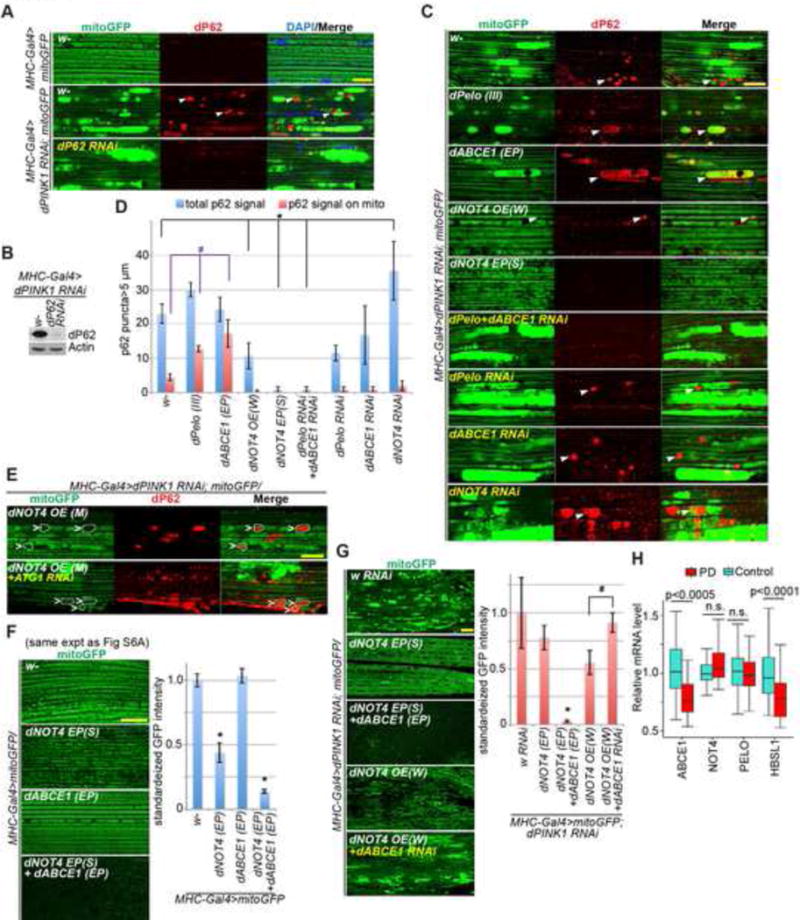
(A) Immunostainings demonstrating recruitment of p62 to damaged mitochondria in Mhc>PINK1 RNAi muscle tissue. p62 RNAi line validates the specificity of p62 immunostaining signals.
(B) Immunoblot analysis assessing the specificity of anti-fly p62 antibody.
(C) Effects of the various genetic manipulations on p62 localization to damaged mitochondria in Mhc>PINK1 RNAi muscle tissue.
(D) Quantification of data shown in C. *, #, p<0.05 in Student’s t-test.
(E) Immunostainings showing promotion of mitophagy by NOT4 overexpression, and the blocking effect of ATG1 RNAi. Dashed white lines outline the damaged mitochondria with p62 recruitment.
(F) Immunostainings showing the effect of overexpression of co-translational quality control factors alone, or co-expression of NOT4 and ABCE1 on mitochondrial mass.
(G) Immunostainings showing the mitophagy-inducing activity of NOT4 and the blockage of such effect by ABCE1 RNAi. Bar graphs show data quantification in F and G. *, p<0.05 in Student’s t-test. Pelo, ABCE1, NOT4, and ATG1 RNAi lines, and corresponding overexpression lines as described in 6A, are used in C-G.
(H) Comparison of relative mRNA expression levels for four co-translational quality control genes in prefrontal cortex between PD cases and healthy control subjects. ABCE1 and HBSL1 show significant differential expression (ABCE1, p<0.0005, FDR < 0.017; HBS1L, p<0.0001, FDR < 0.0073). FDR: false discovery rate.
See also Figure S7.
Downregulation of NGD-Related Quality Control Factors in PD Brain Tissues
We next tested the clinical relevance of our findings to humans. Using published gene expression dataset derived from human brain tissues (Dumitriu et al., 2016), we performed differential expression analysis comparing PD patient vs. healthy control samples. ABCE1 (p<0.001, FDR < 0.017) and HBS1L (p<0.0001, FDR < 0.0073) were found significantly downregulated at the RNA level in PD cases (Figure 7H). HBS1L interacts with Pelo and ABCE1 and participates in NGD (Doma and Parker, 2006). We also examined 22 mitochondrial, familial PD, and autophagy receptor genes, but none of them showed significant changes (Figure S7A–C). The selective downregulation of two genes involved in the NGD pathway highlights the importance of this co-translational quality control process to PD pathogenesis.
DISCUSSION
Our results reveal that on damaged mitochondria, the recruitment of co-translational quality control factors Pelo, ABCE1 and NOT4 to stalled ribosomes results in NOT4-mediated poly-ubiquitination of ABCE1, and that poly-Ub-ABCE1 provides a molecular signal for recruiting autophagy receptors to initiate mitophagy (Figure 4E). This finding advances our understanding of the mechanism of mitochondrial quality control, as it uncovers one of the earliest activating signals in PINK1-directed mitophagy, by linking, for the first time, organelle quality control with co-translational RNA and protein quality control at the molecular level. This conclusion is further supported by our data showing the release of autophagy receptors from MOM by RNase A and EDTA treatments that disrupt mRNP integrity, and the blockage of the abilities of Pelo, ABCE1, and NOT4 to rescue PINK1 mutant phenotypes by knocking down the key autophagy regulator ATG1. Importantly, we found that the expression of two key components of the NGD pathway, ABCE1 and HBS1L, are significantly downregulated in brain samples of PD patients compared to control subjects. The selective downregulation of two genes in the NGD pathway highlights the importance of this co-translational quality control process and further validated the relevance of our findings to human disease.
We provide evidence supporting our hypothesis that the PINK1 pathway is deployed to stimulate translation of nRCC mRNAs on mildly damaged mitochondria to promote nRCC biogenesis and thus mitochondrial repair, but for severely damaged mitochondria that are beyond repair, the PINK1 pathway is used to direct their clearance by mitophagy. Our data suggest that remodeling of the mRNPs on MOM may mediate the shift of PINK1 function from repair to removal of damaged mitochondria. A key event appears to be the damage-dependent ubiquitination of ABCE1 by NOT4, resulting in decreased Pelo/ABCE1 interaction and thus translation, and increased recruitment of autophagy receptors. The molecular events initiated by ABCE1 ubiquitination that leads to eventual mitochondrial removal are likely to be multi-tiered. In addition to its direct recruitment of autophagy receptors, poly-Ub-ABCE1 may attract the proteasome to MOM, resulting in outer membrane rupture and exposure of inner mitochondrial membrane signals such as Prohibitin 2 that could also recruit autophagy machinery (Wei et al., 2017).
Our results also show that many of the effects of PINK1 in activating mitophagy, from recruiting C-I30 mRNP to MOM and co-translational quality control factors and autophagy receptors to MOM-associated mRNPs, to the subsequent activation of mitophagy, can occur in the absence of Parkin. This contrasts the prevailing view that activation of Parkin and subsequent ubiquitination of a battery of Parkin substrates on MOM is essential for PINK1-activated mitophagy. Consistent with previous studies, our results indicate that though not required, the presence of Parkin provides an amplifying mechanism to promote PINK1-directed mitophagy (Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016), as both the recruitment of autophagy receptors and the clearance of damaged mitochondria are accelerated in the presence of Parkin. Our results show that the PINK1-dependent recruitment of Parkin to damaged mitochondria in HeLa/GFP-Parkin cells is compromised by the knockdown of Pelo, ABCE1, or NOT4. Although the detailed mechanism of Parkin recruitment to damaged mitochondria awaits further investigation, our results suggest that MOM-associated mRNPs may play an important role.
Besides mitochondrial dysfunction, the formation of aggregates of misfolded proteins is common to many genetic and sporadic forms of neurodegenerative diseases, e.g., the Aβ and tau aggregates in Alzheimer’s disease (AD) and α-Syn aggregates in PD. It is unknown whether and how mitochondrial dysfunction and proteostasis failure are connected in the disease process. Our results provide mechanistic insights into the connection between mitochondrial dysfunction and proteostasis failure, and implicate ABCE1 as a key mediator. In addition to its ubiquitination by NOT4 in response to MMP drop, ABCE1 may also sense other mitochondrial damages. For example, owing to its dependency on ROS-labile Fe-S clusters as cofactors, ABCE1 has been proposed to be an important target accounting for the cytotoxicity of ROS (Alhebshi et al., 2012). Future studies will test the broader roles of ABCE1 in sensing mitochondrial functional state and in mediating the pathogenic effects of disease-causing insults in other settings.
One immediate implication of our findings is that mitochondrial dysfunction may be a common contributor to the neurodegenerative pathology caused by mutations in the co-translational quality control factors (Chu et al., 2009; Ishimura et al., 2014) or autophagy receptors (Cirulli et al., 2015; Freischmidt et al., 2017; Haack et al., 2016; Maruyama et al., 2010; Rezaie et al., 2002). The mammalian mitochondrial proteome is known to be heterogeneous across cell types, with any two distinct organs sharing only ~ 75% of their proteins and over half of the mitochondrial proteins are currently of unknown function (Calvo and Mootha, 2010). Tissue-or cell type-specific differences in MOM-associated translatomes may underlie the molecular basis of mitochondrial proteome heterogeneity across cell types and developmental or disease states, offering insights into the selective vulnerability associated with neurodegenerative disease and other diseases featuring mitochondrial etiology. A collective of about 1000 or 1500 mitochondrial proteins are present in yeast and mammalian cells, respectively, with the majority of which are encoded by the nuclear genome and many of which are translated on mitochondrial surface (Gehrke et al., 2015). A recent study showed that yeast cells continuously make faulty mitochondrial polypeptides that experience translational stalling on ribosomes but are imported into mitochondria, and can cause cytotoxicity if not properly quality controlled (Izawa et al., 2017). This report is consistent with our finding of the presence of significant amounts of co-translational quality control factors on MOM even in the absence of damage (Figure 2E), and our demonstration of the crucial role of ribosome-associated quality control in PINK1-directed mitochondrial regulation.
A therapeutic implication of our finding of a mechanistic link between mitochondrial dysfunction and proteostasis failure is that mitochondrial diseases, long thought to arise primarily from defective energy production, could instead be treated by targeting the cytosolic proteostasis network. Conversely, the prevalent protein-aggregation diseases like Alzheimer’s, Parkinson’s, Huntington’s, ALS, and Prion disease could be treated by targeting mitochondrial pathways. Since mitochondrial dysfunction and proteostasis failure are intimately associated with aging, mapping the interactive circuitry will help reveal regulatory nodes that can be therapeutically targeted to promote healthy aging.
Limitations of Study
Our work combining mammalian cell culture and in vivo Drosophila models allows us to study the signaling mechanisms linking co-translational quality control and mitophagy and then test their in vivo significance. One caveat of the study is that much of the mammalian studies depended on treatment with CCCP, which, although potent in inducing mitochondrial damage and mitophagy, is nevertheless considered non-physiological. In future studies it would be desirable to test if CCCP-induced effects could be recapitulated in vivo in mammalian neurons. Our analysis of clinical samples on co-translational quality control factor expression is so far limited to transcriptome data. Future studies on patient proteome, especially mitochondria-associated proteome, may uncover more informative disease-associated molecular changes in the key genes studied.
STAR★METHODS
KEY RESOURCES TABLES
| REAGENT or RESOURCE | SOURCE | INDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken anti-GFP | Abcam | ab13970 |
| Rabbit anti-dPelo | Dr. J Han | N/A |
| Rabbit anti-dNOT4 | Dr. E Wahle | N/A |
| Rat anti-HA | Sigma-Aldrich | 11867423001 |
| Rabbit anti-dP62/Ref2P | Abcam | ab178440 |
| Rabbit anti-TH | Pel-Freez | P40101-150 |
| Alexa Fluor® 594-conjugated secondary antibody | Molecular probes | R37117 |
| Alexa Fluor® 488-conjugated secondary antibody | Molecular probes | A-11039 |
| anti-TOM20 | Santa Cruz Biotech | sc-17764, sc-11415 |
| Rabbit anti-TOM40 | Santa Cruz Biotech | sc-11414 |
| Rabbit anti-GST | Sigma-Aldrich | G7781 |
| Mouse anti-GST | GenScript | A00865 |
| Rabbit anti-RpL3 | Santa Cruz Biotech | sc-86828 |
| Rabbit anti-RpL7a | Cell signaling Biotech | 2415S |
| Mouse anti-RpS19 | Santa Cruz Biotech | sc-100836 |
| Mouse anti-RpS6 | Cell signaling Biotech | 2317S |
| Mouse anti-C-IV s.1 | Abcam | ab14705 |
| Mouse anti-C-I 30 | Abcam | ab14711 |
| Rabbit anti-C-I 75 | GeneTex | GTX105270 |
| Mouse anti-CORE2 | Abcam | ab14745 |
| Mouse anti-myc | Cell Signaling biotech | 2272S |
| Mouse anti-puromycin | Millipore | MABE343 |
| Rabbit anti-ABCE1 | Dr. R Hegde | N/A |
| Rabbit anti-ABCE1 | NOVUS biologicals | NB400-116 |
| Rabbit anti-Pelo | Abcam | ab140615 |
| Rabbit anti-NOT4 | Abcam | ab72049 |
| Mouse anti-Ub | Santa Cruz biotech | sc-8017 |
| Rabbit anti-p-S65-Ub | Millipore | ABS1513-I |
| Rabbit anti-K48-Ub | Abcam | ab140601 |
| Rabbit anti-K63-Ub | Millipore | 05-1308 |
| Rabbit anti-OPTN | Abcam | ab23666 |
| Rabbit anti-NDP52 | Abcam | ab68588 |
| Guinea pig anti-p62 | PROGEN | GP62-C |
| Rabbit anti-p-TBK1 | Cell signaling biotech | 5843S |
| Rabbit anti-TBK1 | Cell signaling biotech | 3013S |
| Rabbit anti-LC3B | Cell signaling biotech | 2775S |
| Chemicals, peptides, and Recombinant proteins | ||
| H2O2 | Sigma-Aldrich | H1009-100ML |
| Rotenone | Sigma-Aldrich | R8875-1G |
| CCCP | Sigma-Aldrich | C2759-100MG |
| Cycloheximide | Sigma-Aldrich | C7698-1G |
| Puromycin | Sigma-Aldrich | P7255-100MG |
| Biotin-linked puromycin | Jena Bioscience | NU-925-BIO-S |
| Emetine | Sigma-Aldrich | E2375-500MG |
| 4EGI | Santa Cruz Biotech | sc-202597 |
| homoharringtonine (HTT) | Tocris Bioscience | 1416/10 |
| FuGene 6 | Promega | E2691 |
| Lipofectamine 3000 | Invitrogen | L3000015 |
| Lipofectamine RNAiMAX | Invitrogen | 13778150 |
| Antimycin A | Sigma-Aldrich | A8674-100MG |
| Oligomycin A | Sigma-Aldrich | 75351-5MG |
| TMRM | Invitrogen | T668 |
| Propidium Iodide | Invitrogen | P1304MP |
| Pierce™ Neutravidin™ agarose beads | Pierce | 29200 |
| Percoll | GE Healthcare | 17089101 |
| EDTA | Sigma-Aldrich | E9884-100G |
| EGTA | Sigma-Aldrich | E3889-100G |
| RNase A | Qiagen | 19101 |
| SUPERase• In™ | Invitrogen | AM2694 |
| cOmplete™ Protease inhibitor | Roche | 11697498001 |
| Anti-FLAG M2 affinity gel | Sigma-Aldrich | A2220-5ML |
| Anti-HA magnetic beads | Pierce | 88836 |
| Normal goat serum | Jackson ImmunoResearch | 005-000-121 |
| 3 × FLAG tag peptide | APExBIO | A6001 |
| UBE1 | Boston Biochem | E-305 |
| Ubiquitin | Boston Biochem | E2-616 |
| UbcH5a | Boston Biochem | U-530 |
| Critical Commercial Assays and services | ||
| ATP Bioluminescence Assay Kit HS II | Roche | 11699709001 |
| siRNA-resistant constructs of ABCE1, Pelo, Not4 | GenScript Biotech | N/A |
| ABCE1 K20R mutant | GenScript Biotech | N/A |
| RNeasy Mini kit | Qiagen | 74104 |
| OneStep RT-PCR kit | Qiagen | 210212 |
| NuPAGE™ 4-12% Bis-Tris Protein Gels | Invitrogen | NP0321BOX |
| NuPAGE® MOPS SDS running buffers | Invitrogen | NP0001 |
| Duolink® In Situ Red Starter Kit Mouse/Rabbit | Sigma-Aldrich | DUO92101 |
| Oligonucleotides | ||
| Stealth RNAi™ siRNA of CNOT4 | Invitrogen | HSS107246, HSS107245 |
| Stealth RNAi™ siRNA of Pelo | Invitrogen | HSS122741, HSS122740 |
| Stealth RNAi™ siRNA of ABCE1 | Invitrogen | HSS109286, HSS109286 |
| hC-I 30-RT-N, 5′-TTGCTGCCGGTGAGGCGGG-3′ | This study | N/A |
| hC-I 30-RT-C, 5′-CAGCAGACTCAATGGGCG-3′ | This study | N/A |
| hC-I 30-ms2-RT-N: | This study | N/A |
| 5′-GGAGACAAGAAGCCTGATGC-3′ | ||
| hC-I 30-ms2-RT-C: | This study | N/A |
| 5′-CCGCCAGTGTGATGGATATC-3′ | ||
| hC-IVS 1 -RT-N, 5′-GATATGGCGTTTCCCCGC-3′ | This study | N/A |
| hC-IVS 1-RT-C, 5′-GATCAGACGAAGAGGGGCG-3′ | This study | N/A |
| hb-Actin-RT -N, 5′-ACCACCACGGCCGAGCGGG-3′ | This study | N/A |
| hb-Actin-RT-C, 5′-GGGTACATGGTGGTGCCGCC-3′ | This study | N/A |
| hMfn2-RT-N, 5′-CATCAGTGAGGTGCTGGC-3′ | This study | N/A |
| hMfn2-RT-C, 5′-CATCCTTCAGAAGTGGGC-3′ | This study | N/A |
| dC-I 30-RT-N, 5′-TGTTCCCAAGGCGCCGAC-3′ | This study | N/A |
| dC-I 30-RT-C, 5′-AGCCTAAGAAGGCGGATAAG-3′ | This study | N/A |
| dC-IVS1-RT-N, 5′-CCTGGATTTGGAATAATTTCTC-3′ | This study | N/A |
| dC-IVS1-RT-C, 5′-TCAGAATATCTATGTTCAGCTG-3′ | This study | N/A |
| CG4169-RT-N, 5′-TCGACAATGCCAAAACGGTG-3′ | This study | N/A |
| CG4169-RT-C, 5′-CAAAGAGTCCAGCAGAAGTG-3′ | This study | N/A |
| dPinkl-RT-N, 5′-GAACATGTCGCGCTTTGTTC-3′ | This study | N/A |
| dPink1-RT-C, 5′-TTGATTCTGCAGCAAACGTTC-3′ | This study | N/A |
| Recombinant DNA | ||
| pMS2-bs | Gehrke et al., 2015 | N/A |
| pC-I 30/MS2-bs | Gehrke et al., 2015 | N/A |
| pCMV-FLAG-ABCE1 | Dr. Ramanujan Hegde | N/A |
| pCMV-FLAG-Pelo | This study | N/A |
| pCMV-FLAG-NOT4 | OriGene Inc | RC217418 |
| pCMV-FLAG-rsABCE 1 (siRNA-resistant) | This study | N/A |
| pCMV-FLAG-rsPelo (siRNA-resistant) | This study | N/A |
| pCMV-FLAG-rsNOT4 (siRNA-resistant) | This study | N/A |
| Experimental Models: Drosophila stocks | ||
| See Experimental Model and Subject Details | ||
| Experimental Models: Cell Lines | ||
| See Experimental Model and Subject Details | ||
| Software and Algorithms | ||
| TopHat v2.0.1 | Kim et al., 2013 | |
| HTseq version 0.5.3 | Harrow et al., 2012 | |
| DESeq2 v1.4 | Love et al., 2014 | |
| Source code (required R environment) | This study | Available upon request |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents may be obtained from the Lead Contact, Bingwei Lu (Stanford University; email: bingwei@stanford.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila Stocks
The PINK1B9 mutant line was a gift from Dr. Jongkeong Chung. parkin1 and parkinΔ21 were provided by Dr. Patrik Verstreken and UAS-mito-GFP by Dr. William Saxton. UAS-dPINK1 RNAi, UAS-dPINK1 and UAS-dParkin were described before (Gehrke et al., 2015). TH-Gal4 was a gift from Dr. Serge Birman. dNOT4 EP (#22246), dNOT4 RNAi (#28775), dABCE1-EP (#27954), dABCE1 RNAi (#31601), dPelo RNAi (#34770), dP62 RNAi (#36111 and #33978), ATG1 RNAi (#35177) and other general fly lines were obtained from the Bloomington Drosophila Stock Center, dABCE1-3xHA (F001097) from FlyORF, and dPelo RNAi (v108606) and dNOT4 RNAi (v110472) from the VDRC. UAS-dNOT4-myc (M) and UAS-NOT4 (W) were kindly provided by Dr. Mika Rämet (Gronholm et al., 2012). UASp-Pelo (III), UASp-Pelo (II) and dPelo mutant flies were kindly provided by Dr. Rongwen Xi (Xi et al., 2005).
Unless otherwise indicated in the figure legend, male flies at 2-3 weeks of age were used for the experimental procedures described. Fly culture and crosses were performed according to standard procedures and raised at indicated temperatures. Flies were generally raised at 25 °C and with 12/12 hrs dark/light cycles. Fly food was prepared with a standard receipt (Water, 17 L; Agar, 93 g; Cornmeal, 1,716 g; Brewer’s yeast extract, 310 g; Sucrose, 517 g; Dextrose, 1033 g).
Cell lines
Regular HeLa cells and PINK1 (−/−) HeLa cells (a gift from Dr. Richard Youle, NINDS) were cultured under normal conditions (1× DMEM medium, 8.75% FBS, 5% CO2 37°C). HeLa/GFP-Parkin cells (a gift from Dr. Yuzuru Imai, Juntendo University) were cultured with drug selection (1μg/ml puromycin). HeLa cell line was derived from cervical cancer cells taken from a female patient who died of cancer.
METHOD DETAILS
Drosophila behavior tests and ATP measurement
Fly culture and crosses were performed according to standard procedures and raised at indicated temperatures. For all wing posture assays, male flies at 1-day, 7-day and 14 or 15-day old and aged at 29°C were used. Approximately 30 flies were housed per vial, and the number of flies with held-up or droopy wing postures was counted at the indicated ages. Abnormal wing posture was expressed as the percentage of flies exhibiting abnormal wing posture. Each experiment was repeated at least three times.
Measurements of ATP contents in thoracic muscle were performed as described previously (Gehrke et al., 2015), using a luciferase based bioluminescence assay (ATP Bioluminescence Assay Kit HS II, Roche Applied Science). For each measurement, two muscle thoraces dissected from 3-day old flies were collected. The muscle thoraces were homogenized in 100 μl lysis buffer, boiled for 5 minutes, and centrifuged at 16,000 g for 1 minute. 2 μl of the supernatant was mixed with 98 μl of dilution buffer and 10 μl luciferase/luciferin mixture provided with the kit. The luminescence was immediately measured using a luminometer (Promega). As standard we used various concentrations of ATP alone to convert the measured luciferase activity to the amount of ATP. For each sample at least three independent measurements were prepared.
H2O2, CCCP, and rotenone treatments of flies
Flies were collected after eclosion and divided into separate vials (20~25 flies each vial). Instant fly food (Carolina) was mixed with 1% H2O2, 250 μM rotenone or 100 μM CCCP. Vials were changed every day. Samples were collected for further analyses after 5 days treatment.
Plasmids and molecular cloning
pMS2-bs, pC-I 30/MS2-bs and pMS2-BP/GST plasmids were described before (Gehrke et al., 2015). pCMV-FLAG-ABCE1 was a gift from Dr. Ramanujan Hegde. pCMV-FLAG-Pelo was generated by cloning Pelo cDNA into the pCMV vector via KpnI-Xba I sites from an original plasmid from Dr. Ramanujan Hegde. pCMV-FLAG-NOT4 was obtained from OriGene Inc (TrueORF, RC217418).
RNAi resistant and ABCE1 K20R mutant constructs
The RNA resistant constructs were modified from original pCMV-FLAG-ABCE1, pCMV-FLAG-Pelo and pCMV-FLAG-NOT4 plasmids. For making the siRNA-resistant silent mutations, CCAAGAAGCAGTGGGATAGTGTG was changed to CtAAaAAaCAGTGGGAcA GcGTc, and GGAAAGGCAATTGCTCTCAGCATGA was changed to GaAAgGGgA AcTGtTCcCAGCAcGA in pCMV-FLAG-Pelo plasmid; GCTATCTACGGGTTCAG TTGAT was changed to GCTgTCcACcGGcTCcGTgGAcAAg, and TCAGCAGCAC CGAGCGGTTTATAAT was changed to cCAGCAGCAtCGgGCaGTgTAcAAc in pCMV-FLAG-NOT4 plasmid; GAGTTGCCCTGTGGTTCGGATGGGA was changed to aAGcTGCCCTGTGGTgCGGATGGGc and GCCTTATCAATTGTCAATTTGCCAA was changed to GCtTTgTCcATcGTgAAcTTGCCcA in pCMV-FLAG-ABCE1 plasmid.
For making the ABCE1 K20R mutant, we chose twenty ABCE1 identified ubiquitination sites (K64, K116, K121, K128, K158, K165, K169, K178, K181, K191, K210, K250, K332, K343, K397, K412, K451, K579, K584, K590) from mUbiSiDa and modified them by changing AAG or AAA (K) to aga (R). All mutagenesis was done by GenScript Biotech. Sequencing data of these plasmids and the cloning and mutagenesis procedures are available upon request.
Cell transfection and knock-down
Cell transfections were performed by using FuGENE 6 reagent (Promega), Lipofectamine 3000 reagent (Invitrogen) and knocking-down experiments were performed using Lipofectamine RNAiMAX reagent (Invitrogen), according to instructions from the manufacturers. Stealth RNAi™ siRNAs (Invitrogen) used for the RNAi experiments are: CNOT4 (HSS107246, HSS107245), Pelo (HSS122741, HSS122740), ABCE1 (HSS109286, HSS109285).
For drug treatment, HeLa cells or HeLa/GFP-Parkin cells were cultured and treated with CCCP at the indicated concentrations and durations as described (Narendra et al., 2008). For most experiments, the mitophagy-inducing 20 μM CCCP concentration was used. To induce milder mitochondrial damage, 2 μM and 5 μM CCCP concentrations were used. For Antimycin and oligomycin A treatment, 10 μM of each drug was used. TMRM and Propidium Iodide (PI) staining were performed according to manufacturer’s instructions (Invitrogen). Washout experiment were performed by treating cells with 20 μM CCCP for 2 hrs, removing drug containing medium with fresh medium, re-culture the cells for 2hrs, and then measure MMP with TMRM and ATP level with the ATP Bioluminescence Assay Kit HS II (Roche Applied Science).
Puromycin labeling of NPCs on stalled ribosomes
HeLa/GFP-Parkin cells were cultured and treated with CCCP at the indicated time intervals. Puromycin (100 μM, Sigma) and emetine (200 μM, Sigma) were then added and the cells were incubated at 37 °C for an additional 5 min. Cells were then placed on ice, washed with cold HBS, and subjected to mitochondrial purification. In some experiments, cells were treated with 4EGI (60 μM, Santa Cruz Biotechnology) or homoharringtonine (HTT) (5 μM, Tocris Bioscience) for 10 min to prevent new translation initiation and allow active ribosomes to run off. NPCs on MOM were subjected to western blot or immunoprecipitation analysis.
For in vitro puromycin labeling of NPCs on MOM, purified mitochondria were suspended in 10 mM Tris (pH 7.4), 400 mM KCl, 3mM MgCl2, and 2 μM biotin-linked puromycin (Jena Bioscience). Puromycylation reactions were performed at 37 °C for 90 min, and postreaction, biotin-puromycin labeled NPCs were purified with Pierrce™Neutravidin™ agarose beads and subjected to further analysis. To demonstrate that the newly labeled NPCs are not of mitochondrial matrix origin, the in vitro puromycin labeling reaction was performed in the presence of chloramphenicol, an inhibitor of mitochondrial translation.
Mitochondria purification
Intact mitochondria from in vitro cell cultures and fly muscle tissue samples were purified and quality controlled for the absence of contamination by other organelles as described previously (Gehrke et al., 2015). For analysis of fly samples, male flies at appropriate ages were used for thoracic muscle dissection. To block the release of mRNAs associated with mitochondria, 0.1 mg/ml cycloheximide was applied to all buffer solutions. Samples were homogenized using a Dounce homogenizer. After two steps of centrifugation (1,500g and 13,000g), the mitochondria pellet was washed twice with HBS buffer (5 mM HEPES, 70 mM sucrose, 210 mM mannitol, 1 mM EGTA, 1× protease inhibitor cocktail), then resuspended and loaded onto Percoll gradients. The fraction between the 22% and 50% Percoll gradients containing intact mitochondria were carefully transferred into a new reaction tube, mixed with 1 volume of HBS buffer, and centrifuged again at 20,000g for 20 minutes at 4°C to collect the samples for further analyses.
RT-PCR
For RT-PCR analysis, total RNAs were extracted from the mitochondrial samples of in vitro cell cultures, or fly thoraces using an RNeasy® Mini kit (Qiagen), and one-step RT-PCR was performed using an RT-PCR kit (Qiagen).
The oligonucleotides used in the RT-PCR assays are:
-
hC-I 30: (Primer 1) hC-I 30-RT-N, 5′-TTGCTGCCGGTGAGGCGGG-3′
hC-I 30-RT-C, 5′-CAGCAGACTCAATGGGCG-3′
(Primer 2) hC-I 30-ms2-RT-N: 5′-GGAGACAAGAAGCCTGATGC-3′
hC-I 30-ms2-RT-C: 5′-CCGCCAGTGTGATGGATATC-3′
-
hC-IV s.1: hC-IVS1-RT-N, 5′-GATATGGCGTTTCCCCGC-3′
hC-IVS1-RT-C, 5′-GATCAGACGAAGAGGGGCG-3′
-
hActin: hb-Actin-RT-N, 5′-ACCACCACGGCCGAGCGGG-3′
hb-Actin-RT-C, 5′-GGGTACATGGTGGTGCCGCC-3′
-
hMfn2: hMfn2-RT-N, 5′-CATCAGTGAGGTGCTGGC-3′
hMfn2-RT-C, 5′-CATCCTTCAGAAGTGGGC-3′
-
dC-I 30: dC-I 30-RT-N, 5′-TGTTCCCAAGGCGCCGAC-3′
dC-I 30-RT-C, 5′-AGCCTAAGAAGGCGGATAAG-3′
-
dC-IV s.1: dC-IVS1-RT-N, 5′-CCTGGATTTGGAATAATTTCTC-3′
dC-IVS1-RT-C, 5′-TCAGAATATCTATGTTCAGCTG-3′
-
dUQCR-C2: CG4169-RT-N, 5′-TCGACAATGCCAAAACGGTG-3′
CG4169-RT-C, 5′-CAAAGAGTCCAGCAGAAGTG-3′
-
dPINK1: dPink1-RT-N, 5′-GAACATGTCGCGCTTTGTTC-3′
dPink1-RT-C, 5′-TTGATTCTGCAGCAAACGTTC-3′
Protein release from MOM-associated mRNPs
For release of factors from mRNPs, purified mitochondrial pellets (50μg per sample) from CCCP treated cells were resuspended in 100μl HBS buffer, and EDTA (pH 7.5) or RNase A was supplemented to the samples to reach final concentrations of 25 mM and 1 μg/μl, respectively, to dissociate the ribosome subunits and release the proteins from MOM-associated mRNPs. After incubating on ice for 30 min, the samples were centrifuged (20,000g, 10 min, 4°C) to separate the supernatant and mitochondrial pellet. The non-RNaseA treated control samples were incubates similarly in the base buffer at the same time. The reaction was stopped by adding SDS-PAGE loading buffer and boiling for 5 min.
mRNP purification, immunoprecipitation, and RNA-IP
For C-I 30 mRNP purification, we transiently expressed MS2-BP-GST and MS2-bs-C-I 30 plasmids together with the overexpression or knockdown of the indicated QC factors (ABCE1, Pelo and NOT4). Sixty hours post-transfection, we UV cross-linked the attached cells in the petri dish, and purified mitochondria as described (Gehrke et al., 2015). We homogenized the mitochondria fraction in the lysis buffer [50 mM Tris-HCl, pH7.4, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X-100, 0.1mg/ml cycloheximide, 1× RNase inhibitor, and Complete protease inhibitor cocktail (Roche)], and performed GST pull-down assays using glutathione-sepharose beads (GE Healthcare) at 4°C for 6 hours with gentle shaking. Subsequently, the sepharose beads were washed three times (10 minutes each) at 4°C in lysis buffer, mixed with 2× SDS Sample buffer, and loaded onto SDS-PAGE gels.
For immunoprecipitation, extracts of mitochondrial samples, HeLa cells or fly tissues were prepared by homogenization in lysis buffer. After centrifugation at 10,000g for 5 min, the supernatant was subjected to immunoprecipitation using the indicated antibodies, or affinity gels (Anti-FLAG M2 affinity gel from Sigma-Aldrich or anti-HA magnetic beads from Pierce). The immunocomplexes were analyzed by SDS-PAGE and western blot.
For RNA-IP of quality control factors and autophagy receptors, cells were washed with pre-chilled 1× PBS, UV cross-linked and prepared by homogenizing in lysis buffer with additional 0.4U/μl RNase Inhibitor (SUPERase•In™, Invitrogen, AM2694). Samples were pre-cleaned by incubating with empty beads, and subjected to immunoprecipitation with corresponding antibodies. RNAs were purified with RNeasy® Mini kit (Qiagen), and one-step RT-PCR was performed using RT-PCR kits (Qiagen).
For denature immunoprecipitation of ABCE1, cellular extracts were prepared by homogenization in lysis buffer. The cell extracts were mixed with 2× Laemmli sample buffer (Bio-rad) and denatured in 100°C. The samples then were diluted with pre-chilled lysis buffer at a 1:5 ratio and subjected to immunoprecipitation using Anti-FLAG M2 affinity gel (Sigma).
Mitochondrial ribosomal fraction purification
Briefly, purified mitochondria from fly tissues were lysed with Buffer A (20 mM HEPES, 50 mM KCl and 10 mM MgCl2) with additional 0.1% Triton X-100, 0.1 mg/ml cycloheximide and 1× RNase Inhibitor. The mitochondrial lysate was cleared by centrifugation (20,000g, 10 min, 4°C), and then loaded onto a 25% sucrose cushion (in Buffer A). The ribosome fraction was pelleted via high-speed centrifugation (68,000 rpm, TLA 100.3 Rotor, 20 min, 4°C). After centrifugation, we removed the supernatant and resuspended the ribosomal pellet in Buffer A containing 1% Triton X-100 and analyzed by immunoblotting.
Immunohistochemistry
For immunohistochemical analysis of mitochondrial morphology of adult fly brains and muscle tissues, male flies at around 5 days of age and raised at 29 degree were used. In muscle staining, at least 5 individuals were examined for each genotype and the representative images were presented. For analysis of DA neuron number in the various genetic backgrounds, male flies at around 21 days of age and raised at 29°C degree were used. In DA neuron staining, at least 7 individuals were examined for each genotype. Dissected tissue samples were washed once with 1× PBS and fixed with 1× PBS containing 0.25% Triton X-100 for 10 minutes at room temperature. Fixatives were subsequently blocked with 1× PBS containing 5% serum and incubated for 60 minutes at room temperature followed by incubation with primary antibodies at 4°C overnight. The primary antibodies used were: chicken anti-GFP (1:5,000, Abcam), rabbit anti-dPelo (1:1,000, Dr. J Han) (Wu et al., 2014), anti-dNOT4 (1:500, Dr. E Wahle) (Temme et al., 2010), anti-HA (1:1,000; Sigma), anti-dP62 (1:1000, Abcam) or anti-TH (1:1000, Pel-Freez). After three washing steps with 1× PBS/0.25 % Triton X-100 each for 10 minutes at room temperature, the samples were incubated with Alexa Fluor® 594-conjugated and Alexa Fluor® 488-conjugated secondary antibodies (1:500, Molecular Probes) for 1.5 hours at 37°C and subsequently mounted in Gel/Mount (biomeda). For each genotype, 7-8 flies were assayed and scored.
For immunohistochemical analysis in human cells, the primary antibodies used were chicken anti-GFP (1:5,000, Abcam), mouse anti-TOM20 (1:1,000, Santa Cruz) and rabbit anti-GST (1:1,000, Sigma-Aldrich). The secondary antibodies used were Alexa Fluor® 488 and 594-conjugated antibodies (1:500, Molecular Probes).
In vitro ubiquitin conjugation reaction
For in vitro ubiquitin conjugation reaction, ABCE1-FLAG and ABCE1-K20R mutant were expressed in PINK1(−/−) HeLa cells and NOT4-FLAG was expressed in HEK 293T cells. ABCE1 proteins were purified by denature IP using anti-FLAG M2 affinity gel and NOT4-FLAG was purified by regular IP and released by 3 × FLAG tag peptide (APExBIO). In a total reaction volume of 30 μl, adding 1.5 μl 20× reaction buffer [1 M Tris, 40 mM ATP (from ATP standard sample in ATP Bioluminescence Assay Kit HS II, Roche Applied Science), 100 mM MgCl2, 40 mM DTT, pH7.5], 100 ng E1 (UBE1, Boston Biochem), 200 ng E2 (UbcH5a, Boston Biochem), 200 ng E3 (NOT4-FLAG), 10 μl M2 beads bound with substrate ABCE1-FLAG or ABCE1-K20R mutant (~500 ng) and 5 μg ubiquitin (Boston Biochem). The reactions were incubated at 30°C for 1hour and stopped by adding 10 μl 4× SDS sample buffer. The reaction products were separated by SDS-PAGE and probed by immunoblotting.
Western blot analysis
NuPAGE™ 4-12% Bis-Tris Protein Gels and NuPAGE® MOPS SDS running buffer was applied for SDS-PAGEs and immunoblot analyses. The following antibodies were used for western blot analyses: Anti-RpL3 (Santa Cruz, sc-86828), anti-RpL7a (Cell Signaling, 2415S), anti-RpS19 (Santa Cruz, sc-100836), anti-RpS6 (Cell Signaling, 2317S), anti-C-IV s.1 (Abcam, ab14705), anti-C-I 30 (Abcam, ab14711), anti-C-I 75 (GeneTex, GTX105270), anti-CORE2 (Abcam, ab14745), anti-TOM20 (Santa Cruz, sc-17764, sc-11415), anti-TOM40 (Santa Cruz, sc-11414), anti-PINK1 (Abcam, ab23707), anti-GST (Sigma, G7781; for mRNP immunoblot), anti-GST (GenScript, A00865; for PLA), anti-myc (Cell Signaling, 2272S), anti-HA (Sigma, 11867423001), anti-GFP (Abcam, ab13970), anti-puromycin (Millipore, MABE343), anti-ABCE1 (a gift from Dr. R Hegde), anti-Pelo (Abcam, ab140615), anti-NOT4 (Abcam, ab72049), anti-dNOT4 (a gift from Dr. E Wahle), anti-dPelo (a gift from Dr. J Han), anti-Ub (Santa Cruz, sc-8017), anti-p-S65-Ub (Millipore, ABS1513-I), anti-K48-Ub (Abcam, ab140601), anti-K63-Ub (Millipore, 05-1308), anti-OPTN (Abcam, ab23666), anti-NDP52 (Abcam, ab68588), anti-P62 (PROGEN, GP62-C), anti-dP62/Ref2P (Abcam, ab-178440), anti-p-TBK1 (Cell Signaling, 5843S), anti-TBK1 (Cell Signaling, 3013S), anti-LC3B (Cell signaling, 2775S).
Proximity ligation assay with counterstaining
Duolink® PLA Probes: anti-Rabbit PLUS (DUO92002, Sigma), anti-Mouse MINUS (DUO92004, Sigma); Detection Reagents Orange (DUO92007, sigma) and Mounting medium with DAPI (DUO82040, Sigma) were selected for PLA reactions. Protocol was modified from Duolink® In situ-Fluorescence user guide. The counterstaining steps were applied after the amplification step in user guide. The slides were washed by 1× Wash buffer B [0.2 M Tris and 0.1 M NaCl, pH7.5] for 2 ×10 min, 1 × Wash buffer A [0.01 M Tris, 0.15 M NaCl, 0.05% Tween-20, pH7.4] for 5 min and 1× PBTX (PBS + 0.25% Triton X-100), then blocked with 5% Normal Goat Serum for 30 min. The slides were incubated with counterstaining primary antibody for 2 hours and secondary antibody for 1 hour. After counterstaining, slides were washed with 1× Wash buffer A for 2 × 5 min and 0.01 × Wash buffer B for 5 min, and mounted with mounting medium with DAPI.
Differential gene expression analysis
Original data was from Dumitriu et al (Dumitriu et al., 2016). In brief, RNAseq data were collected from 29 PD cases and 44 controls from prefrontal cortex (Brodmann area 9). RNAseq data were aligned with TopHat v2.0.1 (Kim et al., 2013) to hg19, and integer read counts were generated using HTseq version 0.5.3 using GENCODE annotation v17 (Harrow et al., 2012). DESeq2 v1.4 (Love et al., 2014) were used to performed differential expression analysis on 20,331 protein-coding genes, controlling for post-mortem interval (PMI), age and RNA integrity number (RIN). Neither sex nor ancestry was included as covariates because all brain samples are derived from male individuals of European ancestry. P-values were adjusted using Benjamini and Hochberg (Benjamini and Hochberg, 1995). We plotted the DESeq2 library size normalized gene expression values. The source code (required R environment) will be available upon request.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were performed with EXCEL (Microsoft, USA) and confirmed by MATLAB (MathWorks, USA). Error bars represent standard deviation (SD). Data in Figure 2B, 3B, 3D, 5G and Supplemental Figure S1F, S3G, S4F, S6J were analyzed by Chi-squared test. The analyses of Figure 7H and Supplemental Figure S7A–C were described separately in Differential gene expression analysis section of Methods Details. The rest of the data were evaluated by unpaired two-tailed Student’s t-test. In all statistical comparisons, a p value < 0.05 was considered as a significant difference and sample size/statistical details can be found in the figure legends. No particular method was used to determine whether the data met assumptions of the statistical approach.
Supplementary Material
Highlights.
Enhanced translation of OXPHOS-related mRNAs on mitochondrial surface under stress
Severe mitochondrial damage induces translational stalling on mitochondrial surface
Stalled ribosomes recruit co-translational quality control machinery
Remodeling of stalled ribosome/mRNP generates signals that trigger mitophagy
Acknowledgments
We are grateful to Drs. Ramanujan Hegde and Susan Shao for providing plasmids and antibodies; Drs. Jiahuai Han and Elmar Wahle for antibodies; Drs. Yuzuru Imai, Ming Guo, and Richard Youle for cell lines; Drs. Jongkeong Chung, William Saxton, Serge Birman, Patrick Verstreken, Rongwen Xi, Mika Rämet, the Vienna Drosophila RNAi Center, FlyORF, and the Bloomington Drosophila Stock Center for fly stocks. Special thanks go to J. Gaunce for maintaining flies and providing technical supports and members of the Lu lab for discussions. Supported by the NIH (NS084412, NS083417, and MH080378) (B.Lu), Stanford Computational, Evolutionary, and Human Genetics Fellowship (B.Liu), and National Natural Science Foundation of China (No. 81671252) (Y.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Z.Wu, Y.Wang designed the study, performed the experiments, analyzed data, and contributed equally. J.Lim, B.Liu, Y.Li, R.Vartak, and T. Stankiewicz performed the experiments and analyzed data. S.Montgomery provided resources for analyzing data. Z.Wu, B.Liu, and B.Lu wrote the manuscript. B.Lu conceived and supervised the study, performed experiments, and provided funding.
Declaration of Interests
The authors declare no competing interests.
References
- Alhebshi A, Sideri TC, Holland SL, Avery SV. The essential iron-sulfur protein Rli1 is an important target accounting for inhibition of cell growth by reactive oxygen species. Mol Biol Cell. 2012;23:3582–3590. doi: 10.1091/mbc.E12-05-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210–222. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. doi: 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011a;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011b doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Hong NA, Masuda CA, Jenkins BV, Nelms KA, Goodnow CC, Glynne RJ, Wu H, Masliah E, Joazeiro CA, et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2097–2103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Collart MA, Struhl K. NOT1(CDC39), NOT2(CDC36), NOT3, and NOT4 encode a global-negative regulator of transcription that differentially affects TATA-element utilization. Genes Dev. 1994;8:525–537. doi: 10.1101/gad.8.5.525. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova LN, Kuroha K, Tatematsu T, Inada T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. The Journal of biological chemistry. 2009;284:10343–10352. doi: 10.1074/jbc.M808840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu A, Golji J, Labadorf AT, Gao B, Beach TG, Myers RH, Longo KA, Latourelle JC. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med Genomics. 2016;9:5. doi: 10.1186/s12920-016-0164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29:989–999. doi: 10.1101/gad.262758.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Muller K, Ludolph AC, Weishaupt JH, Andersen PM. Association of Mutations in TBK1 With Sporadic and Familial Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. JAMA Neurol. 2017;74:110–113. doi: 10.1001/jamaneurol.2016.3712. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Wu Z, Klinkenberg M, Sun Y, Auburger G, Guo S, Lu B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell metabolism. 2015;21:95–108. doi: 10.1016/j.cmet.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TE, Hebert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille JC, Sossin WS. Reactivation of stalled polyribosomes in synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16205–16210. doi: 10.1073/pnas.1307747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronholm J, Kaustio M, Myllymaki H, Kallio J, Saarikettu J, Kronhamn J, Valanne S, Silvennoinen O, Ramet M. Not4 enhances JAK/STAT pathway-dependent gene expression in Drosophila and in human cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:1239–1250. doi: 10.1096/fj.11-195875. [DOI] [PubMed] [Google Scholar]
- Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P, Maffezzini C, Lonnqvist T, Suomalainen A, Gorza M, Kremer LS, et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am J Hum Genet. 2016;99:735–743. doi: 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Molecular cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MT. Harringtonine, an inhibitor of initiation of protein biosynthesis. Molecular pharmacology. 1975;11:511–519. [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T, Park SH, Zhao L, Hartl FU, Neupert W. Cytosolic Protein Vms1 Links Ribosome Quality Control to Mitochondrial and Cellular Homeostasis. Cell. 2017;171:890–903. doi: 10.1016/j.cell.2017.10.002. e818. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Lambert TD, Howard J, Plant A, Soffe S, Roberts A. Mechanisms and significance of reduced activity and responsiveness in resting frog tadpoles. The Journal of experimental biology. 2004;207:1113–1125. doi: 10.1242/jeb.00866. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, et al. Parkinson’s Disease-Associated Kinase PINK1 Regulates Miro Protein Level and Axonal Transport of Mitochondria. PLoS Genet. 2012;8:e1002537. doi: 10.1371/journal.pgen.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Acin-Perez R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A. 2011;108:12920–12924. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Bennett EJ. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J Cell Biol. 2014;204:467–476. doi: 10.1083/jcb.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera-Martinez E, Brito Querido J, Valasek LS, Simonetti A, Hashem Y. ABCE1: A special factor that orchestrates translation at the crossroad between recycling and initiation. RNA Biol. 2017;0 doi: 10.1080/15476286.2016.1269993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxidants & redox signaling. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W. Protein import into mitochondria. Annual review of biochemistry. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. The Journal of cell biology. 2008;180:1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Lazarou M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016;26:733–744. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CU, Pestova TV, Pisarev AV. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. The EMBO Journal. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Heon E, Krupin T, Ritch R, Kreutzer D, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. The EMBO Journal. 2012 doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Hegde RS. Reconstitution of a minimal ribosome-associated ubiquitination pathway with purified factors. Molecular cell. 2014;55:880–890. doi: 10.1016/j.molcel.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme C, Zhang L, Kremmer E, Ihling C, Chartier A, Sinz A, Simonelig M, Wahle E. Subunits of the Drosophila CCR4-NOT complex and their roles in mRNA deadenylation. RNA. 2010;16:1356–1370. doi: 10.1261/rna.2145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsunomiya T, Roth JS. Studies on the function of intracellular ribonucleases. V. Ribonuclease activity in ribosomes and polysomes prepared from rat liver and hepatomas. J Cell Biol. 1966;29:395–403. doi: 10.1083/jcb.29.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vilain S, Esposito G, Haddad D, Schaap O, Dobreva MP, Vos M, Van Meensel S, Morais VA, De Strooper B, Verstreken P. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012;8:e1002456. doi: 10.1371/journal.pgen.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, Lavoie MJ, Schwarz TL. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R, Jr, Mishra P, Levine B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell. 2017;168:224–238e210. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4439–4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, He WT, Tian S, Meng D, Li Y, Chen W, Li L, Tian L, Zhong CQ, Han F, et al. pelo is required for high efficiency viral replication. PLoS pathogens. 2014;10:e1004034. doi: 10.1371/journal.ppat.1004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Doan C, Liu D, Xie T. Pelota controls self-renewal of germline stem cells by repressing a Bam-independent differentiation pathway. Development. 2005;132:5365–5374. doi: 10.1242/dev.02151. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.