

Abstract
With advances in neonatal care, preterm neonates are surviving with an evolving constellation of motor and cognitive disabilities that appear to be related to widespread cellular maturational disturbances that target cerebral gray and white matter. Whereas preterm infants were previously at high risk for destructive brain lesions that resulted in cystic white matter injury and secondary cortical and subcortical gray matter degeneration, contemporary cohorts of preterm survivors commonly display less severe injury that does not appear to involve pronounced glial or neuronal loss. Nevertheless, these milder forms of injury are also associated with reduced cerebral growth. Recent human and experimental studies support that impaired cerebral growth is related to disparate responses in gray and white matter. Myelination disturbances in cerebral white matter are related to aberrant regeneration and repair responses to acute death of premyelinating late oligodendrocyte progenitors (preOLs). In response to preOL death, early oligodendrocyte progenitors rapidly proliferate and differentiate, but the regenerated preOLs fail to normally mature to myelinating cells required for white matter growth. Although immature neurons appear to be more resistant to cell death from hypoxia–ischemia than glia, they display widespread disturbances in maturation of their dendritic arbors, which further contribute to impaired cerebral growth. These complex and disparate responses of neurons and pre-OLs thus result in large numbers of cells that fail to fully mature during a critical window in development of neural circuitry. These recently recognized forms of cerebral gray and white matter dysmaturation raise new diagnostic challenges and suggest new therapeutic directions centered on reversal of the processes that promote dysmaturation.
Premature birth is a major public health issue internationally affecting 13 million babies worldwide. In the United States, the rate of preterm birth continues to rise, with prematurity now complicating 1 in 8 deliveries.1 Many neonates delivered preterm require care in a neonatal intensive care unit (NICU). Despite improved NICU therapies that have reduced the mortality of preterm neonates, neurodevelopmental morbidity persists at very high rates.2 Among children born very preterm, 5 to 10% have major motor deficits, including cerebral palsy (CP), and more than half have significant cognitive, behavioral, or sensory deficits, which makes prematurity a leading cause of neurodevelopmental disability in North America.3–13 Despite efforts to improve the brain health and outcomes of children born preterm, there continues to be wide variation in functional disabilities among preterm infants, even those born at the same gestational age.14
The functional consequences of injury to the immature developing brain may involve several domains: motor, cognition, language and behavior, and vision and hearing. Disabilities in these domains often co-occur.15 For example, severe white matter injury (WMI), such as cystic periventricular leukomalacia (PVL), usually results in spastic diplegic CP and visual dysfunction, often with deficits in cognition and learning. There is increasing recognition that following perinatal brain injury, cognitive deficits can occur in the absence of significant motor impairments and cerebral palsy.16 In preterm children with broadly normal IQ, processing deficits include problems in attention and executive functions (eg, cognitive flexibility, inhibitory control, working memory)5,17,18 and visually based information processing and language.19–24 Importantly, cognitive and behavior problems persist to young adulthood.10–12,22,23,25,26 These prevalent neurocognitive impairments point to widely distributed brain abnormalities or problems with brain connectivity.27 Systemic illnesses, such as infections, are common in the very preterm neonate and further affect the normal trajectory of brain maturation.28 The diverse spectrum of neurocognitive and motor outcomes following preterm birth, and the increasing recognition of abnormal brain maturation in this population, underscore the need for refined cellular–molecular mechanisms and high-resolution brain imaging to ascertain what aspects of early brain development are mostly related to long-term outcome.
Until recently, the extensive brain abnormalities in preterm neonates appeared to be related mostly to destructive processes that lead to substantial deletion of neurons, axons, and glia from necrotic lesions in the developing brain. However, advances in neonatal care coincide with a growing body of evidence that the preterm gray and white matter frequently sustain less severe insults, where tissue destruction is the minor component. As discussed below, these milder insults primarily comprise distinctly different forms of pathology in cerebral white and gray matter involving aberrant responses to injury that disrupt the maturation of glial progenitors and neurons. Late oligodendrocyte (OL) progenitors (preOLs) are highly susceptible to early ischemic cell death that triggers a maladaptive regeneration and repair response where the surviving progenitor pool expands but fails to differentiate and myelinate. By contrast, several types of immature projection neurons are resistant to cell death, but nevertheless fail to generate a normal arbor of dendritic processes and spines. These emerging findings suggest that brain injury in the majority of preterm survivors involves a primary cerebral dysmaturation disorder that may ultimately be amenable to strategies directed at promoting brain maturation and improved neurological outcome.
The Spectrum of WMI and Its Relevance to White Matter Dysmaturation
WMI in preterm newborns is linked to ischemia, infections, and inflammation.3,29–32 A broad spectrum of WMI severity is observed in the human preterm infant and in experimental models, which ranges from cystic necrotic lesions to diffuse gliotic lesions that lack overt necrosis. Cystic necrotic WMI has been widely appreciated since the classical descriptions of PVL by Banker and Larroche >50 years ago.33–39 Cystic PVL causes degeneration of all cell types, including glia and axons.40,41 On pathological examination, the cysts are typically >1mm. Whereas cystic PVL was previously the major form of WMI in preterm survivors, the incidence has markedly declined.42–45 In several recent series, focal cystic lesions were detected by magnetic resonance imaging (MRI) in <5% of cases.42–47
Despite the pronounced reduction in cystic PVL, small foci of necrosis continue to be defined by neuropathological examination. These discrete foci of microscopic necrosis (microcysts) typically measure <1mm.48 Similar to cystic PVL, microcysts evolve to lesions enriched in cellular debris, degenerating axons, and phagocytic macrophages (Fig 1A–C).49 The significance of microcysts is an important but clinically inaccessible question, because MRI currently cannot visualize these lesions at clinical field strengths of 3T. The extent to which microcysts contribute to disability or are clinically silent is unclear. Microcysts were visualized by MRI at ultrahigh magnetic field strength (12T) in a model of preterm WMI in fetal sheep (see Fig 1D).50 In this model and recent human autopsy cases including archival and contemporary cases,49 microcysts were observed in ~35% or more of cases. However, in both human and experimental studies, they comprised only ~1 to 5% of total lesion burden (see Fig 1F). Importantly, the overall burden of human necrotic WMI (cystic PVL and microcysts) was decreased by ~10-fold in contemporary cohorts relative to retrospective cases from earlier decades, which underscores the importance of the diffuse component of WMI.49
FIGURE 1.
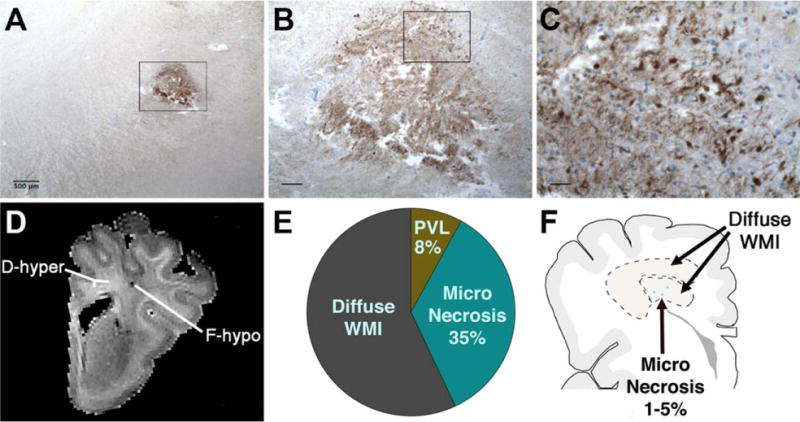
Microscopic necrosis has a high incidence but constitutes a small fraction of preterm cerebral white matter injury (WMI). (A) Typical sparse distribution of human microcysts visualized by staining for β-amyloid precursor protein, a marker of axonal degeneration. Note that axonal degeneration is usually restricted to microcysts and is not visualized in the surrounding regions of diffuse WMI. Sample is from a human autopsy brain at 32 weeks postconception. (B) Detail of the degenerating axons in the microcyst seen in the box in A. Degenerating axons were visualized both in the core and at the periphery of the microcyst. (C) Detail of the degenerating axons in the box in B shows numerous swollen and dystrophic-appearing axons. (D) A microcyst visualized by high-field (12T; T2) ex vivo magnetic resonance imaging as a focal hypointense lesion (F-hypo). Chronic WMI was analyzed 2 weeks after global cerebral ischemia in a fetal sheep model of preterm cerebral injury.50 Note that the surrounding diffuse WMI appeared as a diffuse hyperintense signal (D-hyper). (E) Pie chart showing the approximate relative percentages of human diffuse WMI, cystic periventricular leukomalacia (PVL), and microscopic necrosis, adapted from Pierson et al48 and Buser et al.49 (F) Schematic diagram showing the relative burden of human microcysts (dots) relative to diffuse WMI (within dotted lines), which typically comprises >80% of the total burden of WMI. Scale bars: A, 500μm; B, 100μm; C, 25μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Diffuse WMI is the characteristic pattern of brain injury most frequently observed in contemporary cohorts of premature newborns (see Fig 1F). The extent of these lesions is difficult to define by conventional neuropathology. However, recent quantitative studies of the burden of reactive astrocytes and microglia in chronic human WMI found that these lesions displayed a very diffuse inflammatory reaction that extended considerably beyond foci of microcysts (see Fig 1F).49 As discussed below, the chronic lesions evolve from early WMI, where the human OL lineage is particularly susceptible to oxidative damage51 of a magnitude consistent with hypoxia–ische-mia.52,53 Initially, this diffuse WMI causes selective degeneration of late preOLs, and axons are mostly spared except in necrotic foci.40,41
Diffuse WMI is often readily identified on diagnostic MRI as multifocal lesions that are seen in approximately one-third of preterm newborns at 24 to 32 weeks gestation when scanned in the first weeks of life.3,29 Although MRI-defined focal lesions are associated with an elevated risk of neurocognitive and motor dysfunction, they are likely to underestimate the full extent of injury and do not fully account for the burden of neurodevelopmental disability in this population.3,54–57 These early multifocal lesions are followed by more widespread abnormal microstructural (eg, fractional anisotropy [FA]) and metabolic brain development as premature newborns grow to term age.29,58–60 Importantly, brain dysmaturation observed in the preterm neonate is not transient, but persists through childhood with altered brain structure and connectivity, and is associated with adverse long-term neurodevelopmental outcomes.27,55,61–66
Mechanisms of Abnormal Myelination: Overview and Role of Axonal Injury
The major cell types that may degenerate during the initial phase of WMI are the axon and OL lineage cells. As WMI evolves, the responses of these 2 cell types are quite distinct and are discussed in this and the next section. Essentially complete myelination failure occurs in necrotic foci as a consequence of the degeneration of all cellular elements in these relatively uncommon but clinically significant lesions (Fig 2A, upper pathway). The mechanisms of abnormal myelination in diffuse WMI have received increased attention with the recognition that these are the most extensive lesions in preterm neonates. Disturbances in the normal patterns of myelination in diffuse WMI (see Fig 2A, lower pathway) are initiated by “selective vulnerability” of late preOLs that are enriched in cerebral white matter during restricted windows in development.67
FIGURE 2.
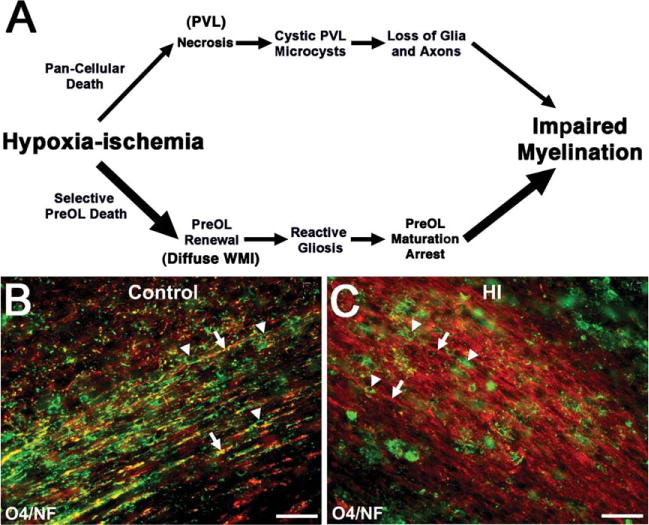
Myelination failure in chronic diffuse white matter injury (WMI) coincides with lesions highly enriched in reactive astrocytes, activated microglia, and oligodendrocyte progenitors (preOLs) that are arrested in their maturation. (A) Distinctly different pathogenetic mechanisms mediate abnormal myelination in necrotic lesions (periventricular leukomalacia [PVL]; upper pathway) versus lesions with diffuse WMI (lower pathway). Hypoxia–ischemia (HI) is illustrated as one potential trigger for WMI. More severe HI triggers white matter necrosis (upper pathway) with pancellular degeneration that depletes the white matter of glia and axons. Severe necrosis results in cystic PVL, whereas milder necrosis results in microcysts. Milder HI (lower pathway) selectively triggers early preOL death. preOLs are rapidly regenerated from a pool of early preOLs that are resistant to HI. Chronic lesions are enriched in reactive glia (astrocytes and microglia/macrophages) generating inhibitory signals that block preOL differentiation to mature myelinating oligodendrocytes. Myelination failure in diffuse WMI thus results from preOL arrest rather than axonal degeneration. The molecular mechanisms that trigger preOL arrest are likely to be multifactorial and related to factors intrinsic and extrinsic to the preOLs. Note that the lower pathway is the dominant one in most contemporary preterm survivors, whereas the minor upper pathway reflects the declining burden of white matter necrosis that has accompanied advances in neonatal intensive care. (B) Typical appearance of normal early myelination of axons in a perinatal rodent at postnatal day 10.80 Axons are visualized by staining for neurofilament protein (NF; red). Early myelination of axons is visualized with the O4 antibody (green). (C) preOL arrest in a chronic white matter lesion where numerous preOLs (green) are seen, but the axons (red) are diffusely unmyelinated. Scale bars = 100μm.
Axonal injury is a prominent feature of WMI where necrosis is present.40,68 Necrotic lesions are a minor component of WMI in both human and experimental models and comprise only about 5% of the total burden of WMI.49,50 Such necrotic lesions often contain dystrophic axons and axonal spheroids, which degenerate during the early phase of coagulative necrosis.33,48,49,69–72
The contribution of axonal injury to diffuse WMI has been more controversial. Axonal injury has been observed in regions of chronic human diffuse WMI that are adjacent to regions of necrosis,40 but these dystrophic axons are likely to be structurally continuous with the degenerating axons in necrotic foci. During the acute phase of diffuse WMI in preterm fetal sheep, acute axonal injury was rarely observed,53 and in chronic diffuse WMI no significant axonal degeneration, axonal loss, or shift in the distribution of axon calibers was observed by quantitative electron microscopy studies.41,50
In vitro studies have defined glutamate-mediated maturation-dependent mechanisms that define the susceptibility of developing axons to oxidative stress and hypoxia–ischemia.73–75 Larger caliber axons, which are preparing to myelinate, are particularly susceptible to injury in contrast to smaller caliber unmyelinated axons, which are more resistant.76 Hence, overt axonal degeneration localizes to discrete foci of necrosis related to severe energy failure. Axonal degeneration does not appear to be a major component of diffuse WMI prior to active myelination. Hence, the major sites of axonal degeneration are necrotic lesions, and axons appear to be mostly intact in diffuse WMI.
Mechanisms of Abnormal Myelination in Diffuse WMI: Degeneration and Dysmaturation of Glial Progenitors
Abnormal myelination in diffuse WMI is initiated by selective degeneration of late preOLs, which extensively populate normal human cerebral white matter throughout the high-risk period for diffuse WMI.77 The timing of appearance and spatial distribution of susceptible OL lineage cells coincides with the magnitude and distribution of acute ischemic injury in several experimental models of WMI. In particular, preOLs are highly susceptible to hypoxia–ischemia and inflammation,78,79 whereas earlier and later OL stages are markedly more resistant.52,53,80 The enhanced susceptibility of preOLs is a cell intrinsic property that is independent of the perinatal age of the animal81 or the location of these cells in the forebrain.53 The increasing developmental resistance of cerebral white matter to hypoxia–ischemia is related to the onset of preOL differentiation to premyelinating OLs that display reduced susceptibility to hypoxia–ischemia.52 The changes in white matter FA seen on MRI with brain maturation correspond closely with maturation of the OL lineage.82,83
Despite the pronounced selective degeneration of preOLs in acute diffuse WMI, abnormal myelination in these lesions is defined by more complex responses of the OL lineage that are related to a cellular dysmaturation process (see Fig 2A, lower pathway). Although it was initially hypothesized that abnormal myelination arises from a persistent loss of pre-OLs,84 subsequent findings have supported an alternative mechanism where myelination disturbances involve a potentially reversible process linked to arrested pre-OL maturation. Depletion of total OL lineage cells has surprisingly not been observed in chronic human or experimental lesions.49,50,72,80,85 Despite substantial acute and delayed preOL degeneration after hypoxia–ischemia, surviving preOLs in preterm-equivalent rats rapidly increased in number to regenerate depleted preOLs.80,86,87 This preOL expansion appeared to be driven mostly by preOLs that proliferated locally at the sites of WMI80 or cortical injury88 rather than from the subventricular zone, where less robust generation of OL lineage cells has been observed.89–91 Hence, regeneration of preOLs from the surviving preOL pool compensates for preOL death, but these newly generated preOLs display persistent arrested differentiation in chronic lesions and fail to myelinate intact axons (see Fig 2B, C). Recently, arrested maturation of preOLs was shown to contribute to myelination failure in diffuse WMI in both preterm fetal sheep and humans,49,50 where lesions were typical of that seen in contemporary cohorts of preterm infants. A robust expansion of human preOLs was also defined in chronic lesions, which was unexpected, given the significant loss of these cells during the acute phase of WMI.51 Hence, chronic diffuse WMI is characterized by an aberrant response to acute injury, which involves a disrupted regeneration and repair process, where preOLs are regenerated but they remain dysmature.
preOL maturation arrest may adversely influence subsequent white matter maturation in several ways. Recent studies support that viable OLs and myelination are critical for axon survival,92 raising the possibility that preOL arrest could also adversely affect the functional integrity of axons in chronic lesions. Chronic WMI may also coincide with an expanded developmental window during which preOL maturation-arrest persists and confers an enhanced risk for recurrent and potentially more severe WMI. In neonatal rat chronic WMI, preOLs with arrested maturation displayed a markedly increased susceptibility to recurrent hypoxia–ischemia that triggered a massive selective apoptotic degeneration of preOLs.80 Serial neuroimaging studies are needed to better define the progression of WMI in human preterm infants at risk for recurrent insults.57,93–95 Prior studies have identified clinical features that identify infants at risk for exacerbation of initial cerebral injury (eg, preterm newborns with postnatal sepsis). Recurrent and systemic illness is an important risk factor that may increase susceptibility to progressively more severe WMI.29,30
Molecular Mechanisms of preOL Maturation Arrest and Abnormal Myelination
There continues to be a significant proportion of preterm infants for whom WMI derives from poorly defined antenatal or perinatal factors. Hence, there is a need for alternative therapies that enhance myelination and promote regeneration and repair of chronic WMI that may not be recognized until after birth. Strategies to reverse myelination failure in multiple sclerosis and other demyelination disorders have been extensively reviewed.96–98 preOL maturation arrest appears to be related to a complex array of intrinsic, extrinsic, and epigenetic factors that regulate OL precursor cell (OPC) cell cycle exit, OL lineage progression, and myelination.99–103 Inhibitors of voltage-activated potassium channels and membrane depolarization block proliferation and differentiation of preOLs.104,105 Sox17 expression regulates cell cycle exit in OPCs, transgenic overexpression promotes OL differentiation,106 and enhanced Sox17 expression occurs in OLs in active remyelinating lesions.107 Numerous genes are activated by oxidative stress, which regulate OL maturation, and oxidative stress promotes global histone acetylation, which can block OL differentiation.108–112 Post-transcriptional control by microRNAs regulates OL differentiation, and OPCs that lack mature microRNAs display arrested maturation.113,114
Multiple molecules likely act in concert with other signals in chronic white matter lesions to prevent preOL maturation and normal myelination. A number of these signals appear to be linked to reactive astrogliosis.115 Diffuse WMI is highly enriched in reactive astrocytes and microglia that overlap with areas of preOL maturation arrest.49 Reactive astrogliosis is the most distinct pathological feature of diffuse WMI.68 From recent quantitative studies, astrogliosis was found to be much more extensive than expected from standard histopathological approaches.49 Hence, diffuse WMI is characterized by a diffuse chronic inflammatory response that may disrupt normal myelination.
In diffuse WMI, the extracellular matrix (ECM) is a rich source of hyaluronic acid (HA) and one of its receptors, CD44.49 During chronic human neonatal WMI,49 CD44-positive reactive astrocytes synthesize high molecular weight HA (ie, >106Da), a nonsulfated, protein-free glycosaminoglycan that accumulates in the ECM.116 Arrest of preOL maturation is stimulated both in vitro and in vivo by high molecular weight forms of HA that are digested to bioactive forms by a central nervous system (CNS) enriched hyaluronidase, PH20 that is glycosyl-phosphatidylinositol anchored and has both neutral and acidic pH optima.117,118 PH20 is expressed by OPCs and reactive astrocytes, and its expression is particularly elevated in demyelinated lesions. Overexpression of PH20 inhibits preOL differentiation in vitro, and HA fragments generated by PH20 block remyelination in vivo. Pharmacological inhibition of PH20 promotes OL maturation in vitro and myelination in vivo, which is accompanied by enhanced nerve conduction.
Reactive astrocytes also increase their expression of bone morphogenetic proteins that inhibit preOL differentiation with concurrent promotion of astrocyte differentiation.119 Similarly, the Notch ligand Jagged1 is elevated on reactive astrocytes in demyelinating lesions and activates Notch signaling on preOLs, preventing their maturation.120 Dysregulation of Wnt/β-catenin signaling in preOLs promotes preOL arrest, delays normal myelination, and disrupts remyelination.121–126 The glycogen synthase kinase 3β (GSK3β) is a component of the Wnt signaling cascade, which also has been implicated in the regulation of preOL maturation, and inhibitors of GSK3β promote remyelination in adult white matter via mechanisms that include decreasing Notch1 signaling.127 Not only does constitutive expression of the epidermal growth factor receptor (EGFR) in neonatal white matter promote pronounced proliferation of pre-OLs,128 but enhanced EGFR signaling stimulates adult CNS myelination and remyelination.129 EGFR activation via an intranasally administered form of EGF reduced OL death from neonatal chronic hypoxia, promoted OL maturation and myelination, and led to functional improvement.130 Other astrocyte-derived growth factors also may contribute to preOL arrest. Insulinlike growth factors promote OL lineage cell survival and myelination and protect against preOL loss in fetal and neonatal models of WMI.131–133 Bone morphogenetic proteins both repress OL differentiation and regulate myelin protein expression.119,134–136
Although neuroimaging studies have defined impaired growth of central white matter pathways in preterm survivors, future studies are needed in relevant experimental models as well as human autopsy studies to define the evolution of cerebral white matter lesions over months to years to identify the relative contributions of dysmyelination and axonal dysfunction to functional disabilities in preterm survivors. Such information is of critical importance to define the period over which chronic WMI may be repaired and to better identify mechanisms to promote additional regeneration and repair of WMI.
Potential Mechanisms of Impaired Cerebral Gray Matter Growth in Preterm Survivors
As proposed by Volpe with the concept of an “encephalopathy of prematurity,” impaired growth of the cerebral gray matter of preterm survivors may involve both destructive and developmental disturbances.32 It is now apparent that premature newborns have more extensive gray matter abnormalities than “injuries,” as identified by signal abnormalities on conventional MRI. Even children and adults born preterm with normal neurocognitive function nonetheless express altered cortical activation and functional connectivity during language and visual processing.27,137–140 Preterm newborns at term age also exhibit reduced functional connectivity between the cortex and thalamus on functional connectivity MRI.141 Altered functional connectivity in children and adolescents born preterm is now recognized as a critical risk factor for adverse neurocognitive outcomes.64,137,138
Several large human neuroimaging studies have identified that preterm survivors display significant reductions in the growth of cortical and subcortical gray matter structures that include the basal ganglia, thalamus, hippocampus, and cerebellum.61,142–145 Impaired growth has been commonly associated with perinatal risk factors, varying degrees of focal or diffuse WMI, and ventriculomegaly without significant overt necrotic WMI in most cases.146–152 As discussed below, at least 2 potentially complementary mechanisms may explain this impaired cerebral growth. The first mechanism involves impaired growth related to widespread primary degeneration of neurons in multiple cortical and subcortical gray matter structures or secondary neuronal degeneration related to axonal injury in foci of white matter necrosis. This may include selective primary loss of subplate neurons (SPNs), a transient cell population required to establish thalamocortical connections.153 SPNs are vulnerable to perinatal hypoxia–ischemia in rodents154,155 and are reduced in association with PVL,156 and thalamocortical connections are disrupted in preterm newborns with WMI, resulting in visual dysfunction.157 A second mechanism involves disturbances in neuronal maturation that disrupt the growth of large distinct populations of neurons in multiple cortical and subcortical gray matter structures.
Factors Related to the Extent of Neuronal Degeneration in Preterm Gray Matter
Cerebral neurons in the full-term neonate are highly susceptible to hypoxic–ischemic degeneration that is mediated via both excitotoxic neuronal necrosis and neuroapoptosis.158–163 However, the susceptibility of preterm neurons to hypoxia–ischemia is more variable and appears to be related to the severity of the underlying insult and the associated severity of WMI. As summarized in Figure 3 (left panel), insults that trigger significant white matter necrosis are accompanied by neuronal degeneration in cerebral gray and white matter. In experimental studies, the magnitude of preterm neuronal degeneration increased with more prolonged hypoxia–ischemia. Widespread neuronal death was triggered by prolonged hypoxia–ischemia that also caused cystic necrotic WMI.53 Significant neuronal loss has also been seen in the human cortex, basal ganglia, thalamus, and cerebellum in association with necrotic WMI,48,156,164,165 but not in cases where diffuse WMI occurred without significant necrosis.48 Secondary neuronal loss appears to principally arise from retrograde axonal degeneration that occurs in association with necrotic WMI (see Fig 3, left panel).40,41
FIGURE 3.
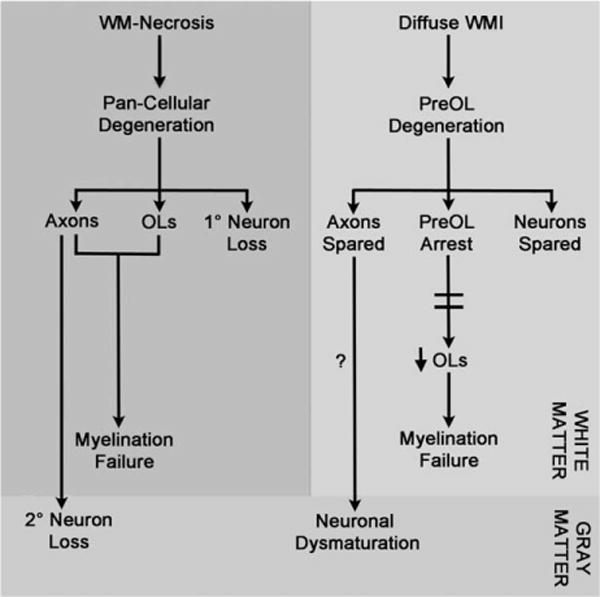
In premyelinated white matter (WM), the cellular mechanisms and extent of neuroaxonal degeneration and myelination failure are distinct for necrotic and diffuse WM injury (WMI). WM necrosis (cystic periventricular leukomalacia/microcysts; left panel) is characterized by loss of all cellular elements in necrotic foci (glia, axons, and interstitial neurons). Degeneration of axons and oligodendrocytes (OLs) both contribute to myelination failure. Retrograde degeneration of axons in necrotic foci contributes to neuronal loss in cerebral gray matter. Diffuse WMI (right panel) involves selective degeneration of OL progenitors (preOLs) with sparing of premyelinating axons and interstitial neurons. Note that recent experimental data support a role for selective vulnerability of larger caliber early myelinating axons that appear later in white matter development as myelination progresses.76 Myelination failure is related to a failure of preOL differentiation (preOL arrest) to OLs. The mechanism of neuronal dysmaturation is unclear and may involve a direct effect of gray matter ischemia on maturation of dendrites and spines as well as axonal factors related to chronic white matter inflammation.
In contrast to the extensive neuronal loss seen in association with white matter necrosis, immature neurons do not appear to significantly degenerate under conditions that primarily generate diffuse WMI (see Fig 3, right panel). As discussed above, diffuse WMI triggers selective preOL degeneration but spares axons. Numerous neurons are also present in regions of diffuse WMI, but do not degenerate under conditions that trigger early preOL degeneration in preterm fetal sheep.53,166 The resistance of the preterm gray matter to neuronal degeneration is further supported by quantitative cerebral blood flow studies in preterm fetal sheep, where the magnitude of global ischemia was very similar in superficial cortex and deeper cerebral structures including the caudate nucleus and periventricular white matter.53,167 This similar degree of ischemia in gray and white matter resulted in diffuse WMI and significant preOL degeneration, but largely spared neurons in the gray and white matter. In human autopsy cases with early diffuse WMI and preOL degeneration, neither the preterm gray matter nor white matter displayed evidence of significant oxidative stress or degeneration that involved neurons or axons.51 The magnitude of oxidative stress in preterm white matter was quite substantial and similar to that sustained by gray matter in term infants diagnosed with severe hypoxic–ischemic encephalopathy. In the preterm gray matter, significant loss of neurons thus appears to be related to more severe ischemia that causes destructive WMI. Neuronal degeneration is not significantly associated with acute diffuse WMI.
Gray Matter Injury and Neuronal Dysmaturation
Reduced growth of cerebral gray matter can also occur in response to conditions that disrupt neuronal maturation without neuronal loss (see Fig 3, right panel). We recently analyzed preterm fetal sheep at ~28 weeks gestation that have cerebral development similar to humans. In response to cerebral ischemia, these animals acquired diffuse WMI, as well as a progressive reduction in cortical growth that was not explained by delayed neuronal degeneration.168 Stereological cell counts of total cortical neurons confirmed that there was no chronic loss of neurons. Cortical volume loss was thus accompanied by an increased packing density of neurons. This unexpected result was explained by detailed analysis of the maturation of the dendritic arbor of pyramidal neurons, the major population of cortical projection neurons. During normal development, pyramidal neurons are highly immature in the preterm cerebral cortex, but in near-term animals the dendritic arbor becomes highly arborized, which coincides with a marked increase in cortical volume (Fig 4). In response to preterm ischemia, cortical growth impairment was accompanied by a significant reduction in the complexity of the dendritic arbor, consistent with the notion that neuronal maturation was disrupted in the setting of cerebral ischemia. Compared to controls, the ischemic animals displayed neuronal dysmaturation that was reflected in a reduction in the total dendritic length as well as the number of branches, branch endings, and branch points. Notably, the dendritic arbor was most simplified closer to the cell body, where synaptic integration occurs.
FIGURE 4.
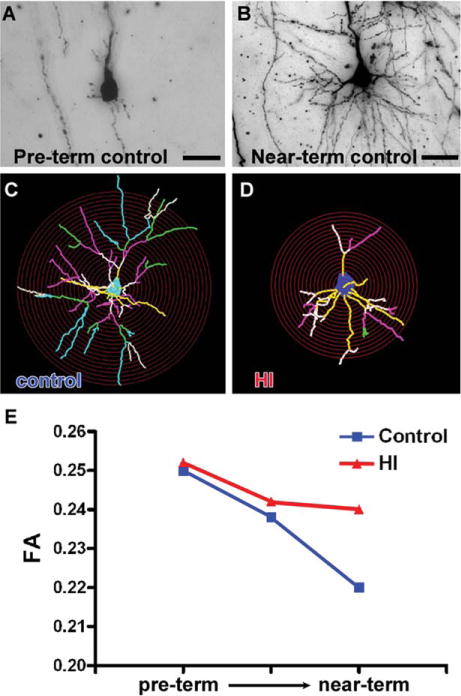
The preterm brain is enriched in immature neurons that do not degenerate in response to ischemia, but are highly susceptible to impaired maturation that manifests as a less mature dendritic arbor with reduced spine density. (A) A typical pyramidal neuron from the preterm cerebral cortex of a control fetal sheep. Note the paucity of processes in contrast to the highly complex dendritic arbor of a pyramidal neuron from a near-term animal (B).168 (C, D) In response to preterm ischemia, cortical pyramidal neurons display disrupted maturation. Note that the typical control cell (C) is more highly arborized in contrast to the response to transient cerebral ischemia that resulted in a more simplified dendritic arbor (D). The relative complexity of the cells can be appreciated from the overlay of the red concentric Scholl rings, which illustrates that the processes of the dysmature neurons intersect less frequently with the rings. The yellow, white, pink, green, and blue lines represent first-, second-, third-, fourth-, and fifth-order branches, respectively, from the soma. Note the overall reduction in the size and complexity of the branching pattern in D. (E) Reductions in cortical growth also manifest as disturbances in cortical anisotropy. Note the normal progressive decline in fractional anisotropy (FA) in controls (blue) between preterm and near-term cortical development, as adapted from Dean et al.168 In response to ischemia, higher cortical anisotropy (more restricted water diffusion) was observed in response to ischemia (red) relative to control (blue), which was related to the reduced complexity of the dendritic arbor of the ischemic neurons (eg, in D) versus controls (eg, in C). Scale bars = 20nm. HI = hypoxia–ischemia.
This neuronal dysmaturation response was not restricted to the cerebral cortex but has been observed in other brain regions. Disturbances in dendritic arborization have also been reported in CA1 pyramidal neurons in the hippocampus in a near-term rodent model of hypoxia–ischemia in which dendritic arborization disturbances occurred as a response to significant necrotic cerebral injury and neuronal loss.169 The caudate nucleus also displayed reduced growth in response to ischemia but with no apparent loss of γ-aminobutyric acidergic (GABAergic) medium spiny projection neurons or interneurons.166 Deletion of GABAergic interneurons has been proposed to occur during their migration through human white matter during the period of high risk for WMI.170 However, reduced growth of the caudate was not explained by loss of GABAergic neurons, but rather by a significant disruption in the dendritic arbor of caudate projection neurons. Neuronal dysmaturation was defined by disrupted maturation of the dendritic arbor. Hence, widespread disturbances in maturation of cortical and caudate projection neurons occurred in association with nondestructive cerebral lesions that had diffuse WMI but lacked significant neuronal degeneration.
Neuronal Dysmaturation and Disturbances in Synaptic Activity
A role for neuronal dysmaturation in cognitive and behavioral disturbances in preterm survivors is suggested by analysis of dendritic spines, the key sites for synaptic activity. In response to ischemia, reduced numbers of spines were observed on the dysmature dendrites of projection neurons in both the cortex168 and caudate,166 which suggests that widespread disturbances in neuronal connectivity may contribute to the global disturbances in neurodevelopment seen in preterm survivors.
Because disturbances in neuronal maturation occur at a critical window in the establishment of neuronal connections, even transient neuronal dysmaturation may have persistent global effects on the subsequent development of CNS circuitry. Consistent with this hypothesis, recent electrophysiological studies in the caudate nucleus employed patch clamp recordings on medium spiny neurons (MSNs) and identified significant abnormalities in excitatory synaptic activity mediated by N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors in fetal ovine survivors at 1 month after preterm hypoxia–ischemia.166 Evoked excitatory postsynaptic currents (eEPSCs) in the ischemic animals were reduced in magnitude consistent with the reduced spine density on MSNs. The eEPSCs displayed a shorter duration and reduced current flow. A shorter duration of the eEPSCs was related to a shorter decay time of the fast AMPA/kainate receptor component of the eEPSC. A reduction in current flow was related to a reduction in the magnitude of the slow NMDA receptor component of the eEPSC.
The aforementioned disturbances in spine density and excitatory synaptic activity of MSNs may have several potential effects on the maturation of neuronal circuitry in the preterm caudate nucleus. A reduction in total afferent excitation of MSNs may occur, as well as a shorter time window for integration of multiple synaptic inputs onto these projection neurons. Moreover, disturbances in NMDA receptor-mediated synaptic activity may disrupt neuronal migration, synapse formation, and dendritic pruning.171–174 As a consequence, a vicious cycle of neuronal dysmaturation may be established that results in further disruption in the normal maturation of neuronal circuitry. Such abnormalities in synaptic activity in the caudate may contribute to a wide variety of neurobehavioral abnormalities in preterm survivors that include motor dyspraxias and transient dystonia as well as disturbances in learning, memory, attention, and executive function.
Imaging Dysmaturation Processes in Preterm WMI
MRI has been widely applied for the safe and reliable diagnosis of injury in the developing brain on conventional T1-weighted, T2-weighted, and diffusion-weighted images.175 As early as the newborn period, advanced imaging methods can now detect several types of developmental abnormalities of brain structure or function that evolve from the acute to recovery phases (Table 1). Recent observations with these advanced magnetic resonance techniques indicate that cerebral injury and some prevalent clinical conditions impede brain maturation in areas that appear normal on conventional MRI.29,58,59,95 Despite the widespread clinical application of diagnostic MRI, it is nonetheless important to recognize the limitations of MRI at regularly used clinical field strengths (eg, 1.5 or 3T). This is particularly relevant for the diagnosis of the full spectrum of WMI, where MRI may not fully define early diffuse WMI and has limited sensitivity for detection of microscopic foci of necrosis.50 Definition of diffuse WMI by MRI is of particular clinical interest, given that these lesions correspond to foci of white matter dysmaturation with preOL maturation arrest and abnormal myelination.
TABLE 1.
Neuroimaging Modalities for Analysis of Different Types of Brain Injury in Preterm Survivors
| Modality | Examples of Specialized Analyses | Types of Data Acquired |
|---|---|---|
| High-resolution MRI | Volumetrics, deformation-based morphometry | Volumes and growth of cortical and subcortical brain structures |
| DTI | Region of interest–based analyses, tract-based spatial statistics, tractography | Brain microstructure, myelination |
| MTR | Myelination | |
| MRSI | Measures of regional brain metabolites reflecting regional brain development and injury including N-acetyl-aspartate and lactate | |
| FCMRI | Map of the developing brain connectome including pathways relevant to specific functional outcomes, such as motor and vision control, as well as “resting state” network |
DTI = diffusion tensor imaging; FCMRI = functional connectivity MRI; MRI = magnetic resonance imaging; MRSI = magnetic resonance spectroscopy and spectroscopic imaging; MTR = magnetization transfer ratio.
To experimentally define cellular mechanisms of MRI-defined WMI in both diffuse WMI and necrotic lesions, registration algorithms were developed that permitted high field (12T) ex vivo MRI data to be aligned at high resolution with the corresponding histopathological data from the same white matter regions.50 WMI was generated by cerebral ischemia in a preterm fetal sheep model that closely reproduces the spectrum of WMI seen in human preterm survivors. At ultra–high-field strengths, 3 classes of MRI-defined chronic WMI were defined during the subacute phase of WMI at 1 and 2 weeks after ischemia. Each lesion type displayed unique astroglial and microglial responses that corresponded to distinct forms of necrotic or diffuse WMI.
Figure 5 compares the MRI characteristics of lesions defined on diagnostic MRI and at 12T. On diagnostic MRI scans, pronounced necrotic WMI is visualized as cystic lesions or manifests as volume loss of major white mater tracts such as the corpus callosum. At 12T, large necrotic lesions were visualized as focal hyperintense signal abnormalities (F-hyper) on T2-weighted images. These subacute large necrotic foci corresponded to histological lesions highly enriched in macrophages and activated microglia but with a progressive reduction in astrocytes. High-field MRI also detected discrete microscopic foci of necrosis <1mm in diameter. As noted above, these microcysts are not detected at clinical MRI field strengths (1.5 or 3T), but have been resolved by microscopic pathology studies.48,49
FIGURE 5.
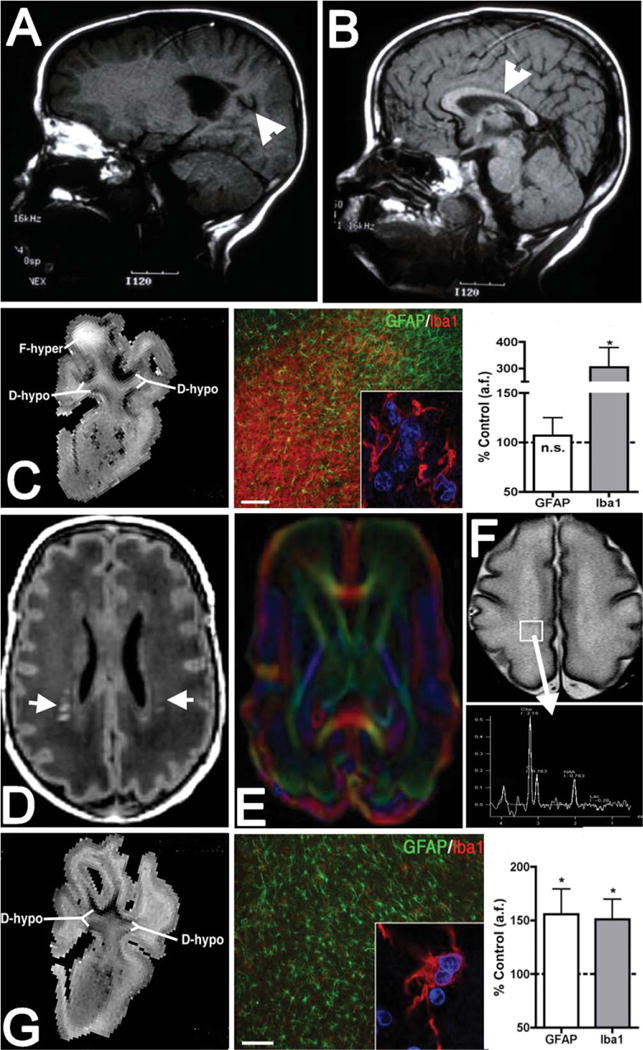
Diagnostic and experimental magnetic resonance imaging (MRI) approaches to define dysmaturation processes related to white matter injury (WMI). (A, B) MRI (1.5T; T1) appearance of marked cystic necrotic human WMI (A; arrowhead) adjacent to the lateral ventricle, which is associated with white matter volume loss that involves the corpus callosum (B; arrowhead). (C) High-field MRI–defined focal necrotic WMI and corresponding histopathological features. WMI was analyzed at 1 or 2 weeks after global cerebral ischemia in 0.65 gestation fetal sheep (adapted from Riddle et al50). At left is a representative T2-weighted image of a large focal hyperintense (F-hyper) lesion detected at 1 week. Note the hypointense diffuse WMI (D-hypo), discussed in G, below. In the center is a typical necrotic lesion defined by focal staining for reactive microglia and macrophages with Iba1 (red and inset) and a paucity of glial fibrillary acidic protein (GFAP)-labeled astrocytes. Nuclei in the inset are visualized with Hoechst 33342 (blue). At right, by 2 weeks after ischemia, necrotic WMI displayed a progressive decrease in GFAP-labeled astrocytes and a pronounced increased in Iba1-labeled macrophages and microglia. *p < 0.05; bar in C = 100μm. n.s. = nonsignificant. (D) Diffuse human WMI on diagnostic MRI (1.5T; T1) has the appearance of bilateral multifocal signal hyperintensities (arrows). (E) Diffusion tensor imaging (DTI) defines the microstructure of white matter tracts and can be used to follow the chronic progression of diffuse WMI. (F) Magnetic resonance spectroscopic imaging (MRSI) can be applied to define biochemical and metabolic abnormalities associated with diffuse WMI. Both DTI and MRSI detect abnormalities beyond the areas of signal abnormality on T1-weighted images. (G) Diffuse fetal ovine WMI defined at 12T as in C.50 At left is a representative image of diffuse hypointense (D-hypo) WMI seen on a T2-weighted image at 1 week after ischemia. In the center are typical histopathological features of diffuse WMI: pronounced astrogliosis defined by staining of reactive astrocytes with GFAP (green) and a less activated population of Iba1-labeled microglia/macrophages (red and inset). At right, quantification of the area fraction (a.f.) of astrocytes and microglia in diffuse WMI at 2 weeks after ischemia is shown. There was significantly elevated GFAP and Iba1 staining, consistent with a diffuse gliotic response to WMI. *p < 0.05; bar in G = 100μm. Images in A and B are courtesy of Dr Patrick Barnes, Stanford University.
The imaging characteristics of diffuse WMI differ substantially at clinical field strengths compared to high field. On diagnostic MRI, WMI without apparent necrosis is indicated by discrete focal or diffuse areas of magnetic resonance signal abnormalities (see Fig 5). Structural or biochemical abnormalities related to diffuse WMI also may be detected by advanced MRI techniques such as diffusion tensor imaging (DTI) and spectroscopic imaging. It is currently unknown whether these advanced imaging modalities can define lesions that correspond to preOL arrest. At 12T, diffuse hypointense signal abnormalities are seen on T2-weighted images. These diffuse hypointense signal changes corresponded to diffuse WMI characterized by reactive astrogliosis and coincided with myelination disturbances related to arrested maturation of preOLs.50 Similar to human autopsy studies, this form of fetal ovine WMI was the major form identified and comprised nearly 90% of the total volume of WMI. The majority of such lesions were large (>2.5mm3) and detected with high sensitivity and specificity. Axonal degeneration was not observed within diffuse WMI except within foci of microscopic necrosis.41
These findings suggest the need for additional studies at clinical MRI field strengths to achieve greater sensitivity to detect early diffuse WMI as well microcysts, and to apply more advanced technologies such as DTI and spectroscopic imaging. The ability to define these lesions would be a major advance to support the earlier diagnostic detection of WMI and provide an early surrogate outcome measure for monitoring during therapeutic trials.
Neuronal Dysmaturation and MRI-Defined Abnormalities in Cortical FA
MRI-defined changes in FA have been extensively studied as a noninvasive means to define the progression of normal cortical maturation. A progressive decline in FA accompanies cortical maturation in human176 and nonhuman primates.177–179 This progressive loss of FA was proposed to relate to the global maturation of the process arbor of cortical neurons during cortical development.176 Support for this hypothesis derives from recent experimental studies that found that progressive maturational complexity of the process arbor of cortical neurons coincides with the decline in FA.180
Disrupted maturation of cortical pyramidal neurons was also recently shown to provide an explanation for FA disturbances arising after global cerebral ischemia. Two recent human studies found that the normal progressive loss of cortical FA was delayed in human preterm survivors with impaired postnatal growth145 or reduced cortical growth.181 We made similar observations in preterm fetal sheep exposed in utero to global cerebral ischemia.168 These animals displayed impaired cortical growth, and MRI measurements of cortical FA were significantly higher in the hypoxic–ischemic animals relative to controls. To explain this observation, a mathematical model was developed to calculate FA based upon the morphology of control and ischemic neurons defined by Golgi stain (eg, Fig 4A, B). The FA values derived from MRI and neuronal morphology were very similar.168 Moreover, the FA values derived from neuronal morphology also demonstrated that the cortex in the hypoxic–ischemic group displayed higher FA values consistent with less random water diffusion along the processes of these more immature neurons (see Fig 4E, red). The decrease in FA in the controls (see Fig 4E, blue) was related to the increased complexity of the dendritic arbor of these neurons related to more random water diffusion. Interestingly, one factor associated with abnormal micro-structural cortical growth in human preterm neonates was impaired somatic growth (weight, length, and head circumference), even after accounting for coexisting brain injuries on MRI (eg, WMI) and other aspects of systemic illness (eg, infection).145 Hence, multiple factors including nutritional status and exposure to cerebral ischemia may contribute to the pathogenesis of neuronal dysmaturation in preterm survivors. Impaired cortical growth and function were also recently related to stressful procedures sustained by preterm neonates during intensive care.140,182,183
Role of Systemic Illness in Brain Growth and Maturation in Premature Neonates
The conditions that influence brain growth and dysmaturation are multifactorial. It is increasingly apparent that postnatal illness severity is a better predictor of brain health than gestational age at birth.67,148 Furthermore, postnatal illness severity is a stronger predictor of brain health than are many prenatal factors.28,29,59,145,184 In preterm neonates, risks of brain injury and adverse outcomes are altered by complex systemic illness. Postnatal infection in preterm newborns is associated with a significantly increased risk of WMI,29,31 including a progressive form of WMI that is more readily evident on MRI scans at term-equivalent age.30 Postnatal infections have been linked to altered development of white matter pathways59 and widespread impairments in brain development.28 Importantly, infections, even without positive cultures, have been associated with impaired neurodevelopmental outcome consistent with neonatal brain imaging findings.3,14,31
Bronchopulmonary dysplasia (BPD), with lung injury and inflammation requiring prolonged treatment with mechanical ventilation and supplemental oxygen, is a strong predictor of cognitive outcome, even after controlling for birth weight and neurological morbidity.185 BPD and days of ventilation have been linked to adverse development of the white matter and cortex.186–188 Recent data indicate that postnatal exposure to corticosteroids, used for the treatment of BPD or low blood pressure, is associated with impaired growth of the cerebellum.189
During a period of rapid brain maturation, preterm infants in the NICU are exposed to multiple painful and stressful procedures. Recently, this procedural pain and stress has also been linked to altered brain maturation that involves gray and white matter structures, as well as impaired brain function.182,183 Procedural pain in preterm neonates has also been associated with impaired postnatal growth,190 a predictor of poor cortical development.145 Importantly and consistent with these neonatal brain imaging observations, pain is associated with poorer neurodevelopment, a relationship moderated by parent–child interaction.191,192 The influence of parent–child interaction on outcomes illustrates that factors outside of the NICU, such as parental stress and anxiety, are also important determinants of neurodevelopmental outcomes in preterm infants.193,194 Preterm infants exposed to a parental intervention demonstrated enhanced maturation and connectivity on MRI at term-equivalent age.195 There are now converging experimental and human studies highlighting the potential of parent–infant interaction to compensate for compromised early brain maturation and adversity, indicating a wide window for optimizing brain development and outcome.192,196–199
Conclusions
Our understanding of the pathogenesis of brain injury in the premature infant has recently undergone significant redefinition, which coincides with advances in neonatal care that have markedly reduced the overall severity and extent of the destructive processes associated with cerebral injury. Significant improvement in the care of premature neonates has also coincided with the emergence and application of improved brain imaging, which has provided better resolution of some of the key features of cerebral injury during the period of most rapid changes in brain growth and maturation.
Surprisingly, what is emerging is the potential that the chronic disabilities in preterm survivors may not arise primarily from irreversible destructive lesions, but rather from a primary diffuse cerebral dysmaturation disorder that may ultimately be amenable to strategies directed at promoting brain maturation and improved neurological outcome. The activation of dysmaturation processes is a key factor that disrupts regeneration and repair in the preterm brain after injury. Glial progenitors respond to WMI by partially but incompletely mounting a repair process that regenerates and expands the preOL pool, which is blocked from maturation. The maturation block may involve both intrinsic and extrinsic factors that prevent OL maturation and myelination. In response to ischemia, some populations of immature projection neurons similarly fail to normally mature. In contrast to mature neurons in the full-term neonate that degenerate from activation of excitotoxic and apoptotic pathways,200 these immature neurons survive with a simplified dendritic arbor that contributes to reduced cerebral growth. Thus, cellular dysmaturation in gray and white matter may disrupt a critical developmental window that coincides with rapid brain growth and enhanced neuronal connectivity related to elaboration of the dendritic arbor, synaptogenesis, and myelination. Consistent with this notion, changes in neuronal gene expression and glial cell commitment to a neuronal fate have been observed in response to chronic hypoxia in mice and were reversible in response to environmental enrichment.201,202 Thus, disturbances in cerebral maturation could be multifactorial and exacerbated by postnatal factors such as intermittent hypoxia related to chronic lung disease.
The timing and nature of future interventions to prevent or reverse cellular dysmaturation may differ for gray and white matter. Factors such as improving infant nutrition, preventing infections, reducing neonatal stress, and implementing earlier behavioral interventions may all play a role in mitigating the impact of neuronal dysmaturation. Pharmacological interventions aimed at blocking the inhibitory pathways that sustain preOL maturation arrest may reverse or prevent myelination failure with the potential for enhanced connectivity of CNS pathways. There are thus a wealth of potential new opportunities to promote enhanced brain maturation and growth that were not feasible even a decade ago, when impaired brain development was largely attributable to irreversible injury related mostly to destructive processes and encephalomalacia.
Acknowledgments
This work was supported by the NIH National Institutes of Neurological Diseases and Stroke (NINDS; 1R01NS054044, R37NS045737-06S1/06S2; S.A.B.), the NIH National Institute of Aging (1R01AG03189; S.A.B.), the American Heart Association (S.A.B.), the March of Dimes Birth Defects Foundation (S.A.B.), the Canadian Institutes of Health Research (MOP-79262; S.P.M.), and NeuroDevNet Network Centres of Excellence. The Neuroscience Imaging Center at Oregon Health and Science University is supported by NINDS grant P30NS061800. S.P.M. is currently the Bloorview Children’s Hospital Chair in Pediatric Neuroscience and was supported by a Tier 2 Canada Research Chair in Neonatal Neuroscience, a Michael Smith Foundation for Health Research Scholar Award, and a Canadian Institutes of Health Research Clinician Scientist award.
We are grateful to Drs E. McClendon and P. Barnes for assistance with the preparation of the figures.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.March of Dimes. Prematurity campaign. Available at: http://www.marchofdimes.com/mission/prematurity-campaign.aspx. Accessed on March 19, 2014.
- 2.Synnes AR, Anson S, Arkesteijn A, et al. School entry age outcomes for infants with birth weight 800 grams. J Pediatr. 2010;157:989–994. doi: 10.1016/j.jpeds.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Miller SP, Ferriero DM, Leonard C, et al. Early brain injury in premature newborns detected with magnetic resonance imaging is associated with adverse early neurodevelopmental outcome. J Pediatr. 2005;147:609–616. doi: 10.1016/j.jpeds.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 4.Vohr BR, Allan WC, Westerveld M, et al. School-age outcomes of very low birth weight infants in the indomethacin intraventricular hemorrhage prevention trial. Pediatrics. 2003;111:e340–e346. doi: 10.1542/peds.111.4.e340. [DOI] [PubMed] [Google Scholar]
- 5.Marlow N, Wolke D, Bracewell MA, Samara M. Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med. 2005;352:9–19. doi: 10.1056/NEJMoa041367. [DOI] [PubMed] [Google Scholar]
- 6.Bodeau-Livinec F, Marlow N, Ancel PY, et al. Impact of intensive care practices on short-term and long-term outcomes for extremely preterm infants: comparison between the British Isles and France. Pediatrics. 2008;122:e1014–e1021. doi: 10.1542/peds.2007-2976. [DOI] [PubMed] [Google Scholar]
- 7.Roberts G, Anderson PJ, Doyle LW. Neurosensory disabilities at school age in geographic cohorts of extremely low birth weight children born between the 1970s and the 1990s. J Pediatr. 2009;154:829–834. doi: 10.1016/j.jpeds.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 8.Walsh MC, Hibbs AM, Martin CR, et al. Two-year neurodevelopmental outcomes of ventilated preterm infants treated with inhaled nitric oxide. J Pediatr. 2010;156:556–561. doi: 10.1016/j.jpeds.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts G, Anderson PJ, Doyle LW. The stability of the diagnosis of developmental disability between ages 2 and 8 in a geographic cohort of very preterm children born in 1997. Arch Dis Child. 2010;95:786–790. doi: 10.1136/adc.2009.160283. [DOI] [PubMed] [Google Scholar]
- 10.Grunau RE, Whitfield MF, Fay TB. Psychosocial and academic characteristics of extremely low birth weight (< or =800 g) adolescents who are free of major impairment compared with term-born control subjects. Pediatrics. 2004;114:e725–e732. doi: 10.1542/peds.2004-0932. [DOI] [PubMed] [Google Scholar]
- 11.Hack M, Flannery DJ, Schluchter M, et al. Outcomes in young adulthood for very-low-birth-weight infants. N Engl J Med. 2002;346:149–157. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- 12.Lindstrom K, Winbladh B, Haglund B, Hjern A. Preterm infants as young adults: a Swedish national cohort study. Pediatrics. 2007;120:70–77. doi: 10.1542/peds.2006-3260. [DOI] [PubMed] [Google Scholar]
- 13.Saigal S, den Ouden L, Wolke D, et al. School-age outcomes in children who were extremely low birth weight from four international population-based cohorts. Pediatrics. 2003;112:943–950. doi: 10.1542/peds.112.4.943. [DOI] [PubMed] [Google Scholar]
- 14.Stoll BJ, Hansen NI, Adams-Chapman I, et al. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292:2357–2365. doi: 10.1001/jama.292.19.2357. [DOI] [PubMed] [Google Scholar]
- 15.Marlow N, Rose AS, Rands CE, Draper ES. Neuropsychological and educational problems at school age associated with neonatal encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2005;90:F380–F387. doi: 10.1136/adc.2004.067520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez FF, Miller SP. Does perinatal asphyxia impair cognitive function without cerebral palsy? Arch Dis Child Fetal Neonatal Ed. 2006;91:F454–F459. doi: 10.1136/adc.2005.092445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond A, Barnett WS, Thomas J, Munro S. Preschool program improves cognitive control. Science. 2007;318:1387–1388. doi: 10.1126/science.1151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson PJ, Doyle LW. Executive functioning in school-aged children who were born very preterm or with extremely low birth weight in the 1990s. Pediatrics. 2004;114:50–57. doi: 10.1542/peds.114.1.50. [DOI] [PubMed] [Google Scholar]
- 19.Grunau RV, Kearney SM, Whitfield MF. Language development at 3 years in preterm children of birth weight below 1000 g. Br J Disord Commun. 1990;25:173–182. doi: 10.3109/13682829009011972. [DOI] [PubMed] [Google Scholar]
- 20.Whitfield MF, Grunau RV, Holsti L. Extremely premature (< or = 800 g) schoolchildren: multiple areas of hidden disability. Arch Dis Child Fetal Neonatal Ed. 1997;77:F85–F90. doi: 10.1136/fn.77.2.f85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grunau RE, Whitfield MF, Davis C. Pattern of learning disabilities in children with extremely low birth weight and broadly average intelligence. Arch Pediatr Adolesc Med. 2002;156:615–620. doi: 10.1001/archpedi.156.6.615. [DOI] [PubMed] [Google Scholar]
- 22.Taylor HG, Minich NM, Klein N, Hack M. Longitudinal outcomes of very low birth weight: neuropsychological findings. J Int Neuropsychol Soc. 2004;10:149–163. doi: 10.1017/S1355617704102038. [DOI] [PubMed] [Google Scholar]
- 23.Saavalainen P, Luoma L, Bowler D, et al. Spatial span in very prematurely born adolescents. Dev Neuropsychol. 2007;32:769–785. doi: 10.1080/87565640701539535. [DOI] [PubMed] [Google Scholar]
- 24.Luu TM, Vohr BR, Schneider KC, et al. Trajectories of receptive language development from 3 to 12 years of age for very preterm children. Pediatrics. 2009;124:333–341. doi: 10.1542/peds.2008-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nosarti C, Giouroukou E, Micali N, et al. Impaired executive functioning in young adults born very preterm. J Int Neuropsychol Soc. 2007;13:571–581. doi: 10.1017/S1355617707070725. [DOI] [PubMed] [Google Scholar]
- 26.Curtis WJ, Lindeke LL, Georgieff MK, Nelson CA. Neurobehavioural functioning in neonatal intensive care unit graduates in late childhood and early adolescence. Brain. 2002;125:1646–1659. doi: 10.1093/brain/awf159. [DOI] [PubMed] [Google Scholar]
- 27.Doesburg SM, Ribary U, Herdman AT, et al. Altered long-range alpha-band synchronization during visual short-term memory retention in children born very preterm. Neuroimage. 2011;54:2330–2339. doi: 10.1016/j.neuroimage.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chau V, Brant R, Poskitt KJ, et al. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatr Res. 2012;71:274–279. doi: 10.1038/pr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chau V, Poskitt KJ, McFadden DE, et al. Effect of chorioamnionitis on brain development and injury in premature newborns. Ann Neurol. 2009;66:155–164. doi: 10.1002/ana.21713. [DOI] [PubMed] [Google Scholar]
- 30.Glass HC, Bonifacio SL, Chau V, et al. Recurrent postnatal infections are associated with progressive white matter injury in premature infants. Pediatrics. 2008;122:299–305. doi: 10.1542/peds.2007-2184. [DOI] [PubMed] [Google Scholar]
- 31.Shah DK, Doyle LW, Anderson PJ, et al. Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J Pediatr. 2008;153:170–175. doi: 10.1016/j.jpeds.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 32.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–224. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banker B, Larroche J. Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol. 1962;7:386–410. doi: 10.1001/archneur.1962.04210050022004. [DOI] [PubMed] [Google Scholar]
- 34.DeReuck J, Chattha A, Richardson E. Pathogenesis and evolution of periventricular leukomalacia in infancy. Arch Neurol. 1972;27:229–236. doi: 10.1001/archneur.1972.00490150037007. [DOI] [PubMed] [Google Scholar]
- 35.Rorke LB. Pathology of perinatal brain injury. New York, NY: Raven Press; 1982. [Google Scholar]
- 36.Leviton A, Gilles F. Acquired perinatal leukoencephalopathy. Ann Neurol. 1984;16:1–10. doi: 10.1002/ana.410160102. [DOI] [PubMed] [Google Scholar]
- 37.Haynes RL, Folkerth RD, Keefe RJ, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 38.Iida K, Takashima S, Ueda K. Immunohistochemical study of myelination and oligodendrocyte in infants with periventricular leukomalacia. Pediatr Neurol. 1995;13:296–304. doi: 10.1016/0887-8994(95)00192-1. [DOI] [PubMed] [Google Scholar]
- 39.Robinson S, Li Q, Dechant A, Cohen M. Neonatal loss of gammaaminobutyric acid pathway expression after human perinatal brain injury. J Neurosurg. 2006;104:396–408. doi: 10.3171/ped.2006.104.6.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haynes RL, Billiards SS, Borenstein NS, et al. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riddle A, Maire J, Gong X, et al. Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke. 2012;43:178–184. doi: 10.1161/STROKEAHA.111.632265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamrick S, Miller SP, Leonard C, et al. Trends in severe brain injury and neurodevelopmental outcome in premature newborn infants: the role of cystic periventricular leukomalacia. J Pediatr. 2004;145:593–599. doi: 10.1016/j.jpeds.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 43.Counsell S, Allsop J, Harrison M, et al. Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.1. [DOI] [PubMed] [Google Scholar]
- 44.Miller SP, Cozzio CC, Goldstein RB, et al. Comparing the diagnosis of white matter injury in premature newborns with serial MR imaging and transfontanel ultrasonagraphy findings. AJNR Am J Neuroradiol. 2003;24:1661–1669. [PMC free article] [PubMed] [Google Scholar]
- 45.Inder TE, Anderson NJ, Spencer C, et al. White matter injury in the premature infant: a comparison between serial cranial sonographic and MR findings at term. AJNR Am J Neuroradiol. 2003;24:805–809. [PMC free article] [PubMed] [Google Scholar]
- 46.Maalouf E, Duggan P, Counsell SJ, et al. Comparison of findings on cranial ultrasound and magnetic resonance imaging in preterm infants. Pediatrics. 2001;107:719–727. doi: 10.1542/peds.107.4.719. [DOI] [PubMed] [Google Scholar]
- 47.Groenendaal F, Termote JU, van der Heide-Jalving M, et al. Complications affecting preterm neonates from 1991 to 2006: what have we gained? Acta Paediatr. 2010;99:354–358. doi: 10.1111/j.1651-2227.2009.01648.x. [DOI] [PubMed] [Google Scholar]
- 48.Pierson CR, Folkerth RD, Billiards SS, et al. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007;114:619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buser J, Maire J, Riddle A, et al. Arrested pre-oligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. 2012;71:93–109. doi: 10.1002/ana.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riddle A, Dean J, Buser JR, et al. Histopathological correlates of magnetic resonance imaging-defined chronic perinatal white matter injury. Ann Neurol. 2011;70:493–507. doi: 10.1002/ana.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Back SA, Luo NL, Mallinson RA, et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58:108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- 52.Back SA, Han BH, Luo NL, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riddle A, Luo N, Manese M, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–3055. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodward LJ, Anderson PJ, Austin NC, et al. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med. 2006;355:685–694. doi: 10.1056/NEJMoa053792. [DOI] [PubMed] [Google Scholar]
- 55.Counsell SJ, Edwards AD, Chew AT, et al. Specific relations between neurodevelopmental abilities and white matter microstructure in children born preterm. Brain. 2008;131:3201–3208. doi: 10.1093/brain/awn268. [DOI] [PubMed] [Google Scholar]
- 56.Krishnan ML, Dyet LE, Boardman JP, et al. Relationship between white matter apparent diffusion coefficients in preterm infants at term-equivalent age and developmental outcome at 2 years. Pediatrics. 2007;120:e604–e609. doi: 10.1542/peds.2006-3054. [DOI] [PubMed] [Google Scholar]
- 57.Chau V, Synnes A, Grunau R, et al. Abnormal brain maturation in preterm neonates associated with adverse developmental outcomes. Neurology. 2013;81:2082–2089. doi: 10.1212/01.wnl.0000437298.43688.b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller SP, Vigneron DB, Henry RG, et al. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. J Magn Reson Imaging. 2002;16:621–632. doi: 10.1002/jmri.10205. [DOI] [PubMed] [Google Scholar]
- 59.Adams E, Chau V, Poskitt KJ, et al. Tractography-based quantitation of corticospinal tract development in premature newborns. J Pediatr. 2010;156:882–888. doi: 10.1016/j.jpeds.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bassi L, Chew A, Merchant N, et al. Diffusion tensor imaging in preterm infants with punctate white matter lesions. Pediatr Res. 2011;69:561–566. doi: 10.1203/PDR.0b013e3182182836. [DOI] [PubMed] [Google Scholar]
- 61.Srinivasan L, Dutta R, Counsell SJ, et al. Quantification of deep gray matter in preterm infants at term-equivalent age using manual volumetry of 3-tesla magnetic resonance images. Pediatrics. 2007;119:759–765. doi: 10.1542/peds.2006-2508. [DOI] [PubMed] [Google Scholar]
- 62.Ment LR, Kesler S, Vohr B, et al. Longitudinal brain volume changes in preterm and term control subjects during late childhood and adolescence. Pediatrics. 2009;123:503–511. doi: 10.1542/peds.2008-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kesler SR, Reiss AL, Vohr B, et al. Brain volume reductions within multiple cognitive systems in male preterm children at age twelve. J Pediatr. 2008;152:513–520. doi: 10.1016/j.jpeds.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullen KM, Vohr BR, Katz KH, et al. Preterm birth results in alterations in neural connectivity at age 16 years. Neuroimage. 2011;54:2563–2570. doi: 10.1016/j.neuroimage.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalpakidou AK, Allin MP, Walshe M, et al. Neonatal brain injury and neuroanatomy of memory processing following very preterm birth in adulthood: an fMRI study. PLoS One. 2012;7:e34858. doi: 10.1371/journal.pone.0034858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pandit AS, Robinson E, Aljabar P, et al. Whole-brain mapping of structural connectivity in infants reveals altered connection strength associated with growth and preterm birth. Cereb Cortex. 2013 Mar 31; doi: 10.1093/cercor/bht086. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 67.Miller SP, Ferriero DM. From selective vulnerability to connectivity: insights from newborn brain imaging. Trends Neurosci. 2009;32:496–505. doi: 10.1016/j.tins.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kinney H, Back S. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- 69.Deguchi K, Oguchi K, Takashima S. Characteristic neuropathology of leukomalacia in extremely low birth weight infants. Pediatr Neurol. 1997;16:296–300. doi: 10.1016/s0887-8994(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 70.Hirayama A, Okoshi Y, Hachiya Y, et al. Early immunohistochemical detection of axonal damage and glial activation in extremely immature brains with periventricular leukomalacia. Clin Neuropathol. 2001;20:87–91. [PubMed] [Google Scholar]
- 71.Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: White matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–235. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 72.Verney C, Pogledic I, Biran V, et al. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J Neuropathol Exp Neurol. 2012;71:251–264. doi: 10.1097/NEN.0b013e3182496429. [DOI] [PubMed] [Google Scholar]
- 73.McCarran W, Goldberg M. White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J Neurosci. 2007;27:4220–4229. doi: 10.1523/JNEUROSCI.5542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- 75.Tekkok SB, Goldberg MP. AMPA/Kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alix JJ, Zammit C, Riddle A, et al. Central axons preparing to myelinate are highly sensitivity to ischemic injury. Ann Neurol. 2012;72:936–951. doi: 10.1002/ana.23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Back SA, Luo NL, Borenstein NS, et al. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. [Google Scholar]
- 78.Back S. Mechanisms of acute and chronic brain injury in the preterm infant. In: Miller S, Shevell M, editors. Acquired brain injury in the fetus and newborn. London, UK: MacKeith Press; 2012. pp. 29–52. [Google Scholar]
- 79.Favrais G, van de Looij Y, Fleiss B, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- 80.Segovia K, McClure M, Moravec M, et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:517–526. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buser J, Segovia K, Dean J, et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J Cereb Blood Flow Metab. 2010;30:1053–1065. doi: 10.1038/jcbfm.2009.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Drobyshevsky A, Song SK, Gamkrelidze G, et al. Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J Neurosci. 2005;25:5988–5997. doi: 10.1523/JNEUROSCI.4983-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Derrick M, Luo NL, Bregman JC, et al. Preterm fetal hypoxia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci. 2004;24:24–34. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev. 1997;3:96–107. [Google Scholar]
- 85.Billiards S, Haynes R, Folkerth R, et al. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 2008;18:153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhiheng H, Liu J, Cheung P-Y, Chen C. Long-term cognitive impairment and myelination deficiency in a rat model of perinatal hypoxic-ischemia brain injury. Brain Res. 2009;1301:100–109. doi: 10.1016/j.brainres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 87.Wright J, Zhang G, Yu T-S, Kernie S. Age-related changes in the oligodendrocyte progenitor pool influence brain remodeling after injury. Dev Neurosci. 2010;32:499–509. doi: 10.1159/000322081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sizonenko SV, Camm EJ, Dayer A, Kiss JZ. Glial responses to neonatal hypoxic-ischemic injury in the rat cerebral cortex. Int J Dev Neurosci. 2008;26:37–45. doi: 10.1016/j.ijdevneu.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 89.Zaidi A, Bessert D, Ong J, et al. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- 90.Felling RJ, Snyder MJ, Romanko MJ, et al. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Z, Levison SW. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience. 2006;139:555–564. doi: 10.1016/j.neuroscience.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 92.Lee Y, Morrison BM, Li Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nossin-Manor R, Card D, Morris D, et al. Quantitative MRI in the very preterm brain: assessing tissue organization and myelination using magnetization transfer, diffusion tensor and T(1) imaging. Neuroimage. 2013;64:505–516. doi: 10.1016/j.neuroimage.2012.08.086. [DOI] [PubMed] [Google Scholar]
- 94.Thompson DK, Lee KJ, Egan GF, et al. Regional white matter microstructure in very preterm infants: predictors and 7 year outcomes. Cortex. 2014;52:60–74. doi: 10.1016/j.cortex.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miller S, McQuillen P, Hamrick S, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357:1971–1973. doi: 10.1056/NEJMoa067393. [DOI] [PubMed] [Google Scholar]
- 96.Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease—can we wrap it up? Brain. 2011;134:1882–1900. doi: 10.1093/brain/awr014. [DOI] [PubMed] [Google Scholar]
- 97.Fancy SP, Chan JR, Baranzini SE, et al. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- 98.Franklin RJ, Gallo V. The translational biology of remyelination: past, present, and future. Glia. 2014 Jan 20; doi: 10.1002/glia.22622. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 99.Barres B, Schmid R, Sendnte M, Raff M. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 100.Emery B, Agalliu D, Cahoy JD, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sizonenko SV, Bednarek N, Gressens P. Growth factors and plasticity. Semin Fetal Neonatal Med. 2007;12:241–249. doi: 10.1016/j.siny.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 102.Silbereis JC, Huang EJ, Back SA, Rowitch DH. Toward improved animal models of neonatal white matter injury associated with cerebral palsy. Dis Model Mech. 2010;3:678–688. doi: 10.1242/dmm.002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fancy SP, Kotter MR, Harrington EP, et al. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp Neurol. 2010;225:18–23. doi: 10.1016/j.expneurol.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 104.Ghiani C, Yuan X, Eisen A, et al. Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors P27 Kip1 and p21CIP1 in glial progenitor cells. J Neurosci. 1999;19:5380–5392. doi: 10.1523/JNEUROSCI.19-13-05380.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Knutson P, Ghiani CA, Zhou JM, et al. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J Neurosci. 1997;17:2669–2682. doi: 10.1523/JNEUROSCI.17-08-02669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ming X, Chew LJ, Gallo V. Transgenic overexpression of sox17 promotes oligodendrocyte development and attenuates demyelination. J Neurosci. 2013;33:12528–12542. doi: 10.1523/JNEUROSCI.0536-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moll NM, Hong E, Fauveau M, et al. SOX17 is expressed in regenerating oligodendrocytes in experimental models of demyelination and in multiple sclerosis. Glia. 2013;61:1659–1672. doi: 10.1002/glia.22547. [DOI] [PubMed] [Google Scholar]
- 108.French HM, Reid M, Mamontov P, et al. Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res. 2009;87:3076–3087. doi: 10.1002/jnr.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baltan S, Murphy SP, Danilov CA, et al. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31:3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dewald LE, Rodriguez JP, Levine JM. The RE1 binding protein REST regulates oligodendrocyte differentiation. J Neurosci. 2011;31:3470–3483. doi: 10.1523/JNEUROSCI.2768-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li W, Zhang B, Tang J, et al. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J Neurosci. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dugas JC, Cuellar TL, Scholze A, et al. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shin D, Shin JY, McManus MT, et al. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sherman L, Back S. A GAG reflex prevents repair of the damaged CNS. Trends Neurosci. 2008;31:44–52. doi: 10.1016/j.tins.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 116.Asher R, Perides G, Vanderhaeghen J, Bignami A. Extracellular matrix of central nervous system white matter: demonstration of an hyaluronan-protein complex. J Neurosci Res. 1991;28:410–421. doi: 10.1002/jnr.490280314. [DOI] [PubMed] [Google Scholar]
- 117.Back S, Tuohy T, Chen H, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;9:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 118.Preston M, Gong X, Su W, et al. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann Neurol. 2013;73:266–280. doi: 10.1002/ana.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y, Cheng X, He Q, et al. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–6058. doi: 10.1523/JNEUROSCI.5524-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.John G, Shankar S, Shafit-Zagardo B, et al. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- 121.Feigenson K, Reid M, See J, et al. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci. 2009;42:255–265. doi: 10.1016/j.mcn.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 122.Fancy SP, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fancy S, Harrington E, Yuen T, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tawk M, Makoukji J, Belle M, et al. Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. 2011;31:3729–3742. doi: 10.1523/JNEUROSCI.4270-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McClain CR, Sim FJ, Goldman SA. Pleiotrophin suppression of receptor protein tyrosine phosphatase-beta/zeta maintains the self-renewal competence of fetal human oligodendrocyte progenitor cells. J Neurosci. 2012;32:15066–15075. doi: 10.1523/JNEUROSCI.1320-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ye F, Chen Y, Hoang T, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Azim K, Butt AM. GSK3beta negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia. 2011;59:540–553. doi: 10.1002/glia.21122. [DOI] [PubMed] [Google Scholar]
- 128.Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci. 2008;28:914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- 130.Scafidi J, Hammond TR, Scafidi S, et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature. 2014;506:230–234. doi: 10.1038/nature12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guan J, Bennet L, George S, et al. Insulin-like growth factor-1 reduces postischemic white matter injury in fetal sheep. J Cereb Blood Flow Metab. 2001;21:493–502. doi: 10.1097/00004647-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 132.Pang Y, Zheng B, Campbell LR, et al. IGF-1 can either protect against or increase LPS-induced damage in the developing rat brain. Pediatr Res. 2010;67:579–584. doi: 10.1203/PDR.0b013e3181dc240f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wood TL, Loladze V, Altieri S, et al. Delayed IGF-1 administration rescues oligodendrocyte progenitors from glutamate-induced cell death and hypoxic-ischemic brain damage. Dev Neurosci. 2007;29:302–310. doi: 10.1159/000105471. [DOI] [PubMed] [Google Scholar]
- 134.See JM, Grinspan JB. Sending mixed signals: bone morphogenetic protein in myelination and demyelination. J Neuropathol Exp Neurol. 2009;68:595–604. doi: 10.1097/NEN.0b013e3181a66ad9. [DOI] [PubMed] [Google Scholar]
- 135.See J, Zhang X, Eraydin N, et al. Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol Cell Neurosci. 2004;26:481–492. doi: 10.1016/j.mcn.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 136.Wu M, Hernandez M, Shen S, et al. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J Neurosci. 2012;32:6651–6664. doi: 10.1523/JNEUROSCI.4876-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gozzo Y, Vohr B, Lacadie C, et al. Alterations in neural connectivity in preterm children at school age. Neuroimage. 2009;48:458–463. doi: 10.1016/j.neuroimage.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schafer RJ, Lacadie C, Vohr B, et al. Alterations in functional connectivity for language in prematurely born adolescents. Brain. 2009;132:661–670. doi: 10.1093/brain/awn353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Narberhaus A, Lawrence E, Allin MP, et al. Neural substrates of visual paired associates in young adults with a history of very preterm birth: alterations in fronto-parieto-occipital networks and caudate nucleus. Neuroimage. 2009;47:1884–1893. doi: 10.1016/j.neuroimage.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 140.Doesburg SM, Chau CM, Cheung TP, et al. Neonatal pain-related stress, functional cortical activity and visual-perceptual abilities in school-age children born at extremely low gestational age. Pain. 2013;154:1946–1952. doi: 10.1016/j.pain.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smyser CD, Inder TE, Shimony JS, et al. Longitudinal analysis of neural network development in preterm infants. Cereb Cortex. 2010;20:2852–2862. doi: 10.1093/cercor/bhq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Keunen K, Kersbergen KJ, Groenendaal F, et al. Brain tissue volumes in preterm infants: prematurity, perinatal risk factors and neurodevelopmental outcome: a systematic review. J Matern Fetal Neonatal Med. 2012;25(suppl 1):89–100. doi: 10.3109/14767058.2012.664343. [DOI] [PubMed] [Google Scholar]
- 143.Tam EW, Ferriero DM, Xu D, et al. Cerebellar development in the preterm neonate: effect of supratentorial brain injury. Pediatr Res. 2009;66:102–106. doi: 10.1203/PDR.0b013e3181a1fb3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nossin-Manor R, Chung AD, Whyte HE, et al. Deep gray matter maturation in very preterm neonates: regional variations and pathology-related age-dependent changes in magnetization transfer ratio. Radiology. 2012;263:510–517. doi: 10.1148/radiol.12110367. [DOI] [PubMed] [Google Scholar]
- 145.Vinall J, Grunau RE, Brant R, et al. Slower postnatal growth is associated with delayed cerebral cortical maturation in preterm newborns. Sci Transl Med. 2013;5:168ra8. doi: 10.1126/scitranslmed.3004666. [DOI] [PubMed] [Google Scholar]
- 146.Inder TE, Huppi PS, Warfield S, et al. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann Neurol. 1999;46:755–760. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 147.Boardman JP, Counsell SJ, Rueckert D, et al. Early growth in brain volume is preserved in the majority of preterm infants. Ann Neurol. 2007;62:185–192. doi: 10.1002/ana.21171. [DOI] [PubMed] [Google Scholar]
- 148.Bonifacio SL, Glass HC, Chau V, et al. Extreme premature birth is not associated with impaired development of brain microstructure. J Pediatr. 2010;157:726–732. doi: 10.1016/j.jpeds.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Inder TE, Warfield S, Wang H, et al. Abnormal cerebral structure is present at term in premature infants. Pediatrics. 2005;115:286–294. doi: 10.1542/peds.2004-0326. [DOI] [PubMed] [Google Scholar]
- 150.Thompson DK, Warfield SK, Carlin JB, et al. Perinatal risk factors altering regional brain structure in the preterm infant. Brain. 2007;130:667–677. doi: 10.1093/brain/awl277. [DOI] [PubMed] [Google Scholar]
- 151.Srinivasan L, Allsop J, Counsell SJ, et al. Smaller cerebellar volumes in very preterm infants at term-equivalent age are associated with the presence of supratentorial lesions. AJNR Am J Neuroradiol. 2006;27:573–579. [PMC free article] [PubMed] [Google Scholar]
- 152.Zubiaurre-Elorza L, Soria-Pastor S, Junque C, et al. Gray matter volume decrements in preterm children with periventricular leukomalacia. Pediatr Res. 2011;69:554–560. doi: 10.1203/PDR.0b013e3182182366. [DOI] [PubMed] [Google Scholar]
- 153.McQuillen PS, Ferriero DM. Perinatal subplate neuron injury: implications for cortical development and plasticity. Brain Pathol. 2005;15:250–260. doi: 10.1111/j.1750-3639.2005.tb00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Failor S, Nguyen V, Darcy DP, et al. Neonatal cerebral hypoxia-ischemia impairs plasticity in rat visual cortex. J Neurosci. 2010;30:81–92. doi: 10.1523/JNEUROSCI.5656-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McQuillen PS, DeFreitas MF, Zada G, Shatz CJ. A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci. 2002;22:3580–3593. doi: 10.1523/JNEUROSCI.22-09-03580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kinney H, Haynes R, Xu G, et al. Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann Neurol. 2012;71:397–406. doi: 10.1002/ana.22612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Counsell SJ, Dyet LE, Larkman DJ, et al. Thalamo-cortical connectivity in children born preterm mapped using probabilistic magnetic resonance tractography. Neuroimage. 2007;34:896–904. doi: 10.1016/j.neuroimage.2006.09.036. [DOI] [PubMed] [Google Scholar]
- 158.Johnston MV. Excitotoxicity in neonatal hypoxia. Ment Retard Dev Disabil Res Rev. 2001;7:229–234. doi: 10.1002/mrdd.1032. [DOI] [PubMed] [Google Scholar]
- 159.Cheng Y, Deshmukh M, D’Costa A, et al. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Han BH, DeMattos RB, Dugan LL, et al. Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat Med. 2001;7:338–343. doi: 10.1038/85487. [DOI] [PubMed] [Google Scholar]
- 161.Holtzman D, Sheldon R, Jaffe W, et al. NGF protects the neonatal brain against hypoxic-ischemic brain injury. Ann Neurol. 1996;39:114–122. doi: 10.1002/ana.410390117. [DOI] [PubMed] [Google Scholar]
- 162.Stone BS, Zhang J, Mack DW, et al. Delayed neural network degeneration after neonatal hypoxia-ischemia. Ann Neurol. 2008;64:535–546. doi: 10.1002/ana.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Andiman SE, Haynes RL, Trachtenberg FL, et al. The cerebral cortex overlying periventricular leukomalacia: analysis of pyramidal neurons. Brain Pathol. 2010;20:803–814. doi: 10.1111/j.1750-3639.2010.00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nagasunder AC, Kinney HC, Bluml S, et al. Abnormal microstructure of the atrophic thalamus in preterm survivors with periventricular leukomalacia. AJNR Am J Neuroradiol. 2011;32:185–191. doi: 10.3174/ajnr.A2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McClendon E, Chen K, Gong X, et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol. 2014 Jan;:3. doi: 10.1002/ana.24100. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.McClure M, Riddle A, Manese M, et al. Cerebral blood flow heterogeneity in preterm sheep: lack of physiological support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab. 2008;28:995–1008. doi: 10.1038/sj.jcbfm.9600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dean J, McClendon E, Hansen K, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med. 2013;5:168ra7. doi: 10.1126/scitranslmed.3004669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhao YD, Ou S, Cheng SY, et al. Dendritic development of hippocampal CA1 pyramidal cells in a neonatal hypoxia-ischemia injury model. J Neurosci Res. 2013;91:1165–1173. doi: 10.1002/jnr.23247. [DOI] [PubMed] [Google Scholar]
- 170.Xu G, Broadbelt KG, Haynes RL, et al. Late development of the GABAergic system in the human cerebral cortex and white matter. J Neuropathol Exp Neurol. 2011;70:841–858. doi: 10.1097/NEN.0b013e31822f471c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–97. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 172.Zhang ZW, Peterson M, Liu H. Essential role of postsynaptic NMDA receptors in developmental refinement of excitatory synapses. Proc Natl Acad Sci U S A. 2013;110:1095–1100. doi: 10.1073/pnas.1212971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Luthi A, Schwyzer L, Mateos JM, et al. NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nat Neurosci. 2001;4:1102–1107. doi: 10.1038/nn744. [DOI] [PubMed] [Google Scholar]
- 174.Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ment LR, Bada HS, Barnes P, et al. Practice parameter: neuroimaging of the neonate: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2002;58:1726–1738. doi: 10.1212/wnl.58.12.1726. [DOI] [PubMed] [Google Scholar]
- 176.McKinstry R, Mathur A, Miller J, et al. Radial organization of developing preterm human cerebral cortex revealed by noninvasive water diffusion anisotropy MRI. Cereb Cortex. 2002;12:1237–1243. doi: 10.1093/cercor/12.12.1237. [DOI] [PubMed] [Google Scholar]
- 177.Kroenke CD, Bretthorst GL, Inder TE, Neil JJ. Modeling water diffusion anisotropy within fixed prenatal primate brain using Bayesian probability theory. Magn Reson Med. 2006;55:187–197. doi: 10.1002/mrm.20728. [DOI] [PubMed] [Google Scholar]
- 178.Kroenke C, Van Essen D, Inder T, et al. Microstructural changes of the baboon cerebral cortex during gestational development reflected in magnetic resonance imaging diffusion anisotropy. J Neurosci. 2007;14:12506–12515. doi: 10.1523/JNEUROSCI.3063-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kroenke CD, Taber EN, Leigland LA, et al. Regional patterns of cerebral cortical differentiation determined by diffusion tensor MRI. Cereb Cortex. 2009;19:2916–2929. doi: 10.1093/cercor/bhp061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jespersen SN, Leigland LA, Cornea A, Kroenke CD. Determination of axonal and dendritic orientation distributions within the developing cerebral cortex by diffusion tensor imaging. IEEE Trans Med Imaging. 2012;31:16–32. doi: 10.1109/TMI.2011.2162099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ball G, Srinivasan L, Aljabar P, et al. Development of cortical microstructure in the preterm human brain. Proc Natl Acad Sci U S A. 2013;110:9541–9546. doi: 10.1073/pnas.1301652110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brummelte S, Grunau RE, Chau V, et al. Procedural pain and brain development in premature newborns. Ann Neurol. 2012;71:385–396. doi: 10.1002/ana.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Smith GC, Gutovich J, Smyser C, et al. Neonatal intensive care unit stress is associated with brain development in preterm infants. Ann Neurol. 2011;70:541–549. doi: 10.1002/ana.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zwicker JG, Grunau RE, Adams E, et al. Score for neonatal acute physiology-II and neonatal pain predict corticospinal tract development in premature newborns. Pediatr Neurol. 2013;48:123–129. doi: 10.1016/j.pediatrneurol.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Short EJ, Klein NK, Lewis BA, et al. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics. 2003;112:e359. doi: 10.1542/peds.112.5.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Anjari M, Counsell SJ, Srinivasan L, et al. The association of lung disease with cerebral white matter abnormalities in preterm infants. Pediatrics. 2009;124:268–276. doi: 10.1542/peds.2008-1294. [DOI] [PubMed] [Google Scholar]
- 187.Kaukola T, Kapellou O, Laroche S, et al. Severity of perinatal illness and cerebral cortical growth in preterm infants. Acta Paediatr. 2009;98:990–995. doi: 10.1111/j.1651-2227.2009.01268.x. [DOI] [PubMed] [Google Scholar]
- 188.Ball G, Counsell SJ, Anjari M, et al. An optimised tract-based spatial statistics protocol for neonates: applications to prematurity and chronic lung disease. Neuroimage. 2010;53:94–102. doi: 10.1016/j.neuroimage.2010.05.055. [DOI] [PubMed] [Google Scholar]
- 189.Tam EW, Chau V, Ferriero DM, et al. Preterm cerebellar growth impairment after postnatal exposure to glucocorticoids. Sci Transl Med. 2011;3:105ra. doi: 10.1126/scitranslmed.3002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vinall J, Miller SP, Chau V, et al. Neonatal pain in relation to postnatal growth in infants born very preterm. Pain. 2012;153:1374–1381. doi: 10.1016/j.pain.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 191.Grunau RE, Whitfield MF, Petrie-Thomas J, et al. Neonatal pain, parenting stress and interaction, in relation to cognitive and motor development at 8 and 18 months in preterm infants. Pain. 2009;143:138–146. doi: 10.1016/j.pain.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tu MT, Grunau RE, Petrie-Thomas J, et al. Maternal stress and behavior modulate relationships between neonatal stress, attention, and basal cortisol at 8 months in preterm infants. Dev Psychobiol. 2007;49:150–164. doi: 10.1002/dev.20204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Perlman JM. Cognitive and behavioral deficits in premature graduates of intensive care. Clin Perinatol. 2002;29:779–797. doi: 10.1016/s0095-5108(02)00051-9. [DOI] [PubMed] [Google Scholar]
- 194.Benzies KM, Magill-Evans JE, Hayden KA, Ballantyne M. Key components of early intervention programs for preterm infants and their parents: a systematic review and meta-analysis. BMC Pregnancy Childbirth. 2013;13(suppl 1):S10. doi: 10.1186/1471-2393-13-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Milgrom J, Newnham C, Anderson PJ, et al. Early sensitivity training for parents of preterm infants: impact on the developing brain. Pediatr Res. 2010;67:330–335. doi: 10.1203/PDR.0b013e3181cb8e2f. [DOI] [PubMed] [Google Scholar]
- 196.Vazquez DM, Lopez JF, Van Hoers H, et al. Maternal deprivation regulates serotonin 1A and 2A receptors in the infant rat. Brain Res. 2000;855:76–82. doi: 10.1016/s0006-8993(99)02307-0. [DOI] [PubMed] [Google Scholar]
- 197.Bagot RC, Zhang TY, Wen X, et al. Variations in postnatal maternal care and the epigenetic regulation of metabotropic glutamate receptor 1 expression and hippocampal function in the rat. Proc Natl Acad Sci U S A. 2012;109(suppl 2):17200–17207. doi: 10.1073/pnas.1204599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.McManus BM, Poehlmann J. Parent-child interaction, maternal depressive symptoms and preterm infant cognitive function. Infant Behav Dev. 2012;35:489–498. doi: 10.1016/j.infbeh.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Brummelte S, Grunau RE, Zaidman-Zait A, et al. Cortisol levels in relation to maternal interaction and child internalizing behavior in preterm and full-term children at 18 months corrected age. Dev Psychobiol. 2011;53:184–195. doi: 10.1002/dev.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 201.Salmaso N, Silbereis J, Komitova M, et al. Environmental enrichment increases the GFAP+ stem cell pool and reverses hypoxia-induced cognitive deficits in juvenile mice. J Neurosci. 2012;32:8930–8939. doi: 10.1523/JNEUROSCI.1398-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Komitova M, Xenos D, Salmaso N, et al. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci. 2013;33:13375–13387. doi: 10.1523/JNEUROSCI.5286-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]