

Abstract
Rapid progress in genome sequencing technology has put us firmly into a postgenomic era. A key challenge in biomedical research is harnessing genome sequence to fulfill the promise of personalized medicine. This Review describes how genome sequencing has enabled the identification of disease-causing biomolecules and how these data have been converted into chemical probes of function, preclinical lead modalities, and ultimately U.S. Food and Drug Administration (FDA)-approved drugs. In particular, we focus on the use of oligonucleotide-based modalities to target disease-causing RNAs; small molecules that target DNA, RNA, or protein; the rational repurposing of known therapeutic modalities; and the advantages of pharmacogenetics. Lastly, we discuss the remaining challenges and opportunities in the direct utilization of genome sequence to enable design of medicines.
Graphical Abstract
1 Introduction
The dissemination of the first draft of the human genome in 2001(1) provided unprecedented amounts of genetic information and, with it, the potential of uncovering the causes of human disease. Indeed, the ability to sequence whole genomes has revolutionized the field of drug discovery and initiated the promise of personalized medicine. In this Review, we discuss how genome sequencing is beginning to fulfill this promise, from the identification of new disease-causing mutations and aberrant gene expression to the development of disease biomarkers and the design of lead therapeutic modalities. The remainder of the Introduction is dedicated to the history of sequencing (section 1.1) and the first examples of disease caused by genetic mutations (section 1.2). We then turn our attention to therapeutic modalities for targeting nucleic acids, using both oligonucleotides (section 2) and small molecules (section 3), as well as proteins (section 4). Lastly, the rational repurposing of known drugs (section 5) and the potential of pharmacogenetics (section 6) are discussed.
1.1 History of Sequencing
Surprisingly, the first biomolecule to be sequenced was RNA, not DNA. RNAs that could be obtained in large quantities from extracts and purified, such as transfer (t)RNAs or ribosomal (r)RNAs, were treated with various ribonucleases (RNases) known to cleave RNA at specific sites. Using this method, Holley and colleagues produced the first sequence of yeast alanine tRNA in 1965.(2) At the same time, Sanger and colleagues developed a two-dimensional fractionation procedure for separating RNA fragments to determine sequence.(3) Using this procedure about a decade later, Fiers and colleagues sequenced the first protein coding RNA, the 3569 nucleotide bacteriophage MS2 RNA.(4)
After these initial sequencing techniques, Sanger and Maxam and Gilbert separately developed novel DNA sequencing procedures using a single separation via polyacrylamide electrophoresis rather than 2D fractionation. Sanger’s first DNA sequencing technique, the plus and minus method, used DNA polymerase to incorporate radiolabeled nucleotides followed by two-second polymerization reactions. The “plus” polymerization reaction contained only a single deoxynucleotide triphosphate (dNTP) while the “minus” reaction contained the other three dNTPs. DNA sequence could then be inferred from extensions ending with the base in the “plus” reaction.(5) This method was used to determine the 5375 nucleotide genome sequence of the ΦX174 bacteriophage in 1977.(6) At the same time, Maxam and Gilbert developed chemical techniques to sequence DNA using reagents such as dimethyl sulfate (DMS) and hydrazine to modify specific bases.(7) Modified bases were then chemically cleaved at phosphodiester bonds, producing fragments that were separated by gel electrophoresis.
Sanger later developed the dideoxy method of sequencing, which uses dideoxynucleotide triphoshpates (ddNTPs) that lack the 3′ hydroxyl group required for extension.(8) Four different reactions, each containing a different individual ddNTP combined with the other three dNTPs, determines a DNA sequence based on chain-termination sites. The human mitochondrial genome was sequenced in this fashion in 1981,(9) and the Sanger dideoxy method became the most common way to sequence DNA with improvements contributed over time. Fluorescence detection soon replaced radiolabeling(10) and capillary electrophoresis(11) replaced other separation methods, allowing for the creation of the first automated DNA sequencers.(12) To sequence large lengths of DNA, shotgun sequencing was developed, where DNA is broken up into smaller fragments and overlapping fragments are reassembled postsequencing.(13) Technologies such as DNA cloning in the 1970’s(14, 15) and polymerase chain reaction (PCR) in the 1980’s(16, 17)further advanced DNA sequencing, and the first commercial dideoxy sequencer, the Applied Biosystems (ABI) Prism, was introduced in 1986.(18) On the basis of Leroy Hood’s work, this instrument enabled the sequencing of the yeast(19) and worm(20) genomes in 1992 and 1994, respectively.
Perhaps the most important advances in sequencing technologies have occurred in the past decade, particularly with the development of next-generation sequencing (NGS) which enabled massively parallel DNA sequencing. Next-generation sequencing methods begin with a DNA library formed by ligation of library-specific DNA adapters onto the ends of the DNA fragments to be sequenced. The library fragments are then amplified, although the amplification surface and method is different for each platform. These platforms include the use of pyrosequencing (Roche/454) or chemically blocked fluorescently labeled dNTPs (Illumina and ABI SOLiD).(21–23)Because of their higher output per run, next-generation sequencers have reduced the cost of sequencing per genome to ~$1,000 (Figure 1).(24) Next-generation methods to sequence RNA (RNA-seq) and corresponding bioinformatics approaches to analyze sequencing data have greatly expanded the data that can be obtained from a single sequencing run.
Figure 1.

Cost of sequencing has decreased dramatically in the past 15 years, and it now only costs ~$1,000 to sequence a genome. Data were obtained from the Genome Sequencing Program of the National Human Genome Research Institute (NHGRI).
1.2 Sequencing and the Promise of Personalized Medicine
Before DNA sequencing was developed, many diseases, such as sickle cell disease,(25, 26) were known to be genetically inherited; however, the exact genetic cause of the disease was largely unknown. One of the first human diseases that could be linked to a specific defective gene was Huntington’s disease (HD), an ultimately fatal neurological disorder that causes progressive deterioration of both movement and cognition. In 1983, a polymorphic DNA marker on chromosome 4 was discovered to be the location of the genetic defect that causes HD.(27) It took another 10 years for the exact nature of the genetic defect to be defined as an expanded CAG repeat located within the open reading frame (ORF) of the huntingtin (HTT) gene.(28) Around the same time, other triplet-repeat mutations contributing to disease were discovered, including a CGG expansion in the 5′ untranslated region (UTR) of the fragile X mental retardation 1 gene [fragile X syndrome (FXS) and fragile X-associated tremor ataxia syndrome (FXTAS)],(29, 30) a CAG expansion in the ORF of the androgen receptor [spinal and bulbar muscular atrophy (SMBA)],(31) and a CTG expansion in the 3′ UTR of the dystrophia myotonica protein kinase (DMPK) gene [myotonic dystrophy type 1 (DM1)].(32) In 1989, the most common genetic mutation (p.F508del) that causes cystic fibrosis was identified,(33–35) and other mutations causing disease have since been identified.(36) More recently, whole-genome sequencing revealed genetic variants in rare diseases, including Charcot-Marie-Tooth Disease,(37) Miller syndrome,(38, 39)and Kabuki syndrome.(40) Although identifying the causative agents of these diseases has not yet led to a treatment, these discoveries have already enabled a better understanding of disease pathology and potential targets for therapeutic intervention.
One example in which the discovery of a genetic abnormality led to an approved therapeutic for a disease is chronic myeloid leukemia (CML) and the Philadelphia chromosome. The Philadelphia chromosome is generated by a reciprocal translocation of genetic material between chromosomes 9 and 22, resulting in a fusion gene between the breakpoint cluster region (BCR) gene and Abelson leukemia virus (ABL) gene; the encoded fusion protein is an unregulated tyrosine kinase.(41) Imatinib (Gleevec), which will be discussed in detail in section 4.1, inhibits the tyrosine kinase, providing an effective treatment for CML.(42)
Perhaps one of the most interesting aspects of the Human Genome Project was the revelation that the majority of the human genome is transcribed into RNA but does not encode protein.(1)Later studies on these “non-coding” (nc) RNAs revealed that they have important implications in disease. The discovery that short ncRNAs, including microRNAs (miRNAs), regulate gene expression(43, 44) and that their aberrant expression contributes to many diseases(45) has provided an entirely new class of drug targets. Collectively, advances in genome sequencing have broadened our understanding of complex diseases and opened new pathways for treatment of these diseases.
2 Oligonucleotide Therapeutics
Seminal work studying oligonucleotides, including the first use of complementary DNA to decrease production of viral proteins,(46) the discovery of RNA interference (RNAi),(43) and the development of aptamers,(47–49) suggested their potential as therapeutics. Indeed, oligonucleotide-based modalities have recently made their way into the clinic and many more are in clinical trials. There are many advantages to oligonucleotides, including their ease of design by using simple Watson–Crick base pairing rules, their ability to recruit endogenous cellular machinery to induce cleavage of the target mRNA, and available modifications that increase metabolic stability and/or increase thermal stability of the oligonucleotide-target complex. Because oligonucleotides must form a complex with the RNA target for activity, they are most effective when targeting unstructured regions.(50)
Despite their promise, oligonucleotides suffer from various limitations including their metabolic instability, high molecular weights, and anionic charge that reduce cellular and tissue permeability, as well as their nonspecific stimulation of the immune system.(51) Thus, much energy has been invested in improving metabolic stability and in creating efficient oligonucleotide delivery systems. Naked or unmodified oligonucleotides are quickly degraded in vivo by cellular nucleases. Various modifications to the phosphodiester backbone and ribose moieties have been developed(52) to improve stability, including phosphorothioate backbones,(53) locked nucleic acids (LNAs),(54) 2′ modifications,(55) and morpholino oligomers(56) (Figure 2A). Phosphorothioate backbones confer metabolic stability by binding to plasma proteins, preventing renal filtration and facilitating tissue uptake,(57) while also maintaining the ability to recruit endogenous nucleases to cleave the RNA target. Gapmer oligonucleotides, which often contain a stretch of phosphorothioate nucleotides flanked on both sides by nucleotides with 2′ modifications, are often employed to further increase metabolic and thermal stability.(58)
Figure 2.

Oligonucleotide modifications and delivery strategies. (A) Common oligonucleotide modifications. (B) Lipid nanoparticle delivery systems. (C) GalNAc conjugated oligonucleotides for targeted delivery to the liver.
Various oligonucleotide delivery systems have also been developed, including cationic lipids, such as cholesterol, which form lipid nanoparticles (LNPs) that are often coated with a neutral polymer such as polyethylene glycol (PEG) (Figure 2B).(59, 60) The nanoparticles are usually trafficked to the liver and thus have been an effective delivery system for liver-specific therapies.
Other targeted delivery approaches for oligonucleotide therapies have also been developed,(61, 62) including conjugation to antibodies(63) or receptor-targeting small molecules (Table 1).(64)Perhaps the most successful and well-developed targeted delivery system is conjugation with multivalent N-acetylgalactosamine (GalNAc) for targeted delivery to the liver (Figure 2C).(65, 66)Extensive reviews of oligonucleotide modifications and delivery systems can be found elsewhere.(67–70)
Table 1.
Oligonucleotide Therapeutics in the Clinic Discussed in This Review
| therapeutic | class/type | target | disease | clinical trial status | refs |
|---|---|---|---|---|---|
| Vitravene | antisense | cytomegalovirus mRNA | AIDS patients with CMV | approved | 76–78 |
| Mipomersen | antisense | Apolipoprotein B mRNA | homozygous familial hypercholesterolemia | approved | 79–87, 91, 92 |
| Eteplirsen | antisense | dystrophin Splice site | Duchenne muscular dystrophy | provisional approval | 99–103 |
| Nusinersen | antisense | SMN2 intron 7 | spinal muscular atrophy | approved | 110–115 |
| NCT02341560 | siRNA | caspase-2 | nonarteritic ischemic optic neuropathy (NAION) | III | 134, 135 |
| Patisiran | siRNA | transthyretin | familial ammyloidotic polyneuropathy due to transthyretin amyloidosis (ATTR) | III | 139, 140 |
| FANG | siRNA | furin | ovarian cancer | III | 141 |
| TKM-PLK1 | siRNA | polo-like kinase | multiple cancers | II | 142 |
| siG12D LODER | siRNA | mutant Kirsten ras oncogene | pancreatic cancer | III | 143 |
| TD101 | siRNA | mutant keratin K6a | pachyonychia congentia | I | 144 |
| RXI-109 | siRNA | connective tissue growth factor | scar healing | II | 145 |
| CEQ508 | siRNA | β-Catenin | familial adenomatous polyposis | I | 146 |
| Miravirsen | antagomiR | miR-122 | hepatitis C (HCV) | II | 169, 170 |
| RG-101 | antagomiR | miR-122 | hepatitis C (HCV) | Ib | 171 |
| MRG-106 | antagomiR | miR-155 | cutaneous T-cell lymphoma | I | 175 |
| RG-125 | antagomiR | miR-103/107 | nonalcoholic steatohepatitis | 177 | |
| MesomiR-1 | miRNA mimic | miR-16 | malignant pleural mesothelioma and nonsmall-cell lung cancer | I | 190, 191 |
| MRX34 | miRNA mimic | miR-34 | nonsmall-cel lung cancer | halted | 195–197 |
| Pegaptanib | aptamer | vascular endothetial growth factor | wet age-related macular degeneration | approved | 223–227 |
| REG1 | aptamer | coagulation factor IX a | coronary artery disease | terminated in phase III | 228, 229, 233–239 |
| ARC1779 | aptamer | von Willebrand factor | coronary artery disease | I | 240, 242–247 |
| NOX-A12 | aptamer | stromal cell-derived factor 1 | multiple myeloma | II | 217, 251, 252 |
| NOX-E36 | aptamer | monocyte chemoattractant protein-1 | diabetic nephropathy | II | 219, 255–257 |
| OZ1 | ribozyme | HIV-1 | HIV | terminated | 276, 280–283 |
| Angiozyme | ribozyme | VEGF | metastatic breast cancer | terminated in phase II | 271, 273, 285–287 |
2.1 Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) were first discovered when Zamecnick and colleagues found that complementary, or antisense, DNA oligonucleotides inhibited translation of Rous sarcoma viral proteins.(−46, 71) Subsequent studies revealed that ASOs form DNA-RNA hybrids that recruit RNase H, resulting in cleavage of the RNA strand. The cleaved fragments are then degraded by the cell’s RNA surveillance/quality control system, in particular members of the XNR (5′-3′ exoribonuclease) and Dis3 (exosome endoribonuclease and 3′-5′ exoribonuclease) families.(72)ASOs can target a disease-causing transcript by one of two different mechanisms, RNase H-mediated cleavage, as discovered by Zamecnick (Figure 3A) and RNase H-independent hybridization to the RNA target (Figure 3B).
Figure 3.

Mechanisms of action of antisense oligonucleotides (ASOs). (A) ASOs can affect gene expression by recruitment of RNase H, resulting in cleavage and degradation of the RNA target (B) ASOs with backbone or sugar modifications that prevent recruitment of RNase H can regulate expression by steric blocking of the ribosome and hence translational repression. (C) ASOs that target splice sites in pre-mRNAs can alter pre-mRNA alternative splicing.
Many modifications to the phosphodiester backbone and deoxyribose moieties often change the overall structure of the oligonucleotide:mRNA complex, such that it is no longer recognized by RNase H. In such cases, the antisense oligonucleotide acts as a steric block for repressing translation of the message. In 1993, Kole and colleagues expanded the use of steric-blocking antisense oligonucleotides to inhibit the spliceosomal machinery (Figure 3C).(73) In this case, a 2′-OMe RNA (Figure 2A) ASO was used to block a cryptic splice site caused by a mutation in human β-globin gene, restoring proper pre-mRNA splicing patterns.(74, 75)
Four ASOs have been granted FDA approval, with many more in early and late stage clinical trials. The first antisense oligonucleotide to be FDA-approved was Vitravene (fomivirsen) in 1998 to treat cytomegalovirus (CMV) retinitis in immunocompromised patients.(76–78) CMV is among the most common opportunistic infections in immunocompromised patients, particularly those with acquired immunodeficiency syndrome (AIDS), and can result in blindness. Vitravene, administered by intraocular injection, is a 21-nucleotide phosphorothioate (Figure 2A) ASO that is complementary to UL123 viral mRNA, which encodes intermediate early protein 2 (IE2). Although a successful treatment for CMV, Vitravene was discontinued in 2006 as CMV was no longer a significant complication of AIDS due to improved antiretroviral AIDS therapies.
It took another 15 years before the FDA approved another ASO, Mipomersen (Kynamro), which treats homozygous familial hypercholesterolemia, a genetic disorder characterized by high cholesterol levels. Mipomersen is a 20-nucleotide gapmer ASO with phosphorothioate and MOE (2′-O-methoxyethyl) modifications (Figure 2A) that targets apolipoprotein B (ApoB) mRNA.(79–87)ApoB is a structural component of low-density lipoprotein (LDL) and its metabolic precursor, very-low-density lipoprotein (VLDL).(88) As such, ApoB is a desirable target to lower LDL cholesterol levels, which are difficult to reduce using standard care, such as statins, in patients with homozygous familial hypercholesterolemia.(89, 90) Although this drug is approved for use in the United States, it was rejected twice by the European Medicines Agency over concerns about liver toxicity and cardiovascular effects.(91, 92) The market for Mipomersen diminished significantly following approval of the small molecule drug lomitapide.(93, 94)
The two most recently approved ASOs regulate the splicing of pre-mRNAs (Figure 3C) associated with Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA), two rare diseases. DMD, which is characterized by progressive muscle degeneration and weakness, is caused by mutations in the dystrophin gene, resulting in a complete loss of dystrophin production or production of a dysfunctional dystrophin protein that causes destabilization of muscle fibers during contraction.(95–97) Although these mutations vary across patients, they usually cause frame shifting or premature truncation of protein. Despite the broad differences in mutations, DMD phenotype is largely uniform due to nonsense-mediated decay of the mutated transcript.(98) In a later-onset and more mild form of muscular dystrophy, Becker muscular dystrophy, mutated transcripts are not degraded by nonsense mediated decay, and thus, a partially functional dystrophin is produced. One therapeutic strategy for DMD is to take advantage of the normal cellular process of alternative pre-mRNA splicing; that is, if an exon of a proper size can be skipped to restore the proper reading frame, a partially functional dystrophin could be produced and alleviate disease. Indeed, the morpholio (Figure 2C) ASO Eteplirsen treats DMD caused by deletion of exons 45–50 by inducing skipping of dystrophin exon 51 and reframing the transcript to produce a partially functional dystrophin. As only a partially functional dystrophin is produced, DMD symptoms are not completely reversed but are rather mitigated to resemble those observed in the less severe Becker muscular dystrophy.(99) Interestingly, the clinical trials and eventual approval of Eteplirsen were quite controversial. Clinical efficacy, as measured by the 6 min walk test, was questioned as the treated cohort was compared to a group that began as placebo for 24 weeks but switched to treatment at week 25.(100) Further, increases in dystrophin expression as measured by immunohistochemistry and Western blotting were quite small and not observed in all patients (15.9–29.0% increase in dystrophin positive muscle fibers in patients treated with 30 mg/kg over 24 weeks).(100, 101) It is unclear if a small increase in dystrophin levels would lead to an improved clinical outcome. Thus, the drug has been granted provisional approval pending the results of a larger, ongoing phase III clinical trial.(102) Only ~13–14% of DMD cases are caused by the exon 45–50 deletion;(103) however, up to 80% of DMD patients have deletions that cause frame shifting that could be helped by such an approach.
Spinal muscular atrophy (SMA) is caused by mutations in the survival motor neuron 1 (SMN1) gene, resulting in a loss of protein function.(104) The major clinical features are muscle weakness and atrophy.(105) A second SMN gene, SMN2, is present in the human genome; however, it differs from SMN1 by a single nucleotide. This single nucleotide change results in exon 7 exclusion in the majority of SMN2 transcripts.(106, 107) The lack of exon 7 decreases the half-life of SMN2 protein by ~2-fold and thus it is unable to substitute functionally for SMN1.(108) Disease severity (four grades) varies with the number of copies of the SMN2 gene, where more copies result in a less severe form of SMA.(109) Type 0 SMA is the most severe with a life expectancy of less than 6 months. Infants with SMA type 1 usually develop respiratory failure prior to two years of age. Children with SMA type 2 are never able to walk on their own, while patients with SMA type 3 are able to walk unassisted at some point during their lifetime. One therapeutic strategy, first put forward by Adrian Krainer’s laboratory,(110) to treat SMA is to use ASOs to direct alternative splicing to include exon 7 in SMN2 transcripts (Figure 4).(111, 112) Indeed, Nusinersen, a 2′-MOE phosphorothioate ASO (Figure 2A) administered via lumbar puncture, binds a region of intron 7 in SMN2 to induce exon 7 inclusion.(113, 114) Phase III trials of Nusinersen showed that 40% of treated patients achieved a motor milestone response. The results from this interim analysis led to accelerated FDA approval in late 2016 while the clinical study is ongoing.(115) Ongoing use of the drug will fully reveal the effectiveness of Nusinersen for treating SMA; however, this ASO shows promise for treating an incurable rare disease.
Figure 4.

ASO therapy for SMA. (A) In a healthy individual, the SMN1 gene produces a functional SMN1 protein. The SMN2 gene has a C to T mutation in exon 7, which results in exon 7 exclusion and a less stable SMN protein. (B) In SMA, mutations in the SMN1 gene result in loss of SMN protein leading to disease. Nusinersen targets a region of intron 7 in the SMN2 pre-mRNA to include exon 7 in the mature mRNA, resulting in production of a stable SMN protein.
2.2 RNAi Therapeutics
In 1998, Fire and Mello reported the silencing of an endogenous mRNA by a double-stranded (ds)RNA, dubbed RNA interference or RNAi (Figure 5A).(43) In RNAi, a long dsRNA activates the cytoplasmic nuclease Dicer,(116) which cleaves it into 21–22 nucleotide fragments with 3′ overhangs, as discovered by Tuschl and colleagues.(117, 118) These fragments, or small interfering (si)RNAs, are separated into a passenger (degraded) and guide strands, the latter of which is loaded into the RNA-induced silencing complex (RISC). Although the exact composition of RISC has yet to be fully defined, it is known that the Argonaute (Ago) protein family is essential for function. Using the guide strand, RISC identifies complementary mRNAs and cleaves them via Ago2.(119, 120) MicroRNAs (miRNAs), which will be discussed in detail in section 2.3, are also an important component of the endogenous RNAi pathway (Figure 5B). Indeed, the RNAi pathway is critical for developmental timing(121) and immune response against viral RNAs.(122, 123)
Figure 5.

Gene regulation by small noncoding RNAs. (A) The RNAi pathway. In RNAi, dsRNAs are cleaved in the cytoplasm by the Dicer-TRBP complex. The short fragments are then incorporated into the RISC complex, which cleaves complemenatry mRNAs. The RNAi pathway has been exploited therapeutically, by introducing exogenous shRNAs, typically produced from a DNA vector. These shRNAs are processed to double-stranded RNAs by Dicer before incorporation into the RISC complex. Likewise, siRNAs (double-stranded RNAs) can be exogenously introduced and incorporated into the RISC complex without processing. (B) Endogenous miRNAs are processed by Drosha (nucleus) and Dicer (cytoplasm) to produce a double-stranded RNA, where one strand is loaded into RISC to induce target mRNA cleavage or translational repression.
The discovery of the RNAi pathway provided an additional therapeutic strategy to eliminate RNAs that encode dysfunctional proteins. The feasibility of RNAi therapeutics has primarily been investigated using exogenously administered siRNAs (Figure 5A), however, short hairpin RNAs (shRNAs) that are transcribed from exogenously administered gene vectors can also be used. Although RNAi therapeutics suffer from the same limitations as other oligonucleotide-based modalities, one advantage of siRNAs is that the active strand is stable within RISC, albeit diluted with every cell division.(124) Thus, the same siRNA molecule can target multiple transcripts in a nondividing cell, limiting the number of siRNAs that are needed for efficient knockdown per cell. Therapeutically, siRNAs are chemically modified to protect from nuclease digestion, prevent immune response, and limit off-target effects. Like antisense oligonucleotides, siRNAs can be modified at the 2′-position of the ribose, or modified to locked nucleic acids (LNAs; Figure 2A).(125, 126) Structural modifications, however, are limited to those that allow incorporation into RISC.
There are multiple siRNAs currently in phase I–III clinical trials for ocular diseases, liver diseases, dermal conditions, diseases affecting the gastrointestinal tract, cancer, and infectious diseases.(127–129) Ocular diseases are particularly favorable for an RNAi approach, as siRNAs can be administered naked (no modifications) and locally without major off-target effects or inflammatory response.(130) There are currently four therapies in clinical trials for ocular diseases. Targets for these therapies include transient receptor potential vanilloid 1 (TRPV1) to treat chronic dry eye,(131) adrenoceptor beta 2 (ADRB2) to reduce intraocular pressure associated with optic nerve degeneration in glaucoma,(132) apoptosis stress-response gene (RTP801/REDD1) for wet age-related macular degeneration,(133) and caspase-2 to reduce retinal ganglion apoptosis in glaucoma and nonarteritic ischemic optic neuropathy (NAION).(134) The most advanced therapeutic is the siRNA targeting caspase-2 for NAION, which is currently in phase III trials (NCT02341560). In the phase I/IIa study, the siRNA, which is administered via intravitreal injection, was well-tolerated with no serious adverse events, and 52% of patients had improved best-corrected visual acuity.(135)
Liver disease is another attractive therapeutic target because of the ease of delivery of siRNAs to the liver with lipid nanoparticle delivery systems and GalNAc chemistry (Figure 2C). In particular, phase I trials for siRNAs that treat hypercholesterolemia,(136) hemophilia,(137) and hepatic fibrosis(138) are currently underway. Patisiran (Alnylam) is in phase III clinical trials for treatment of familial amyloidotic polyneuropathy due to transthyretin amyloidosis (ATTR), a rare genetic disease causing autonomic dysfunction for which the only therapeutic option is liver transplant or management of symptoms. ATTR is caused by a mutation in the transport protein transthyretin (TTR) which is mainly synthesized in the liver and misassembles into amyloid fibrils.(139) Patisiran is a LNP-siRNA that is administered subcutaneously to silence defective TTR.(140) In phase II trials, Patisiran knocked down ~80% of TTR over 9 months, and patients saw improved neuropathy impairment scores.(139)
Because of limited treatment options and the high rate of mortality of some cancers, RNAi therapeutics have garnered much interest, the most advanced of which is FANG (phase III) for treatment of ovarian cancer. FANG is unique from other RNAi therapeutics in that plasmid DNA drives expression of two shRNAs.(141) These shRNAs target furin, a protease required for maturation of transforming growth factor beta (TGF-β) that is overexpressed in cancer cells. The plasmid is delivered to tumor resections by electroporation and then reintroduced into the patient. In phase I trials for treatment of ovarian cancer, patients survived more than double the time span compared to patients with other treatments. Phase II trials for ovarian cancer are ongoing, and a phase III trial for high-risk ovarian cancer has been initiated.
Other RNAi therapeutics that have passed phase I trials include TKM-PLK1 and siG12D LODER. TKM-PLK1 is an siRNA against polo-like kinase (PLK) to reduce cell division in multiple types of cancers. TKM-PLK1 is delivered via LNP and is currently in phase II clinical trials.(142) SiG12D LODER is currently undergoing phase II/III trials for pancreatic cancer and is unique in that it is administered via an implanted matrix embedded with RNAi triggers that release the naked siRNA over time.(143) The siRNA in siG12D LODER targets a mutant of the Kirsten ras oncogene (KRASG12D), which is associated with increased cell proliferation of pancreatic cancer cells and is coadministered with chemotherapeutics.
RNAi has also been an attractive therapeutic option for skin disorders as therapeutics could be administered intradermally. The first of two RNAi therapeutics in the clinic for skin disorders is TD101 in phase I trials for pachyonychia congentia, an orphan disease in which a keratin mutation disrupts the organization of keratin filaments, resulting in a painful thickening of nails and skin.(144) The second, RXI-109, is in phase II trials to improve scar healing. RXI-109 targets connective tissue growth factor, which is associated with keloid and hypertrophic scar formation.(145) Both are administered via intradermal injection.
siRNAs are also in clinical trials for infectious diseases and diseases of the gastrointestinal tract. One siRNA therapeutic, CEQ508, has been granted fast-track status for familial adenomatous polyposis, an orphan disease caused by an adenomatous polyposis coli (APC) mutation that leads to the formation of adenomatous polyps, or abnormal growths, in the epithelium of the large intestine.(146) Infectious diseases are another popular target of RNAi therapeutics, with an shRNA therapeutic in phase I trials to target hepatitis C virus (HCV)(147) and two siRNAs in phase I/II clinical trials to target hepatitis B virus (HBV).(148, 149) Interestingly, there is also an siRNA in development to combat Ebola.(150) Clinical studies of this siRNA are under fast-track status and fall under the animal rule where clinical effectiveness is measured in animals, but safety studies are conducted in humans. The Ebola siRNA was administered to emergency patients and was well-tolerated; however, phase I safety studies have been halted after higher doses caused high cytokine levels.(62)
With many RNAi therapeutics progressing in clinical trials, it will be interesting to see if the first RNAi therapeutic is approved in the next few years. While there have been some successes in clinical trials, issues with delivery technology and stability in order to optimize maximum therapeutic dose still need to be addressed. Despite these issues, RNAi therapeutics offer promise for treating genetic disease and are invaluable tools for studying disease pathology.
2.3 MicroRNA-Targeting Therapeutics
In 1993, Victor Ambros and colleagues discovered a small, single-stranded, nonprotein-coding regulatory RNA molecule in Caenorhabditis elegans named lin-4.(44) In 2000, let-7, another C. elegans small RNA regulatory molecule, was discovered by the Ruvkun lab.(151, 152) As let-7 is conserved in many species, including vertebrates, it is considered to be the first mammalian miRNA discovered. Extensive studies based on these discoveries unveiled a new mechanism of gene regulation by small noncoding RNAs called microRNAs (miRNAs) (Figure 5B). The miRNAs bind to mRNAs that have complementary sites in their 3′ UTRs, which mediates translational repression or promotes degradation of targeted mRNAs.(153) The biogenesis of miRNAs begins with transcription by RNA polymerase II (RNAP II), affording primary miRNAs (pri-miRNAs). The pri-miRNAs are then processed by the type III RNase Drosha in complex with cofactor protein DiGeorge Syndrome Critical Region Gene 8 (DGCR8) in the nucleus, generating 70–80 nucleotide-long precursor miRNAs (pre-miRNAs). The exportin 5 complex regulates movement of these pre-miRNAs from the nucleus into the cytoplasm through nuclear pores. Once in the cytoplasm, the terminal loops of the pre-miRNAs are cleaved by the Dicer-TAR RNA binding protein (TRBP) complex, forming miRNA duplexes. The miRNA duplexes then enter RISC and bind the 3′ UTRs of complementary mRNAs.(154) In recent years, the scientific community has witnessed the critical importance of miRNAs in genetics, molecular biology, and physiology.
MiRNAs constitute only 1–5% of the human genome, yet they regulate the expression of at least 30% of all genes.(155, 156) Therefore, it is not surprising that mutations in miRNAs or in the mRNAs they regulate as well as the aberrant expression of miRNAs can cause disease.(157–160)Extensive studies on the roles of miRNAs in cancer has revealed that they function as both oncogenes, or oncomiRs, (overexpression) or tumor suppressors (under-expression). Below, we discuss antagomiR therapeutics used to knock down miRNAs that are overexpressed in disease, as well as miRNA mimics to supplement levels of under-expressed endogenous miRNAs.
2.3.1 Targeting Overexpressed miRNAs with AntagomiR Therapeutics
The discovery and development of miRNA-related therapeutics has involved numerous steps and years of effort. Analyzing patient samples and confirming the downstream target of the miRNA, combined with the use of genomic and proteomic data in public databases, has helped to identify potential miRNA candidates and establish their relevance in diseases. One way to manipulate the expression of miRNAs is through the use of antagomiRs,(161) short single-stranded RNAs complementary to the mature miRNA sequence. These oligonucleotides are designed to bind the mature miRNA and prevent its effect on its mRNA target. AntagomiRs have been useful tools to study miRNA expression and function in disease and have recently advanced as therapeutics in clinical trials.
The first example of an antagomiR in clinical trials is anti-miR-122, which is associated with hepatitis C viral infections (Figure 6). HCV is a small virus containing single-stranded, positive-sense RNA enveloped by a protein shell(162) and causes chronic liver infection, leading to approximately 400 000 deaths each year.(163) The miR-122 is highly tissue-specific, constituting 70% of the total miRNA population in the liver.(164) Unlike other miRNAs, which usually bind to the 3′ UTR of an mRNA, miR-122 enhances HCV viral RNA genome replication by binding to the 5′ and 3′ end of the viral RNA noncoding region (NCR), thereby promoting its stability and protecting it from degradation via Xrn1 exoribonuclease.(165, 166) In 2008, Sakari Kauppinen and colleagues developed an unconjugated LNA-antagomiR oligonucleotide to target miR-122.(166)This antagomiR silenced miR-122 in both mouse and nonhuman primate models.(167) Anti-miR-122 was used to treat HCV genotype 1 infection in chronically infected chimpanzees, leading to significant suppression of HCV viremia with no viral resistance or side effects.(168)
Figure 6.

Mechanism of action of Miravirsen. Miravirsen, an LNA-antagomiR, sequesters mature miR-122 in a highly stable heteroduplex and represses HCV viral RNA replication. In Miravirsen, the uppercase letters indicate LNA modifications and the lowercase letters indicate DNA nucleotides.
This strategy of using an LNA-antagomiR to target miR-122 was further developed to afford SPC3649, or Miravirsen, which contains seven deoxyriboses and eight LNA residues, and entered phase I clinical trials in 2009 (Santaris Pharma, acquired by Roche in 2014) (Figure 6). With no adverse effects in phase I trials, phase IIa clinical trials began in 2010.(169, 170) The safety and efficacy of Miravirsen were evaluated in 36 patients with chronic HCV genotype 1 infection, and five weekly subcutaneous injections of Miravirsen over 29 days resulted in significant reductions in HCV RNA levels in patients. A safety and efficacy study in a 3-year, long-term extension phase II study on Miravirsen was initialized in 2014. Interestingly, Miravirsen can bind to both pri- and pre-miR-122 and inhibit miR-122 biogenesis by preventing Drosha and Dicer cleavage, respectively.(170)
Another miR-122-targeting antagomiR currently in clinical trials is RG-101, a hepatocyte-targeted GalNAc conjugated oligonucleotide (Figure 2C) developed by Regulus Therapeutics that recently passed a phase 1b clinical trial.(171) It was administered as a single subcutaneous injection to patients with genotype 1, 3, and 4 chronic HCV infection, the most common HCV infection genotypes. Treatment with RG-101 resulted in reduction of viral load in patients within 4 weeks and sustained virological response in three patients after 76 weeks with no discernible adverse side effects.
Clinical trials have also been conducted on antagomiRs that target miR-155, an oncomiR that operates in pancreatic cancer, lymphoma, and many other types of aggressive cancers. In 2007, Dusetti and colleagues identified tumor protein p53 inducible nuclear protein 1 (TP53INP1), a proapoptotic stress-induced p53 activated protein, as a direct, downregulated target of miR-155 in vivo.(172) The miRNA also directly targets and downregulates Src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP1) mRNA, which leads to myeloproliferative disorder (MPD), as characterized by increased granulocyte/monocyte (GM) populations and decreased B-lymphocyte numbers.(173) Another target of miR-155 is wee1 kinases. The miR-155-induced downregulation of these kinases facilitates G2/M transition and allows cells to skip DNA repair and proceed directly to mitosis, resulting in accumulated mutations in inflammatory-induced cancers.(174)MRG-106 (developed by MiRagen Therapeutics) is an LNA-based antimiR-155 that entered phase I clinical trials in 2015 for patients suffering from cutaneous T-cell lymphoma (CTCL) of the mycosis fungoides (MF) subtype.(175)
Another miRNA family that is the target of clinical therapeutics is miR-103/107. This family of miRNAs plays a critical regulatory role in type 2 diabetes by targeting caveolin-1 (CAV-1). CAV-1 activates insulin signaling by stabilizing caveolae and associated insulin receptors, and derepression of CAV-1 in diabetes patients leads to an overall improvement in glucose metabolism.(176) RG-125, a GalNAc-conjugated (Figure 2C) antagomiR targeting miR-103/107, is currently in a phase I study to treat nonalcoholic steatohepatitis (NASH) in patients with type 2 diabetes/prediabetes in which high expression of miR-103/107 in liver cells is observed.(177)
In addition to these antagomiRs currently in clinical trials, many preclinical studies have also shown promising results for modulating miRNA expression with antagomiRs. One example is that of miR-10b, which is highly expressed in metastatic breast cancer cells.(178) To affect this miRNA, a miR-10b LNA-antagomiR was delivered by conjugation to dextran-coated magnetic nanoparticles with thiols.(179) This nanodrug, combined with a low dose of doxorubicin, led to a more prominent repression of metastatic cancer.(179) Another notable antagomiR used in preclinical studies is an LNA-modified antagomiR targeting miR-221 and miR-222. These miRNAs are oncomiRs in many cancers including hepatocellular carcinoma (HCC).(180–182) In a mouse model of HCC, a cholesterol-modified anti-miR-221 reduced tumor cell proliferation, induced apoptosis, and increased mouse survival.(183)
2.3.2 Alleviating miRNA Under-Expression with miRNA Mimics
In many diseases, miRNA levels are too low; thus, therapeutic interventions consist of introduction of miRNA mimics to supplement them.(158) These miRNA mimics are synthetic double-stranded small RNAs that function like naturally occurring miRNAs. One cancer that has been targeted with a miRNA mimic is chronic lymphocytic leukemia (CLL). Chromosomal deletions and translocations in chromosome 13q14 occur in about 65% of B cell CLL patients, and the most frequently deleted genomic region encodes miR-15/16.(184, 185) This miRNA functions as a tumor suppressor and binds B-cell lymphoma 2 (BCL2) mRNA; BCL2 protein plays a critical role in increasing cellular survival. BCL2 acts on the mitochondrial membrane to promote permeabilization and the release of cytochrome C and reactive oxygen species (ROS), important messengers in cellular apoptosis.(186) Croce and colleagues transfected a vector containing a genomic region encoding miR-15/16 into the leukemic cell line MEG-01 to more comprehensively understand the functions of this miRNA cluster.(187)
In addition to CLL, the miR-15/16 family is also downregulated in nonsmall-cell lung cancer (NSCLC) and malignant pleural mesothelioma (MPM). In NSCLC, downregulation of miR-15/16 prevents cell cycle arrest through inactivation of the retinoblastoma (Rb) gene.(188) In MPM, a consistent downregulation of the miR-15/16 family was observed across all tumors and cell lines, as compared with normal mesothelium or mesothelial cell lines.(189) Scientists in the Asbestos Diseases Research Institute (Sydney, Australia) used epidermal growth factor receptor (EGFR)-targeted nanocells to deliver miR-16 mimics to MPM xenografted tumors, which inhibited lung tumor growth in vivo.(190) Furthermore, they collaborated with EnGeneIC to develop a second-generation EGFR-targeted EnGeneIC Delivery Vehicle (EDV)-packaged miR-16-based mimic therapeutic called MesomiR-1, which has entered phase I clinical trial (NCT02369198) to treat MPM and advanced NSCLC.(191)
Many cancers including colon, prostate, and hepatocellular carcinoma (HCC) have been associated with lower expression of miR-34a, a direct transcriptional target of p53 tumor suppressor protein.(192–194) Bader and colleagues from Mirna Therapeutics demonstrated that a miR-34 mimic, encapsulated in lipid nanoparticles, could block lung tumor growth in a NSCLC mouse model through downregulation of cyclin-dependent kinase 4 (CDK4), c-Met, Bcl-2 proteins, and others.(195, 196) This miR-34 mimic, MRX34, was approved for human trials (NCT01829971). Unfortunately, Mirna Therapeutics decided to halt the MRX34 study after a fifth, immune-related serious adverse event (severe grade 4 cytokine release syndrome) in one of their clinical sites.(197)
In systemic sclerosis, which is characterized by joint pain and chronic tightening of the skin, miR-29 is strongly downregulated.(198–200) Indeed, a miR-29 mimic, MRG-201, is currently being tested in a phase I clinical trial (NCT02603224; MiRagen Therapeutics) to treat systemic sclerosis by mimicking the activity of miR-29, decreasing expression levels of collagen and other proteins that are involved in scar formation, thereby limiting the formation of fibrous scar tissue.(201) A cholesterol conjugated miR-29 duplex displays enhanced cellular uptake and allows for longer circulation lifetimes in serum, due to the greater stability gained from interactions with lipoproteins or albumin.
Although there are only a few examples of miRNA mimics in clinical trials, miRNA mimics have been effective in animal models of disease and have shown therapeutic potential. The miR-200 family is underexpressed in many cancers, and ectopic overexpression of miR-200 causes reduced motility of cancer cells.(202) Welsh and colleagues developed a miR-200c mimic to regulate intracellular reactive oxygen species (ROS) generation and increase cellular radiosensitivity.(203) The miR-200c mimics could potentially be used in combination with radiation therapy by targeting and downregulating several oxidative stress response proteins. Other miR-200 family mimics have been delivered with DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) lipid nanoparticles to inhibit tumor angiogenesis by targeting interleukin-8 (IL-8) and chemokine motif ligand 1 (CXCL1) in several experimental models.(204)
In ovarian cancer, miR-506 is a tumor suppressive miRNA that regulates epithelial-to-mesenchymal transition (EMT) by induction of E-cadherin expression and mesenchymal marker suppression. Nanoparticle delivery of miR-506 mimics in an orthopic mouse model of ovarian cancer resulted in reduced tumor growth.(205) Like miR-506, miR-520d is tumor suppressive in ovarian cancer and targets the mRNA of ephrin type-A and B receptor 2 proteins (EphA2 and EphB2).(206) EphA2, which is overexpressed in ovarian cancer and is associated with increased tumor growth and angiogenesis, can be partially silenced using an siRNA in an ovarian cancer mouse model.(207) Treatment with a combination of a miR-520d mimic and EphA2-targeting siRNA incorporated in DOPC nanoliposomes showed a greater suppression of tumor growth than siRNA alone, indicating that a combination of miRNA mimics and siRNAs can result in efficient silencing of a message.(205, 206) In addition, Mendell and colleagues used a self-complementary adeno-associated virus vector (scAAC) delivery system to boost miR-26a expression in a mouse model of hepatocellular carcinoma.(208) In HCC, miR-26a targets the cell cycle controllers cyclin D2 and cyclin E2 and induces a G1 arrest, resulting in tumor-specific apoptosis and inhibition of cancer cell proliferation.(209)
Collectively, miRNA-based therapeutics are complicated by delivery to the desired tissues;(67)however, they hold great potential as more data about miRNAs involved in disease are collected.
2.4 Aptamer Therapeutics
In 1990, the Gold, Szostak, and Joyce laboratories independently reported a process for selecting and amplifying single-stranded nucleic acids that selectively bound to target molecules with high affinity, or aptamers.(47–49) The target molecules of these aptamers were dye molecules, a single-stranded DNA sequence and bacteriophage T4 DNA polymerase.(47–49) Perhaps not surprisingly in retrospect, aptamers exist in nature as riboswitches that regulate gene expression.(210)Aptamers fold into secondary and tertiary structures depending on sequence, allowing them to bind to their targets with high specificity and affinity, with dissociation constants in the nanomolar to picomolar range.(211) For these reasons, they have been compared to antibodies.
Aptamers are generally selected using systematic evolution of ligands by exponential enrichment (SELEX) (Figure 7).(211) In this method, a random library containing 1013 to 1016 single-stranded DNA or RNA sequences is assembled and mixed with a ligand of interest.(211) Bound DNA or RNA sequences are then separated from unbound sequences by affinity chromatography or nitrocellulose filtration.(211) Bound nucleic acids are then eluted and amplified by PCR (DNA) or RT-PCR (RNA) to generate a new pool of sequences for another selection cycle.(211) The high selectivity of aptamers for their targets, combined with the powerful SELEX process used to identify them, has made them attractive therapeutic candidates.
Figure 7.
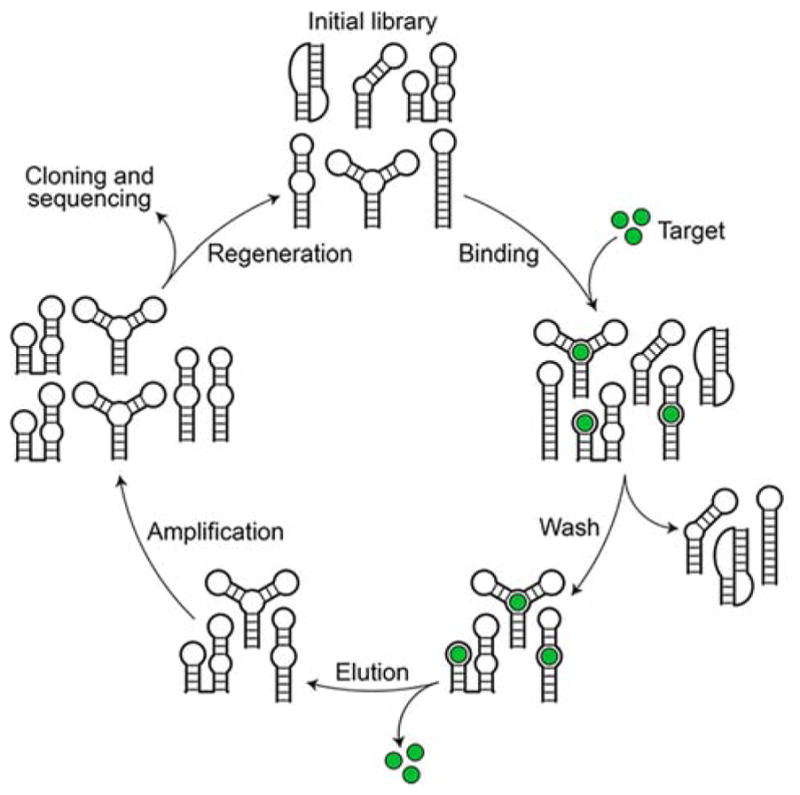
Flowchart of the SELEX process. The process begins with a random library of DNA or RNA sequences that is mixed with a ligand of interest. The bound sequences are separated from unbound sequences, eluted, and amplified to generate a new pool of sequences for another selection cycle
Aptamers have several advantages over antibodies for diagnostic as well as therapeutic applications. Aptamers can be identified with an in vitro process that allows them to target virtually any protein, whereas antibodies require animals or cells for production, wherein the selection process can be complicated by toxins, low immunogenic molecules, or restrictive in vivo conditions.(211–213) Further, aptamers can be produced in large batches by chemical synthesis with high purity and accuracy, and a wide variety of chemical modifications may also be incorporated into aptamers for different functions. Finally, aptamers have a longer shelf life than antibodies and refold into their original conformation after denaturation.
Aptamers can also have several drawbacks when compared to other therapeutics. Under the in vitro SELEX conditions used to select aptamers, their targets may adopt a different structure(s) than that observed in vivo. To overcome these potential inconsistencies, Mi et al. reported an in vivo process for selecting RNA aptamers, as applied to a hepatic tumor target.(214) First, a random library of RNA sequences was injected into mice-bearing hepatic tumors. Tumors were then harvested, and the RNA was extracted and amplified by RT-PCR. The resulting pool of RNA sequences was then reinjected for another round of selection, with increasing affinity for the tumor in successive rounds.
Although such selection methods can help overcome some limitations, aptamers still suffer from many of the same issues as other oligonucleotide therapeutics. Aptamers are too large to penetrate cellular membranes,(215) and although there have been examples of aptamers that can be internalized by cells upon binding to their cognate receptor on the surface,(216) they have mostly been targeted toward extracellular molecules or surface proteins of cells present in the blood or interstitial fluids.(217) Aptamers are also prone to degradation by nucleases and rapid renal filtration, leading to short bioavailability.(217, 218) As such, they are often modified to protect the reactive 2′ position of RNA. Despite the wide range of chemical modifications available to aptamers, they usually cannot be completely modified without the loss of binding affinity, so some degree of in vivo instability remains.(219) One way to circumvent problems associated with bioavailability, aptamers are often conjugated to PEG polymers.(218) Anti-PEG antibodies may be induced in treatments with PEGylated drugs, however, reducing their efficacy and causing allergic reactions.(220) To address this problem, Qi et al. demonstrated a PEG-like brush polymer, poly[oligo(ethylene glycol)methyl ester methacrylate] (POEGMA), which improved in vivo half-life and eliminated antibody binding.(220)
Importantly, in 1992 Bock et al. reported an aptamer that bound to and inhibited human thrombin, with potential therapeutic application as an anticoagulant.(221) Thrombin is responsible for production of fibrin (via cleavage of fibrinogen) that, along with platelets, forms clots. Structural studies of the thrombin aptamer have revealed that the aptamer is sandwiched between two positively charged regions of thrombin, the fibrinogen recognition exosite and the heparin binding site (binding of heparin inhibits pro-coagulation functions of thrombin).(222) Since this discovery, aptamers have been used for a variety of therapeutically relevant targets, including small inorganic ions, organic molecules, peptides, proteins, and intact cells.(211) In this section, several examples of aptamer therapeutics are discussed.
Pegaptanib, an Aptamer to Treat Wet Age-Related Macular Degeneration
Wet age-related macular degeneration (AMD) is a leading cause of blindness worldwide(223) and is characterized by choroidal neovascularization (CNV), in which new blood vessels form and leak fluid within the macula, leading to damage of photoreceptors and loss of central vision.(223, 224)Vascular endothelial growth factor (VEGF) has been associated with CNV, making anti-VEGF therapies an attractive treatment strategy for wet AMD.(224) NeXstar Pharmaceuticals developed Pegaptanib (Macugen; Figure 8A), a 28-nucleotide RNA injectable aptamer, to target VEGF isoform 165 (VEGF165) that was approved by the U.S. FDA in 2004.(223) Like two other RNA-based drugs that succeeded in clinical trials, the aptamer targets the immune privileged eye; that is, inflammatory immune responses generally do not occur in the eye and thus lowers the likelihood of an immune-related adverse effect.(225)
Figure 8.

Secondary structures of aptamers. (A) Pegaptanib, (B) REG1 (left) and REG1 with oligonucleotide antidote (right), (C) ARC1779, (D) N0X-A12, and (E) NOX-E36. iT denotes deoxythymidine.
Pegaptanib was selected using SELEX on libraries of random sequences to identify those that bind to VEGF165.(226) Photo-cross-linking experiments and computational modeling suggest that the aptamer binds to VEGF165 via an interaction between uridine-14 of the aptamer and cysteine-137 of VEGF165.(226) Indeed, Pegaptanib inhibits VEGF165 binding to VEGF receptors, reducing the formation of blood vessels in the eye and vascular leakage.(226) Importantly, 2′-fluoro modifications were added to pyrimidines to improve in vivo stability, and a 5′-PEG moiety prolonged in vivo half-life.(226)
In clinical trials, 70% of patients given a 0.3 mg dose of Pegaptanib could not distinguish 15 letters or fewer in a visual acuity test, compared to 55% of patients given a sham injection.(223)However, patients treated with Pegaptanib still continued to experience visual decline, likely because Pegaptanib targets only one isoform of VEGF.(223, 227) Meanwhile, treatments with anti-VEGF antibodies bevacizumab and ranibizumab improved visual acuity, thus are the most commonly used therapeutics to treat wet AMD.(227)
REG1, an Anticoagulation Aptamer
REG1 is an anticoagulation aptamer used for treatment of coronary artery disease (Figure 8B).(228) In particular, REG1 targets coagulation factor IX a (FIXa), which is activated by cleavage of its zymogen, factor IX (FIX).(229) FIXa initiates a signaling cascade that results in thrombin formation and, in turn, facilitates fibrin and blood clot formation.(229, 230) Thus, inhibition of FIXa may be a therapeutic anticoagulation strategy. Current anticoagulation strategies include administration of heparin followed by its antidote, protamine, once blood coagulation profiles have normalized. Excess heparin can induce an immune response termed heparin-induced thrombocytopenia, and protamine administration is also associated with life-threatening side effects.(231) Thus, inhibition of FIXa by an aptamer and control of this inhibition through a complementary oligonucleotide antidote offers an alternative anticoagulation strategy. Indeed, Sullenger and colleagues developed such an approach to control anticoagulation in plasma samples and in a porcine model of systemic anticoagulation.(228, 232)
REG1 consists of an injectable 31-nucleotide PEGylated aptamer anticoagulation factor (RB006, or pegnivacogin) and a 15-nt antidote oligonucleotide (RB007, or anivamersen) that forms Watson–Crick base pairs with the aptamer to modulate its activity.(228) REG1 was selected using SELEX to screen a library of 1014 sequences for those that bind to FIXa.(228) The resulting sequence bound with a KD of 0.65 ± 0.2 nM.(228) The in vivo stability was improved by addition of 2′ fluoropyrimidine modifications and an inverted deoxythymidine (iT) at the 3′ end.(228)
A phase 1a trial to characterize the safety profile and pharmacodynamic responses of RB006 in healthy individuals produced no significant bleeding in the subjects.(233) Next, a phase 1b trial was carried out in patients with stable coronary artery disease. In the study subjects, RB006 produced a statistically significant dose-dependent increase in activated partial thromboplastin time (aPTT), a measurement of blood clotting time, which RB007 then reversed with a median time of 1 min after injection.(234, 235) The pharmacodynamics of REG1 were then verified in a phase 2a and phase 2b trial of patients undergoing percutaneous coronary intervention, as well as those with with acute coronary syndromes, respectively. Although three patients in the phase 2b study had allergic-like reactions, REG1 proceeded to phase 3 trials.(236, 237) A phase 3 trial was terminated early because of severe allergic reactions in 1% of patients, which were partially attributed to anti-PEG antibodies.(238, 239)
ARC1779, an Antithrombotic Aptamer
ARC1779 (Archemix) is a 40-nt antithrombotic, PEGylated DNA/RNA aptamer (Figure 8C) that targets von Willebrand factor (VWF), without significant anticoagulation, for treatment of coronary artery disease.(240) Acute coronary syndrome is most commonly caused by rupture of an atherosclerotic plaque.(241) The high shear rates at the rupture site results in activation of VWF, followed by its binding to platelets and subsequent blood clot formation.(241) Activation of VWF leads to a structural change that allows its A1 domain to bind platelets,(241) thus neutralizing VWF is a therapeutic strategy to combat clot formation in coronary artery disease.
The anti-VWF aptamer was selected using a SELEX screen of 1014 random nucleic acid sequences for those that bind to the A1 domain of VWF as well as to intact VWF.(242) ARC1779 was derived from ARC1772 by adding modifications to prevent nuclease degradation.(243)ARC1779 contains 13 unmodified 2′-deoxynucleotides, 26 2′-O-methyl modified nucleotides, and one inverted deoxythymidine at the 3′ end to minimize endonuclease and exonuclease digestion, respectively.(244) A phosphorothioate linkage between nucleotides mG20 and dT21 enhances affinity for VWF.(244) Collectively, ARC1779 binds avidly to the A1 domain of VWF with a KD of 2 nM.(242) A crystal structure of the first generation aptamer, ARC1772, and the VWF A1 domain demonstrated that A8-G11 and G21–C32 participate in interactions with VWF.(243) Multiple cation−π interactions account for 65% of the binding interface between the A1 domain of VWF and the aptamer.(243)
A phase 1 clinical trial produced no serious adverse events or spontaneous bleeding in healthy individuals.(244) Furthermore, ARC1779 significantly reduced platelet adhesion in blood taken from patients with coronary artery disease.(245) ARC1779 was also used to treat patients with VWF-related disorders. In von Willebrand disease, which is characterized by deficient VWF, patients often receive desmopressin infusion to increase VWF, which can lead to hyperactive VWF and low platelet counts. In these patients, ARC1779 blocked hyperactive VWF and reversed desmopressin-induced drops in platelet counts.(246) In a separate study, ARC1779 increased platelet counts in patients with thrombotic thrombocytopenic purpura (TTP), where ultralarge VWF multimers aggregate platelets and cause organ damage.(247)
NOX-A12, an Aptamer to Treat Multiple Myeloma
NOX-A12 (olaptesed pegol, NOXXON Pharma) is a 45-nucleotide aptamer (Figure 8D) that targets stromal cell-derived factor 1 (SDF-1 or CXCL12), a chemokine implicated in multiple myeloma (MM).(217) Chemokines are small proteins defined by the feature of conserved cysteine residues at the N-terminus.(248, 249) These proteins activate G-protein coupled receptors and induce migration of cells through a concentration gradient.(248, 249) The CXCR4/CXCL12 axis, which consists of the CXCR4 receptor and CXCL12 ligand, plays a significant role in cancer biology and progression.(249) For instance, in acute myeloid leukemia (AML), CLL, and MM, malignant cells expressing CXCR4 migrate toward bone marrow cells expressing CXCL12.(248, 249) Adhesive interactions between the malignant cells and bone marrow stromal cells confer drug resistance.(248, 249) Thus, a therapeutic strategy is inhibition of CXCR4 expressed on malignant cells from binding CXCL12 expressed on bone marrow cells.
In particular, NOX-A12 is a Spiegelmer, a highly stable oligonucleotide consisting of non-natural l-nucleotides to protect them from nucleases in biological fluids.(219) The l-aptamers are selected using SELEX to screen a library of d-aptamers for those that bind to an enantiomer of a target molecule.(219, 250) Identified sequences are then synthesized using l-nucleotides and should bind to the natural target on the basis of shape complementarity.(219, 250) In functional assays with CLL cell lines, NOX-A12 significantly reduced CLL cell migration toward CXCL12 when administered at a very low concentration of 3 nM.(251) Bone marrow stromal cells pretreated with NOX-A12 to remove CXCL12 were significantly more chemosensitized than those that were not pretreated.(251) In a phase 2 study, NOX-A12 significantly increased the levels of myeloma cells in circulation (that is, they are not bound to bone marrow cells), making the cells more susceptible to the effects of chemotherapeutic drugs, and enhanced the clinical activity of bortezomib and dexamethasone in relapsed or refractory MM patients.(252)
NOX-E36, an Aptamer to Treat Diabetic Nephropathy
Diabetic nephropathy is a kidney disease that often leads to end-stage renal disease and mortality.(253) High glucose levels have been shown to stimulate production of monocyte chemoattractant protein-1 (MCP-1 or CCL2) in kidney cells.(253) MCP-1 promotes recruitment of macrophages by binding to its receptor, C–C chemokine receptor type 2 (CCR2), and thereby contributes to progression of disease by promoting inflammation.(254) NOX-E36 (emapticap pegol, NOXXON Pharma) is a 40-nucleotide PEGylated Spiegelmer (Figure 8E) that targets MCP-1and neutralizes the chemokine’s activity in diabetic nephropathy.(219, 255) In a crystal structure of the CCL2-NOX-E36 complex, binding was shown to be mediated through hydrogen bonds, electrostatic interactions, and at least one cationic–π interaction involving 11 nucleotides and 10 amino acid residues.(255)
In a phase 2 study, NOX-E36 reduced blood monocyte count by 15–20% within 1 week after treatment was initiated.(256) Patients treated with NOX-E36 experienced up to 40% reduction in the albumin to creatinine ratio (ACR), a measurement of albuminuria, which is a biomarker for renal disease.(256, 257)
2.5 Ribozyme Therapeutics
In the 1980’s, the Cech and Altman laboratories independently discovered that RNAs can serve as a catalyst, and these RNAs were dubbed ribozymes.(258–260) Since then, seven naturally occurring classes of ribozymes have been identified, all of which catalyze cleavage or ligation of the RNA backbone.(258) Cleavage reactions generally occur by acid–base or two-metal ion catalyzed transesterification, where a 2′ oxygen nucleophile attacks the phosphate of the 3′ nucleotide, forming a 2′-3′-cyclic phosphate and a 5′ hydroxyl group as cleavage products.(258)Ribozymes are potentially advantageous as drugs because of their catalytic activity, in that each ribozyme molecule may cleave multiple target RNAs at single-nucleotide precision.(261)
Two ribozymes used in therapeutic applications are the hammerhead and hairpin ribozymes, both of which are 50–100 nucleotide RNAs that undergo self-splicing by acid–base catalysis.(258, 262)The hammerhead ribozyme consists of three variable helices and three single-stranded regions containing highly conserved nucleotides (Figure 9A).(258, 262) Cleavage occurs on the 3′ side of a 5′ NUH triplet, where N is any nucleotide and H is any nucleotide except for G, although the most effective triplet is 5′ GUC.(258, 262) The hairpin, or paperclip, ribozyme consists of four variable helices and two internal loops with highly conserved nucleotides (Figure 9B).(258, 262, 263) Cleavage reactions occur at the * site of a 5′RYN*GUC sequence, where R is A or G, and Y is C or U.(258, 262) These ribozymes may be converted from cis-acting (on the same RNA molecule) to trans-acting (on a different RNA molecule) by splitting the catalytic core from the substrate sequence.(263) For hammerhead ribozymes, the 5′ NUH triplet should be retained and ribozyme sequences complementary to those flanking the cleavage site should be designed for the target of interest.(264) For hairpin ribozymes, the junction between the two domains may be reduced to a hinge.(265) Ribozyme therapeutics have most commonly consisted of hammerhead or hairpin domains, which have been developed extensively for trans-cleavage and whose structures are well-understood.(263, 266)
Figure 9.

Secondary structures of ribozymes. (A) The hammerhead ribozyme/substrate model and (B) the hairpin ribozyme/substrate model. Red arrows denote the cleavage site.
Ribozymes may be rationally designed or subjected to an in vitro selection process(266, 267) (see description of SELEX in section 2.4, Aptamer Therapeutics) to generate new or improved functions.(268, 269) When designing ribozymes, careful consideration must be given to potential ribozyme misfolding, which may cause loss of catalytic activity,(269) the accessibility of the target site (RNA–protein interactions or extensive secondary or tertiary structures),(263, 269, 270) and the stability of the ribozyme-RNA complex. Weaker binding of the ribozyme to the target site could make the ribozyme more sensitive to the effects of mismatches, resulting in better sequence specificity, whereas a ribozyme-target complex that is too stable could eliminate catalytic activity.(271, 272) To overcome potentially reduced activity due to highly structured target sites, computational or experimental methods are often employed. Free-energy minimization algorithms are used to find single-stranded regions of mRNA, but these programs cannot predict tertiary structures or long-range interactions.(263, 269, 270) Experimentally, readily accessible sites can be identified by hybridizing complementary oligodeoxynucleotides to potential ribozyme cleavage sites within an RNA target and then incubating with RNase H.(273) The percentage of the substrate cleaved at each position can then be quantified to identify the most accessible sites and to inform ribozyme design.(273) Zinnen et al. reported an example of this process to identify a potentially therapeutic ribozyme containing a hammerhead domain.(274)
While in vitro cleavage of target RNA may be used to screen ribozymes for activity in different regions, the kinetics observed in these studies may not be predictive of ribozyme activity in vivo.(270) Furthermore, in vivo parameters are typically not known and cannot be predicted due to too many unknown variables.(275) Therefore, empirical testing is required once a ribozyme and target pair have been determined to have catalytic activity in vitro.(275)
The major challenges with ribozyme therapeutics are delivery and production of the intended therapeutic effects.(261) Ribozymes may be delivered to cells by exogenous delivery in RNA form or via endogenous expression in a viral or plasmid vector.(261) Although exogenous delivery is relatively easy and rapid, endogenous expression allows the ribozyme to be continuously expressed, thereby knocking down its target RNA over a longer period of time.(261) Antiretroviral ribozymes could thus target several stages in the viral life cycle, inhibiting the generation of drug-resistant viruses.(263, 276) Endogenous delivery through a vector is also advantageous as exogenously delivered ribozymes are prone to problems with cellular uptake and degradation.(261, 263) To extend their lifespans and stabilities, ribozymes are often chemically modified.(261, 263) Residues in the core, however, must remain unmodified to maintain catalytic activity, and phosphorothioate linkages can promote nonspecific binding to other biomolecules.(271) It is also important to note that the effectiveness of the ribozyme may be weakened if target genes are not critical to disease progression, and cells, particularly cancer cells, circumvent target gene inhibition by using alternate pathways.
Anti-HIV Hairpin Ribozymes
The human immunodeficiency virus (HIV)-1 genome consists of a single RNA sequence that encodes 15 proteins.(277) The U5 region contains the 5′ cap, and the pol region encodes the Pol polyprotein.(275, 277) Together, these sequences are essential for viral replication. Anti-HIV hairpin ribozymes are 59-nt ribozymes that target the U5 and pol regions of HIV-1 and were the first ribozymes to be approved for human clinical trials, via expression from a retroviral MY-2 vector.(275, 278, 279) Cleavage occurs at the * sites of 5′C*GUC and 5′U*GUC motifs in highly conserved sequences within U5 and pol, respectively.(275) In a phase 1 clinical trial of anti-HIV ribozyme gene therapy, three HIV-1 patients were infused with cluster of differentiation 4 positive (CD4+) transduced T-cells.(278, 279) MY-2 vector was detected in only one patient after infusion, and no Pol ribozyme driven by a murine leukemia virus (MLV) promoter was expressed.(278, 279)However, the U5 ribozyme driven by a tRNAVal promoter was detected in cells at five and seven months after infusion.(278, 279) Despite the low efficacy of the vectors due to challenges with expression, the study demonstrated the safety and feasibility of ribozyme therapy against HIV-1.(278, 279)
OZ1 (RRz2, Johnson & Johnson) is a 39-nt hammerhead ribozyme that targets a region (named Rz2) of the human immunodeficiency virus type 1 (HIV-1) genome in the overlapping trans-activator of transcription (tat) and viral protein R (vpr) reading frames (Figure 9).(280, 281) In HIV-1, Tat enhances the processivity of RNA polymerases and rate of transcription initiation, and Vpr is required for localization of viral RNA in nondividing cells.(277) Interestingly, the Rz2 target region of OZ1 is highly conserved in almost all naturally occurring HIV-1 isolates, thus mutations that result in resistance to the ribozyme may not be well-tolerated by the virus.(276, 282) Pluripotent CD34+-expressing hematopoietic progenitor cells give rise to mature myeloid and lymphoid cells that can be infected by HIV-1.(283) For this reason, transducing CD34+ with OZ1 could generate myeloid and lymphoid cells that express the ribozyme gene and thus inhibit HIV-1 replication.(283)
The OZ1 ribozyme gene is expressed in the 3′ UTR of the neoR gene (which provides resistance to G418 antibiotic) in the Moloney murine leukemia virus (MMLV) retroviral LNL6 vector.(281)Cleavage occurs on the 3′ side of a 5′ GUA triplet, after the adenine.(280, 281) In a phase 1 clinical trial, mutations at the −4 and −1 positions relative to the 5′ GUA triplet were detected in patients, but they were also present before treatment, and OZ1 was active against these mutations in vitro.(281) Naïve T lymphocytes and myeloid precursor cells expressing resistance to the G418 antibiotic were detected for up to two years after infusion of CD34+ cells transduced with LNL6 and OZ1 vectors, indicating expression of the neoR gene.(281) In a phase 2 study, HIV-1 viral load was lower in patients who received OZ1 than in those receiving a placebo at weeks 47 and 48, but the difference was not statistically significant.(280) Throughout the 100 weeks of treatment, mature CD4+ T cell counts were higher in the OZ1 group than the placebo group.(280)A lack of resistance to OZ1 in the phase 2 study may be attributed to a low percentage of OZ1 gene-marked cells, production of less fit viruses due to mutation(s), or both.(280) OZ1 did not proceed to further clinical trials due to its lack of efficacy.
Angiozyme, a Ribozyme Targeting Vascular Endothelial Growth Factor Receptor-1
Angiozyme (RPI.4610, Merck) is a 35-nt injectable anticancer ribozyme that targets vascular endothelial growth factor receptor-1 (VEGFR-1 or FLT-1) mRNA.(271) VEGFR-1 signaling has been implicated in tumor angiogenesis, which is critical for tumor growth and metastasis.(284)Activation of VEGFR-1 by VEGF-A stimulates multiple signaling networks that result in endothelial cell migration and survival.(284) The synthetic hammerhead ribozyme contains 30 2′-OMe (Figure 2A) nucleotides, four phosphorothioate linkages at the 5′ end, and an inverted 2′-deoxyabasic cap at the 3′ end to protect it from nucleases in vivo.(271) The core contains five unmodified purine nucleotides to maintain catalytic activity.(271) This ribozyme was selected from an RNase H cleavage assay.(273) In phase 1 studies, angiozyme was well-tolerated and had biological activity against solid tumors.(285, 286) However, a phase 2 study of patients with metastatic breast cancer failed to establish clinical efficacy, suggesting that the ribozyme was unable to inhibit its target.(287)
3 Small Molecules Targeting Nucleic Acid Sequence
Oligonucleotides have been successfully used to target specific disease-causing genetic sequences; however, their delivery and tissue distribution hamper their therapeutic utility. As an alternative, great effort has been invested in the development of small molecules, which generally have better cellular and broader tissue distribution in vivo than oligonucleotides. Further, broad chemical space is available for small molecule optimization via medicinal chemistry. In the next sections, we discuss the design of small molecules targeting a specific genetic sequence beginning with DNA targeting agents. Following is a discussion of methods to identify small molecules that target specific RNAs from sequence and how these small molecules have been designed to target disease-causing RNAs in cells and in vivo. For a broader discussion of nucleic acid-targeting molecules, outside of sequence-based design, please see the following reviews.(288–292)
3.1 Small Molecules Targeting DNA
In the late 1950’s and early 1960’s, two polyamides with antimicrobial and antiviral activity were discovered from Streptomyces netropsis, netropsin and distamycin (Figure 10A).(293, 294) Nearly two decades later, details about their mode of action, particularly their ability to sequence-specifically recognize DNA, began to emerge from seminal biochemical and biophysical studies by Dickerson, Shultz, Dervan, Wemmer, Patel, Wartell, and others.(295–299) In particular, netropsin, a pyrrole amidine (two N-methyl pyrrole units (Py); Figure 10A) binds the minor groove of AT-rich DNA stretches by forming hydrogen bonds with A and T residues on opposite strands and van der Waals contacts with adenine C2 hydrogens.(298) Netropsin has a natural twist that is mirrored by the minor groove of AT stretches, and thus, the small molecule is preorganized to bind. From these structural studies, Dickerson hypothesized that substitution of Py with imidazole (Im) could afford selective recognition of GC base pairs.(298) Distamycin is structurally related to netropsin but is comprised of three Py units (Figure 10A). Perhaps unexpectedly, Wemmer’s laboratory showed that two copies of natural product distamycin bound to the minor groove of a 5′-AAATT-3′ segment of DNA.(300) Collectively, these studies indicated that polyamide recognition of DNA might be programmable, allowing small molecule readout of sequence and perhaps modulation of gene function. Indeed, Dervan’s pioneering work in this area led to his eponymous rules for the sequence-specific targeting of the DNA minor groove (Figure 10, panels B and C).(301)
Figure 10.

Design of polyamides to target DNA sequence. (A) Structures of the naturally occurring polyamides netropsin and distamcyin and (B) Watson—Crick hydrogen-bonding patterns in the DNA minor groove. The black circles represent lone electron pairs, and circles containing an H represent the 2-amino group of guanine. R represents the sugar backbone of DNA; (C) binding model between ImHpPyPy-γ-mHpPyPy-β-Dp and a 5′-TGTACA-3′/3′-TGTACA-5′ sequence. Hydrogen bonds are shown as dashed lines.
Interestingly, another class of naturally occurring antitumor antibiotics that target DNA, pyrrolobenzodiazepines (PBDs), was discovered in Streptomyces around the same time as netropsin (Figure 11A).(302, 303) The first PBD isolated and characterized was anthramycin, and Kohn and colleagues were the first to suggest that the compound forms a covalent bond with DNA, in particular at positions 9 and 11 on the PBD ring system, and ruled out an intercalative binding mode (Figure 11A).(304–306) In 1978, a limited clinical trial of anthramycin was conducted for reduction of anxiety(307) followed by a study two years later in mice to study the effects on the central nervous system, in particular depression.(308) Shortly thereafter, Hurley & Thurston showed that anthraymcin’s position 11 forms adducts with the exocyclic amine of guanosine residues in the dsDNA minor groove and may have sequence selectivity.(309) A detailed structure–activity relationship (SAR) study by Thurston indeed revealed sequence selectivity of PBDs for the DNA minor groove,(310) culminating in the discovery that PBDs form a covalent bond with the exocyclic amine of guanosine in the minor groove, preferring R-G-R triples (where R is A or G) and indicating that the small molecule binding site spans three base pairs.(310, 311)Collectively, this foundational work set the stage for the rational design of PBD dimers that react with DNA in a sequence specific manner and has led to the development of an anticancer therapeutic that completed a phase I clinical trial in patients with solid tumors(312, 313) and antibody-drug conjugates (ADCs) with PDB dimer payloads in various stages of clinical trials.(314–318)
Figure 11.
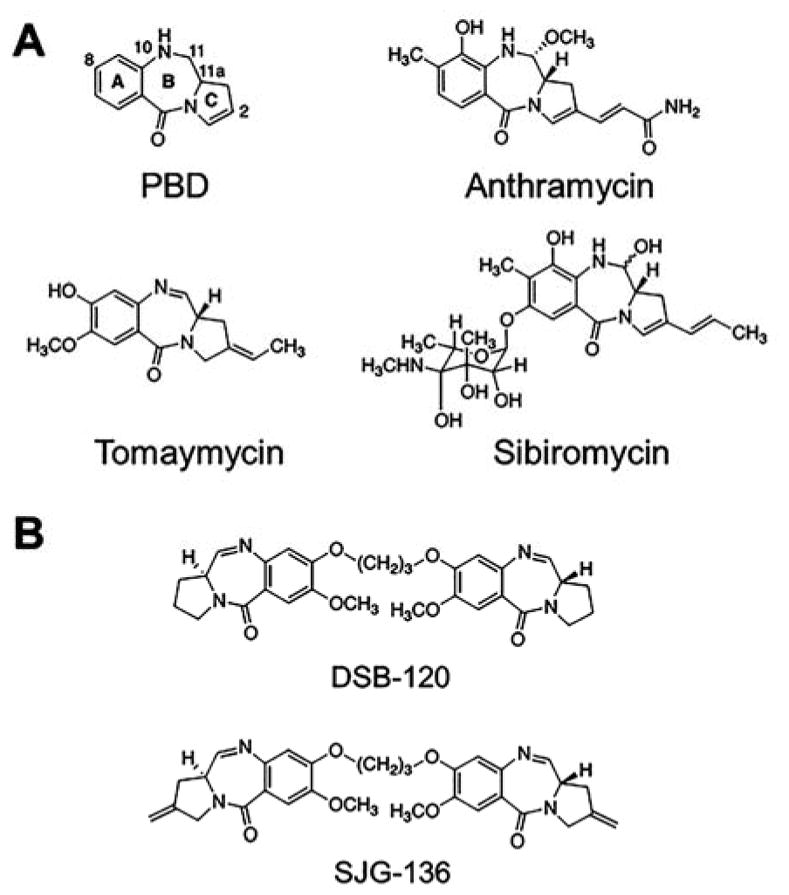
Chemical structures of the DNA minor groove binding pyrrolobenzodiazepines (PBDs). (A) Structures of PBDs. Anthramycin tomaymycin, and sibiromycin were discovered in the 1960’s and function as chemotherapeutics by forming covalent bonds with DNA (exocylic amine of guanosine), (B) The PBD dimer SJG-136 conjugated to antibodies has shown promise in clinical trials.
Other natural products that target DNA have been extensively studied for their sequence specificity and have provided inspiration for DNA binding agents. These natural products include duocarmycin, bleomycin, and others that have been reviewed elsewhere.(319, 320) The duocarmycins are a class of potent antitumor agents with sequence specific minor groove alkylation.(321) These small molecules exhibit AT-rich binding selectivity similar to that of distamycin.(321) The duocarmycins are unique alkylating agents in that they are unreactive toward conventional nucleophiles at pH 7, however, upon binding to DNA, a conformational change in the molecule activates the cyclopropane for nucleophilic attack.(322, 323) Bleomycin is an FDA-approved anticancer antibiotic that causes DNA single and double strand breaks.(324, 325) Both SAR studies and NMR studies revealed that bleomycin interacts with DNA in a sequence specific manner.(324, 325) Specifically, the C-terminal bithiazole group interacts with DNA via intercalation while the valerate-threonine linker lies in the minor groove. The N-terminal metal-binding pyrimidine core forms hydrogen bonds with G, which results in cleavage at the 5′-GC and 5′GT sites.
3.1.1 Targeting the DNA Minor Groove: Polyamides
An important pairing rule for polyamides was defined after Dervan and colleagues unexpectedly found that the three unit polyamide, ImPyPy, binds DNA as an antiparallel 2:1 dimer.(326, 327) A 1:1 stoichiometry with a G·C base pair was predicted based on the 1:1 complex formed between netropsin and the DNA minor groove.(298) These studies enabled the development of modules that selectively recognize 5′C·G3′ base pairs (Py/Im) and 5′G·C3′ (Im/Py) and a module that recognizes both 5′A·T3′ and 5′T·A3′ pairs (Py/Py). To distinguish between 5′A·T3′ and 5′T·A3′, Dervan and colleagues introduced N-methyl-3-hydroxypyrrole (Hp), affording the Hp/Py pair to recognize a 5′T·A3′ and Py/Hp to bind selectively to 5′A·T3′.(298) Finally, these compounds were developed into an eight-ring hairpin-like polyamide that bind a DNA sequence with extremely high affinity (Figure 10C).(328)
Because Hp has less stability in biological environments, particularly acidic conditions, Dervan explored and found hydroxybenzimidazole bicycle (Hz), the first heterocyclic ring pairing element that can be used within polyamides. Hz is more effective at differentiating between T and A when paired with Py and within different sequence contexts compared with Hp.(329) To increase selectivity and recognition between the target sequence and base pair mismatches, a Py in an eight ring hairpin can be replaced by β-alanine, a flexible residue, which can reset the curvature to optimize hydrogen bonding.(330) On the basis of this sequence-selective strategy, Sugiyama and colleagues developed a series of sequence-specific DNA-alkylating agents by conjugation to Py/Im polyamides.(331–334) They also synthesized dimers as DNA interstrand cross-linking agents with potential application of inhibition of both DNA replication and gene expression.(335, 336)
The first example of the use of polyamides is the DNA-binding of an eight-ring hairpin polyamide that recognizes a transcription factor (TF) IIIA binding site within Xenopus 5S rRNA gene and represses its transcription.(337) These studies were the first proof of concept that Py/Im polyamides are cell-permeable and can regulate gene expression. Other studies of Dervan polyamides showed that they permeate human cells and inhibit transcription of HIV-1 viral genome. The HIV-1 viral promoter contains binding sites for various human transcription factors including TATA-box binding protein (TBP), lymphoid-enhancer binding factor 1 (LEF-1), and cell-encoded proteins upstream stimulatory factor (Ets-1), which then recruit RNAP II. Polyamides were designed to bind sequences adjacent to each protein binding site, which are unique to HIV.(338) That is, polyamides that bind transcription factor binding sites could interfere with transcription of human genes and cause undesired side effects. Binding of the polyamides in concert effectively obstructs the RNAP II promoter and inhibits transcription of the HIV-1 genome.(338)
Interestingly, Py/Im polyamides can be used as artificial transcription activators when conjugated to an activation domain (AD) and a linker domain (LD).(339) A polyamide-peptide conjugate was designed to sequence-specifically recruit the developmental regulator Exd to a cognate DNA site with higher efficiency than its natural Hox protein partner.(340) These examples demonstrate that structure-based modular design is a valid strategy toward artificial transcriptional activators.
Sequence-specific polyamides have also been used to regulate disease-causing aberrant gene expression. Dervan and colleagues designed a polyamide to inhibit binding of the heterodimeric hypoxia-inducible factor 1 (HIF-1)/aryl hydrocarbon receptor nuclear translocator (ARNT) heterodimer to its cognate genomic DNA sequence, hypoxia response element (HRE), to downregulate the expression of VEGF and other hypoxia-inducible genes in HeLa cells.(341)Another polyamide was used to target a DNA abnormality common in Friedreich’s ataxia (FRDA), an unstable hyperexpansion of a GAA/TTC triplet repeat in frataxin (FXN) gene. The hyperexpansion causes decreased transcription of FXN and reduced levels of FXN protein by forming triplexes that block RNAP II transcription.(342, 343) A β-alanine-linked polyamide was used to target these repeats, which disrupted GAA/TCC triplex formation, induced a chromatin opening, and increased transcription of the FXN gene.(29)
Polyamides also have therapeutic potential to treat cancers. Dervan and colleagues demonstrated that a designed polyamide that targets the sequence 5′-WGWWCW-3′ (W = A/T) found in the androgen response element (ARE) disrupts androgen receptor (AR)-regulated gene expression including prostate-specific antigen (PSA) expression in cultured prostate cancer cells.(344)Further studies showed that the polyamide perturbed multiple DNA-dependent cellular processes including interference with RNAP II elongation, resulting in degradation of the RNAP II large subunit (RPB1), activation of p53, and p53-mediated apoptosis in a prostate tumor xenograft model.(345) Py/Im polyamides can also be utilized for targeting the 5′-WGGWWW-3′ and 5′-GGGWWW-3′ (W = A/T) binding site of transcription factor NF-κB, which regulates various aspects of cell death, differentiation, and the immune response by regulating NF-κB-driven genes including IL6 and IL8.(346) Additional studies demonstrated its efficacy in vivo, in particular in mouse xenograft models.(347)
3.1.2 Targeting the DNA Minor Groove: Pyrrolobenzodiazepines
The discovery of anthramycin in 1965 spurred interest not only in the identification of other naturally occurring PBDs but also elucidation of its mechanism of action. One of the first SAR studies was completed by comparing anthramycin, sibiromycin, and tomaymycin (Figure 11A).(348) Sibiromycin, which lacks anthramycin’s amide group and is glycosylated at position 7, reacts much faster than anthramycin and tomaymycin and competes for their binding sites.(348)Tomaymycin (Figure 11A), however, does not compete with anthramycin for binding the same sites in DNA.(348) An SAR study of 15 PBDs was completed by Thurston and co-workers in 1990, indicating structural features required for PBD binding, including that reverse stereochemistry at position C2 and substitution of the N10–C11 with lactam are not well-tolerated.(349) Additional studies have investigated the role of the stereochemistry of C11a/C11, functionalization of the A ring, unsaturation, and substitution of the C-ring, and other modifications (Figure 11A). For a comprehensive review of PBD SAR, please see ref 350.
Suggs and co-workers were the first to dimerize anthramycin in an effort to increase its sequence selectivity and reactivity in 1988.(351) Although modest reactivity was observed, these studies and the SAR studies completed shortly thereafter(349) laid the foundation for the development of the PBD dimer DSB-120 by Thurston and colleagues (Figure 11B).(352) DSB-120 is a C8–C8′-linked PBD that was one of the most efficient DNA cross-linking agents identified to date with significant cellular activity.(352) The structure of a DNA-DSB-120 adduct showed that the dimer spanned 6 base pairs in the minor groove and formed a symmetric cross-link between guanine residues as expected, governed by formation of hydrogen bonds and van der Waals and electrostatic interactions.(353) Unfortunately, preclinical studies of DSB-120 revealed poor antitumor activity likely due to a combination of lack of tumor selectivity, poor uptake and/or significant drug metabolism, or reaction/binding of plasma proteins.(354) Therefore, second generation molecules were designed, affording SJG-136, a C2-exo-methylene PBD dimer that is more cytotoxic against cancer cell lines, reacts more efficiently than DSB-120,(355) and shows superior antitumor activity in various mouse xenograft models.(356)
Preliminary in vitro pharmacology studies of SJG-136 (Figure 11B) showed that the dimer forms interstrand cross-links between two guanosine residues separated by two base pairs,(357) with a preference for 5′-R-GATC-Y-3′ (where R is A or G and Y is T or C).(358) More recent studies have shown that SJ-136 can react with 5′-R-GAATC-Y-3′ to form longer interstrand cross-links;(359)intrastrand cross-links in the context of 5′-R-GATG-Y-3′ and 5′-R-GAATG-Y-3′;(359) and shorter inter- and intrastrand cross-links with 5′-R-GAC-Y-3′ and 5′-R-GAG-Y-3′.(360) Although SJ-136 did not progress past phase I clinical trials,(312, 313) it served as a basis for the development various ADCs with PDB dimer payloads. An SJG-136-antibody conjugate, ADCT-402, recently completed a phase I clinical trial for patients with relapsed or refractory non-Hodgkin’s lymphoma(318) and is currently being evaluated in a phase I clinical trial for acute lymphoblastic leukemia (ALL).(315) Another PBD dimer ADC SC16LD6.5 (StemcentRx) is currently in phase III clinical trials for treatment of small cell lung cancer,(316) while phase III clinical trials for a third PBD dimer ADC, SGN-CD33A (Seattle Genetics), for treatment of acute myeloid leukemia(317)were recently halted by the FDA.
3.1.3 Targeting the DNA Minor Groove: Diamidines
Diamidines (Figure 12A), such as stilbamidine and pentamidine, have been used clinically since the late 1930’s to treat a wide variety of maladies, in particular parasitic infections [human African trypanosomiasis (HAT); African sleeping sickness, leishmaniasis, and malaria] and cancer.(361–365) In 1980, Boykin and co-workers reported the antitrypanosomal activity of a series of bis-guanyl compounds and several cyclic guanyl derivatives.(366) These early studies into the SAR of diamidines revealed that diphenyl oxazole diamidines, diphenyl thiadiazole diamidines, and diphenyl pyridazine diamidines were of similar potency to stilbamidine, hydroxystilbamidine, and pentamidine.(366) The cyclic guanyl derivatives as well as the diphenyl oxadiazole diamidines were much less active.(366) Although the mechanism of action of pentamidine is still not completely clear, its structure and its similarities to netropsin (Figure 10), suggest that aromatic diamidines bind the minor groove of DNA.
Figure 12.

Chemical structures of the DNA minor groove binding pyrrolobenzodiazepines (PBDs). (A) The structures of pentamidine and stibamidine, diamidines used clinically since the late 1930’s and early 1940’s to treat a wide variety of diseases. (B) Boykin and Wilson discovered that diamidines bind AT-rich regions in DNA. SAR and structural studies revealed important features of molecular recognition and allowed for the rational design of diamidines that selectively bind GC base pairs. DB75 has shown promise as an anti- trypanosomiasis agent. (C) Dimers, such as RT533 and DB2232, target mixed DNA sequences.
Indeed, from the early 1980’s onward, the Boykin and Wilson laboratories sought to identify the DNA binding preferences of diamidines in an effort to develop compounds with improved antimicrobial properties. This large body of work elucidated the preference of aromatic diamidines for stretches of AT base pairs(367–375) and revealed important features of selective molecular recognition that were confirmed by the many crystal structures of diamidine-DNA complexes from the Niedle laboratory.(358) Effective binding of diamidines is promoted by closely mirroring the shape of the minor groove of AT stretches, a degree of flexibility to optimize interactions within the minor groove whether hydrogen bonds or van der Waals contacts and, unsurprisingly, based on their dicationic nature, electrostatic interactions.(367–374, 376)
One particularly interesting diamidine with antimicrobial activity developed from these studies is a dibenzimidamide, dubbed DB75 (Figure 12B).(377, 378) A SAR study of DB75 revealed simple substitution of one of its phenyl rings with benzimidazole (DB293; Figure 12B) afforded affinity for DNA sequences with a single GC base pair.(379, 380) Thermal melting, surface plasmon resonance, and NMR spectral studies revealed that DB293 binds a 5′-ATGA-3′ oligomer as a stacked dimer, suggesting recognition of both strands containing the GC base pair.(379, 380)DB293 does retain its affinity for AT pairs in classic minor groove binding mode.(379, 380) These studies in conjunction with structural studies of DB75 and its analogs to AT-rich DNA by the Niedle group(358) provided means to rationally design diamidines that selectively recognize stretches of GC base pairs. Such compounds must exploit the differences in the minor grooves of AT and GC base pairs, particularly that the minor grooves of GC pairs are wider, have decreased electronegative potential, and present guanosine’s exocyclic amine. DB1242 (Figure 12B) was the first nonpolyamide compound identified to bind selectively to GC base pairs and binds as a highly cooperative, stacked dimer.(381) Molecular modeling studies revealed that the complex is driven primarily by formation of hydrogen bonds.(381) More recently, a preorganized N-methylbenzimidazole thiophene module was developed to bind GC pairs.(382) Armed with diamidines selective for both AT and GC base pairs, the modules have been linked together to target longer stretches of DNA in a sequence-selective manner (RT533 and DB2232 shown in Figure 12C, for example), and studies are ongoing to determine the proper linker length between binding modules.(382–386)
3.1.4 Targeting DNA G-Quadruplexes
In addition to duplexes, DNA can form other secondary structures, such as Z-DNA, Holliday junctions, triplexes, and G-quadruplexes.(387, 388) Among them, G-quadruplexes have emerged as targets of therapeutics.(389–392) G-quadruplexes, also called G4 structures, are four-stranded structures formed by four guanine repeats through Hoogsteen hydrogen bonds in the presence of monovalent cations such as Na+ or K+ (Figure 13A).(393–395) DNA G-quadruplexes are classified as antiparallel, parallel, or mixed parallel-antiparallel form comprised of three G-tetrads (Figure 13B).(396–398) The variety of G-quadruplex structures results from the strand direction and the orientation of the external loop configuration.(396, 398, 399)
Figure 13.

DNA G-quadruplex binding ligands. (A) Structure of DNA G-quadruplex. (B) Different DNA strand directions, antiparallel, mixed-type, and parallel, result in different G-quadruplex structures. (C) Structure of BSU1051 which targets human telomeric G-quadruplex. (D) Structure of TMPyP4 which targets the promoter region of c-Myc. (E) Structure of PDS which targets telomeric G-quadruplex and proto-oncogene tyrosine-protein kinase Src. (F) Structure of quarfloxin which targets the rDNA G-quadruplex and inhibits Pol I transcription.
The therapeutic potential of targeting G-quadruplexes was first realized when it was discovered that human telomeres are comprised of a guanine-rich sequence TTAGGG(3–7) that can form a G-quadruplex and inhibit telomerase activity.(400–403) Thus, the human telomeric G-quadruplex is a promising target for an anticancer strategy as human telomerase is overexpressed in a variety of cancers. In general, most ligands that interact with G-quadruplexes do so via end-stacking binding modes including π–π interactions and electrostatic interactions with the phosphate backbone of the loop, despite the structural differences described above.
In 1997, Neidle and Hurley first used a flat aromatic small molecule, 2,6-diamidoanthraquinone (BSU1051) (Figure 13C), as a ligand to target the human telomeric G-quadruplex.(404) This anthraquinone derivative recognized the G-quadruplex structural motif and inhibited extension by human telomerase.
In addition to telomeres, DNA G-quadruplexes are also found in gene promoters. Ineed, Hurley and co-workers demonstrated that G-quadruplexes can be stabilized by small molecules and opened up the possibility of targeting other G-quadruplexes in the human genome to affect gene expression. In particular, they discovered that a G-quadruplex motif within the promoter region of the oncogene c-Myc (overexpressed in up to 80% of solid tumors) can be stabilized with a porphyrin derivative, TMPYP4 (Figure 13D), thereby suppressing the downstream expression of c-Myc.(405) This early work showed that G-quadruplexes can be targeted with small molecules. Since these initial discoveries, many novel G-quadruplex ligands have been designed to target both human telomere and oncogene promoters.(390, 406–411) Balasubramanian and colleagues developed a small molecule, PDS (Pyridostatin)(412) (Figure 13E), that not only binds to human telomeric G-quadruplex and disrupts telomere binding but also induces DNA damage in cancer cells.(412, 413) Chromatin immunoprecipitation (ChIP)-seq of the DNA damage marker γ-H2AX was performed to map PDS-induced DNA damage sites in the genome.(414) This study found that there are more than 20 independent G-quadruplex motifs targeted by PDS and identified that proto-oncogene tyrosine-protein kinase Src is the predominant target of PDS.
Importantly, the G-quadruplex ligand Quarfloxin (CX-3543) entered phase II clinical trials for treatment of carcinoid and neuroendocrine tumors in 2008 (Figure 13F).(415) Quarfloxin was originally derived from fluoroquinophenoxazines, which are dual-targeting topoisomerase II inhibitors and G-quadruplex binders.(416) Cylene Pharmaceuticals optimized this lead compound to yield Quarfloxin, a selective G-quadruplex binder with no topoisomerase II inhibitory activity. Quarfloxin not only targets the rDNA G-quadruplex and inhibits Pol I transcription but also disrupts complex formation between rDNA G-quadruplex and nucleolin, leading to relocalization of nucleolin to the nucleoplasm and induction apoptosis in cancer cells.(416)
Recent Next-Gen sequencing and bioinformatics analyses has identified over 700000 potential G-quadruplex-forming sequences in the human genome.(391, 417–420) Many of these G-quadruplexes are located on oncogene promoters, such as c-KIT,(421) K-Ras,(422) Bcl-2,(423)VEGF,(424) hTERT,(425) and WNT1,(426) where binding to these promoters could suppress transcriptional activity. Some potential G-quadruplex forming sequences are noncanonical, such as two G-tetrads, bulge, and long loop fashion (>7 nt), implying that diverse G-rich motifs can form G-quadruplexes.(420) G-quadruplexes have also become attractive targets for drug targeting human pathogens,(427) such as neisseria gonorrheae(428) and HIV.(429)
3.2 Small Molecules Targeting RNA
Although antisense oligonucleotides have been used to target successfully unstructured regions of RNA via Watson–Crick base-pairing(46, 71, 430) and Dervan polyamides have been used to successfully target specific DNA sequences, sequence-based design of small molecules that target structured regions of RNA is still in its infancy. Historically, small molecules have been used to target RNAs with complex tertiary folds such as the bacterial ribosome(431–433) or bacterial riboswitches.(434) In the case of bacterial riboswitches, small molecule ligands have been designed by mimicking the structure of the endogenous ligand. Targeting other cellular RNAs, whether mRNAs or noncoding RNAs, has been much more difficult as they do not adopt significant tertiary structure, comprise very little of the total cellular content,(435) and do not have endogenous ligands upon which to base drug design.
Drugging RNAs, however, could be highly advantageous as RNA often lies upstream of a defective protein in disease pathomechanisms. Further, unlike DNA, RNA folds into diverse structures composed of base pairs and noncanonically paired regions such as hairpins, internal loops, and bulges (Figure 14). Targeting structure, not sequence, could improve the selectivity of small molecules and combat off-target effects. Importantly, secondary structure can be annotated and predicted from the sequence of an RNA.(436–438) This work was largely started by studies on the thermodynamic stability of RNA base pairs and of noncanonically paired regions by Doty, Tinoco, Turner, and others.(439–442) The resulting data set of free-energy increments of RNA pairs was used by Zuker to construct algorithms to predict RNA structure from sequence.(443)Subsequent work by Turner and Zuker improved parameters for RNA secondary structure prediction,(444, 445) but it became clear that prediction alone would not suffice to deduce the structure of some RNAs, particularly those >400 nucleotides. Free energy minimization and alignment protocols were developed along with the ability to incorporate chemical modification and other experimental parameters to restrain folding.(446) These modification restraints can be obtained in vitro with reagents originally used for Maxam–Gilbert sequencing(447, 448) or by selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE).(449) As RNA folding in vivo can be very different than in vitro, secondary structure mapping methods have been developed in cells using DMS(437, 450–454) and various SHAPE reagents.(455, 456)
Figure 14.

RNA is a single-stranded biomolecule, the structure of which can be predicted accurately from sequence. RNA adopts conformations that include Watson—Crick base pairs, internal loops, bulges, multibranch loops, and hairpin loops.
Notably, some small molecules that target RNA have been identified without structural information by high-throughput screening, including drugs developed by PTC Therapeutics, Roche, and Novartis to target muscular dystrophy and spinal muscular atrophy.(457–459) Below we discuss approaches that have been developed to target RNA from sequence using small molecules.
3.2.1 Methods to Target RNA from Sequence
Given the ability to predict secondary structure from sequence, Disney and colleagues envisioned that small molecules could be designed to drug RNA if information was known about: (i) a small molecule’s binding preferences in terms of the secondary structures it selectively recognizes and (ii) the presence of those secondary structural elements, or motifs, in disease-causing RNA. To target the myriad of disease-causing RNAs, a large data set of small molecule binding preferences, with broad chemical diversity, is required. An approach called two-dimensional combinatorial screening (2DCS) was developed to identify ligands that bind to RNA secondary structural motifs with high affinity and selectivity in a high-throughput manner.(460, 461) The 2DCS platform uses agarose-coated glass slides to create small molecule microarrays. Agarose can be easily modified to display various functional groups that can be used to site-specifically immobilize small molecules (provided they have an orthogonal chemical handle). Importantly, the positions where small molecules have been immobilized can be easily removed by manual excision. The small molecule microarray is incubated with a radiolabeled RNA library with a randomized region displaying a discrete secondary structural element under conditions of high oligonucleotide stringency. That is, oligonucleotides that mimic regions common to all library members are included at high concentrations to restrict small molecule binding to the randomized region. The randomized region is kept intentionally small (1–7 base pairs) so that it is likely that the selected RNA structures are present in the transcriptome. The arrays are imaged, bound RNA is excised, and the bound RNAs are identified by RT-PCR and subcloning or RNA-seq.(462)
To validate 2DCS, the binding preferences of aminoglycoside derivatives that were acylated at their 6′ positions with 5′-hexynoic acid, particularly neamine and kanamycin A, were studied.(463)Aminoglycosides are well-known RNA binders; however, acylation of the 6′ amine ablates binding to the bacterial A-site, a main mode of aminoglycoside resistance.(464) The neamine and kanamycin A derivatives were site-specifically immobilized on an agarose-coated microarray functionalized with azides via a Huisgen 1,3 dipolar cycloaddition reaction. The small molecule array was then incubated with an RNA library displaying randomized nucleotides in a 3 × 3 nucleotide internal loop pattern. Interestingly, the A-site was not a privileged binding site for the modified aminoglycosides. Rather, 6′-N-5-hexynoate kanamycin A prefers pyrimidine-rich internal loops and A/C internal loops,(463, 465) while 6′-N-5-hexynoate neamine prefers loops G/A internal loops.(466) Indeed, 2DCS has been used to identify the RNA-binding capacity of a variety of small molecules including bis-benzimidazoles,(467) amino-benzimidazoles,(468)aminoglycosides,(460) and others.(461) 2DCS has also been merged with in-solution high-throughput screening methods to study diverse chemical space, and it was determined that indole, 2-phenyl indole, 2-phenyl benzimidazole, and pyridinium chemotypes were privileged for binding RNA.(461)
To score the affinity and relative selectivity of selected RNA motif–small molecule interactions from 2DCS studies, a statistical method named structure–activity relationships through sequencing (StARTS) was developed.(466, 468) StARTS identifies privileged trends within RNAs that bind a given small molecule (such as G/A internal loops or GC steps) by completing a pooled population comparison. In brief, a pooled population comparison compares the frequency of occurrence of a trend in selected RNAs to the frequency of occurrence of the same trend in all RNAs present in the library. Statistical significance is dependent upon difference in this frequency and the population sizes of the selected RNAs and the starting RNA library. This analysis affords Zobs, a measure of statistical significance that can have positive (contributes to binding affinity) or negative (detracts from binding) values. An RNA motif may have more than one statistically significant feature; thus a sum of all Zobs, or ΣZobs, is computed. Importantly, the affinity of the small molecule for an RNA scales with ΣZobs.(466, 468) A plot of ΣZobs as a function of affinity can be fit to define a scoring function for the entire RNA library. Normalization of ΣZobs values across a given selection affords a Fitness Score. Comparing the Fitness Score of an RNA motif for various small molecules provides insight into the selectivity. A variant of StARTS, dubbed high-throughput structure–activity relationships through sequencing (HiT-StARTS) was recently developed to complete analyses directly from RNA-seq data.(462) In HiT-StARTS, both the starting library and the RNAs selected to bind a small molecule are subjected to RNA-seq. Completing RNA-seq on the starting library is imperative to account for biases that occur during transcription and sequencing. Rather than identifying trends or submotifs, HiT-StARTS analyzes the RNA sequence directly.
The selected RNA motif–small molecule interactions and their associated meta data such as Fitness Scores and measured dissociation constants comprise a database, called Inforna, that can inform drug design for disease-causing RNAs.(469, 470) Using Inforna, the secondary structure of a target RNA, whether determined by experiment or computation, can be compared to a database of privileged RNA motif–small molecule interactions to identify lead compounds (Figure 15). Importantly, Inforna identifies the preferred RNA target for a small molecule. Thus, this strategy is not focused on a specific RNA target but rather is “target agnostic”.
Figure 15.
Inforna facilitates the design of lead small molecules targeting a disease-causing RNA, Small molecules are identified by comparison of the secondary structural motifs in an RNA target to an annotated database of RNA motif-small molecule interactions.
3.2.2 Small Molecules Targeting miRNA Precursors
The Inforna approach was validated by targeting disease-associated miRNA precursors.(469) As previously discussed, miRNAs have diverse roles in biology and their dysregulation can lead to cancer, immune diseases, and a variety of other disorders. The secondary structures of miRNAs can be easily predicted from sequence, and their Drosha and Dicer processing sites, which can also restrain secondary structure prediction, can be easily inferred from RNA-seq studies.(471) It was hypothesized that a small molecule that binds to a Drosha or Dicer processing site could affect cellular function by inhibiting miRNA biogenesis. Using Inforna, the secondary structures of all human miRNAs were compared to the database of privileged RNA motif-small molecule interactions. These lead interactions were further refined by restricting them to (i) Drosha and Dicer processing sites and (ii) disease-causing miRNAs. This analysis through Inforna identified 29 lead interactions that could potentially inhibit the biogenesis of disease-causing miRNAs. The levels of 12 of the desired miRNAs were reduced, affording a hit rate of 41%.(469)
The most avid interaction identified from these studies was between a substituted benzimidazole (Figure 16) and the Drosha site in miR-96 hairpin precursor.(469) The small molecule contains bulky t-butyl groups located on the benzene ring that ablate binding to the DNA minor groove. The Drosha site of the miR-96 precursor was predicted via StARTS and confirmed via experiment to be the highest affinity RNA motif for the small molecule from the entire 4096-member RNA motif library from which it was selected. As miR-96 is upregulated in triple negative breast cancer and silences Forkhead Box O1 (FOXO1), a transcription factor involved in apoptosis, it is an attractive target to potentially treat breast cancer.(472)
Figure 16.

Lead optimization of a lead compound that targets the Drosha site of miR-96 to afford Targaprimir-96, a dimeric small molecule that targets the Drosha site and an adjacent 1 × 1 GG internal loop.
Indeed, the small molecule inhibited the production of mature miR-96 at 10 μM but did not affect levels of other miRNAs that are produced from the same transcript (pri-miRNA) as miR-96.(469)The compound also derepressed the expression of FOXO1 and triggered apoptosis only in breast cancer cells. To probe if apoptosis was triggered via the miR-96-FOXO1 circuit, an siRNA was used to ablate FOXO1, and the compound’s ability to trigger apoptosis was reduced by 75%.(469)The selectivity of the interaction between the benzimidazole and the Drosha site of miR-96 was probed by measuring its effect on the levels of all miRNAs with sufficient expression in MCF-7 cells by RT-qPCR. The only miRNA that was significantly affected upon compound treatment was miR-96. The small molecule’s selectivity rivaled or exceeded that of an antagomiR against miR-96.(469)
Although the initial study with the miR-96-targeting small molecule showed that small molecules could selectively inhibit the biogenesis of a single miRNA, the low micromolar activity in cells was not sufficiently potent to translate to in vivo models. Thus, a method was sought to lead optimize the original small molecule to increase potency and selectivity. Inforna was used to find a small molecule that would bind to a motif in the miR-96 hairpin precursor that was adjacent to the Drosha site to inform design of a dimeric small molecule. Inforna identified a bis-benzimidazole with high affinity for a 1 × 1 nucleotide GG internal loop adjacent to the Drosha site in the miR-96 hairpin precursor (Figure 16).(473) Using rules to design a linker that spans the distance of two base pairs,(474) the compound Targaprimir-96 was developed. This dimeric compound had nanomolar affinity for the miRNA hairpin precursor while binding was ablated when the target motifs were mutated to base pairs. The small molecule is also RNA-selective as saturable binding to an AT-rich DNA was not observed despite the fact that the bis-benzimidazole is a known DNA binder.
In cells, Targaprimir-96 decreased mature miR-96 levels at concentrations as low as 30 nM. The small molecule also derepressed FOXO1 expression, enhancing protein levels 2-fold and inducing apoptosis with 50 nM treatment of Targaprimir-96. Because of its nanomolar potency, Targaprimir-96 was further investigated in an in vivo study. The concentration of Targaprimir-96 in plasma of mice after intraperitoneal (i.p.) injection (2 or 7 mg/kg of drug) was measured to determine a sufficient dose. These studies showed that the concentration of Targaprimir-96 in serum is greater than 1 μM 48 h post IP injection of either dose, well above the concentration needed to trigger apoptosis in cell culture. Targaprimir-96 inhibited tumor growth in a xenograft mouse model of triple negative breast cancer upon i.p. injection of 10 mg/kg. Analysis of resected tumors showed a decrease in the amount of mature miR-96 and a boost in FOXO1 protein. These studies show that Inforna can be used to lead optimize small molecules to create selective dimeric compounds with nanomolar potency. Collectively, these studies show that small molecules targeting miRNAs can inhibit disease biology in cellular and animal models.
Another miRNA target identified by Inforna was miR-10b.(475) miR-10b overexpression has been implicated in numerous cancers and contributes to invasion and metastasis.(178) 2DCS identified that guanidinylated neomycin B (G-Neo-B) binds to the 5′A/3′A internal loop in the Drosha processing site of the miR-10b precursor.(475) G-Neo-B bound to this loop with a Kd of 417 ± 60 nM, inhibited processing of the mature miRNA in cells at 100 μM as measured by RT-qPCR, and boosted protein levels of the downstream target Homeobox D10 (HOXD10). Importantly, G-Neo-B did not affect the biogenesis of a miRNA to which the compound was not predicted to bind.
Another disease-causing RNA that was identified as a target by Inforna is miR-525.(469) MiR-525 is overexpressed in 60% of liver cancer tissues, indicates poor prognosis, and is associated with invasive properties in hepatocellular carcinoma (HCC) cells.(476) 5″-Azido-neomycin B (Neo-N3) was predicted to bind to the Drosha processing site of miR-525 and was found to reduce levels of mature miRNA at concentrations as low as 6.25 μM.(477) By inhibiting miR-525, Neo-N3derepressed miR-525’s downstream target zinc finger protein 395 (ZNF395) and inhibited invasion. Interestingly, the parent aminoglycoside, neomycin was also able to inhibit miR-525 biogenesis in HCC cells. Since neomycin B is already used clinically to treat hepatic encephalopathy,(478) this study suggests that neomycin B or Neo-N3 could be used at low doses to work synergistically with HCC therapeutics.
While the two compounds mentioned above were both aminoglycoside derivatives, other small molecule scaffolds have been identified for selective binding to RNA motifs. Two potent small molecules have recently been identified to modulate miRNAs that are upregulated under hypoxic stress,(479, 480) a characteristic of many advanced tumors. In response to hypoxia, miR-544 upregulates ataxia telangiectasia mutated (ATM) serine/threonine kinase (a direct modulator of hypoxia-inducible factor 1α [HIF-1α]) and downregulates mechanistic target of rapamycin (mTOR), thus modulating tumor cell growth.(481) Inforna identified two heterocyclic compounds that reduced mature miR-544 levels in cells at a concentration of 20 nM.(479) The best compound, a substituted naphthyridine, decreased ATM and HIF-1α transcript levels and increased mTOR transcript levels, resulting in inhibition of proliferation and induction of apoptosis in hypoxic cells only. NOD/Scid mice were treated with a single intraperitoneal injection of the small molecule 24 h after implantation of MDA-MB-231 cells stably expressing luciferase. Three weeks post treatment, tumors were resected, revealing a dramatic decrease in tumor size. Importantly, the resected tumors exhibited reduced levels of mature miR-544 and HIF-1α, and increased levels of mTOR as analyzed by RT-qPCR.(479)
The second small molecule that was identified to modulate a hypoxia-upregulated miRNA is a bis-benzimidazole targeting miR-210.(480) This miRNA directly represses the glycerol-3-phosphate dehydrogenase 1-like (GPD1L) enzyme, which contributes to the suppression of prolyl hydroxylase (PHD) activity.(482) PHD is responsible for the degradation of HIF-1α; as a result of miR-210 affecting GPD1L, HIF-1α is able to heterodimerize with hypoxia-inducible factor 1-beta (HIF-1β) and activate hypoxic responses that contribute to a cancerous phenotype.(483, 484) By binding to the 5′ACU3′/3′UCA5′ loop in miR-210s Dicer site, the small molecule, Targapremir-210 decreased mature miR-210 levels in MDA-MB-231 cells cultured under hypoxic conditions with an IC50 of 200 nM. Furthermore, treatment with Targapremir-210 derepressed GPD1L mRNA levels and decreased HIF-1α mRNA levels. This reversion toward a normoxic state resulted in apoptosis of hypoxic cells treated with Targapremir-210. Importantly, miR-210 is the only hypoxic-associated miRNA affected by Targapremir-210.
One of the many advantages of Inforna is that each small molecule has a Fitness Score assigned for each targetable RNA motif. Thus, Inforna has the ability to predict other miRNAs that may be affected by a small molecule. The 2DCS selection of Targapremir-210 revealed that it binds other RNA motifs; however, these motifs have lower Fitness Scores than the binding site in pre-miR-210. Interestingly, the mature level of miRNAs containing lower Fitness Score motifs were not affected by Targapremir-210 treatment. Like the small molecule targeting miR-544, TargapremiR-210 inhibited miR-210 levels in a mouse xenograft model of hypoxic triple negative breast cancer, decreasing tumor growth, increasing GPD1L mRNA levels, and decreasing HIF-1α mRNA levels, as compared to untreated tumors. Both of these studies reveal that Inforna can identify small molecules that inhibit miRNAs associated with complex hypoxic pathways and can affect these pathways in mouse models of disease.
Another miRNA that was targeted by a small molecule identified by Inforna was miR-18a. The miRNA is part of the oncogenic cluster miR-17/92, which is comprised of miR-17, −18a, −20a, −19a, −19b-1, and −92a-1. Interestingly, miR-17/92 was the first cluster identified to be involved in oncogenesis.(485) Indeed, miR-18a was previously indicated in multiple cancers, including prostate cancer.(486) Targapremir-18a, a benzimidazole derivative, was identified to bind 5′G_U/3′CUA, the Dicer processing site common to pre-miR-17, −18a, and −20a.(462)TargapremiR-18a inhibited Dicer processing in vitro with an IC50 of ~10 μM. There was ~50% knockdown of mature miR-17, −18a, and -20a upon treatment of DU145 cells (prostate cancer) with 10 μM Targapremir-18a. Interestingly, as expected, Targapremir-18a did not affect the levels of miR-19b-1 and -92a-1 as they do not have predicted binding sites. The small molecule also reverses downstream effects. MiR-18a represses the levels of serine/threonine protein kinase (STK4), a tumor suppressor.(486) Treatment of DU-145 cells with Targapremir-18a derepressed STK4 and induced apoptosis.
The selectivity of Targapremir-18a was assessed via RT-qPCR of 33 miRNAs with potential binding sites for Targapremir-18a as predicted by Inforna. No significant effect was observed on any of the miRNAs studied. Taken together, these studies identified factors that contribute to bioactivity of small molecules that target miRNAs, including miRNA expression level and the location of the small molecule binding site, which must be in a Drosha or Dicer processing site.
3.2.3 Small Molecules Targeting RNA Repeat Expansions
Inforna has not only been used to target precursor miRNAs but also has been used to identify compounds that bind expanded repeating RNAs [denoted by a sequence followed by “exp” or r(sequence)exp]. Expanded RNA repeats are associated with many diseases including myotonic dystrophy types 1 and 2 (DM1 and DM2, respectively),(487, 488) C9ORF72 amyotrophic lateral sclerosis and frontal temporal dementia (c9ALS/FTD),(489, 490) Huntington’s disease (HD),(27)fragile X-associated tremor ataxia syndrome (FXTAS),(491) and spinocerebellar ataxia 10 (SCA10).(492) Repeating transcripts cause disease by a variety of mechanisms including sequestration of proteins that regulate alternative splicing (DM1, DM2, FXTAS, and SCA10) or translation of a toxic protein (HD, c9ALS/FTD, and FXTAS) (Figure 17).
Figure 17.

Repeating RNA transcripts contribute to disease by multiple mechanisms. Repeating transcripts often fold into hairpins that display internal loops. The loops sequester RNA-binding proteins, causing disease via an RNA gain-of-function mechanism. Repeating transcripts can also undergo repeat-associated non-ATG (RAN) translation, generating toxic proteins.
Using Inforna, many small molecules have been developed to target these RNA repeating transcripts.(493, 494) The potency of these compounds has been increased by using the same multivalent approach described above for the design of Targaprimir-96.(474, 494–499) An approach to create multivalent compounds in cellulis by using a proximity induced click reaction with the RNA repeat acting as a catalyst has also been developed.(497, 500)
A noncoding repeat expansion, r(CGG)exp, located in the 5′ UTR of the fragile X mental retardation 1 gene (FMR1) causes FXTAS, a neurological condition characterized by difficulties in motor function and cognition.(491) The RNA folds into a hairpin structure that displays GG internal loops that bind and sequester proteins, including DGCR8, Src-Associated substrate during mitosis of 68 kDa (Sam68), and heterogeneous nuclear ribonucleoprotein (hnRNP).(491) Sequestration of these proteins leads to pre-mRNA splicing defects and the formation of nuclear foci. In FXTAS, r(CGG)exp can also cause disease through repeat associated non-ATG (RAN) translation, where a repeating RNA is translated without the use of an AUG start codon and produces toxic polymeric proteins.(501, 502) Two small molecules that target r(CGG)exp have been identified by Inforna. One is a modularly assembled small molecule displaying two bis-benzimidazole RNA binding motifs, 2H-5.(503) The other is a hydroxyellipticine derivative (1a) that was later used to create a dimeric compound (2HE-5NMe).(494, 496) All three compounds improve pre-mRNA splicing defects and reduce the number of nuclear foci in a FXTAS cellular model. 2HE-5NMe completely restored Sam68-dependent pre-mRNA splicing patterns to wild-type levels and inhibited the production of RAN proteins without affecting canonical translation. Its inhibitory effect on RAN translation was traced to reducing the number of polysomes loaded on the repeats.(494, 496)Interestingly, oligonucleotides that target r(CGG)exp in cells do affect downstream ORF translation,(503) suggesting that a small molecule approach to inhibiting RAN translation could have functional benefits. Collectively, these studies showed that small molecules designed to target r(CGG)exp can affect two different modes of RNA repeat toxicity, gain-of-function, and RAN translation.
An expanded hexanucleotide repeat of G4C2exp in the C9ORF72 gene is the most common genetic cause of ALS/FTD.(489, 490) The RNA repeat expansion causes toxicity through the formation of nuclear foci caused by protein sequestration (causing alternative pre-mRNA splicing(504) and nuclear trafficking defects(505, 506)) and toxic “c9RAN proteins” generated by RAN translation that form neuronal inclusions throughout the central nervous system.(507, 508)Interestingly, r(CGG)exp and the repeat expansion that causes c9ALS/FTD, r(G4C2)exp, fold into similar structures, displaying 1 × 1 GG loops. Therefore, it was studied whether 1a and structurally similar compounds could inhibit RAN translation of r(G4C2)exp in a cellular model. Three small molecules, including 1a, inhibited RAN translation at low micromolar concentrations and two decreased the number of nuclear foci. Importantly, 1a ameliorates both defects in induced neurons (iNeurons) derived from c9ALS/FTD patients.
SCA10 is caused by an RNA gain-of-function mechanism in which r(AUUCU)exp, located in intron 9 of ataxin 10 mRNA (ATX10), binds and sequesters proteins such as heterogeneous nuclear ribonucleoprotein K (hnRNP K), causing formation of nuclear foci, translocation of protein kinase C-δ (PKCδ) in mitochondria resulting in dysfunction, and activation of caspase 3 which induces apotosis.(492) The SCA10 repeat folds into an array of 3 × 3 pyrimidine-rich loops closed by AU pairs (5′UCU3′/3′UCU5′).(509) The small molecule designed to target this RNA is unique in that it binds the AU base pairs rather than the loop region.(499) The RNA-binding module was identified by screening for selective binding of AU or GC base pairs using a fluorescence-based binding assay. The screen identified two small molecules that bound AU pairs more selectively than GC pairs as determined by measuring an EC50 for each RNA. Binding to the AU pairs in the r(AUUCU)exp was confirmed using a nuclease protection assay. The more selective small molecule was appended with an azide to make a modularly assembled dimeric small molecule, 2 AU-2.(499) In SCA10 patient-derived fibroblasts, 2 AU-2 reduced caspase-3 activity to healthy levels at 50 and 100 nM. 2 AU-2 also reduced mitochondrial abundance of PKCδ and reduced the number of nuclear foci at 50 nM.(499) This study showed that small molecules can be identified to selectively target RNA base pairs, and this information can be used to target disease-causing RNAs rich in these base-paired sequences.
Two other RNA repeats that have been potently targeted are r(CUG)exp and r(CCUG)exp, the RNAs causative of DM1 and DM2, respectively.(487, 488) Both repeats cause disease via a gain-of-function mechanism in which the RNA repeat folds into a hairpin structure and binds and sequesters proteins. Some of these proteins, in particular muscleblind-like protein 1 (MBNL1), regulate alternative pre-mRNA splicing, and splicing defects are thus a hallmark of the myotonic dystrophies.(488, 510, 511) Sequestration of MBNL1 also leads to the formation of nuclear foci and decreased nucleo-cytoplasmic transport of the message.(512–514) DM1 is caused by an expanded r(CUG) repeat in the 3′ UTR of the dystrophia myotonica protein kinase gene (DMPK),(488) while DM2 is caused by r(CCUG)exp in intron 1 of the zinc finger 9 (ZNF9) mRNA.(487) A small molecule aminoglycoside derivative, K-Ak, improved DM2-associated splicing defects in a cellular model.(495) Importantly, since this RNA-binding module was equipped with an alkyne handle, it could be used to make a modularly assembled dimer and trimer displayed on a peptoid backbone (2K-4 or 3K-4).(495) The trimeric compound, 3K-4, improved DM2-associated splicing defects at concentrations as low as 2.5 μM.(495)
Over 20 monomeric small molecules that target r(CUG)exp have been developed using a variety of methods, including the use of Inforna and rationally designed small molecules.(497, 498, 515–517)The DM1 repeat, r(CUG)exp, has been most potently targeted using the dimeric compound 2H–K4NMeS.(497) 2H–K4NMeS displays two H RNA-binding module that were identified by Inforna to bind 1 × 1 UU internal loops on an N-methyl peptide backbone. The dimer improved DM1-associated splicing defects at concentrations as low as 100 nM in DM1 patient-derived fibroblasts, disrupted nuclear foci, and increased nucleocytoplasmic transport of DMPK message. Collectively, we have found that multivalent compounds most potently and selectively target RNA repeat expansions.
In some cases, the cellular potency of multivalent compounds decreases as a function of valency as their higher molecular weights decrease cellular permeability. Therefore, an approach to synthesize multivalent compounds on-site, catalyzed by the disease-causing RNA in a cell was developed. To do so, RNA-binding modules were appended with alkyne and azide handles that, upon binding to the target RNA, would be in close enough proximity to undergo a Huisgen 1,3-dipolar cycloaddition reaction. In this way, the RNA repeat would act as a catalyst for a reaction of two otherwise unreactive groups to form a stable triazole.(518) This approach had previously been used in vitro to target acetylcholine esterase and the DNA minor groove but had not been translated to a cellular system.(518–520) RNA repeat expansions provide an ideal target for this approach as there are many binding sites and thus many possibilities to create potent oligomers.
This approach was first employed against r(CCUG)exp using the small molecule, K-Ak.(500) A structural study of r(CCUG) repeats enabled modeling of the binding of dimeric compound 2K-4. Analysis of this model showed that an azido group at the 6″ position (N3–K) and an alkyne at the 6′ position (K-Ak) could be within close enough proximity to react upon binding to adjacent 2 × 2 nucleotide internal loops.(500) Thus, N3-K and K-Ak could react to form a dimer, and a compound appended with both reactive modules (N3-K-Ak) could react to form oligomers (Figure 18).
Figure 18.

Small molecules that bind to RNA repeats can be appended with azide and alkyne functional groups which, when bound to the RNA repeat, are in close enough proximity to react and form potent oligomers in disease-aflfected cells.
Previous studies have shown that electron-deficient alkyne reaction rates are faster than their alkyl counterparts(520) and therefore may be advantageous for the in situ, RNA-catalyzed click approach. Therefore, an alkyne-deficient kanamycin (K-Aak) and its dually functionalized counterpart (N3-K-Aak) were synthesized. Indeed, when incubated with r(CCUG)12, but not in its absence, K-Ak + N3-K reacted to afford a dimer while N3-K-Ak and N3-K-Aak formed dimers and other oligomers as determined by mass spectrometry.(500) The RNA-catalyzed click reaction was then tested in cellulis using a cellular model in which r(CCUG)300 is expressed. Cells were cotreated with N3-K and either N3-K-Ak or N3-K-Aak. N3-K was used as a terminator for the templated click reaction to limit the molecular weights of the products to allow for mass spectral analysis. Analysis of reaction products purified from cell lysates indicated that dimeric and trimeric reaction products were observed in cells treated with the compound and that oligomerization was not observed in cells that did not express r(CCUG)300.(500) N3-K-Aak improved DM2-associated splicing defects when cells were treated with as little as 100 nM compound, which corresponds to a 100-fold improvement over the modularly assembled dimer (2K-4) and a 1000-fold improvement over the corresponding monomers (K-Ak and N3-K).
To further confirm that r(CCUG)300 was the catalyst for oligomerization in cells, an approach called Chemical Reactivity and Binding Isolated by Pull-down (ChemReactBIP) was developed (Figure 19).(500) In this method, a kanamycin derivative containing a biotin module and an azide module was used to terminate the polymerization reaction in cells and to isolate the oligomerized compounds and their bound cellular targets using capture with streptavidin beads. In agreement with the mass spectral data from cell lysates, oligomerized products were only formed in cells expressing r(CCUG)300. ChemReactBIP also allowed for the identification of the RNA targets of the templated reaction by using RT-qPCR. Importantly, the amount of r(CCUG)300 pulled down using ChemReactBIP increased as a function of potency, with N3-K-Aak being the most potent.
Figure 19.

Chemical reactivity and binding isolated by pull-down (ChemReactBIP) is a method to confirm the reaction “clickable” small molecules and to identify the RNA that served as a catalyst for the reaction. ChemReactBIP uses a small molecule appended with a biotin tag to terminate the click reaction and to allow purification of both reaction products and bound RNAs with streptavidin beads.
This in situ click chemistry approach has also been used to create potent modulators of r(CUG)exp. In this case, 2H–K4NMeS, was appended with azide and alkyne handles at the appropriate distance, as determined by in vitro experiments.(497) These compounds (2H-K4NMeS-Aak and N3-2H-K4NMeS) formed a tetramer in vitro only in the presence of r(CUG)exp as determined by mass spectral analysis. The dual functionalized compound, N3-2HK4NMeS-Aak, improved DM1-associated splicing defects at 100 pM with an IC50 of about 10 nM,(497) a 100-fold more potent than the unreactive, parent dimer. ChemReactBIP confirmed the presence of oligomerized compounds due to reaction of N3-2HK4NMeS-Aak in DM1 patient-derived fibroblasts, which were absent from healthy fibroblasts. Further, analysis of the RNAs isolated from ChemReactBIP studies in DM1 patient-derived fibroblasts showed that the DMPK mRNA, not other transcripts containing short r(CUG) repeats of nonpathogenic length, was the cellular catalyst for this oligomerization.(497)
Taken together, these studies show that RNA repeat expansions can catalyze the synthesis of potent, multivalent compounds in cells. The ability to synthesize oligomers in cells from monomeric subunits offers the ability to more potently and selectively target a desired RNA without increasing the molecular weight of the compound and potentially affecting cellular permeability. This approach could be especially beneficial when developing compounds to target RNA repeat expansions that cause brain dysfunction by increasing their potential to cross the blood-brain barrier.
3.2.4 Structure-Based Design of Small Molecules that Target RNA
Structure-based design has emerged as another method of target disease-causing RNA. These small molecules are not designed in a target agnostic fashion but rather focus on modulating specific targets through a known secondary or tertiary structure or by mimicking a known ligand. Indeed structure-based design has enabled the development of bioactive small molecules that target viral RNAs, miRNAs, RNA repeats, and others. Druglike properties of these small molecules targeting RNA, including the ones discussed above, have previously been analyzed and were shown to be similar to known drugs.(521)
Various groups have employed structure-based design to target HIV Tat-transactivating response element (TAR) RNA, enabled by NMR spectral studies of the RNA alone and complexed with trans-activator of transcription (tat) protein.(522) The structure of the Tat-TAR complex enabled design of synthetic peptides that bound TAR RNA, inhibited the Tat-TAR RNA interaction in vitro, and inhibited viral replication in primary human lymphocytes.(523–525) Identification of other small molecules that bind TAR RNA has employed in silico docking. Al-Hashimi and co-workers docked 51000 small molecules into a dynamic ensemble of TAR RNA structures generated by NMR data and computational molecular dynamics.(526) Six commercially available small molecules were identified to bind TAR RNA and inhibit the Tat-TAR complex. One compound, netilmicin, inhibited HIV replication in cells. James and colleagues also used a four-step docking method to analyze >180000 compounds for binding to the TAR RNA.(527, 528) Eleven of these compounds inhibited the Tat-TAR interaction.(528)
Ligands that bind miRNA precursors and inhibit their processing in cells have been identified by various screening approaches. Through a screen of aminoglycosides, Maiti and colleagues identified that streptomycin decreased levels of oncogenic miR-21 in cells.(529) Varani and colleagues also discovered a cyclic peptide that bound miR-21 and inhibited its biogenesis in cells.(530)
Various small molecules have also been developed to target RNA repeat expansions. As mentioned in section 3.2.3, DM1 is caused by r(CUG)exp. Zimmerman, Baranger, and colleagues used an X-ray crystal structure of a short r(CUG) repeat to design a small molecule that inhibited the r(CUG)exp-MBNL1 complex.(531) The small molecule is comprised of an acridine DNA intercalator and a triaminotriazine unit that recognizes the U–U pairs through Janus-wedge hydrogen bonding. Molecular modeling of this small molecule suggested that the ligand could target CUG as a “stacked intercalator” through minor or major groove interactions.(531)Zimmerman and colleagues have used this small molecule to design other compounds that target r(CUG)exp and improve splicing defects in a DM1 cellular model.(532) Miller and colleagues identified a small molecule through resin-bound dynamic combinatorial screening that improved DM1-associated splicing defects in a mouse model.(533) In another important study, Nakamori and colleagues discovered that erythromycin can inhibit the r(CUG)-MBNL1 complex and that oral dosing can improve DM1-associated splicing defects in a mouse model.(534) Artero and colleagues have also identified a peptide that reduces CUG-induced toxicity in fly and mouse models.(535) Small molecules that target r(CCUG)exp, the RNA repeat causative of DM2, and disrupt r(CCUG)exp-MBNL1 foci in DM2 model cells have also been reported by Zimmerman and colleagues.(536) Importantly, r(G4C2)exp biology has also been manipulated with a small molecule that destabilizes RNA G-quadruplex structures. Pearson and colleagues discovered that the G-quadruplex binder TMPyP4, a cationic porphyrin, bound r(G4C2)8 and inhibited the binding of r(G4C2)exp-binding proteins.(537) Rothstein and colleagues further discovered that TMPyP4 suppressed G4C2-mediated neurodegeneration in a Drosophila model.(538)
Riboswitches are another example of a class of structured RNAs amenable to small molecule structure-based design. First discovered in 2002, riboswitches are conformational switches within mRNAs that regulate gene expression by binding metabolites.(434, 539, 540) Since riboswitches have an endogenous ligand, efforts to target riboswitches with small molecules have involved structural-mimicry in which analogs of the endogenous ligand are probed for binding. Such an approach has afforded novel small molecule inhibitors for the lysine,(541) cyclic diguanylate monophosphate,(542) purine,(543, 544) and glmS(545) riboswitches. Brenk and colleagues used RNA-ligand docking to identify small molecules that bind the purine riboswitch.(546) As riboswitches are primarily found in bacteria, small molecule inhibitors are potential antibacterials.(547)
Other RNA targets that have benefitted from structure-based design include the P2b region in telomerase RNA(547) and the RNA pseudoknot in the −1 ribosomal frameshifting site of severe acute respiratory syndrome (SARS)-coronavirus.(548) The HCV internal ribosome entry site (IRES) RNA(549) and RNA G-quadruplexes have also been targeted with small molecules.(550)Complete reviews of structure-based design of small molecules targeting RNA can be found elsewhere.(290, 292, 551–553)
3.2.5 Small Molecule Target Validation Methods
One major challenge in the field of RNA chemical biology and drug discovery is target validation, particularly in cells. Various methods have been developed to identify compound binding sites in vitro, including the use of Maxam–Gilbert type sequencing reagents such as DMS and kethoxal, as demonstrated by the Noller group to identify binding sites of aminoglycosides within the bacterial ribosome.(554, 555) The positions of modification and protection by small molecules were determined using reverse transcriptase and primer extension, which is inhibited by chemical modification. Nuclease protection assays have also been used as a method of determining binding sites of a small molecule in vitro.(469, 556) In-line probing identifies ligand binding sites by studying changes in cleavage patterns upon incubation with the small moelcule.(557) Identifying the cellular RNA target of a small molecule, however, is more challenging. To overcome this challenge multiple approaches have been developed to measure direct target engagement of a small molecule and an RNA target in cellulis (Figure 20).
Figure 20.

Methods to validate the cellular targets of small molecules. (A) Chemical cross-linking and isolation by pulldown (Chem-CLIP) involves a small molecule appended with a biotin tag and nucleic acid cross-linking agent. The cellular RNA targets are captured with biotin and analyzed using qRT-PCR. (B) Chem-CLIP-Map has been used to confirm the binding site of a small molecule within an RNA by digesting fragments of the RNA with antisense oligonucleotides and RNase H and analyzing the bound fragments using qRT-PCR (C) A cleavage approach can also be used for target validation where a small molecule is attached to bleomycin a5, a natural product that can cleave nucleic acids. RNA target are analyzed by target depletion by qRT-PCR.
One of the first methods to identify the cellular targets of small molecules used a classic pull-down experiment.(558) To pull-down the RNA targets of small molecules, a bioactive compound was appended with a biotin tag. Cellular RNA targets were then pulled down using streptavidin beads and identified by RT-qPCR. This approach has been used to confirm small molecule binding to a desired targeting in cell lysates including the aforementioned small molecules designed to target r(AUUCU)exp and r(CGG)exp.(499)
An in celluis pull-down approach, Chemical Cross-linking and Isolation by Pull-down (Chem-CLIP; Figure 20A) has also been developed.(559) In Chem-CLIP, a bioactive small molecule is appended with the nucleic acid reactive module chlorambucil (CA) and biotin for a purification tag. The CA warhead reacts with the small molecule’s cellular targets, via a proximity-based reaction, which are then isolated using the biotin module and streptavidin beads. The captured targets are then analyzed by RT-qPCR or RNA-seq To control for nonselective effects of the cross-linking warhead, a method called competitive chem-CLIP (C-Chem-CLIP) was developed in which cells are cotreated with the parent small molecule and the small molecule Chem-CLIP probe. In this competitive method, targets of the noncovalent compound are depleted in the pulled-down fraction. Chem-CLIP and C-Chem-CLIP have been used to validate the targets of various small molecules including those that target miR-96, miR-210, and RNA repeat expansions (all those to which Chem-CLIP has been applied).(473, 480, 560)
A recent Chem-CLIP study has revealed important aspects of this target validation method and drugging RNA. The power of Inforna is that it identifies all privileged RNA motifs that bind a given small molecule with high affinity or RNA isoforms. Chem-CLIP was used to study the occupancy of Targapremir-210 for the RNA isoforms predicted by Inforna. These studies showed that cellular occupancy (binding) was governed by affinity of the RNA motif-small molecule complex and the expression level of the RNA in which the motif is found. In particular, Chem-CLIP revealed that four other miRNAs are bound by Targapremir-210 in MDA-MB-231 cells: miR-497, miR-1273c, miR-3174, and miR-107.(480) Thus, the combination of Inforna and Chem-CLIP are effective methods to identify plausible and actual cellular binding sites. Despite binding to five different miRNAs in cells, Targapremir-210 has no statistically significant effect on the expression level of these miRNAs; that is, binding and bioactivity cannot be equated. Thus, additional factors are in play that govern bioactivity. Insight can be gained by the case of miR-497, which contains the exact same motif as the miR-210s Dicer site. The motif is located outside Dicer or Drosha processing sites, however. Cellular activity therefore is influenced by affinity, expression level, and if the small molecule binding site is a functional site in the RNA target. Interestingly, the cross-linking compounds used in Chem-CLIP are often almost 1000-fold more potent than the unreactive parent compound.(497) These results suggest that if reactivity could be controlled to limit the off-target effects of the cross-linking probe, covalent modifiers could be used to more potently target disease-causing RNAs.
Since Chem-CLIP allowed for the pull-down of RNA targets in cells, an approach to determine the site of reaction, Chem-CLIP-Map was also developed (Figure 20B).(560) In Chem-CLIP-Map, cells are treated with a small molecule appended with CA and biotin and total RNA is harvested. Total RNA is then subjected to RNase H cleavage using oligonucleotides complementary to the RNA of interest. The cleaved RNA fragments are captured by streptavidin beads and quantified by RT-qPCR to identify the region(s) that reacted with the compound and hence the binding site. Chem-CLIP-Map has been used to identify compound binding sites in transcripts containing RNA repeat expansions, which confirmed that compound binding is localized to regions containing the repeat and not other segments of the message.(497, 560)
Other covalent methods have been developed to identify the cellular RNA targets of the anticancer drug cisplatin. Using an azide-modified version of cisplatin, DeRose and colleagues were able to identify Pt-RNA adducts by fluorescent labeling using Cu-free click chemistry, revealing that the ribosome is a target of the anticancer compound.(561–563)
In addition to covalent cross-linking, cleavage-based experiments have been used to validate small molecule RNA targets in cells (Figure 20C). This approach was first developed by appending a modularly assembled r(CUG)exp-targeting compound (2H-4) with N-hydroxylthiopyridine (HPT), which generates hydroxyl radicals that cleaves nucleic acids upon photochemical irradiation.(564)Cells treated with this compound and irradiated had reduced levels of r(CUG)exp as determined by RT-qPCR, validating this binding site.
This approach was further developed into a targeted cleavage method by conjugating an RNA-targeting small molecules to bleomycin a5, a natural product that has been shown to cleave RNA in vitro.(565) Attachment of bleomycin a5 to the r(CUG)exp-targeting dimer 2H–K4NMeS allowed for cleavage of r(CUG)exp both in vitro and in DM1-affected cells.(497) Importantly, targeted cleavage allowed for the evaluation of allele selectivity of the small molecule. Using allele-specific primers for RT-qPCR, it was shown that 2H-K4NMeS-Bleomycin only cleaved the mutant allele containing the expanded r(CUG) repeat. Similar to C-Chem-CLIP, competitive cleavage experiments can be completed by cotreating with the parent compound and the small molecule cleaver to determine bona fide targets of the small molecule.
Collectively, the methods described above allow for direct target identification of small molecules in cells. These methods not only can be used for target validation but also can be used to more potently inhibit a disease-causing RNA. If these approaches can be further optimized to limit the off-target effects of the reactive probe then the corresponding compounds might more potent and selective than the unreactive compounds from which they were derived.
4 Protein Targeted Therapeutics
Most drugs in the clinic target proteins,(566) and while the number of protein drug targets is estimated to be around 5000, fewer than 500 distinct proteins are the targets of currently available therapeutics.(566) The ten highest grossing drugs in the U.S. benefit relatively few of the patients who take them, and some drugs may even be harmful to patients.(567) The sequencing of the human genome, however, has the potential to revolutionize personalized medicines that target specific malfunctioning proteins that are effective and better tolerated in patient groups with specific genetic profiles.(566, 567) Below, we discuss examples of drugs that specifically target disease-associated proteins caused by genetic variations.
4.1 Imatinib and the Philadelphia Chromosome
The field of personalized medicines largely began with the identification of the Philadelphia (Ph) chromosome that causes chronic myeloid leukemia and the discovery of Imatinib to treat it. CML is a blood cancer characterized by uncontrolled growth of hematopoietic stem cells, resulting in an increased number of myeloid cells in the blood and bone marrow.(568) The average age at diagnosis for CML was 64 years between 2007 and 2011, but the disease affects all age groups, including children and adults. CML usually presents with fatigue, weight loss, anorexia, and/or enlarged spleen, although about 40% of patients have no symptoms.(568) The disease does not appear to be curable, but patients may achieve remission with current drug therapies.
CML is diagnosed based upon detection of the Ph chromosome, which is present in 95% of patients.(568) It is typically identified by cytogenetic analysis, fluorescence in situ hybridization (FISH), or quantitative real time-polymerase chain reaction (qRT-PCR).(569) The Ph chromosome was first identified by P. C. Nowell and D. Hungerford in 1960, who noticed an abnormally short chromosome in cells from patients with CML.(41) In the 1970’s, it was determined with improved cytogenetic techniques to be a translocation between chromosomes 9 and 22.(41) The result is a shortened chromosome 22 and fusion of the breakpoint cluster region (BCR) gene on chromosome 22 and Abelson leukemia virus (ABL) gene on chromosome 9 (Figure 21A).(42, 568)
Figure 21.

Philadelphia chromosome and Imatinib. (A) The Philadelphia chromosome results from a translocation between chromosome 9 and chromosome 22, resuling in a BCR-ABL fusion gene. (B) BCR-ABL encodes a hyperactive tyrosine kinase, causing cells to uncontrollably divide in CML. Imatinib binds to the ATP-binding site and inhibits cell signaling and the progression of CML. (C) Chemical structure of Imatinib. (D) Hydrogen-bonding interactions between c-ABL and Imatinib.
The Ph chromosome encodes the BCR-ABL fusion protein, a cytoplasmic tyrosine kinase consisting of the ABL tyrosine kinase and the N-terminal domain of BCR.(42, 568) The molecular weight of the fusion protein can range from 185 (p185BCR-ABL) to 230 kDa. Nearly all patients with chronic-phase CML express the 210 kDa isoform (p210BCR-ABL), while patients with Ph-positive acute lymphoblastic leukemia (ALL) phase express either a 190 kDa or a 210 kDa isoform.(568)The N-terminal oligomerization domain of BCR facilitates formation of BCR-ABL dimers, which associate into a tetramer.(570) The BCR-ABL oligomerization constitutively activates ABL kinase activity and stimulates autophosphorylation of the kinase, resulting in activation of downstream signaling pathways, such as the mitogen-activated kinase/extracellular signal-regulated kinase (MAPK/ERK), the Janus kinase/signal transducer and activator of transcription (JAK/STAT), and phosphatidylinositol-3-kinase/AKT serine/threonine kinase (PI3K/AKT; AKT is also known as protein kinase B [PKB]) pathways (Figure 21B).(42, 570) Upregulation of these pathways leads to increased cell growth and proliferation, decreased apoptosis, and altered adhesion to the bone marrow stroma.(571)
Imatinib (Gleevec, or STI-571, Novartis) is a drug for treatment of CML. The chemical precursor of Imatinib, a phenylaminopyrimidine derivative, was identified from an in vitro screen of compounds that inhibited protein kinase C (PKC) (Figure 21C).(572) The ATP-competitive inhibitor was lead-optimized using structure–activity relationships to inhibit the activity of tyrosine kinases.(572)Attachment of an N-methylpiperazine group to enhance solubility and bioavailability yielded STI-571.(572) A crystal structure showed that Imatinib binds and stabilizes an inactive form of the ABL kinase.(572, 573) The pyridine and pyrimidine rings occupy the region where the adenine ring of ATP normally binds (P-loop), while the rest of the compound lies further in the hydrophobic core of the kinase, between the activation loop (A-loop) and helix αC of the kinase domain.(573) The compound forms six hydrogen bonds with the kinase, including two that involve the N-methylpiperazine group (Figure 21D).(572, 573) Imatinib was shown to inhibit autophosphorylation of viral ABL (v-ABL), p210BCR-ABL, and p185BCR-ABL (IC50 of 0.1–0.3 μM, 0.25 μM, and 0.25 μM, respectively) in cell lines.(42, 572) In mice implanted with KU812 human cells expressing BCR-ABL, treatment with 160 mg/kg day of Imatinib over 11 consecutive days was associated with blockage of p210BCR-ABL and resulted in tumor-free survival.(42, 572)
Imatinib was evaluated in clinical studies for treatment of CML. Clinical outcomes were measured by hematological responses, which results from a normalization of blood counts; cytogenetic responses, which results from a reduction in the number of Ph-positive chromosomes; and molecular responses, which results from a reduction in BCR-ABL transcripts.(574) A phase 2 study was carried out on 260 CML patients, 229 of whom had blast crisis, a phase in which malignant cells rapidly proliferate and is usually fatal within three to six months of onset.(575)Imatinib treatment of oral daily doses of 400 mg or 600 mg induced hematologic responses in 52% of patients and major cytogenetic responses in 16% of patients, with a median overall survival (OS) time of 6.9 months.(575) A separate phase 2 study was conducted on 454 patients with late chronic phase CML in which previous therapy with interferon alfa (contains several interferon-α subtypes) had failed. Daily treatment with 400 mg of Imatinib led to complete hematologic responses for 95% of patients and major cytogenetic responses for 60% of patients. A phase 3 study was then conducted on 1106 patients with newly diagnosed CML to compare the efficacy of Imatinib to combination therapy of interferon alfa and cytarabine.(576) Patients who received Imatinib achieved complete hematological responses at a higher overall rate than those who received the combination therapy (95.3% vs 55.5%, respectively) and more rapidly (within 1 month vs 2.5 months, respectively).(576) The rate of complete cytogenetic responses was also significantly higher in the Imatinib group than the combination therapy group (85.2% vs 22.1%, respectively).
Unfortunately, resistance to Imatinib has been observed in patients with advanced-stage CML who relapsed after therapy with the drug.(577) The occurrence of point mutations in BCR-ABL is the most common cause of acquired resistance(574) and usually occur in the Imatinib binding site, the P-loop, the A-loop, and the catalytic domain.(578, 579) The most common mutation in the Imatinib binding site is the T315I mutation and confers resistance to Imatinib by disrupting the hydrogen bond from threonine to Imatinib, sterically interfering with drug binding.(42, 574, 578, 580) T315 is termed the “gatekeeper” residue because it controls access of ATP-competitive inhibitors to the active site.(581) Mutations in the ATP-binding P-loop, such as G250E, Q252H/R, and E255K, or the activation A-loop, such as H396R, may destabilize the inactive kinase conformation required for Imatinib binding.(574, 578) Substitution of F359 with valine in the catalytic domain may disrupt van der Waals interactions with the piperazine ring of Imatinib.(578) Identification of mutations that confer resistance has prompted a search for drugs that can overcome them. Nilotinib binds to the inactive form of BCR-ABL with a 20-fold greater potency than Imatinib, and many Imatinib-resistant mutations such as L248V, G250E, Q252H, and F317L are sensitive to it.(574, 578)Dasatinib binds to the active form of BCR-ABL and is effective against most Imatinib-resistant mutations. Nilotonib and Dasatinib, however, are inactive against the T315I mutation. Another inhibitor, Ponatinib, was later developed to combat resistance to the T315I mutation and is discussed below.(574, 578)
Ponatinib (Iclusig, or AP24534, ARIAD Pharmaceuticals) is a drug approved for treatment of CML and Ph-positive ALL (Table 2).(582) The tyrosine kinase inhibitor (TKI) was developed with a structure-guided design strategy for targeting the BCR-ABL protein with a T315I mutation.(583) X-ray crystallography showed that Ponatinib binds to and stabilizes an inactive conformation of BCR-ABL (Asp-Phe-Gly [DFG]-out).(583) The conformation of the DFG motif is controlled by the protonation state.(584) In the active, DFG-in conformation, the D381 residue of DFG lies in the polar environment of the ATP-binding site and interacts with a catalytic Mg2+ ion that coordinates ATP.(584) Protonation of D381 causes DFG to flip to the DFG-out conformation and release ADP.(584) Ponatinib forms five hydrogen bonds with the protein: the imidazo[1,2b]pyridazine lies in the adenine (ATP) pocket; the methylphenyl group occupies the hydrophobic pocket behind the mutated I315 residue; and the ethynyl linkage interacts with I315 through van der Waal interactions.(583) Ponatinib inhibited native ABL and ABL-T315I (IC50 of 0.37 nM and 2.0 nM, respectively), in in vitro kinase assays.(583) In a mouse xenograft model of BCR-ABL-T315I, Ponatinib inhibited tumor growth in a dose-dependent manner.(583)
Table 2.
Chemical Structures and Targets of Personalized Cancer Therapeutics Discussed Herein
| Therapeutic | Structure | Target | Disease | References |
|---|---|---|---|---|
| Imatinib |
|
BCR-ABL | Chronic myeloid leukemia | 572–580 |
| Ponatinib |
|
BCR-ABL | Chronic myeloid leukemia | 582–584 |
| Cetuximab | Antibody | EGFR | EGFR-positive colorectal cancer | 594–600 |
| Panitumumab | Antibody | EGFR | EGFR-positive colorectal cancer | 601–604 |
| Gefitinib |
|
EGFR | Non-small-cell lung cancer | 605–610 |
| Erlotinib |
|
EGFR | Non-small-cell lung cancer | 606, 611–613 |
| Afatinib |
|
EGFR | Non-small-cell lung cancer | 590, 614–618 |
| Osimertinib |
|
T790M mutated EGFR | Non-small-cell lung cancer | 619–620 |
| Trastuzumab | Antibody | HER2 | HER2-positive breast cancer | 623–627 |
| Lapatinib |
|
EGFR/HER2 | HER2-positive breast cancer | 590, 628–631 |
| Pertuzumab | Antibody | HER2 | HER2-positive breast cancer | 635–640 |
| Olaparib |
|
PARP | BRCA mutated cancers | 641–645 |
| Rucaparib |
|
PARP | BRCA mutated Ovarian cancer | 642, 647–650 |
| Vemurafenib |
|
BRAF-V600E | BRAF-V600E mutated melanoma | 655–661 |
| Dabrafenib |
|
BRAF-V600E | BRAF-V600E mutated melanoma | 662–665 |
| Trametinib |
|
MEK1/2 | BRAF-V600E mutated melanoma | 666–667 |
| Crizotinib |
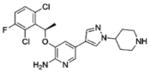
|
ALK | Non-small-cell lung cancer | 595, 672–679 |
| Pembrolizumab | Antibody | PD-L1 | Mismatch repair deficient cancers | 684–695 |
The clinical activity of Ponatinib in patients with CML was reported in a phase 1 and a phase 2 clinical study. In the phase 1 study, Ponatinib treatment elicited a hematological response in for 100% of patients with chronic-phase CML and T315I mutations, a cytogenetic response in 92% of patients, and a major molecular response in 67% of patients.(582) Among chronic CML patients with non-T315I mutations, 93% had a complete hematological response, 67% had a major cytogenic response, and 53% had a major molecular response.(582) In a phase 2 study, 70% of patients with chronic-phase CML and T315I mutations had a major cytogenetic response and 56% had a major molecular response. In summary, these studies show that Ponatinib has clinical activity in patients with CML.
These remarkable achievements catalyzed the field of personalized medicine. One of the goals of the human genome project is to accelerate the drug development process. The timeline from understanding the Ph-chromosome and its role in disease to the discovery of Imatinib was 32 years. From there, another 9 years were required to obtain FDA approval.(110) Advances in whole genome sequencing are key to advancing drugs to the clinic.
4.2 Personalized Cancer Medicines
The development of high-throughput technologies to examine the cancer genome has enabled the identification of specific genes and biomarkers associated with tumor development or affect patient response to particular treatments (i.e., molecular profiles).(585) Oncologists use these molecular profiles to tailor treatments to the unique characteristics of cancers in individuals, such as by matching drugs to specific mutations or targeting molecular pathways essential for cell growth and proliferation.(585) Traditional chemotherapy with nonspecific cytotoxic drugs damages both tumor and normal cells, leading to side effects that reduce the quality of life of patients.(585–588) In this section, examples of drugs used in personalized cancer therapies is discussed. Many of these personalized cancer therapies are accompanied by FDA-approved companion diagnostic devices to determine the presence of an overexpressed gene or a mutation in a gene.(589)
The erythroblastosis oncogene B (ErbB also known as human epidermal growth factor receptor [HER]) is a family of four receptors (EGFR/HER1/ErbB1, HER2/ErbB2/NEU (neuro/glioblastoma derived oncogene), HER3/ErbB3, and HER4/ErbB4) that control cellular functions through a network of pathways, including PI3K/AKT, MAPK/ERK, and phospholipase C-γ (PLCγ).(590) Upon binding of a growth factor, such as epidermal growth factor (EGF), the kinase domains of the receptors dimerize and phosphorylate tyrosine residues, which creates docking sites for proteins that act in downstream signaling.(590) The PI3K/AKT pathway is involved in regulation of cell survival, while MAPK/ERK and PLCγ pathways have important roles in cell proliferation.(590)Mutations or increased expression of ErbB proteins are associated with several cancers via signaling through these pathways; mutations tend to promote formation of the active state of the kinases.(590)
Targeting EGFR Overexpression or Mutations
Therapeutics targeting EGFR have been developed for cancers in which EGFR is overexpressed or mutated. In-frame exon 19 deletions (typically L747 to E749) and an exon 21 point mutation (T to G point mutation that results in L858R) comprise 90% of all EGFR (EGFR)-activating mutations.(591) In the inactive state, L858 lies in a hydrophobic pocket in the activation (A-) loop of EGFR(592) and its substitution with a positively charged arginine side chain disrupts the hydrophobic interaction and locks EGFR into an active state.(593) The exon 19 deletions may also destabilize the inactive conformation of EGFR.(590, 593) At the same time, these mutations decrease the affinity of EGFR for ATP, rendering them susceptible to inhibition by TKIs.(590)Resistance to TKIs is most often observed when a gatekeeper mutation in exon 20, T790M, occurs concurrently with EGFR-L858R; the T790M mutation occurs in 50–60% of patients with drug resistance.(590) The double mutant has increased affinity for ATP, making it more difficult for small molecules to compete for binding to ATP-binding pocket. The overall result is decreased potency of the drugs.(590) Several inhibitors that target specific mutations of EGFR have been developed, which are discussed below.
Cetuximab (Erbitux, or IMC-C225, Bristol-Myers Squibb) is a monoclonal antibody against EGFR approved by the U.S. FDA for treatment of EGFR-positive colorectal cancer.(594) Expression of EGFR is confirmed prior to treatment using an FDA-approved qualitative immunohistochemical (IHC) kit.(595) The antibody was identified by inoculating mice with A431 (epidermoid carcinoma) cells, which overexpress EGFR, and screening for antibodies that inhibit epidermal growth factor (EGF) binding.(594) The resulting antibodies inhibited growth of A431 carcinoma cells in vitro and in vivo.(594) A human/mouse chimeric version of one of these antibodies, mAb 225, was later developed.(594) A crystal structure of the antigen binding (Fab) fragment of Cetuximab in complex with the soluble extracellular region of EGFR showed that Cetuximab binds to domain III, sterically blocking the EGF binding site.(594) Binding of Cetuximab to EGFR introduces steric clashes between Fab and domain I, preventing receptor dimerization and activation.(594) Inhibition of EGFR by Cetuximab results in disruption of many processes regulated by EGFR, including cell cycle progression, tumor cell motility and invasion, and tumor angiogenesis.(596)
In EGFR mutation-positive metastatic colorectal cancer patients, Cetuximab in combination with Irinotecan, Fluorouracil, and Leucovorin chemotherapy (FOLFIRI) reduced the risk of progression by 15% and increased the response rate by 10% relative to FOLFIRI alone, primarily in patients with wild-type Kirsten ras oncogene (KRAS).(597) Pooled analysis of patient data from two other studies of mCRC treatment demonstrated significant improvements in patients with wild-type KRAS when Cetuximab was added to FOLFIRI.(598) The association of KRAS mutation with disease progression was demonstrated to be significant in a study of 59 mCRC patients who received Cetuximab and chemotherapy.(599) Thus, Cetuximab was not indicated for treatment of patients who test positive for KRAS mutations.(600)
Panitumumab (Vectibix, Amgen, Inc., Thousand Oaks, CA) is another FDA-approved monoclonal antibody against EGFR used to treat EGFR-expressing colorectal cancer.(601) Panitumumab is a fully human anti-EGFR monoclonal antibody that inhibits phosphorylation and activation of EGFR-associated kinases.(601) The binding site of Panitumumab partially overlaps with the EGF binding site and prevents EGFR dimerization.(602) The binding of Panitumumab is very similar to that of cetuximab, and its inhibition of EGFR results in similar downstream effects.(602) In phase III clinical trials in patients with mCRC, patients treated with Panitumumab combined with Fluorouracil, Leucovorin, and Oxaliplatin (FOLFOX4) had a significantly improved progression-free survival compared to FOLFOX4 treatment alone.(603) Like Cetuximab, Panitumumab is not effective in patients with KRAS mutations.(603) In 2014, a phase III open-label, noninferiority study showed that Panitumumab is noninferior to Cetuximab and that the two drugs provide similar overall survival benefit to patients with EGFR-expressing colorectal cancer.(604)
There are multiple EGFR-targeting therapeutics in the clinic to treat nonsmall cell lung carcinoma (NSCLC) including Gefitinib, Erlotinib, Afatinib, and Osimertinib. These drugs target specific EGFR mutations that are detected by FDA-approved companion diagnostics, including real-time PCR and high throughput sequencing.(595)
Gefitinib (Iressa, AstraZeneca Pharmaceuticals) is a TKI that was briefly approved in 2003 for treatment of patients with advanced NSCLC after treatment with chemotherapy. Gefitinib is an anilinoquinazoline small molecule that is a potent, reversible inhibitor of EGFR (IC50 of 9 nM) (Table 2).(605) Other 4-anilinoquinazolines such as Gefitinib have been shown to inhibit EGFR through binding to the site occupied by ATP during phosho-transfer.(606) Gefitinib forms a single hydrogen bond with Met793 in the “hinge” region of EGFR with other hydrophobic interactions in the ATP biding cleft.(607) Binding of Gefitinib to EGFR blocks EGF-stimulated phosphorylation and slows EGF-stimulated tumor growth.(608) Gefitinib was voluntarily withdrawn from the market after confirmatory trials failed to verify clinical benefit and an alternate drug (Erlotinib, discussed below) was approved.(609) In 2015, Gefinitib was approved for the first-line treatment of patients with metastatic NSCLC with exon 19 deletions or exon 21 L858R mutations as detected by an FDA-approved companion diagnostic.(610) Interestingly, Gefinitinb binds 20 times more tightly to the L858R mutant than wild-type EGFR.(607) In clinical trials with patients with these mutations, 50% of patients treated with Gefitinib saw a decrease in tumor size over an average time of six months.(610)
Erlotinib (Tarceva, Astellas Pharma Inc.) is a TKI that was first approved by the FDA in 2004 for advanced or metastatic NSCLC. The drug is a reversible inhibitor of EGFR tyrosine kinase (IC50 of 2 nM) that reduces EGFR autophosphorylation in intact tumor cells (IC50 of 20 nM) (Table 2).(611)A crystal structure of erlotinib and EGFR shows key hydrogen-bonding interactions between N1 of the quinazoline and Met769 and a solvent-bridged interaction between the other quinazoline nitrogen and Thr766.(606) The binding mode and mechanism of action of Erlotinib is very similar to that of Gefitinib. Erlotinib was initially approved for NSCLC patients who had received at least one prior chemotherapy regimen based on an increase in median overall survival in clinical trials (6.7 months in patients treated with Erlotinib, compared to 4.7 months in the placebo group).(612)It was later approved in 2013 for the first-line treatment of patients with metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 (L858R) mutations after patients with these mutations saw a significant increase in progression-free survival.(613)
Afatinib (Gilotrif, or BIBW2992, Boehringer Ingelheim) is a second-generation TKI drug, indicated for treatment of NSCLC with exon 19 deletions or exon 21 mutation of EGFR.(590) The drug is an ATP-competitive anilinoquinazoline derivative containing an electrophilic acrylamide group that acts as a Michael acceptor (Table 2).(590, 614) Afatinib irreversibly binds to receptor tyrosine kinases EGFR, HER2, and ErbB4 to inhibit tyrosine autophosphorylation.(590, 614) Specifically, it forms a covalent bond with cysteine residues in the kinase domains of EGFR (Cys773), HER2 (Cys805), and ErbB4 (Cys803) via a Michael addition.(590, 614) Afatinib inhibits wild-type EGFR (IC50 of 0.5 nM), EGFR-L858R (0.4 nM), HER2 (14 nM), and ErbB4 (1 nM).(590, 614) Additionally, it inhibits the EGFR-L858-T790M double mutant with an IC50 of 10 nM, which may be attributed to its irreversible nature of inhibition.(590, 614)
In clinical trials, Afatinib significantly improved progression-free survival (PFS) of patients with the L858R mutation and exon 19 deletions (13.6 months) over cisplatin and Pemetrexed (6.9 months)(615) and also demonstrated significant improvements in PFS of patients who received Afatinib compared to those who received cisplatin and Gemcitabine (11.0 months vs 5.6 months).(616)However, Afatinib did not improve OS in patients who had disease progression after treatment with Erlotinib or Gefitinib.(617) Afatinib resistance was also found among NSCLC patients, and the major mechanism was reported to be a T790M mutation.(618)
Osimertinib (Tagrisso, or AZD9291, AstraZeneca) is a third generation TKI drug approved by the U.S. FDA to treat EGFR mutant NSCLC. The drug was developed in response to the failure of second-generation TKIs to overcome T790M resistance in patients. Osimertinib selectively targets the T790M mutation, the exon 19 deletion, and the L858R mutation, while sparing wild-type EGFR.(619) The drug is a monoanilino-pyrimidine that covalently binds to the conserved Cys797 residue in the ATP-binding site via the acrylamide group (Table 2).(619) Consequently, through inhibition of EGFR, several downstream pathways, such PI3K/AKT and MAPK/ERK, are affected.(619) In enzymatic assays, Osimertinib inhibited EGFR-L858R (IC50 of 12 nM) and L858R/T790M (1 nM) more potently than wild-type EGFR (184 nM).(619) In clinical trials, patients with a confirmed T790M mutation receiving Osimertinib therapy yielded a significantly higher response rate than platinum-Pemetrexed therapy (71% vs 31%, respectively).(620) Patients in the Osimertinib group also had significantly longer median progression-free survival (PFS) than the platinum-pemetrexed group (10.1 months vs 4.4 months).(620) In summary, these findings show that Osimertinib is more effective than platinum-based chemotherapy for treatment of patients with T790M-positive NSCLC.(620)
Targeting HER2
Human epidermal growth factor receptor 2 (HER2/ErbB2) has no known ligands and is activated when expressed at high levels by forming homo- or heterodimers with another ErbB family member.(621) HER2 overexpression occurs in 20% to 30% of breast cancers and results from amplification of the HER2 gene, located at chromosome 17q12, increasing HER2 mRNA and protein levels.(622) HER2-positive breast cancers, which are correlated with poor prognosis,(590, 621, 622) can have 25–50 copies of the HER2 gene, a 40–100-fold increase in HER2 protein, and up to two million receptors on a tumor cell.(621) FDA-approved methods to measure HER2 expression in patient samples include immunohistochemistry and in situ hybridization.(595)
HER2 overexpression causes resistance to apoptosis and hence increased cellular proliferation.(621, 622) For example, HER2 and ErbB3 heterodimerization stimulates cell proliferation and survival via the MAPK/ERK pathway and antiapoptosis via the PI3K/AKT pathway.(621, 622) One hypothesis is that these pathways result in phosphorylation of BCL2-associated agonist of cell death protein (BAD), preventing it from neutralizing antiapoptotic proteins BCL-2 and BCL-xL.(621) Meanwhile, phosphorylation of Bcl-2-interacting mediator of cell death (BIM) by ERK silences the apoptotic activity of BIM.(622) Multiple drugs have been approved for cancers that overexpress HER2.
The first therapeutic developed to target HER2 is Trastuzumab (Herceptin, Genentech Inc.). Trastuzumab is a recombinant monoclonal antibody against HER2.(623) Trastuzumab was FDA-approved in 2006 to treat HER2-overexpressing breast cancer. The interaction of Trastuzumab with HER2 is mediated by two loop regions in HER2 that form electrostatic interaction (a loop formed by residues 557–561 and a portion of a loop formed by residues 593–603) and one loop region that makes hydrophobic contacts (a loop formed by residues 570–573).(624) By binding to HER2, Trastuzumab interferes with HER2 dimerization, thus inhibiting its activation.(625) This results in downstream suppression of PI3K/AKT signaling and reduction in cell growth and survival. There is also evidence that Trastuzumab’s mechanism of action is through antibody-dependent cellular cytotoxicity.(626) In clinical trials, patients with HER2-overexpressing breast cancer were treated with chemotherapy and Trastuzumab or chemotherapy alone, and Trastuzumab reduced the risk of recurrence, secondary primary cancer, or death by 52%.(627)Trastuzumab combined with chemotherapy is used as first-line treatment of HER2-positive breast cancer.
Lapatinib (Tykerb, or GW572016, GlaxoSmithKline) is a TKI approved by the U.S. FDA for second-line treatment of HER2-positive breast cancer (Table 2).(590) The ATP-competitive inhibitor simultaneously inhibits EGFR and HER2.(590) X-ray crystallography showed that the drug binds the ATP-binding pocket of EGFR, and its aniline substituent lies deep in the back pocket and makes predominantly hydrophobic interactions with the protein.(628) The slow dissociation rate of the drug may be attributed to the conformational change in EGFR required to facilitate the interactions with the aniline group or alternatively to tight binding affinity of the drug to EGFR.(628) The drug would likely bind to HER2 in a similar fashion as to EGFR since the proteins have similar catalytic domains (88% identical).(590, 628) In in vitro kinase assays, Lapatinib inhibited EGFR and HER2 with IC50’s of 10.2 and 9.8 nM, respectively.(628, 629) Lapatinib may also stabilize formation of HER2 dimers, resulting in receptor accumulation and subsequent attack by anti-ErbB antibodies.(630) By binding to EGFR and HER2, Lapatinib inhibits the activation of three main downstream signaling pathways, MAPK, PI3K-AKT, and PLCγ.(631)
In clinical trials of HER2-positive breast cancer, addition of Lapatinib to Capecitabine chemotherapy significantly increased median time to progression (TTP) from 4.4 months to 8.4 months in patients.(632) Addition of Lapatinib to Letrozole hormone therapy also significantly increased median PFS from 3.0 months to 8.2 months over Letrozole alone in HER2-positive patients.(633) As expected, HER2-negative patients had no improvement in PFS.(633) Addition of Lapatinib to Trastuzumab significantly median-improved PFS over Lapatinib alone (11.1 weeks vs 8.1 weeks) in patients with HER2-positive metastatic breast cancer whose disease had progressed during prior treatment with Trastuzumab.(634) In summary, these studies demonstrate that Lapatinib has clinical activity in HER2-positive breast cancer.
Pertuzumab (Perjeta, or mAb 2C4, Genentech) is a humanized monoclonal antibody for treatment of HER2-positive breast cancer.(635) The antibody was developed using stably transfected NIH 3T3 (mouse fibroblast) cells that express the HER2 gene.(635) 2C4, a murine IgG1κ antibody, was found to bind to the extracellular domain of HER2.(635) A humanized variant of this antibody was later developed.(635) A crystal structure of Pertuzumab in complex with HER2 showed that it binds to domain II, in a region that overlaps with the binding site for the dimerization hairpin of a heterodimer partner.(636) Therefore, the antibody sterically inhibits heterodimerization of HER2 with EGFR or ErbB3 and subsequent transphosphorylation of the receptors.(636) This inhibition affects key downstream signaling pathways including MAPK and PI3K/AKT.(637) Since Pertuzumab binds to the extracellular domain or HER2, it can also affect tumor growth through antibody-dependent cell-mediated cytotoxicity.(638) In clinical trials of HER2-positive breast cancer, addition of Pertuzumab to Trastuzumab and Docetaxel therapy significantly improved median PFS from 12.4 to 18.7 months.(639) Altogether, these studies demonstrate that Pertuzumab and Trastuzumab have activity against HER2-positive breast cancer.(640)
Personalized Medicines for Treatment of Breast Cancer 1 (BRCA1) and Breast Cancer 2 (BRCA2) Gene Mutations
Inherited BRCA1 or BRCA2 mutations confer up to 85% lifetime risk of developing breast cancer.(641) The BRCA1 and BRCA2 proteins are involved in DNA repair of double strand breaks (DSBs) and collapsed replication forks by homologous recombination (HR).(641) HR is an conservative DNA repair mechanism that restores the original DNA sequence at the site of damage.(641)Carriers of BRCA1/2 mutations are heterozygous and can carry out HR repair in cells but loss of the remaining wild-type allele renders them defective in HR repair.(641) Cells that lose HR use alternative mechanisms to repair DSBs, which may increase genetic variation and lead to tumor formation.(641) One such mechanism utilizes poly(ADP-ribose) polymerase (PARP), an enzyme that repairs single strand breaks (SSBs).(641) Loss of PARP function in cells lacking BRCA1/2 results in cell death, a condition termed synthetic lethality.(641) Since inhibition of PARP does not affect normal cells, which have functioning HR, PARP inhibitors selectively target tumor cells.(641)Therefore, one strategy to treat breast cancers with BRCA1/2 mutations is to inhibit PARP and induce synthetic lethality.
Olaparib (Lynparza, or AZD2281, AstraZeneca) is a small molecule drug for treatment of breast and ovarian cancers with BRCA mutations by selectively inhibiting PARP1 and PARP2 catalytic activity (Table 2).(641) These BRCA mutations can be detected using the FDA-approved companion diagnostic, BRACAnalysis CDx (NGS-based).(595) A crystal structure of Olaparib bound to PARP2 showed that the small molecule formed several hydrogen bonds in the catalytic domain including Arg444 and a water-mediated hydrogen bond with Asp339.(642) Binding of Olaparib to PARP1 and PARP2 prevents the formation of PAR polymers and blocks the binding of NAD+ at the site of DNA damage, preventing the cell from overcoming DNA-dependent damage.(643) In clinical trials, patients with BRCA mutations in several types of cancers who had received prior treatment, Olaparib treatment resulted in a tumor response rate of 31% for ovarian cancer and 13% for breast cancer, with stable disease rates (defined as neither progression of disease nor response to treatment(644)) in 40% and 47% of patients with those cancers, respectively, after 8 weeks.(645) In a phase 3 clinical trial of patients with HER2-negative metastatic breast cancer and a BRCA mutation, Olaparib monotherapy significantly increased median PFS (7.0 months vs 4.2 months) and response rate (59.9% vs 28.8%) over standard therapy and decreased risk of disease progression or death by 42%.(646) Altogether, these studies demonstrate that Olaparib provides benefits for treatment of ovarian and breast cancers with BRCA mutations.
Rucaparib (Rubraca, Clovis Oncology Inc.) is a PARP inhibitor designed to treat ovarian cancer patients with BRCA gene mutations who have previously received two or more types of chemotherapy treatment (Table 2). Rucaparib is a three-ring heterocyclic small molecule that greatly improved chemosensitization in preclinical studies.(647) A crystal structure of Rucaparib in contrast with PARP1 suggests that the flexible terminal secondary amine may facilitate different modes of action depending on the environment.(642) Rucaparib inhibits PARP1, PARP2, and PARP3 which leads to inhibition of single- and double-strand break repair pathways leading to tumor death.(648) Mice that were treated with a combination of chemotherapy and Rucaparib took 33 days for tumor volumes to reach four times the size of its initial volume, compared to 16 days for chemotherapy only mice.(647) In clinical trials, 54% of patients who received Rucaparib experienced complete or partial shrinkage of their tumors lasting a median of 9.2 months.(649, 650)
Targeting BRAF Mutations
The MAPK/ERK pathway is an intracellular signaling pathway that transmits signals from the cell surface to the nucleus to regulate genes involved in cell proliferation, differentiation, motility, survival, and angiogenesis.(651) Activation of the cascade follows binding of a ligand to a receptor tyrosine kinase, inducing autophosphorylation of the receptor.(651) Adaptor proteins bind to the phosphorylated receptors and recruit guanine nucleotide exchange factors, which activate the membrane-bound GTPase Ras, by converting GDP to GTP.(651) Ras recruits Raf kinases (A-Raf, B-raf, or Raf-1) to the plasma membrane and activates them.(651) Raf proteins activate MAPK/ERK kinases 1 and 2 (MEK1 and MEK2, respectively) which activate Erk1 and Erk2 kinases.(651) Erk1 and Erk2 translocate to the nucleus and phosphorylate substrates, including other kinases and transcription factors that regulate gene expression.(651)
Mutations among Ras and Raf are prevalent in multiple cancers, including pancreatic and colon cancers.(651) The most common mutation in the BRAF kinase, V600E, is caused by substitution of a GTG codon (valine) with a GAG codon (glutamic acid).(652) The replacement of the hydrophobic side chain of V600 with the larger, charged side of E600 disrupts the hydrophobic interaction between V600 and F467 of the P-loop that stabilizes the inactive conformation of the DFG motif, thereby stabilizing the catalytically preferred conformation.(653) This results in constitutive activation of BRAF-V600E and MEK/ERK signaling in tumor cells, allowing the cell to become self-sufficient in growth signals.(653, 654) The discovery of mutations in members of the MAPK pathway has led to interest in developing anticancer therapeutics that target MAPK signaling.
Vemurafenib (Zelboraf, or PLX4720, Plexxicon/Roche) is a RAF inhibitor approved by the U.S. FDA to treat patients with the BRAF-V600E mutation (Table 2).(655) The BRAF-V600E mutation is found in about half of all melanomas in addition to various other cancers.(656, 657) Vemurafenib was identified in a fragment-based screen of 20000 small molecules and found that 7-azaindoles bound to the ATP-binding site of another kinase, Pim-1, in a cocrystal structure.(655) Lead optimization afforded 3-aminophenyl-7-azaindole, 3(3-methoxybenzyl)-7-azaindole, and finally Vermurafenib, which inhibited BRAF-V600E at 10-fold lower concentrations than wild-type BRAF.(655) The drug preferentially binds to the active conformation of wild type and V600E BRAF, with the “DFG-in” conformation in which the phenylalanine side chain of the DFG motif in the activation loop is buried inside and away from the ATP-binding pocket.(655, 658) Binding of Vemurafenib to mutant BRAF leads to inhibition of phosphorylation and reduced signaling through MEK and ERK, leading to decreased cell proliferation.(659) In preclinical studies, Vemurafenib inhibited ERK activity and induced cell cycle arrest and apoptosis in a V600E melanoma cell lines.(655) In clinical trials, Vemurafenib produced improvements in OS and PFS over Dacarbazine in patients with metastatic melanoma with the V600E mutation.(657) Addition of Cobimetinib, a MEK1 inhibitor, to Vemurafenib therapy significantly improved PFS over Vemurafenib alone in melanoma patients but with some increase in toxicity.(660, 661)
Dabrafenib (Tafinlar capsules, GlaxoSmithKline, LLC) is a small molecule BRAF inhibitor used to treat BRAF V600E mutated metastatic melanoma (Table 2). Upon approval of Dabrafenib, the FDA also approved the THxID BRAF assay (bioMerieux, Inc.) to detect BRAF mutations.(595)Dabrafenib is a sulfonamide-containing small molecule that inhibits BRAF V600E with an IC50 of 0.8 nM and growth of a BRAF V600E SKMEL28 melanoma cell line with an IC50 of 3 nM.(662)Dabrafenib’s pyrimidin-2-amine group forms hydrogen bounds in the ATP binding site of BRAF and its sulfonamide group extends to the subpocket near the C-α helix of BRAF.(663) By binding to mutant BRAF, Dabrafenib inhibits MEK and ERK, leading to cell cycle arrest and apotosis.(664)In clinical trials, patients treated with Dabrafenib saw a 52% increase in objective response rates compared to a 17% increase in patients treated with Dacarbazine.(665)
Trametinib (Mekinist tablet, GlaxoSmithKline, LLC) is a small molecule inhibitor of MEK1 and MEK2 approved for the treatment of metastatic melanoma with the BRAF V600E mutation (Table 2). As stated above, the MEK/ERK signaling pathway is activated by mutated BRAF, thus inhibition of MEK is another strategy to treat BRAF mutated cancers. Trametinib is a hetertocyclic small molecule that inhibited MEK-dependent ERK1/2 phosphorylation on Ser217 in BRAF V600E SKMEL28 melanoma cell line with an IC50 of 0.8 nM.(666) Trametinib inhibition of MEK results in cell cycle arrest. In clinical trials, patients treated with Trametinib saw a statistically significant prolongation of progression-free survival compared to traditional chemotherapy (4.8 and 1.5 months, respectively).(667) Objective response rates were 22% for patients treated with Trametinib and only 8% for patients treated with chemotherapy.(667) In 2014, the FDA approved Trametinib and Dabrafenib for use in combination to treat patients with the BRAF V600E mutation. Combination of the two therapies resulted in a 76% objective response rate over 10.5 months compared to single-agent treatment where the response rate was 54% over 5.6 months. Interestingly, this combination therapy has also recently been approved for BRAF V600E mutation-positive metastatic NSCLC.
Targeting ALK and ROS1 Fusion Proteins
Anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) are receptor tyrosine kinases encoded by the ALK and ROS1 genes, respectively.(668) ALK is located on chromosome 2p23 and commonly forms a fusion gene with echinoderm microtubule-associated protein-like 4 (EML4) through a small inversion within chromosome 2p that joins intron 13 of EML4 to intron 19 of ALK.(668, 669) The result is a fusion protein containing the intracellular catalytic domain of ALK and the basic domain of EML4; as the basic domain of EML4 replaces the extracellular and transmembrane domains of ALK, the fusion protein is relocalized to the cytoplasm.(669) The basic domain from EML4 promotes dimerization, resulting in constitutive ALK kinase activity and activation of downstream pathways that control cell proliferation and survival.(668, 669) ROS1 is located on chromosome 6q22 and forms fusion genes with a variety of 5′ fusion partners, although the oncogenic role of the fusion partner is uncertain.(668) The resulting fusion protein retains the ROS1 kinase domain, and its activation leads to signaling through the PI3K/AKT, JAK/STAT, and MAPK/ERK pathways.(668) Interestingly, ROS1 and ALK have high homology in the kinase domain (49%) and ATP-binding site (77%).(670) Inhibition of these kinases suppresses these signaling pathways, leading to growth arrest and apoptosis.(671) For these reasons, these fusion proteins have been targeted in anticancer therapies.(670) ALK and ROS1 translocate and are identified with a break-apart FISH assay, where two differently colored probes that hybridize to sequences on either side of the translocation breakpoint are split in the presence of a chromosomal rearrangement, resulting in separation of the two colors.(671)
Crizotinib (Xalkori, or PF-02341066, Pfizer) is a drug for treatment of NSCLC(672, 673) with ALKand ROS1 fusions (Table 2).(595) The drug was synthesized as an inhibitor of mesenchymal-epithelial transition factor (MET), another kinase that is upregulated in NSCLC.(672, 673) In cell assays of more than 120 kinases, Crizotinib demonstrated potent inhibition of c-MET and ALK.(672) In a crystal structure of Crizotinib and ALK, a leucine residue stabilizes 2-aminopyrimidine and 3-benzyoloxy groups of Crizotinib via hydrophobic interactions and anchors the compound in an L-shape.(673) Crizotinib was found to inhibit ALK and ROS1 with high potency and selectivity (IC50 of 4.5 and 1.7 nM, respectively).(674) Inhibition of ALK and MET by Crizotinib results in upregulation of proapoptotic proteins and induction of apoptosis.(675)
In clinical trials comparing first-line Crizotinib with chemotherapy in patients with ALK-positive NSCLC, Crizotinib significantly increased median progression-free survival PFS from 7.0 months to 10.9 months and response rate from 45% to 74%.(676) Among patients with advanced ALK-positive NSCLC, Crizotinib also significantly increased median PFS (from 3.0 to 7.7 months) and response rate (from 20% to 65%) relative to chemotherapy.(677) Crizotinib therapy for patients with ROS1-positive advanced NSCLC resulted in an overall response rate of 72% and 80% in phase 1 studies of 50 and 32 patients, respectively.(678, 679) In summary, these studies show that Crizotinib is highly active for treatment of NSCLC with ALK or ROS1 rearrangements.
Targeting the Programmed Death-1 Receptor (PD-1)
The anticancer immune response is initiated when antigens released from cancer cells are captured by dendritic cells.(680) Dendritic cells prime and activate T cells with the antigens, which traffic to the tumor and infiltrate the tumor bed where they bind and kill the cancer cells.(680)Dying cancer cells release additional antigens, propagating the immune response.(680) The programmed death-1 (PD-1) pathway suppresses anticancer immune responses, leading to progression of tumors.(680–682) Programmed death-ligand 1 (PD-L1) is a ligand mainly expressed on the surface of various solid tumor cells.(681) Its receptor, PD-1, is a checkpoint molecule that is expressed on T cells.(681) PD-L1 expression is caused by a constitutive or induced mechanism.(681) Constitutive expression may result from amplification of chromosome 9, which encodes PD-L1, or an increase in PD-L1 mRNA transcripts stabilized by disruption of the 3′ UTR, which is involved in post-transcriptional regulation of mRNA decay rate.(682, 683) In the induced mechanism, tumor-infiltrating T cells secrete interferon-γ (IFNγ), which upregulates expression of PD-L1 on tumor cells.(681, 682) Binding of PD-L1 to PD-1 induces apoptosis of T cells and causes adaptive immune resistance.(681, 682) Since normal tissues rarely express PD-L1, therapies that block the PD1 pathway may benefit cancer patients.(681)
Pembrolizumab (Keytruda, or MK-3475, Merck) is an immunotherapy drug for treatment of solid tumors, such as melanoma, overexpressing PD-L1.(684, 685) PD-L1 expression can be determined using PD-L1 IHC 22c3 pharmDx, an FDA-approved immunohistochemical assay.(595)The drug contains a variable region against human PD-1 in a human IgG4 antibody; the antibody contains an S228P mutation in the fragment crystallization (Fc) region to prevent antigen binding fragment (Fab) arm exchange, which can reduce therapeutic efficacy.(686, 687) The drug binds to PD-1 (EC50 of 0.1 to 0.3 nM), preventing it from interacting with PD-L1 which results in the restoration of a T cell antitumor immune response.(684, 685) A crystal structure of Permbrolizumab in complex with PD-1 revealed that the Pembrolizumab epitope overlaps with binding regions for PD-L1 with key hydrogen-bonding interactions with Gln75-Lys78 and Pro84-Gly90 of PD-1.(688)
The efficacy of Pembrolizumab for cancer treatment was evaluated in clinical trials. Results of the first in-human study showed that Pembrolizumab had antitumor activity against multiple solid tumors.(689) A phase 2 study showed that patients with mismatch repair deficient cancers were more responsive to anti-PD1 therapy with Pembrolizumab than mismatch repair proficient cancers.(690) A separate phase 2 study was later carried out to evaluate Pembrolizumab therapy in 86 patients with MMR deficient cancers across 12 different tumor types.(691) Objective responses were reported in 53% of patients, and complete responses were achieved in 21% of patients.(692) The response of MMR-deficient cancers to Pembrolizumab may be attributed to the production of large amounts of cancer neoantigens by mutations that result from MMR, which can be recognized by the immune system upon immune checkpoint blockade.(690, 691) In a phase 3 study of patients with advanced melanoma, 80.5% of whom had PD-L1-positive tissue samples, Pembrolizumab significantly improved PFS and response rate over Ipilimumab antibody, with lower risk of adverse events.(693) Pembrolizumab therapy resulted in significantly prolonged median PFS (10.3 vs 6.0 months) and OS at six months (80.2% vs 72.4%) compared to chemotherapy in two phase 3 studies of patients with advanced PD-L1 positive NSCLC.(694, 695)In summary, these studies demonstrate that Pembrolizumab therapy is effective against multiple PD-L1 positive solid tumors.
The implementation of a personalized therapy strategy has improved treatment outcomes and reduced nonspecific cytotoxicity associated with chemotherapy.(585–588) Repositioning of noncancer drugs may be an effective strategy for developing future anticancer drugs since most noncancer drugs have few or tolerable side effects in humans.(586, 696) Meanwhile, the increasing power of “omics” technologies has yielded new information about mutations, targets, and tumor vulnerabilities for the development of current and next generation of molecular cancer therapeutics.(588)
4.3 Ivacaftor and Cystic Fibrosis
Another disease that has recently seen approval of a therapeutic for a specific genetic mutation is cystic fibrosis (CF). Cystic fibrosis affects nearly 70000 people worldwide, primarily in Caucasian populations.(697) Most cystic fibrosis patients start to have symptoms by 2 years of age. The underlying cause of cystic fibrosis is deficient or dysfunctional CF transmembrane conductance regulator (CFTR) protein due to genetic mutations in the CFTR gene, resulting in loss of epithelial chloride transport.(36, 698) The CFTR protein, an ATP-gated ion channel, contains 1480 amino acids and regulates transport of chloride ions across epithelial cell membranes to regulate salt, water, and pH balance in multiple tissues and organs, such as the lungs, gastrointestinal tract, pancreas, and sweat glands.(36, 697, 698) Correspondingly, loss of CFTR function leads to accumulation of thick, sticky mucus in the lungs and intestines, impaired pancreatic function, and high levels of sweat chloride.(698, 699) Collectively, this results in breathing problems, frequent respiratory infections, malnutrition, slow weight gain, and eventually chronic lung disease. Death is primarily caused by respiratory complications. Available therapies generally treat downstream disease processes associated with CF rather than correct for the loss of CFTR function.(700) The discovery of the CFTR gene, however, has led to efforts to identify therapies that target underlying causes of CF.(697, 701)
Almost 2000 mutations have been associated with CF and can be classified according to the mechanism by which they cause disease.(36, 698, 699, 702, 703) The most common mutation is a deletion of phenylalanine residue 508 (F508del) and accounts for approximately 70% of defective CF alleles.(36, 698, 699, 702, 703) Mutant F508del blocks post-translational processing of CFTR in the endoplasmic reticulum (ER) into its mature form, causing aberrant folding and premature degradation.(36, 698, 699, 703–706) About 5 to 10% of CFTR mutations are caused by nonsense mutations.(36, 703) Among these is G542X, the second most common mutation, which causes a reduction in mRNA levels and absence of the CFTR protein.(36, 699, 703)Approximately 4–5% of patients have the G551D mutation, which is the third most common CFTR mutation worldwide and the most common gating mutation.(697, 698, 707) The CFTR protein with the G551D mutation is fully processed and found at normal levels in the plasma membrane but has little to no chloride transport activity due to abnormal channel function.(36, 698, 699, 703, 707, 708)
The CFTR protein has two transmembrane domains (TMDs) that form the channel pore, two nucleotide binding domains (NBD1 and NBD2), and a regulatory domain with phosphorylation sites (Figure 22A).(698, 709) The F508del mutation is caused by deletion of three nucleotides (CTT), which consists of the last nucleotide (C) of isoleucine 507 (Ile507ATC) and first two nucleotides (TT) of phenylalanine 508 (Phe508TTT), from the CFTR gene.(36, 699, 710) This results in the loss of Phe508 and a synonymous substitution for Ile507 (Ile507ATT).(710) The consequent misfolding of NBD1 results in degradation of most of the protein in the ER and aberrant channel gating for the small amount that is delivered to the plasma membrane.(698, 699, 703–706, 711) Bartoszewski et al. proposed that the F508del mutation generates folded CFTRmRNA structures with larger hairpin loops near the mutation site than wild-type CFTR mRNA.(710)This elongates pause cycles during translation and contributes to misfolding of F508del-CFTR.(710) Weakening of the NBD1-NBD2 dimerization interface accounts for the F508del-CFTR gating defect.(698, 703, 711, 712) The destabilization of NBD1 and misfolding of F508del CFTR was supported by (i) a 6 to 7 °C drop in melting temperature of purified NBD1 from F508del CFTR; and (ii) reversal of functional defects by increasing NBD1 stability via suppressor mutations.(713, 714)
Figure 22.

Cystic fibrosis and therapeutics. (A) Structure of the CFTR membrane. (B) Ivacafor acts on a channel gate of defective CFTR (C) Chemical structures of Ivacaftor and Lumacaftor.
In CFTR, channel opening requires phosphorylation of the regulatory domain by cAMP-activated protein kinase A (PKA) and binding of ATP by the NBDs.(698, 699, 708, 709) Upon dimerization of the NBDs, two ATP binding pockets (ABP1 and ABP2) form, with ATP molecules sandwiched at the interface, leading to ABP2-catalyzed opening of the channel.(707–709, 715) Hydrolysis of ATP and bound at ABP2 destabilizes the dimer and closes the channel.(707–709, 715) Each NBD contains a signature sequence, specifically in ABP2 of NBD1 and ABP1 of NBD2.(707) The G551D mutation, located in ABP2 of NBD1, consists of a single nucleotide polymorphism at codon 551, where a GGT codon (glycine) is converted to a GAT codon (aspartic acid).(707, 708, 716) A longer amino acid side chain results, which may reduce the affinity between the two NBDs during dimerization.(717, 718) This mutation abolishes the function of ABP2, resulting in a 100-fold lower open probability than that of wild-type CFTR, with residual activity of the mutant CFTR possibly resulting from ATP-independent channel openings.(707, 708) A high-affinity ATP analog, P-ATP, was shown to increase open time of G551D-CFTR upon binding to ABP1.(708) Since the G551D mutation may not affect the function of ABP1, ABP1 may serve as a therapeutic target for CFTR potentiators.(708)
Ivacaftor (VX-770, or Kalydeco, Vertex Pharmaceuticals) is a drug for treatment of patients aged 6 years old and older with a G551D-CFTR mutation in at least one allele.(697, 719) As stated above, the G551D-CFTR is fully processed but is not able to transport chloride ions. Ivacaftor increases chloride ion flow by affecting a gating defect in the G551D mutant protein. Recent studies to investigate Ivacaftor’s mechanism of action suggest that it binds to CFTR’s transmembrane domain and stabilizes the open conformation of the channel gate.(720, 721) The CFTR potentiator is the first drug to be approved to target an underlying cause of CF (Figure 22, panels B and C). Ivacaftor was identified from a high-throughput screen of 228000 chemically diverse compounds using a cell-based fluorescence membrane potential assay.(700, 722)Optimization of compounds for potency, selectivity, and chemical tractability led to the identification of VX-770.(700)
Loss of CFTR function and CF lung disease in the lungs may be explained by dehydration of the airway surface.(723) Normal airway surfaces balance Na+ absorption out of with Cl− secretion into the surface liquid.(700, 723) Epithelial cells on CF airway surfaces fail to secrete Cl−, while not regulating Na+ absorption.(700, 723) Since water moves in response to net transport of NaCl, CF airway surfaces become dehydrated, leading to accumulation of mucus.(700, 723) Ivacaftor increased cilia beating frequency and apical fluid height in in vitro experiments in cultures of human bronchial epithelial cells by reducing excessive Na+ and fluid absorption.(700) Ivacaftor increased chloride secretion only after stimulation of the PKA signaling pathway, suggesting that it acted directly on CFTR and increased its open probability (PO) while working in the background of normal cAMP/PKA signaling pathways.(700) Additional studies have shown that Ivacaftor opens the channel gate of defective CFTR in an ATP-independent manner but requires phosphorylation of the protein (Figure 22, panels B and C).(721, 724)
The efficacy and safety of Ivacaftor on patients with at least one G551D allele was evaluated in two placebo-controlled phase 3 clinical trials. The primary efficacy end point in both studies was the absolute change in percent of forced expiratory volume in 1 s (FEV1), a measurement of airway obstruction, from baseline through week 24.(697, 719, 724, 725) Enrolled patients were aged 12 years or older (n = 161, mean age of 25.5 years, and FEV1 of 63.6%) in the first study and 6 to 11 years (n = 52, mean age of 8.9 years, and FEV1 of 84.2%) in the second study.(697, 719, 724) In both studies, patients were randomly assigned in a 1:1 ratio to receive a 150 mg dose of Ivacaftor or a placebo orally every 12 h, over 48 weeks.(697, 719, 724) Patients receiving Ivacaftor demonstrated a 10.6% and a 12.5% increase in FEV1 in the first and second study, respectively, over those receiving a placebo.(697, 719, 724) Patients in the Ivacaftor group gained 2.7 kg more than in the placebo group in the first study and 3.7 kg more than the placebo group in the second study at week 24. Scores for quality of life on the CF Questionnaire-Revised (CFQ-R) were higher for the Ivacaftor group than the placebo group at weeks 24 and 48 in both studies, indicating improvements in respiratory symptoms.(697, 719, 724) Sweat chloride concentrations dropped below the diagnostic threshold for CF (60 mmol/L) by day 15 in the Ivacaftor group in both studies, and the mean drop from baseline was maintained at 50 to 60 mmol/L through week 48, indicating improved CFTR function.(697, 719, 724) Altogether, Ivacaftor produced statistically significant improvements in pulmonary function, weight, and CFTR function in patients aged 6 years and older carrying a G551D mutation in these studies.(697, 719, 724)
Ivacaftor was also evaluated separately in a phase 3 clinical study carried out on 39 CF patients aged 6 years or older with non-G551D gating mutations. These mutations include G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, and G1349D, which jointly account for 1% of CF mutations.(726) Throughout 8 weeks of treatment, Ivacaftor was associated with a treatment effect of 7.5% in FEV1, in addition to statistically significant improvements from baseline in sweat chloride, body mass index (BMI), and CFQ-R, similar to benefits observed in patients with a G551D mutation.(697, 719, 724, 726) Serious adverse events were not increased by Ivacaftor, compared to placebo.(697, 719, 726) In summary, these studies demonstrate that Ivacaftor improved CFTR function in patients with the G551D and other CFTR gating mutations.(697, 719)
Lumacaftor/Ivacaftor (VX-809/VX-770, or Orkambi, Vertex Pharmaceuticals) is a combination drug used to treat CF patients homozygous for the F508del CFTR mutation. The drug consists of Lumacaftor, a CFTR-folding chaperone, and Ivacaftor, a CFTR potentiator.(697, 727) Lumacaftor corrects defective F508del post-translational processing and increases the epithelial delivery of CFTR protein, while Ivacaftor potentiates surface-localized F508del-CFTR.(728, 729) Lumacaftor was identified from a screen of 164000 compounds using a cell-based assay to identify those that increased F508del-CFTR-mediated chloride transport.(706) A search for compounds on the basis of potency, efficacy, and other druglike properties led to the identification of VX-809.(706)
In studies completed in human embryonic kidney (HEK)-293 cells expressing F508del-CFTR, Lumacaftor suppressed folding defects, increased F508del-CFTR exit from the ER, and caused a fraction of F508del-CFTR in the ER to fold into a protease-resistant conformation.(706) In human bronchial epithelial (HBE) cells expressing F508del-CFTR, Lumacaftor increased chloride transport, which was further enhanced after addition of Ivacaftor.(706) These results suggest that Lumacaftor increased the conformational stability and improved the folding of F508del-CFTR.(706) Experimental and modeling data suggest that Lumacaftor binds at the interface of NBD1 and intracellular loop (ICL4) of transmembrane domain 2 (TMD2) to fill the space left empty by the F508 deletion.(712)
The safety, pharmacodynamics, and efficacy of Lumacaftor/Ivacaftor were evaluated in two phase 3 clinical trials on patients who were homozygous for the F508del mutation.(728, 729) Enrolled patients were aged 12 years or older (n = 1108, FEV1 = 61%) and 6 to 11 years (n = 58, FEV1 = 91.4%) in the second study.(728, 729) In the first study, patients were randomly assigned in a 1:1:1 ratio to one of three groups and either received oral doses of 600 mg of Lumacaftor once daily with 250 mg of Ivacaftor every 12 h, 400 mg of Lumacaftor with 250 mg of Ivacaftor every 12 h, or Lumacaftor-matched placebo with Ivacaftor-matched placebo every 12 h over 24 weeks.(728) In the second study, all enrolled patients received 200 mg of Lumacaftor with 250 mg of Ivacaftor every 12 h over 24 weeks.(729)
In the first study, the difference between Lumacaftor/Ivacaftor and placebo with respect to the mean absolute change in percent predicted FEV1 was statistically significant and ranged from 2.6 to 4.0% in all study groups.(728) No significant effect on percent predicted FEV1 was observed in the second study, although that may be explained by a smaller sample size and milder lung disease in the study group.(729) Significant increases in BMI were observed over 24 weeks in both studies, specifically by 0.24 to 0.28 kg/m2 in the first study and to 0.64 kg/m2 in the second study.(728, 729) Treatment differences for CFQ-R between both Lumacaftor dose groups and placebo were nominally significant in the first study, but CFQ-R improved significantly in the second study.(728, 729) Lumacaftor/Ivacaftor had a smaller effect on sweat chloride concentrations in the first study than that observed in patients with the G551D mutation who were treated with Ivacaftor alone, but a substantial decrease was observed in the second study.(728, 729) Overall, the safety profile of Lumacaftor/Ivacaftor was consistent with those in patients treated with placebo.(728, 729) In summary, these studies show that the combination therapy Lumacaftor/Ivacaftor improved CFTR protein function in patients with the F508del mutation.(728, 729)
5 Rational Repurposing of Drugs
The process of drug discovery and development has traditionally consisted of identifying new molecular entities, which continues to be risk-laden, time-consuming, and costly.(586, 696) A single compound identified from a screen of compound libraries has an 8% chance of passing through clinical trials and requires an average of 12–15 years for product launch.(696) The average cost to develop a new drug now exceeds $2.5 billion.(730) Drug repositioning consists of the application of existing drugs to treat diseases other than its clinical indication.(586)Repositioned drug candidates can enter clinical trials at phase 2 since they have been approved or shown to be safe, and reduce the research and discovery (R&D) process by 3–5 years.(696)Genome sequencing in disease can further accelerate this process by identifying genetic similarities between two different diseases. For example, a drug may be approved to treat one disease based on a genetic abnormality; this genetic abnormality may be present in other diseases as well, and thus, the drug can be repurposed to treat the other disease. In this section, examples of drugs that have been repurposed for indications other than the disease that they were approved to treat are discussed.
Repurposing Drugs Targeting Tyrosine Kinases
Protein tyrosine kinases (PTKs) are a large family of enzymes involved in cellular signaling pathways.(731) Many drugs that target these kinases are approved for the treatment of cancer. PTKs catalyze the transfer of the γ phosphate of ATP to tyrosine residues in substrate proteins and regulate a variety of cellular processes(731) and are subdivided into families of receptor tyrosine kinases (RTKs) and nonreceptor tyrosine kinases (NRTKs).(731) Whereas the RTKs are transmembrane proteins, the NRTKs are cytoplasmic proteins and include the JAK family.(731)
The JAK/STAT signaling pathway transduces extracellular signals to the nucleus to control expression of genes involved in cellular proliferation, immunity, differentiation, and apoptosis.(732, 733) The JAK/STAT pathway is also implicated in the pathogenesis of cancers and autoimmune and inflammatory diseases.(732, 733) JAKs are activated downstream by extracellular cytokines, which are small secreted proteins that immune cells use to communicate and modulate immune responses.(732) Upon binding of a cytokine to its JAK-associated receptor, the receptors dimerize and the two JAKs phosphorylate themselves and the receptors, creating docking sites for signal transducer and activator of transcription (STAT) proteins, a family of transcription factors.(732)STATs are then activated by phosphorylation, oligomerize, and translocate to the nucleus, where they bind to DNA and regulate gene expression.(732)
The JAK family has four members, all of which have four structural domains consisting of seven homologous regions.(732) The JH1 domain is the catalytic kinase domain and target of JAK inhibitors, while JH2 regulates JH1 function.(732, 733) The STAT family has seven members in humans, within which the SH2 domain binds to phosphorylated tyrosine residues and is the target of most STAT inhibitors.(732, 733) Different JAKs and STATs act on different cytokine receptors, without any relationship between which JAK family members activate which STAT family members.(732, 733)
Tofacitinib (Xeljanz, Pfizer) is a JAK inhibitor originally approved by the U.S. FDA to treat rheumatoid arthritis (RA) and repurposed to treat psoriasis and ulcerative colitis.(734, 735) RA is a chronic autoimmune disease characterized by infiltration of inflammatory cells into the synovial membrane, the connective tissue that lines synovial joints.(735) JAK-3 is highly expressed in synovial tissue and cells of the immune system,(736, 737) and thus modulation of JAK-3 is a potential therapeutic for RA. Cytokine signaling via JAK pathways leads to further recruitment of inflammatory cells, resulting in joint damage.(735) Tofacitinib was identified in a high-throughput screen, followed by cellular assays, as an ATP competitive inhibitor primarily against JAK1 and JAK3, although it inhibits all JAKs.(734, 735, 738) It suppresses production of IL-6 and IL-8, which contribute to joint damage, by inhibiting production of IL-17 and IFNγ, possibly through the IL-2-mediated JAK-STAT signaling pathway.(735, 739) In a mouse model, Tofacitinib interfered with the generation of pathogenic T helper (Th) cells, both Th1 and Th17 cells, which is regulated by the JAK-STAT pathway.(739, 740) In a phase 3 clinical trial on RA patients, Tofacitinib significantly reduced signs and symptoms of RA and progression of joint damage(741) and was later investigated for use in treating several other diseases as discussed below.
Psoriasis is a chronic inflammatory skin disease that affects about 25 million people in North America and Europe.(742) Psoriasis is triggered by an activated immune system and characterized by infiltration of the skin with several types of immune cells, leading to hyperproliferation and aberrant differentiation of keratinocytes.(742) The dysregulated keratinocytes produce antimicrobial peptides, chemokines, and cytokines, which in turn recruit additional immune cells that feed back into the pathogenic cycle.(743) This causes thick, silvery plaques with inflammatory infiltrate containing intermixed T cells and dendritic cells.(742)Inflammation can also occur in the nail unit, resulting in nail psoriasis.(744) JAK3 is primarily expressed in hematopoietic cells, especially T cells.(732, 745) In psoriasis, JAK3 expression is enhanced(746) and thus treatment with a JAK inhibitor is an attractive therapeutic option.(747)Tofacitinib inhibits JAK-dependent cytokines such as IL-7 and IL-15, which promote survival of resident T cells and dendritic cells, leading to a reduction in their numbers in psoriatic lesions.(743) Tofacitinib inhibits differentiation of Th17 cells by inhibiting expression of IL-23 receptors, resulting in a reduction in IL-17 expression.(740, 743) Tofacitinib reduces expression of IL-19, IL-20, and IL-24, which are produced by IL-17- and IL-22-stimulated keratinocytes and signal in a JAK-dependent manner to recruit additional inflammatory cells to lesion sites.(743) At the same time, a reduction in IFNγ expression, which contributes to activation and proliferation of keratinocytes, may result from inhibition of JAK signaling required for Th1 cell differentation.(743)
In clinical trials of patients with moderate to severe plaque psoriasis, significantly more patients who received 5 or 10 mg of Tofacitinib twice daily achieved at least a 75% reduction in Psoriasis Area and Severity Index score (PASI75) and a Physician’s Global Assessment (PGA) score of 0 (“clear”) or 1 (“almost clear”) by the end of the study periods than those who received placebo.(748, 749) In the third study, significantly more patients who received Tofacitinib (5 mg or 10 mg twice daily) achieved at least a 50%, 75%, or 100% (complete) reduction from baseline in the Nail Psoriasis Severity Index (NAPSI) score than placebo.(744) In summary, these studies demonstrated efficacy and safety of Tofacitinib for treatment of moderate to severe plaque or nail psoriasis.(748, 749)
Ulcerative colitis (UC) is a chronic inflammatory disease of the colon.(750) It is characterized by mucosal inflammation, rectal bleeding, and diarrhea.(750) Although the underlying mechanism of UC is uncertain, one hypothesis is that genetic, environmental, and immunological factors cause overly aggressive T cell immune responses to bacteria of the intestines.(751) The JAK-STAT pathway has been implicated in the pathogenesis of UC.(752) Th17 cells, which were reported to be involved in the pathogenesis of UC, differentiate in response to STAT3 activation by cytokines such as IL-6, IL-21, and IL-23.(752) Inflammation in UC patients is mediated by Th2 cells, which release cytokines such as IL-4, IL-5, and IL-13.(752)
In clinical trials of patients receiving induction therapy with Tofacitinib, 18.5% of patients reached remission at 8 weeks versus 8.2% in the placebo group.(738) In a longer study, 34.3% patients receiving 5 mg of Tofacitinib and 40.6% of patients receiving 10 mg of Tofacitinib reached remission at 52 weeks versus 11.1% in the placebo group. In all studies, significantly more patients in the Tofacitinib groups experienced mucosal healing than in the placebo group. In summary, Tofacitinib was more effective than placebo in treating patients with moderate to severe UC.
Ruxolitinib (Jakafi, Incyte) is a JAK inhibitor approved by the U.S. FDA for treating primary myelofibrosis (PMF) and polycythemia vera (PV) which has been repurposed to treat alopecia areata (AA).(753) The compound was identified for inhibition of JAK targets from in vitro JAK kinase and cellular assays.(754) PMF and PV are myeloproliferative neoplasms (MPNs) that result from gain-of-function JAK2 mutations.(753) The JAK2 V617F mutation, present in more than 95% of PV and 50% of PMF patients, is caused by substitution of a GTC codon (valine) with a TTC codon (phenylalanine) at position 617.(755) The result is constitutive kinase activity and hyperproliferation of hematopoietic cells, leading to overproduction of red blood cells, leukocytes, and platelets.(755, 756) Symptoms of both diseases may include weakness, fatigue, an enlarged spleen, and anemia.(757, 758) Ruxolitinib selectively targets JAK1 and JAK2 and spares JAK3, which is critical for lymphopoiesis.(753) In preclinical studies on mice inoculated with JAK2-V617F expressing cells, Ruxolitinib reduced circulating levels of inflammatory cytokines such as IL-6 and tumor necrosis factor α (TNF-α), which contribute to symptoms seen in MF patients.(753)
Alopecia areata (AA) is an inflammatory disease caused by T cell attack on hair follicles.(759, 760)The disease typically presents with patchy hair loss, most commonly on the scalp.(760) Although hair regrowth is generally observed in patients who receive steroid treatments, the disease may progress to complete loss of scalp or body hair.(760) AA is characterized by the presence of infiltrating inflammatory cells around the hair follicle bulb.(759, 760) Cytotoxic (CD8+) T cells are the dominant cell type within the infiltrate and necessary to the pathogenesis of AA.(759) Within AA hair follicles, receptors for IL-2 and IL-15 were shown to be upregulated.(759) IL-2 and IL-15 are associated with cytotoxic activity of IFN-γ-producing CD8+ cells.(759) IFN-γ and IFN-γcreceptors signal through the JAK-STAT pathway, in particular JAK1/2 and JAK1/3, respectively.(759) These signaling pathways involved in AA can be modified through JAK inhibitors. In preclinical studies on AA mice, topical Ruxolitinib therapy reversed the disease and resulted in complete hair regrowth.(759, 761)
In clinical trials of AA patients, 20 mg of oral Ruxolitinib twice daily caused complete hair regrowth within 3–5 months.(759, 761) In a separate trial, nine of the 12 patients receiving Ruxolitinib reached the primary end point of at least 50% hair regrowth and reported a significant decrease in the mean baseline severity of alopecia tool (SALT) score, a quantitative measurement of scalp hair loss.(762) Regrowth was observed in the group as soon as 4 weeks after start of treatment. In summary, these studies show that Ruxolitinib therapy is effective for treating AA patients through inhibition of JAK-STAT signaling.
Repurposing BRAF Inhibitors
As discussed in section 4.2, personalized cancer medicines, Vemurafenib is a RAF inhibitor used to treat melanoma patients with the BRAF-V600E mutation. Vemurafenib has been repurposed to treat other cancers with this RAF mutation. The BRAF-V600E mutation is relatively rare in MM but associated with an increased incidence of extramedullary disease in patients compared to wild-type BRAF.(656) In one clinical study, a MM patient with the V600E mutation achieved remission after receiving Vemurafenib therapy.(763) In two other cases of treatment of refractory myeloma with the BRAF V600E mutation, short durations of response was observed in two patients and a durable response in the third.(656, 764, 765) In a patient with refractory MM and extramedullary manifestations, combined Vemurafenib and Cobimetinib therapy resulted in a complete response. In summary, Vemurafenib has clinical activity against MM with the BRAF V600E mutation.
Repurposing VEGF-Targeting Therapeutics
The VEGF family consists of six glycoproteins.(284) VEGF-A, or VEGF, is alternatively spliced into four isoforms of 121, 165, 189, and 206 amino acids, among which VEGF165 is the predominant form and commonly overexpressed in solid tumors.(284) Bevacizumab (Avastin, Genentech) is a monoclonal antibody approved by the U.S. FDA for treatment of metastatic colorectal cancer which has been repurposed for the treatment of wet AMD.(766) The drug is an angiogenesis inhibitor that binds all isoforms of VEGF-A, preventing it from binding to its receptors.(284, 766, 767) VEGF is expressed in over 50% of colorectal cancers.(768) In a phase 3 clinical trial of patients with metastatic colon cancer, therapy with Bevacizumab in addition to Irinotecan, fluorouracil, and Leuocovorin (IFL) significantly improved OS and PFS over IFL and placebo.(769)
Bevacizumab is commonly prescribed off-label for treatment of wet AMD.(770) In wet AMD, VEGF contributes to the development of intraocular neovascularization, leading to blindness if untreated.(771) In the first prospective study of treatment of wet AMD with Bevacizumab, patients who received three monthly injections of the drug experienced improvements in visual acuity and decreased central retinal thickness.(772) A noninferiority clinical trial was carried out to compare the efficacy of Bevacizumab and Ranibizumab (Lucentis, Genentech), an antibody approved by the U.S. FDA to treat wet AMD that neutralizes all active forms of VEGF-A.(773) After one year, 1.25 mg Bevacizumab and 0.50 mg Ranibizumab administered monthly produced equivalent gains in visual acuity and decreased central retinal thickness.(774) However, Ranibizumab alone eliminated fluid more often, and there was a higher proportion of serious adverse events occurred in the Bevacizumab group than the Ranibizumab group.(774) Altogether, these studies demonstrate that Bevacizumab, an anticanter VEGF therapy, can be used to treat wet AMD and has similar effects on visual function compared to Ranibizumab, but at a lower cost per dose.(770, 773)
Repurposing Thioredoxin Reductase Inhibitors
In all organisms, thioredoxin reductase (TrxR) catalyzes the reduction of thioredoxin by transferring electrons from nicotinamide adenine dinucleotide phosphate (NADPH).(775) Through its oxidoreductase activities, thioredoxins reduce disulfides of transcription factors, such as NF-κB and activator protein 1 (AP-1), to either activate or inactivate them.(775) Thioredoxins protect cells from oxidative stress by controlling intracellular levels of reactive oxygen species (ROS) directly or indirectly, such as by donating electrons to thioredoxin peroxidases or peroxiredoxins, which catalyze the reduction of H2O2.(775) Thioredoxin-(SH)2 forms a complex with apoptosis signaling kinase 1 (ASK1), which activates apoptosis when thioredoxin becomes oxidized.(775) In synovial fibroblasts, thioredoxin facilitates IL-17- and TNF-α-induced activation of NF-κB, which controls transcription of IL-6 and IL-8.(739, 776) Thioredoxin levels were demonstrated to be significantly increased in RA patients over healthy patients.(777) Clinical studies have demonstrated that Auranofin significantly reduced disease activity of RA compared to placebo.(778)
Auranofin (Ridaura) is a drug approved by the U.S. FDA for treating RA which has been repurposing for treating cancer.(779) It consists of a gold(I)-thiol complex stabilized by a triethylphosphine group.(779) The compound was identified from a series of gold complexes as having good bioavailability and antiarthritic activity in rats.(779) These properties were attributed to the nature of the phosphine ligand attached to the gold atom.(779) The main mechanism of Auranofin is release of gold(I) that binds to the thiol groups of redox enzymes such as thioredoxin reductase (TrxR), inhibiting electron transfer to the substrate thioredoxin (Trx).(780)
Auranofin inhibition of mitochondrial TrxR leads to a membrane permeability transition and release of cytochrome c into the cytoplasm and apoptotic cell death.(781, 782) Cancer cells overexpress redox enzymes such as TrxR, which protect the cells from DNA damage by reducing ROS.(783)Thus, inhibition of TrxR by Auranofin can lead to increased production of ROS that trigger apoptosis.(781) Preclinical studies on cell lines of several cancers demonstrated the activity of Auranofin. Auranofin induced early apoptosis in gastrointestinal stromal tumor cells.(781) In CML cells, Auranofin inhibited proteasome-associated deubiquintinases in imatinib-resistant, activating caspase and apoptosis.(784) Auranofin inhibited TrxR activity, increased ROS levels, and induced apoptosis in primary CLL cells, and significantly improved survival of an in vivo mouse model of CLL.(785) In addition to inhibiting NF-κB, Auranofin inhibited IL-6-induced activation of the JAK-STAT pathway in human multiple myeloma cells, reducing levels of the antiapoptotic protein MCL-1 and leading to apoptosis.(782) Altogether, these studies demonstrate the potential of Auranofin as a therapeutic against a variety of cancers.
Repurposing Thalidomide
Thalidomide is a drug approved by the U.S. FDA for treatment of MM and leprosy. Thalidomide and its derivatives, lenalidomide and pomalidomide, are known as immunomodulatory drugs (IMiDs).(786) It was initially prescribed as a sedative and antiemetic for morning sickness in pregnant women.(787) The drug is a synthetic glutamic acid analogue and consists of a racemic mixture of S(−) and R(+) enantiomers that interconvert under physiological conditions.(787) The R(+) form acts as a sedative while the S(−) form is a teratogen.(787) The mechanism of thalidomide’s teratogenic effects has not been established. Current theories, however, are that it damages embryos by inhibiting the ubiquitin ligase cereblon (CRBN), inhibits angiogenesis, and generates ROS that induce apoptosis in cells.(787) Thalidomide also inhibits the inflammatory response by inhibiting production of TNF-α.(787) This, in addition to its antiangiogenic and immunomodulatory properties, has led to interest in developing the drug as an anticancer therapeutic.(788)
Multiple myeloma (MM) is a blood cancer that develops in plasma cells.(789) Genetic and microenvironment changes cause malignant cells to infiltrate the bone marrow, causing lesions in the bone.(789–791) These cells release abnormal monoclonal paraproteins into the bloodstream that can cause kidney damage.(789, 792, 793) At the same time, a reduction in the number of normal blood cell results in anemia, a decrease in antibody production, and predisposition to infections.(792) In MM, IL-6 causes proliferation of MM cells and inhibits apoptosis.(794)Angiogenic cytokines VEGF, FGF-2, and IGF-2 recruit hematopoietic stem cells into the tumor microenvironment, where they differentiate into MM endothelial cells and participate in formation of new blood vessel walls.(793) NF-κB plays roles in the growth and survival of MM cells, in addition to regulating expression of many other proteins.(793) Proflammatory cytokine TNF-α regulates production of VEGF, activates secretion of IL-6, and upregulates adhesion molecules on MM cells and bone marrow stromal cells.(793) Thus, Thalidomide could be repurposed to inhibit TNF-α and affect VEGF and IL-6 levels. Thalidomide downregulates NF-κB, inducing caspase-8-mediated apoptosis, and inhibits expression of TNF-α, thereby reducing MM cell adhesion and drug resistance.(788, 793) Thalidomide also induces T cell proliferation by enhancing production of IL-2 while stimulating proliferation of natural killer cells that target MM cells.(795) Binding of Thalidomide to CRBN promotes the ubiquitination of transcription factors IKZF1 and IKZF3, resulting in their degradation and decreased viability of MM cells.(796)
Single-agent Thalidomide was used to treat relapsed MM patients in a phase 2 clinical trial.(797) A response rate of 32% occurred in patients, in 78% of whom reductions in serum or urine paraprotein levels occurred within two months.(797) In a separate phase 2 study, a response rate (defined as a drop in urine or serum paraprotein by 50% or higher) of 66% was reported in patients, with a progression-free survival rate of 80% at one year and 63% at two years.(798) In a trial in combination with dexamethasone, the response rate for thalidomide plus dexamethasone was significantly higher than for dexamethasone alone (63% and 41%, respectively).(799)Altogether, these studies showed that thalidomide has activity in MM.
Leprosy is a chronic infectious disease of the skin and peripheral nerves caused by Mycobacterium leprae.(800) It commonly presents with skin lesions, numbness, weakness, and eye pain.(800) The disease may be cured with a multidrug therapy.(800) One major complication of leprosy is erythema nodosum leprosum (ENL), which is an inflammatory reaction that may be caused by the release of M. leprae antigens from macrophages.(800, 801) The released antigens may then induce production of TNF-α, which can result in tissue and nerve damage.(802)Thalidomide inhibition of TNF-α could reduce this nerve damage in leprosy. In a clinical study on 14 patients with ENL, doses of 300 mg/day thalidomide resulted in a 50% to 80% decrease in monocyte TNF-α secretion.(803) A separate study showed that thalidomide reduced levels of activated NF-κB and TNF-α in monocytes obtained from 13 leprosy patients.(803) Altogether, these studies demonstrate that thalidomide can control inflammation in leprosy.
In this section, an overview of target-based drug repositioning was discussed. All of these drugs have undergone clinical trials or are approved for their new indications. The evolution of personalized medicine has transitioned patients to consumers and led to a demand for cheaper, safer, and more effective medicines.(696) Drug repositioning efforts may be applied to rare or neglected diseases where no therapeutic efficacy had previously existed, while providing better medicines that treat a broad spectrum of diseases.(696) Because of these important successes, many screening campaigns also seek to repurpose known drugs.(804)
6 Pharmacogenetics
The ability to sequence genomes has also greatly evolved the fields of pharmacogenomics and pharmacogenetics. The terms pharmacogenetics and pharmacogenomics are generally used interchangeably; however, pharmacogenetics generally focuses on single drug–gene interactions, while pharmacogenomics has more recently emerged as the study of the effect of a drug on a whole genome. Pharmacogenomics focuses on the identification of genes and gene variants that can alter the pharmacokinetics or pharmacodynamics of a drug. For example, genetic variants can alter the absorption of a drug, toxicity, or ability to achieve the desired pharmacological affect.
The earliest example of pharmacogenetics was in 1956 when it was discovered that a deficiency in glucose-6-phosphate dehydrogenase (G6PD) predisposes patients to hemolysis when taking several medications.(805) Transformative studies in the fields of pharmacogenetics involved studies of antipyrine (a pain reliever) metabolism in identical and fraternal twins.(806) Vesell and Page showed that the metabolism of antipyrine is consistent in identical twins but not as consistent in fraternal twins, implying that responses to drugs could be inherited.(806) Other early studies included observations that different racial groups had a different metabolism profile and different side effects to the tuberculosis drug Isoniazid.(807) Later studies showed that this was due to genetic variation in NAT2 gene which encodes N-acetyltransferase 2 enzyme.(808)
In addition to the tuberculosis drug mentioned above, codeine is another example of a drug whose metabolic profile is affected by gene variants. In particular, patients with three or more copies of cytochrome P450 gene CYP2D6 rapidly metabolize codeine to toxic levels of morphine causing severe adverse events.(809, 810) Other genetic variants in CYP2D6 can lead to reduced enzyme function or deletion, leading to poor metabolism of the drug and decreased analgesic effect.(811)Since these findings, the U.S. FDA has revised the codeine label to warn individuals who may have these severe adverse events or who will not benefit from therapy.(812) These discoveries as well as other early work revealed classic pharmacogenetic traits, such as NAT2, TPMT, and CYP2D6, that are involved in metabolism of drugs and identified inherited genetic polymorphisms that alter drug effectiveness. Genetic polymorphisms and their effects on drug metabolism have been reviewed elsewhere.(813–815) Below we discuss the use of pharmacogenetics in prescribing the anticoagulant Warfarin, the anti-HIV drug Abacavir, and Carbamazepine, an anticonvulsant.
Warfarin, an Anticoagulant
One of the most well-studied cases of pharmacogenomics is the anticoagulant drug Warfarin. Warfarin is used for prevention and treatment of venous thromboembolism and acts by inhibiting vitamin K epoxide reductase, decreasing the amount of vitamin K available for the synthesis of coagulation factors. Warfarin is highly effective, but incorrect dosing can lead to many severe adverse effects such as bleeding or thrombosis.(816) Cytochrome P450 family 2 subfamily C Member 9 (CYP2C9) is a common drug metabolizing enzyme and is the primary metabolizer of Warfarin. Individuals who are homozygous for CYP2C9 have a “normal” metabolism phenotype. Patients with one or more specific single-nucleotide polymorphisms in CYP2C9 have a greater risk of bleeding(817) and require a lower dose to achieve appropriate levels of anticoagulation.(818)
Genetic variants in the gene that encodes vitamin K epoxide reductase, VKORC1, also accounts for variations in Warfarin dosing.(818, 819) One of the common VKORC1 genetic variants (G3673A, commonly referred to as −1639G > A) is associated with warfarin sensitivity.(820) This VKORC1 polymorphism located in the promoter region alters a transcription factor binding site and leads to lower protein expression.(820) Patients with the 1639A variant have lower expression of VKORC1 and require a lower dose of warfarin compared to 1639G carriers.(821)
Since the discovery of pharmacogenetic factors that contribute to warfarin dosing, the FDA has updated the warfarin label to include recommendations for initial dosing ranges for patients with different combinations of CYP2C9 and VKORC1 genotypes. The Clinical Pharmacogenetics Implementation Consortium (CPIC) also has recommended dosing based on genotype.(822)
Abacavir, an Anti-HIV Drug
More recent studies have revealed other genetic abnormalities that can cause adverse side effects to drugs such as Abacavir for HIV infection. Abacavir is a reverse transcriptase inhibitor used in a number of treatment combinations to treat HIV infection. After its approval, many patients experienced hypersensitivity syndrome from Abacavir therapy,(823) and this hypersensitivity was later linked to the presence of a gene variant in the human leukocyte antigen complex, HLA-B*5701.(824, 825) A prospective clinical trial called PREDICT-1 was organized to determine selectivity and specificity for the gene variant–drug interaction.(824) The study found very high predictive values where 78% of patients with abacavir hypersensitivity had HLA-B*5701, and only 2% of the abacavir tolerant patients had this gene.(824) The results were initially reported in July 2007, and within a month, the number of physician-requested tests for HLA-B*5701 doubled.(824)The use of HLA-B*5701 screening has allowed physicians to accurately predict which patients will experience adverse effects to the drug. The ability to predict these effects has made Abacavir one of the preferred antiretroviral therapies.
Carbamazepine, an Anticonvulsant
Identification of other HLA gene variants has reduced the side effects of Carbamazepine. Carbamazepine is an anticonvulsant used to treat a variety of seizure disorders but can cause severe adverse reactions including hypersensitivity syndrome, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN), two types of severe skin reactions resulting in sheetlike skin and mucosal loss.(826) Patients with HLA-B*1502 variant are at a higher risk for SJS/TEN,(827)while patients with HLA-A*31:01 allele are at a higher risk of developing hypersensitivity.(828)Pharmacogenetic testing of these variants has been especially helpful for Asian populations where the frequency of the HLA-B*1502 variant is high (ranging from 1 to 10%).(829, 830)
Summary and Outlook for Pharmacogenetics
Thus, far, pharmacogenetics has been successful in identifying genetic factors contributing to side effects and effectiveness of drugs. As more and more information about genome sequence becomes available, the potential for pharmacogenetic understanding increases. Targeted therapies in cancer could become more specific if pharmacogenetics could be used to predict the effectiveness of a cancer drug.
One of the major challenges that remains in the field of pharmacogenetics is translating the science in to the clinic.(831) Currently, CPIC creates standardized guidelines on how to use genomic data to inform prescribing, although only a few gene-drug pairs are clinically actionable. These clinically actionable genes have at least one high-risk gene variant for which clinical action is recommended. Although many drug labels contain genetic information, only 16 human genes are considered to be clinically actionable based on pharmacogenomics.(832) As more sequencing data become available, the challenge will be to discover new drug–gene interactions with actionable clinical outcomes. With the few successes thus far and new sequencing technology, the field of pharmacogenetics has a promising future in helping to identify more precise and personalized medicines.
7 Conclusion and Outlook
It is clear that human genome sequencing has had an immense impact on disease biology and designing personalized therapeutics. Nevertheless, genome sequencing has not yet reached its full potential, and the field of designing drugs based on genome sequence is only in its infancy. With improved methods of sequencing, it will become easier and less expensive to sequence genomes of many patients. As more genetic information becomes available, it will also become easier to correlate this genetic information with a clinical outcome. Collecting a large data set of genome sequences will help to elucidate genetic abnormalities in diseases that currently have no known genetic causes. Identification of these underlying genetic variants will help develop therapeutics designed to target these specific genes as evidenced by the examples discussed herein.
The expansion of the field of personalized medicine has allowed new therapeutic modalities with new targets to enter the clinic. Advances in oligonucleotide therapies offer promise of regulating gene expression in disease by a variety of mechanisms. Alternatively, small molecules that target specific RNA sequences and secondary structures in disease is an emerging field that will continue to develop as more genetic information is made available. Multiple personalized cancer medicines have been developed, beginning with the discovery of the Philadelphia chromosome and the development of Imatinib. Sequencing cancer genomes has already led to the development of more precise and effective medicines and offers the promise of many more. The field of pharmacogenetics will greatly expand with more sequencing data and allow clinicians to determine the effectiveness and side-effects of therapeutics once sequencing data correlate with clinical outcomes. Lastly, rational repurposing of known drugs could potentially allow for faster drug approval and decrease the cost of drug development. Advances in genome sequencing have already transformed the study of diseases and the drug development process. In the years to come, the accumulation of more genome sequencing data and sequencing technology will revolutionize the treatment and understanding of complex diseases.
Acknowledgments
We thank Amanda Graves for helpful comments regarding therapies that target proteins. This work was funded by the National Institutes of Health (R01 GM097455, DP1 NS096898, and P01 NS099114 to M.D.D.).
Biographies
Alicia J. Angelbello attended Villanova University where she graduated with a B.S. in Chemistry in 2015. She joined the Disney laboratory in 2015 as a graduate student where she works on developing small molecules to target RNA repeat expansions.
Jonathan L. Chen completed undergraduate studies at the University of Rochester. He earned a M.S. in chemistry at the University of Southern California under the supervision of Professors G. K. Surya Prakash and Golam Rasul, followed by a Ph.D. in Chemistry at the University of Rochester under Professor Douglas H. Turner. He joined the lab of Professor Matthew D. Disney as a postdoctoral researcher in 2015.
Jessica L. Childs-Disney received her B.S. in Chemistry from Messiah College and Ph.D. in Biophysical Chemistry from the University of Rochester under the tutelage of Prof. Douglas H. Turner. She spent four years as an Assistant Professor at Canisius College before moving to The Scripps Research Institute as a staff scientist.
Peiyuan Zhang obtained his B.S. in Chemistry in 2016 from Nanjing University. He joined the Disney Laboratory as a graduate student in August 2016 and currently works on high-throughput screening to identify novel small molecules targeting RNAs of interest.
Zi-Fu Wang completed his undergraduate studies at National Taiwan Normal University in 2007 and received his M.S. and Ph.D. in Chemistry at National Taiwan University under the supervision of Professor Ta-Chau Chang in 2013. He joined the lab of Professor Matthew D. Disney as a visiting scholar in 2016, where he works on targeting RNA G4C2 repeat expansions in ALS/FTD.
Matthew D. Disney received his B.S. in Chemistry from the University of Maryland (College Park) and his M.S. and Ph.D. degrees in Physical Chemistry at the University of Rochester. He completed postdoctoral work at the Massachusetts Institute of Technology and the Swiss Federal Institute of Technology (ETH) Zurich, after which he began his independent research program. The Disney Laboratory is focused on developing precision medicines for disease-causing human RNAs, long-considered “undruggable” targets for small molecules. He is a Full Professor in the bicoastal Department of Chemistry at the Scripps Research Institute, Jupiter, Florida.
Footnotes
The authors declare no competing financial interest.
References
This article references 832 other publications.
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. Initial Sequencing and Analysis of the Human Genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Holley RW, Apgar J, Everett GA, Madison JT, Marquisee M, Merrill SH, Penswick JR, Zamir A. Structure of a Ribonucleic Acid. Science. 1965;147:1462–1465. doi: 10.1126/science.147.3664.1462. [DOI] [PubMed] [Google Scholar]
- 3.Sanger F, Brownlee GG, Barrell BG. A Two-Dimensional Fractionation Procedure for Radioactive Nucleotides. J Mol Biol. 1965;13:373–IN374. doi: 10.1016/S0022-2836(65)80104-8. [DOI] [PubMed] [Google Scholar]
- 4.Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A. Complete Nucleotide Sequence of Bacteriophage Ms2 Rna: Primary and Secondary Structure of the Replicase Gene. Nature. 1976;260:500–507. doi: 10.1038/260500a0. [DOI] [PubMed] [Google Scholar]
- 5.Sanger F, Coulson AR. A Rapid Method for Determining Sequences in DNA by Primed Synthesis with DNA Polymerase. J Mol Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 6.Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, Hutchison CA, Slocombe PM, Smith M. Nucleotide Sequence of Bacteriophage Phi X174 DNA. Nature. 1977;265:687–695. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- 7.Maxam AM, Gilbert W. A New Method for Sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanger F, Nicklen S, Coulson AR. DNA Sequencing with Chain-Terminating Inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F. Sequence and Organization of the Human Mitochondrial Genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 10.Prober JM, Trainor GL, Dam RJ, Hobbs FW, Robertson CW, Zagursky RJ, Cocuzza AJ, Jensen MA, Baumeister K. A System for Rapid DNA Sequencing with Fluorescent Chain-Terminating Dideoxynucleotides. Science. 1987;238:336–341. doi: 10.1126/science.2443975. [DOI] [PubMed] [Google Scholar]
- 11.Swerdlow H, Gesteland R. Capillary Gel Electrophoresis for Rapid, High Resolution DNA Sequencing. Nucleic Acids Res. 1990;18:1415–1419. doi: 10.1093/nar/18.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunkapiller T, Kaiser RJ, Koop BF, Hood L. Large-Scale and Automated DNA Sequence Determination. Science. 1991;254:59–67. doi: 10.1126/science.1925562. [DOI] [PubMed] [Google Scholar]
- 13.Anderson S. Shotgun DNA Sequencing Using Cloned Dnase I-Generated Fragments. Nucleic Acids Res. 1981;9:3015–3027. doi: 10.1093/nar/9.13.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen SN, Chang AC, Boyer HW, Helling RB. Construction of Biologically Functional Bacterial Plasmids in Vitro. Proc Natl Acad Sci U S A. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson DA, Symons RH, Berg P. Biochemical Method for Inserting New Genetic Information into DNA of Simian Virus 40: Circular Sv40 DNA Molecules Containing Lambda Phage Genes and the Galactose Operon of Escherichia Coli. Proc Natl Acad Sci U S A. 1972;69:2904–2909. doi: 10.1073/pnas.69.10.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic Amplification of Beta-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 17.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 18.Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent SB, Hood LE. Fluorescence Detection in Automated DNA Sequence Analysis. Nature. 1986;321:674–679. doi: 10.1038/321674a0. [DOI] [PubMed] [Google Scholar]
- 19.Oliver SG, van der Aart QJ, Agostoni-Carbone ML, Aigle M, Alberghina L, Alexandraki D, Antoine G, Anwar R, Ballesta JP, Benit P. The Complete DNA Sequence of Yeast Chromosome Iii. Nature. 1992;357:38–46. doi: 10.1038/357038a0. [DOI] [PubMed] [Google Scholar]
- 20.Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, Bonfield J, Burton J, Connell M, Copsey T, Cooper J. 2.2 Mb of Contiguous Nucleotide Sequence from Chromosome Iii of C. Elegans. Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- 21.Mardis ER. A Decade/’S Perspective on DNA Sequencing Technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 22.Shendure J, Ji H. Next-Generation DNA Sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 23.Metzker ML. Sequencing Technologies [Mdash] the Next Generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 24. [accessed June 21];DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) www.genome.gov/sequencingcostsdata.
- 25.Neel JV. The Inheritance of Sickle Cell Anemia. Science. 1949;110:64–66. doi: 10.1126/science.110.2846.64. [DOI] [PubMed] [Google Scholar]
- 26.Pauling L, Itano HA. Sickle Cell Anemia a Molecular Disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 27.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY. A Polymorphic DNA Marker Genetically Linked to Huntington’s Disease. Nature. 1983;306:234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- 28.The Huntington’s Disease Collaborative Research Group. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 29.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP. Identification of a Gene (Fmr-1) Containing a Cgg Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 30.Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, Tassone F, Brown WT, Greco CM, Hagerman PJ. Fragile X Premutation Carriers: Characteristic Mr Imaging Findings of Adult Male Patients with Progressive Cerebellar and Cognitive Dysfunction. Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- 31.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen Receptor Gene Mutations in X-Linked Spinal and Bulbar Muscular Atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 32.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T. Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (Ctg) Repeat at the 3′ End of a Transcript Encoding a Protein Kinase Family Member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 33.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the Cystic Fibrosis Gene: Genetic Analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 34.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 35.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 36.Rowe SM, Miller S, Sorscher EJ. Cystic Fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 37.Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DCY, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA. Whole-Genome Sequencing in a Patient with Charcot–Marie–Tooth Neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M. Analysis of Genetic Inheritance in a Family Quartet by Whole Genome Sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA. Exome Sequencing Identifies the Cause of a Mendelian Disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin M, Gildersleeve H, Beck AE, Tabor HK, Cooper GM, Mefford HC. Exome Sequencing Identifies Mll2Mutations as a Cause of Kabuki Syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowell PC. Discovery of the Philadelphia Chromosome: A Personal Perspective. J Clin Invest. 2007;117:2033–2035. doi: 10.1172/JCI31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deininger M, Buchdunger E, Druker BJ. The Development of Imatinib as a Therapeutic Agent for Chronic Myeloid Leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 43.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and Specific Genetic Interference by Double-Stranded Rna in Caenorhabditis Elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 44.Lee RC, Feinbaum RL, Ambros V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small Rnas with Antisense Complementarity to Lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 45.Garofalo M, Condorelli G, Croce CM. Micrornas in Diseases and Drug Response. Curr Opin Pharmacol. 2008;8:661–667. doi: 10.1016/j.coph.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Zamecnik PC, Stephenson ML. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellington AD, Szostak JW. In Vitro Selection of Rna Molecules That Bind Specific Ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 48.Robertson DL, Joyce GF. Selection in Vitro of an Rna Enzyme That Specifically Cleaves Single-Stranded DNA. Nature. 1990;344:467–468. doi: 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- 49.Tuerk C, Gold L. Systematic Evolution of Ligands by Exponential Enrichment: Rna Ligands to Bacteriophage T4 DNA Polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 50.Vickers TA, Wyatt JR, Freier SM. Effects of Rna Secondary Structure on Cellular Antisense Activity. Nucleic Acids Res. 2000;28:1340–1347. doi: 10.1093/nar/28.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frazier KS. Antisense Oligonucleotide Therapies: The Promise and the Challenges from a Toxicologic Pathologist’s Perspective. Toxicol Pathol. 2015;43:78–89. doi: 10.1177/0192623314551840. [DOI] [PubMed] [Google Scholar]
- 52.Agrawal S. Importance of Nucleotide Sequence and Chemical Modifications of Antisense Oligonucleotides. Biochim Biophys Acta, Gene Struct Expression. 1999;1489:53–68. doi: 10.1016/S0167-4781(99)00141-4. [DOI] [PubMed] [Google Scholar]
- 53.Eckstein F. Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014;24:374–387. doi: 10.1089/nat.2014.0506. [DOI] [PubMed] [Google Scholar]
- 54.Campbell MA, Wengel J. Locked Vs. Unlocked Nucleic Acids (Lna Vs. Una): Contrasting Structures Work Towards Common Therapeutic Goals. Chem Soc Rev. 2011;40:5680–5689. doi: 10.1039/c1cs15048k. [DOI] [PubMed] [Google Scholar]
- 55.Prakash TP, Bhat B. 2′-Modified Oligonucleotides for Antisense Therapeutics. Curr Top Med Chem. 2007;7:641–649. doi: 10.2174/156802607780487713. [DOI] [PubMed] [Google Scholar]
- 56.Järver P, O’Donovan L, Gait MJ. A Chemical View of Oligonucleotides for Exon Skipping and Related Drug Applications. Nucleic Acid Ther. 2014;24:37–47. doi: 10.1089/nat.2013.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu RZ, Kim TW, Hong A, Watanabe TA, Gaus HJ, Geary RS. Cross-Species Pharmacokinetic Comparison from Mouse to Man of a Second-Generation Antisense Oligonucleotide, Isis 301012, Targeting Human Apolipoprotein B-100. Drug Metab Dispos. 2006;35:460–468. doi: 10.1124/dmd.106.012401. [DOI] [PubMed] [Google Scholar]
- 58.Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of Antisense Oligonucleotides Stabilized by Locked Nucleic Acids. Nucleic Acids Res. 2002;30:1911–1918. doi: 10.1093/nar/30.9.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang L, Liu Y. In Vivo Delivery of Rnai with Lipid-Based Nanoparticles. Annu Rev Biomed Eng. 2011;13:507–530. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Miao L, Satterlee A, Huang L. Delivery of Oligonucleotides with Lipid Nanoparticles. Adv Drug Delivery Rev. 2015;87:68–80. doi: 10.1016/j.addr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winkler J. Oligonucleotide Conjugates for Therapeutic Applications. Ther Delivery. 2013;4:791–809. doi: 10.4155/tde.13.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ming X, Laing B. Bioconjugates for Targeted Delivery of Therapeutic Oligonucleotides. Adv Drug Delivery Rev. 2015;87:81–89. doi: 10.1016/j.addr.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P. Antibody Mediated in Vivo Delivery of Small Interfering Rnas Via Cell-Surface Receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 64.Nakagawa O, Ming X, Huang L, Juliano RL. Targeted Intracellular Delivery of Antisense Oligonucleotides Via Conjugation with Small-Molecule Ligands. J Am Chem Soc. 2010;132:8848–8849. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crooke ST, Graham MJ, Zuckerman JE, Brooks D, Conklin BS, Cummins LL, Greig MJ, Guinosso CJ, Kornbrust D, Manoharan M. Pharmacokinetic Properties of Several Novel Oligonucleotide Analogs in Mice. J Pharmacol Exp Ther. 1996;277:923–937. [PubMed] [Google Scholar]
- 66.Prakash TP, Yu J, Migawa MT, Kinberger GA, Wan WB, Ostergaard ME, Carty RL, Vasquez G, Low A, Chappell A. Comprehensive Structure-Activity Relationship of Triantennary N-Acetylgalactosamine Conjugated Antisense Oligonucleotides for Targeted Delivery to Hepatocytes. J Med Chem. 2016;59:2718–2733. doi: 10.1021/acs.jmedchem.5b01948. [DOI] [PubMed] [Google Scholar]
- 67.Juliano RL. The Delivery of Therapeutic Oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watts JK, Corey DR. Silencing Disease Genes in the Laboratory and the Clinic. J Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma VK, Watts JK. Oligonucleotide Therapeutics: Chemistry, Delivery and Clinical Progress. Future Med Chem. 2015;7:2221–2242. doi: 10.4155/fmc.15.144. [DOI] [PubMed] [Google Scholar]
- 70.Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol. 2017;57:81–105. doi: 10.1146/annurev-pharmtox-010716-104846. [DOI] [PubMed] [Google Scholar]
- 71.Stephenson ML, Zamecnik PC. Inhibition of Rous Sarcoma Viral Rna Translation by a Specific Oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keller W, Crouch R. Degradation of DNA Rna Hybrids by Ribonuclease H and DNA Polymerases of Cellular and Viral Origin. Proc Natl Acad Sci U S A. 1972;69:3360–3364. doi: 10.1073/pnas.69.11.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sierakowska H, Gorman L, Kang SH, Kole R. Antisense Oligonucleotides and Rnas as Modulators of Pre-Mrna Splicing. Methods Enzymol. 2000;313:506–521. doi: 10.1016/S0076-6879(00)13032-0. [DOI] [PubMed] [Google Scholar]
- 74.Dominski Z, Kole R. Restoration of Correct Splicing in Thalassemic Pre-Mrna by Antisense Oligonucleotides. Proc Natl Acad Sci U S A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suwanmanee T, Sierakowska H, Fucharoen S, Kole R. Repair of a Splicing Defect in Erythroid Cells from Patients with Beta-Thalassemia/Hbe Disorder. Mol Ther. 2002;6:718–726. doi: 10.1006/mthe.2002.0805. [DOI] [PubMed] [Google Scholar]
- 76.The Vitravene Study Group. A Randomized Controlled Clinical Trial of Intravitreous Fomivirsen for Treatment of Newly Diagnosed Peripheral Cytomegalovirus Retinitis in Patients with Aids. Am J Ophthalmol. 2002;133:467–474. doi: 10.1016/S0002-9394(02)01327-2. [DOI] [PubMed] [Google Scholar]
- 77.The Vitravene Study Group. Randomized Dose-Comparison Studies of Intravitreous Fomivirsen for Treatment of Cytomegalovirus Retinitis That Has Reactivated or Is Persistently Active Despite Other Therapies in Patients with Aids. Am J Ophthalmol. 2002;133:475–483. doi: 10.1016/S0002-9394(02)01326-0. [DOI] [PubMed] [Google Scholar]
- 78.The Vitravene Study Group. Safety of Intravitreous Fomivirsen for Treatment of Cytomegalovirus Retinitis in Patients with Aids. Am J Ophthalmol. 2002;133:484–498. doi: 10.1016/S0002-9394(02)01332-6. [DOI] [PubMed] [Google Scholar]
- 79.Waldmann E, Vogt A, Crispin A, Altenhofer J, Riks I, Parhofer KG. Effect of Mipomersen on Ldl-Cholesterol in Patients with Severe Ldl-Hypercholesterolaemia and Atherosclerosis Treated by Lipoprotein Apheresis (the Mica-Study) Atherosclerosis. 2017;259:20–25. doi: 10.1016/j.atherosclerosis.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 80.Geary RS, Baker BF, Crooke ST. Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (Kynamro®): A Second-Generation Antisense Oligonucleotide Inhibitor of Apolipoprotein B. Clin Pharmacokinet. 2015;54:133–146. doi: 10.1007/s40262-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kastelein JJ, Wedel MK, Baker BF, Su J, Bradley JD, Yu RZ, Chuang E, Graham MJ, Crooke RM. Potent Reduction of Apolipoprotein B and Low-Density Lipoprotein Cholesterol by Short-Term Administration of an Antisense Inhibitor of Apolipoprotein B. Circulation. 2006;114:1729–1735. doi: 10.1161/CIRCULATIONAHA.105.606442. [DOI] [PubMed] [Google Scholar]
- 82.Yu RZ, Geary RS, Flaim JD, Riley GC, Tribble DL, van Vliet AA, Wedel MK. Lack of Pharmacokinetic Interaction of Mipomersen Sodium (Isis 301012), a 2′-O-Methoxyethyl Modified Antisense Oligonucleotide Targeting Apolipoprotein B-100 Messenger Rna, with Simvastatin and Ezetimibe. Clin Pharmacokinet. 2009;48:39–50. doi: 10.2165/0003088-200948010-00003. [DOI] [PubMed] [Google Scholar]
- 83.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S. Mipomersen, an Apolipoprotein B Synthesis Inhibitor, for Lowering of Ldl Cholesterol Concentrations in Patients with Homozygous Familial Hypercholesterolaemia: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 84.Akdim F, Stroes ES, Sijbrands EJ, Tribble DL, Trip MD, Jukema JW, Flaim JD, Su J, Yu R, Baker BF. Efficacy and Safety of Mipomersen, an Antisense Inhibitor of Apolipoprotein B, in Hypercholesterolemic Subjects Receiving Stable Statin Therapy. J Am Coll Cardiol. 2010;55:1611–1618. doi: 10.1016/j.jacc.2009.11.069. [DOI] [PubMed] [Google Scholar]
- 85.Akdim F, Visser ME, Tribble DL, Baker BF, Stroes ES, Yu R, Flaim JD, Su J, Stein EA, Kastelein JJ. Effect of Mipomersen, an Apolipoprotein B Synthesis Inhibitor, on Low-Density Lipoprotein Cholesterol in Patients with Familial Hypercholesterolemia. Am J Cardiol. 2010;105:1413–1419. doi: 10.1016/j.amjcard.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 86.McGowan MP, Tardif JC, Ceska R, Burgess LJ, Soran H, Gouni-Berthold I, Wagener G, Chasan-Taber S. Randomized, Placebo-Controlled Trial of Mipomersen in Patients with Severe Hypercholesterolemia Receiving Maximally Tolerated Lipid-Lowering Therapy. PLoS One. 2012;7:e49006. doi: 10.1371/journal.pone.0049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, Chin W, Tribble DL, McGowan M. Apolipoprotein B Synthesis Inhibition with Mipomersen in Heterozygous Familial Hypercholesterolemia: Results of a Randomized, Double-Blind, Placebo-Controlled Trial to Assess Efficacy and Safety as Add-on Therapy in Patients with Coronary Artery Disease. Circulation. 2012;126:2283–2292. doi: 10.1161/CIRCULATIONAHA.112.104125. [DOI] [PubMed] [Google Scholar]
- 88.Davis RA. Cell and Molecular Biology of the Assembly and Secretion of Apolipoprotein B-Containing Lipoproteins by the Liver. Biochim Biophys Acta, Mol Cell Biol Lipids. 1999;1440:1–31. doi: 10.1016/S1388-1981(99)00083-9. [DOI] [PubMed] [Google Scholar]
- 89.Raal FJ, Santos RD. Homozygous Familial Hypercholesterolemia: Current Perspectives on Diagnosis and Treatment. Atherosclerosis. 2012;223:262–268. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 90.Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, Marais AD. Reduction in Mortality in Subjects with Homozygous Familial Hypercholesterolemia Associated with Advances in Lipid-Lowering Therapy. Circulation. 2011;124:2202–2207. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 91.Sjouke B, Balak DM, Beuers U, Ratziu V, Stroes ES. Is Mipomersen Ready for Clinical Implementation? A Transatlantic Dilemma. Curr Opin Lipidol. 2013;24:301–306. doi: 10.1097/MOL.0b013e328362dfd9. [DOI] [PubMed] [Google Scholar]
- 92.Kling J. Safety Signal Dampens Reception for Mipomersen Antisense. Nat Biotechnol. 2010;28:295–297. doi: 10.1038/nbt0410-295. [DOI] [PubMed] [Google Scholar]
- 93.Davis KA, Miyares MA. Lomitapide: A Novel Agent for the Treatment of Homozygous Familial Hypercholesterolemia. Am J Health-Syst Pharm. 2014;71:1001–1008. doi: 10.2146/ajhp130592. [DOI] [PubMed] [Google Scholar]
- 94.Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D. Efficacy and Safety of a Microsomal Triglyceride Transfer Protein Inhibitor in Patients with Homozygous Familial Hypercholesterolaemia: A Single-Arm, Open-Label, Phase 3 Study. Lancet. 2013;381:40–46. doi: 10.1016/S0140-6736(12)61731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koenig M, Monaco AP, Kunkel LM. The Complete Sequence of Dystrophin Predicts a Rod-Shaped Cytoskeletal Protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 96.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: The Protein Product of the Duchenne Muscular Dystrophy Locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 97.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin Protects the Sarcolemma from Stresses Developed During Muscle Contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kerr TP, Sewry CA, Robb SA, Roberts RG. Long Mutant Dystrophins and Variable Phenotypes: Evasion of Nonsense-Mediated Decay? Hum Genet. 2001;109:402–407. doi: 10.1007/s004390100598. [DOI] [PubMed] [Google Scholar]
- 99.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An Explanation for the Phenotypic Differences between Patients Bearing Partial Deletions of the Dmd Locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 100.Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J. Eteplirsen for the Treatment of Duchenne Muscular Dystrophy. Ann Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 101.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ. Exon Skipping and Dystrophin Restoration in Patients with Duchenne Muscular Dystrophy after Systemic Phosphorodiamidate Morpholino Oligomer Treatment: An Open-Label, Phase 2, Dose-Escalation Study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dowling JJ. Eteplirsen Therapy for Duchenne Muscular Dystrophy: Skipping to the Front of the Line. Nat Rev Neurol. 2016;12:675–676. doi: 10.1038/nrneurol.2016.180. [DOI] [PubMed] [Google Scholar]
- 103.Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T. The Treat-Nmd Dmd Global Database: Analysis of More Than 7,000 Duchenne Muscular Dystrophy Mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D. Genetic Mapping of Chronic Childhood-Onset Spinal Muscular Atrophy to Chromosome 5q11.2–13.3. Nature. 1990;344:540–541. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 105.Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015;33:831–846. doi: 10.1016/j.ncl.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the Sma Gene Smn1 from the Copy Gene Smn2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 107.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A Single Nucleotide in the Smn Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burnett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of Smn Protein Stability. Mol Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Watihayati MS, Fatemeh H, Marini M, Atif AB, Zahiruddin WM, Sasongko TH, Tang TH, Zabidi-Hussin Z, Nishio H, Zilfalil BA. Combination of Smn2 Copy Number and Naip Deletion Predicts Disease Severity in Spinal Muscular Atrophy. Brain Dev. 2009;31:42–45. doi: 10.1016/j.braindev.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 110.Cartegni L, Krainer AR. Correction of Disease-Associated Exon Skipping by Synthetic Exon-Specific Activators. Nat Struct Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 111.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a Critical Exon of Human Survival Motor Neuron Is Regulated by a Unique Silencer Element Located in the Last Intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G. Antisense Oligonucleotides Delivered to the Mouse Cns Ameliorate Symptoms of Severe Spinal Muscular Atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. Results from a Phase 1 Study of Nusinersen (Isis-Smn(Rx)) in Children with Spinal Muscular Atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E. Treatment of Infantile-Onset Spinal Muscular Atrophy with Nusinersen: A Phase 2, Open-Label, Dose-Escalation Study. Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 115. [accessed June 21];FDA approves first drug for spinal muscular atrophy. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm534611.htm.
- 116.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer Functions in RNA Interference and in Synthesis of Small Rna Involved in Developmental Timing in C. Elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Elbashir SM, Lendeckel W, Tuschl T. Rna Interference Is Mediated by 21- and 22-Nucleotide Rnas. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-Nucleotide Rnas Mediate Rna Interference in Cultured Mammalian Cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 119.Hammond SM, Bernstein E, Beach D, Hannon GJ. An Rna-Directed Nuclease Mediates Post-Transcriptional Gene Silencing in Drosophila Cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 120.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a Link between Genetic and Biochemical Analyses of Rnai. Science. 2001;293:1146–1150. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 121.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. Genes and Mechanisms Related to Rna Interference Regulate Expression of the Small Temporal Rnas That Control C. Elegans Developmental Timing. Cell. 2001;106:23–34. doi: 10.1016/S0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 122.Hu WY, Myers CP, Kilzer JM, Pfaff SL, Bushman FD. Inhibition of Retroviral Pathogenesis by Rna Interference. Curr Biol. 2002;12:1301–1311. doi: 10.1016/S0960-9822(02)00975-2. [DOI] [PubMed] [Google Scholar]
- 123.Mourrain P, Beclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, Jouette D, Lacombe AM, Nikic S, Picault N. Arabidopsis Sgs2 and Sgs3 Genes Are Required for Posttranscriptional Gene Silencing and Natural Virus Resistance. Cell. 2000;101:533–542. doi: 10.1016/S0092-8674(00)80863-6. [DOI] [PubMed] [Google Scholar]
- 124.Bartlett DW, Davis ME. Insights into the Kinetics of Sirna-Mediated Gene Silencing from Live-Cell and Live-Animal Bioluminescent Imaging. Nucleic Acids Res. 2006;34:322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Behlke MA. Chemical Modification of Sirnas for in Vivo Use. Oligonucleotides. 2008;18:305–319. doi: 10.1089/oli.2008.0164. [DOI] [PubMed] [Google Scholar]
- 126.Rettig GR, Behlke MA. Progress toward in Vivo Use of Sirnas-Ii. Mol Ther. 2012;20:483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bobbin ML, Rossi JJ. Rna Interference (Rnai)-Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- 128.Wittrup A, Lieberman J. Knocking Down Disease: A Progress Report on Sirna Therapeutics. Nat Rev Genet. 2015;16:543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zuckerman JE, Davis ME. Clinical Experiences with Systemically Administered Sirna-Based Therapeutics in Cancer. Nat Rev Drug Discovery. 2015;14:843–856. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 130.Thakur A, Fitzpatrick S, Zaman A, Kugathasan K, Muirhead B, Hortelano G, Sheardown H. Strategies for Ocular Sirna Delivery: Potential and Limitations of Non-Viral Nanocarriers. J Biol Eng. 2012;6:7. doi: 10.1186/1754-1611-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gonzalez V, Moreno-Montañés J, Sádaba B, Ruz V, Jímenez AI. Syl1001 for Treatment of Ocular Discomfort in Dry Eye: Safety and Tolerance (Phase I Study) Invest Ophthalmol Visual Sci. 2012;53:575. [Google Scholar]
- 132.Martinez T, Gonzalez MV, Roehl I, Wright N, Paneda C, Jimenez AI. In Vitro and in Vivo Efficacy of Syl040012, a Novel Sirna Compound for Treatment of Glaucoma. Mol Ther. 2014;22:81–91. doi: 10.1038/mt.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee DU, Huang W, Rittenhouse KD, Jessen B. Retina Expression and Cross-Species Validation of Gene Silencing by Pf-655, a Small Interfering Rna against Rtp801 for the Treatment of Ocular Disease. J Ocul Pharmacol Ther. 2012;28:222–230. doi: 10.1089/jop.2011.0116. [DOI] [PubMed] [Google Scholar]
- 134.Solano EC, Kornbrust DJ, Beaudry A, Foy JW, Schneider DJ, Thompson JD. Toxicological and Pharmacokinetic Properties of Qpi-1007, a Chemically Modified Synthetic Sirna Targeting Caspase 2 Mrna, Following Intravitreal Injection. Nucleic Acid Ther. 2014;24:258–266. doi: 10.1089/nat.2014.0489. [DOI] [PubMed] [Google Scholar]
- 135. [accessed June 21];Quark QPI-1007. http://quarkpharma.com/?page_id=23.
- 136.Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J. Effect of an Rna Interference Drug on the Synthesis of Proprotein Convertase Subtilisin/Kexin Type 9 (Pcsk9) and the Concentration of Serum Ldl Cholesterol in Healthy Volunteers: A Randomised, Single-Blind, Placebo-Controlled, Phase 1 Trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. [accessed June 21, 2017];Alnylam Pharmacueticals, Hemophilia. http://www.alnylam.com/patients/hemophilia/
- 138. [accessed June 21];New Anti-fibrosis Drug with Molecular Targeting DDS Completed Phase-1a Dose Escalation. https://www.nitto.com/us/en/press/2014/0422.jsp.
- 139.Updates from Patisiran and Revusiran. [accessed June 21, 2017];Development for the Treatment of hATTR Amyloidosis. http://www.alnylam.com/2016/07/01/updates-from-patisiran-and-revusiran-for-hattr-amyloidosis/
- 140.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E. Safety and Efficacy of Rnai Therapy for Transthyretin Amyloidosis. N Engl J Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 141.Nemunaitis J, Barve M, Orr D, Kuhn J, Magee M, Lamont J, Bedell C, Wallraven G, Pappen BO, Roth A. Summary of Bi-Shrna/Gm-Csf Augmented Autologous Tumor Cell Immunotherapy (Fang) in Advanced Cancer of the Liver. Oncology. 2014;87:21–29. doi: 10.1159/000360993. [DOI] [PubMed] [Google Scholar]
- 142.Arbutus. [accessed June 21, 2017];TKM-PLK1. http://arbutusbio.com/portfolio/tkm-plk1.php.
- 143.Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, Domb A, Harari G, David EB, Raskin S. Rnai Therapy Targeting Kras in Combination with Chemotherapy for Locally Advanced Pancreatic Cancer Patients. Oncotarget. 2015;6:24560–24570. doi: 10.18632/oncotarget.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Leachman SA, Hickerson RP, Schwartz ME, Bullough EE, Hutcherson SL, Boucher KM, Hansen CD, Eliason MJ, Srivatsa GS, Kornbrust DJ. First-in-Hum. Mutat. -Targeted Sirna Phase Ib Trial of an Inherited Skin Disorder. Mol Ther. 2010;18:442–446. doi: 10.1038/mt.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. [accessed June 21];RXI-109. http://www.rxipharma.com/technology/rxi-109/
- 146. [accessed June 21, 2017];Marina Biotech’s CEQ508 Granted FDA Fast Track Designation for Familial Adenomatous Polyposis. http://www.marinabio.com/files/5014/3880/5803/15-03-30_-_Marina_Biotech_Announces_FDA_Fast_Track_Designation_for_CEQ508.pdf.
- 147.Benitec Biopharma. [accessed August 18, 2017];In-house programs detail: 1. HepatitisC. 2015 http://www.benitec.com/pipeline/in-house-programs-detail#hepc.
- 148.Pol S, Lampertico P. First-Line Treatment of Chronic Hepatitis B with Entecavir or Tenofovir in ’Real-Life’ Settings: From Clinical Trials to Clinical Practice. J Viral Hepat. 2012;19:377–386. doi: 10.1111/j.1365-2893.2012.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Arbutus. [accessed August 18th, 2017];Tekmira initiates PhaseI clinical trialof TKM-HBV. 2015 http://www.sec.gov/Archives/edgar/data/1447028/000117184315000285/newsrelease.htm.
- 150.Geisbert TW, Lee AC, Robbins M, Geisbert JB, Honko AN, Sood V, Johnson JC, de Jong S, Tavakoli I, Judge A. Postexposure Protection of Non-Human Primates against a Lethal Ebola Virus Challenge with Rna Interference: A Proof-of-Concept Study. Lancet. 2010;375:1896–1905. doi: 10.1016/S0140-6736(10)60357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Müller P. Conservation of the Sequence and Temporal Expression of Let-7 Heterochronic Regulatory Rna. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 152.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-Nucleotide Let-7 Rna Regulates Developmental Timing in Caenorhabditis Elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 153.Bartel DP. Micrornas: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 154.Bartel DP. Micrornas: Target Recognition and Regulatory Functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M. Combinatorial Microrna Target Predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 156.Lewis BP, Burge CB, Bartel DP. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are Microrna Targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 157.Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. Mirna Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discovery. 2016;6:235–246. doi: 10.1158/2159-8290.CD-15-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rupaimoole R, Slack FJ. Microrna Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat Rev Drug Discovery. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 159.Ha M, Kim VN. Regulation of Microrna Biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 160.Lin S, Gregory RI. Microrna Biogenesis Pathways in Cancer. Nat Rev Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of Micrornas in Vivo with ‘Antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 162.Hoofnagle JH. Course and Outcome of Hepatitis C. Hepatology. 2002;36:s21. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 163.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of Tissue-Specific Micrornas from Mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/S0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 164.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of Hepatitis C Virus Rna Abundance by a Liver-Specific Microrna. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 165.Thibault PA, Huys A, Amador-Cañizares Y, Gailius JE, Pinel DE, Wilson JA. Regulation of Hepatitis C Virus Genome Replication by Xrn1 and Microrna-122 Binding to Individual Sites in the 5′ Untranslated Region. J Virol. 2015;89:6294–6311. doi: 10.1128/JVI.03631-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjärn M, Hansen JB, Hansen HF, Straarup EM. Antagonism of Microrna-122 in Mice by Systemically Administered Lna-Antimir Leads to up-Regulation of a Large Set of Predicted Target Mrnas in the Liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U. LNA-Mediated Microrna Silencing in Non-Human Primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 168.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. Therapeutic Silencing of Microrna-122 in Primates with Chronic Hepatitis C Virus Infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y. Treatment of Hcv Infection by Targeting Microrna. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 170.Gebert LF, Rebhan MA, Crivelli SE, Denzler R, Stoffel M, Hall J. Miravirsen (Spc3649) Can Inhibit the Biogenesis of Mir-122. Nucleic Acids Res. 2014;42:609–621. doi: 10.1093/nar/gkt852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.van der Ree MH, de Vree JM, Stelma F, Willemse S, van der Valk M, Rietdijk S, Molenkamp R, Schinkel J, van Nuenen AC, Beuers U. Safety,Tolerability, and Antiviral Effect of Rg-101 in Patients with Chronic Hepatitis C: A Phase 1b, Double-Blind, Randomised Controlled Trial. Lancet. 2017;389:709–717. doi: 10.1016/S0140-6736(16)31715-9. [DOI] [PubMed] [Google Scholar]
- 172.Gironella M, Seux M, Xie M-J, Cano C, Tomasini R, Gommeaux J, Garcia S, Nowak J, Yeung ML, Jeang K-T. Tumor Protein 53-Induced Nuclear Protein 1 Expression Is Repressed by Mir-155, and Its Restoration Inhibits Pancreatic Tumor Development. Proc Natl Acad Sci U S A. 2007;104:16170–16175. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol Phosphatase Ship1 Is a Primary Target of Mir-155. Proc Natl Acad Sci U S A. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tili E, Michaille J-J, Wernicke D, Alder H, Costinean S, Volinia S, Croce CM. Mutator Activity Induced by Microrna-155 (Mir-155) Links Inflammation and Cancer. Proc Natl Acad Sci U S A. 2011;108:4908–4913. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Foss FM, Querfeld C, Porcu P, Kim YH, Pacheco T, Halwani AS, DeSimone J, William BM, Seto AG, Ruckman J, Landry M, Jackson AL, Pestano LA, Dickinson BA, Sanseverino M, Rodman DM, Rubin P, Gordon GS, Marshall WS. Phase 1 Trial Evaluating Mrg-106, a Synthetic Inhibitor of MicroRNA-155, in Patients with Cutaneous T-Cell Lymphoma (Ctcl). Proceedings of the 2017 Asco Annual Meeting; Chicago, Illinois. June 5, 2017; Abstract 7564. [Google Scholar]
- 176.Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. Micrornas 103 and 107 Regulate Insulin Sensitivity. Nature. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- 177.RG-125(AZD4076), a microRNA Therapeutic Targeting microRNA-103/107 Being Developed for the Treatment of NASH in Patients with Type 2 Diabetes/Pre-Diabetes, Enters Phase I Clinical Development. [accessed July 14, 2017]; http://ir.regulusrx.com/releasedetail.cfm?releaseid=947579.
- 178.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour Invasion and Metastasis Initiated by Microrna-10b in Breast Cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 179.Yoo B, Kavishwar A, Ross A, Wang P, Tabassum DP, Polyak K, Barteneva N, Petkova V, Pantazopoulos P, Tena A. Combining Mir-10b–Targeted Nanotherapy with Low-Dose Doxorubicin Elicits Durable Regressions of Metastatic Breast Cancer. Cancer Res. 2015;75:4407–4415. doi: 10.1158/0008-5472.CAN-15-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM, Dejean A. Mir-221 Overexpression Contributes to Liver Tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA. Regulation of the P27kip1 Tumor Suppressor by Mir-221 and Mir-222 Promotes Cancer Cell Proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Garofalo M, Di Leva G, Romano G, Nuovo G, Suh S-S, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P. Mir-221&222 Regulate Trail Resistance and Enhance Tumorigenicity through Pten and Timp3 Downregulation. Cancer Cell. 2009;16:498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 183.Park J-K, Kogure T, Nuovo GJ, Jiang J, He L, Kim JH, Phelps MA, Papenfuss TL, Croce CM, Patel T. Mir-221 Silencing Blocks Hepatocellular Carcinoma and Promotes Survival. Cancer Res. 2011;71:7608–7616. doi: 10.1158/0008-5472.CAN-11-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M. Mir-15 and Mir-16 Induce Apoptosis by Targeting Bcl2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pekarsky Y, Croce CM. Role of Mir-15/16 in Cll. Cell Death Differ. 2015;22:6–11. doi: 10.1038/cdd.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hardwick JM, Soane L. Multiple Functions of Bcl-2 Family Proteins. Cold Spring Harbor Perspect Biol. 2013;5:a008722. doi: 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI. Mir-15a and Mir-16–1 Cluster Functions in Human Leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L, Kappeler A, Brunner T, Vassella E. Mir-15a and Mir-16 Are Implicated in Cell Cycle Regulation in a Rb-Dependent Manner and Are Frequently Deleted or Down-Regulated in Non-Small Cell Lung Cancer. Cancer Res. 2009;69:5553–5559. doi: 10.1158/0008-5472.CAN-08-4277. [DOI] [PubMed] [Google Scholar]
- 189.Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, Williams M, Wright C, Edelman JJ, Vallely MP. Restoring Expression of Mir-16: A Novel Approach to Therapy for Malignant Pleural Mesothelioma. Ann Oncol. 2013;24:3128–3135. doi: 10.1093/annonc/mdt412. [DOI] [PubMed] [Google Scholar]
- 190.Reid G, Williams M, Kirschner MB, Mugridge N, Weiss J, Brahmbhatt H, MacDiarmid J, van Zandwijk N. Targeted Delivery of a Synthetic microRNA-based Mimic as an Approach to Cancer Therapy. Proceedings of AARC 106th Annual Meeting; Philadelphia, PA. 2015. [Google Scholar]
- 191.VanZandwijk N, Pavlakis N, Kao S, Clarke S, Lee A, Brahmbhatt H, MacDiarmid J, Pattison S, Leslie F, Huynh Y. P1. 02mesomir 1: A Phase I Study of Targomirs in Patients with Refractory Malignant Pleural Mesothelioma (Mpm) and Lung Cancer (Nsclc) Ann Oncol. 2015;26:ii16–ii16. doi: 10.1093/annonc/mdv090.2. [DOI] [Google Scholar]
- 192.He L, He X, Lim LP, De Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D. A MicroRNA Component of the P53 Tumour Suppressor Network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-Suppressive Mir-34a Induces Senescence-Like Growth Arrest through Modulation of the E2f Pathway in Human Colon Cancer Cells. Proc Natl Acad Sci U S A. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ. Transactivation of Mir-34a by P53 Broadly Influences Gene Expression and Promotes Apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S. The Microrna Mir-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing Cd44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG. Development of a Lung Cancer Therapeutic Based on the Tumor Suppressor Microrna-34. Cancer Res. 2010;70:5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, Smith S, Bader AG, Kim S, Hong DS. Phase I Study of Mrx34, a Liposomal Mir-34a Mimic, Administered Twice Weekly in Patients with Advanced Solid Tumors. Invest New Drugs. 2017;35:180–188. doi: 10.1007/s10637-016-0407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M. Micro-Rna Profiling Reveals a Role for Mir-29 in Human and Murine Liver Fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 199.Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH. Microrna-29, a Key Regulator of Collagen Expression in Systemic Sclerosis. Arthritis Rheum. 2010;62:1733–1743. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- 200.Van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of Micrornas after Myocardial Infarction Reveals a Role of Mir-29 in Cardiac Fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gallant-Behm C, Piper J, Hutnick K, Beatty X, Montgomery R, Yu G, Kaminski N, Van Rooij E, Dalby C, Jackson A. Regulation of Ecm Production and Fibrosis by Mrg-201, a Mimic of Microrna Mir-29. Wound Repair and Regeneration. 2016;24:A9. doi: 10.1111/wrr.12405. [DOI] [Google Scholar]
- 202.Park SM, Gaur AB, Lengyel E, Peter ME. The Mir-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-Cadherin Repressors Zeb1 and Zeb2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cortez MA, Valdecanas D, Zhang X, Zhan Y, Bhardwaj V, Calin GA, Komaki R, Giri DK, Quini CC, Wolfe T. Therapeutic Delivery of Mir-200c Enhances Radiosensitivity in Lung Cancer. Mol Ther. 2014;22:1494–1503. doi: 10.1038/mt.2014.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, Wu S, Han H-D, Shah MY, Rodriguez-Aguayo C. Tumour Angiogenesis Regulation by the Mir-200 Family. Nat Commun. 2013;4 doi: 10.1038/ncomms3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot CV, Zhao Y, Reynolds S, Cheng H, Rupaimoole R. Integrated Analyses Identify a Master Microrna Regulatory Network for the Mesenchymal Subtype in Serous Ovarian Cancer. Cancer Cell. 2013;23:186–199. doi: 10.1016/j.ccr.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nishimura M, Jung E-J, Shah MY, Lu C, Spizzo R, Shimizu M, Han HD, Ivan C, Rossi S, Zhang X. Therapeutic Synergy between MicroRNA and SiRNA in Ovarian Cancer Treatment. Cancer Discovery. 2013;3:1302–1315. doi: 10.1158/2159-8290.CD-13-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK. Therapeutic Epha2 Gene Targeting in Vivo Using Neutral Liposomal Small Interfering Rna Delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 208.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang H-W, Chang T-C, Vivekanandan P, Torbenson M, Clark KR. Therapeutic Microrna Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chen L, Zheng J, Zhang Y, Yang L, Wang J, Ni J, Cui D, Yu C, Cai Z. Tumor-Specific Expression of Microrna-26a Suppresses Human Hepatocellular Carcinoma Growth Via Cyclin-Dependent and -Independent Pathways. Mol Ther. 2011;19:1521–1528. doi: 10.1038/mt.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mairal T, Cengiz Özalp V, Lozano Sánchez P, Mir M, Katakis I, O’Sullivan CK. Aptamers: Molecular Tools for Analytical Applications. Anal Bioanal Chem. 2008;390:989–1007. doi: 10.1007/s00216-007-1346-4. [DOI] [PubMed] [Google Scholar]
- 211.Iliuk AB, Hu L, Tao WA. Aptamer in Bioanalytical Applications. Anal Chem. 2011;83:4440–4452. doi: 10.1021/ac201057w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Jayasena SD. Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin Chem. 1999;45:1628. [PubMed] [Google Scholar]
- 213.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: An Emerging Class of Therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 214.Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA, Clary BM. In Vivo Selection of Tumor-Targeting Rna Motifs. Nat Chem Biol. 2010;6:22–24. doi: 10.1038/nchembio.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jain B, Jain A. Taming Influenza Virus: Role of Antisense Technology. Curr Mol Med. 2015;15:433–445. doi: 10.2174/1566524015666150630124300. [DOI] [PubMed] [Google Scholar]
- 216.Hernandez LI, Flenker KS, Hernandez FJ, Klingelhutz AJ, McNamara J, Giangrande PH. Methods for Evaluating Cell-Specific, Cell-Internalizing Rna Aptamers. Pharmaceuticals. 2013;6:295–319. doi: 10.3390/ph6030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sundaram P, Kurniawan H, Byrne ME, Wower J. Therapeutic Rna Aptamers in Clinical Trials. Eur J Pharm Sci. 2013;48:259–271. doi: 10.1016/j.ejps.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 218.Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C, McCauley TG. Pharmacokinetics and Biodistribution of Novel Aptamer Compositions. Pharm Res. 2004;21:2234–2246. doi: 10.1007/s11095-004-7676-4. [DOI] [PubMed] [Google Scholar]
- 219.Vater A, Klussmann S. Turning Mirror-Image Oligonucleotides into Drugs: The Evolution of Spiegelmer® Therapeutics. Drug Discovery Today. 2015;20:147–155. doi: 10.1016/j.drudis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 220.Qi Y, Simakova A, Ganson NJ, Li X, Luginbuhl KM, Ozer I, Liu W, Hershfield MS, Matyjaszewski K, Chilkoti A. A Brush-Polymer/Exendin-4 Conjugate Reduces Blood Glucose Levels for up to Five Days and Eliminates Poly(Ethylene Glycol) Antigenicity. Nat Biomed Eng. 2016;1:0002. doi: 10.1038/s41551-016-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of Single-Stranded DNA Molecules That Bind and Inhibit Human Thrombin. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 222.Padmanabhan K, Padmanabhan KP, Ferrara JD, Sadler JE, Tulinsky A. The Structure of Alpha-Thrombin Inhibited by a 15-Mer Single-Stranded DNA Aptamer. J Biol Chem. 1993;268:17651–17654. doi: 10.2210/pdb1hut/pdb. [DOI] [PubMed] [Google Scholar]
- 223.Gragoudas ES, Adamis AP, Cunningham ETJ, Feinsod M, Guyer DR. Pegaptanib for Neovascular Age-Related Macular Degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 224.Ambati J, Fowler BJ. Mechanisms of Age-Related Macular Degeneration. Neuron. 2012;75:26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Reautschnig P, Vogel P, Stafforst T The Notorious RNA. In the Spotlight - Drug or Target for the Treatment of Disease. RNA Biol. 2017;14:651–668. doi: 10.1080/15476286.2016.1208323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ng EWM, Shima DT, Calias P, Cunningham ET, Guyer DR, Adamis AP. Pegaptanib, a Targeted Anti-Vegf Aptamer for Ocular Vascular Disease. Nat Rev Drug Discovery. 2006;5:123–132. doi: 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- 227.Emerson MV, Lauer AK. Current and Emerging Therapies for the Treatment of Age-Related Macular Degeneration. Clin Ophthamol. 2008;2:377–388. doi: 10.2147/opth.s1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TL, Monroe D, Sullenger BA. Rna Aptamers as Reversible Antagonists of Coagulation Factor Ixa. Nature. 2002;419:90–94. doi: 10.1038/nature00963. [DOI] [PubMed] [Google Scholar]
- 229.Rallapalli PM, Kemball-Cook G, Tuddenham EG, Gomez K, Perkins SJ. An Interactive Mutation Database for Human Coagulation Factor Ix Provides Novel Insights into the Phenotypes and Genetics of Hemophilia B. J Thromb Haemostasis. 2013;11:1329–1340. doi: 10.1111/jth.12276. [DOI] [PubMed] [Google Scholar]
- 230.Mosesson MW. Fibrinogen and Fibrin Structure and Functions. J Thromb Haemostasis. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 231.Kelton JG. Heparin-Induced Thrombocytopenia: An Overview. Blood Rev. 2002;16:77–80. doi: 10.1054/blre.2001.0189. [DOI] [PubMed] [Google Scholar]
- 232.Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G, Jr, Scardino E, Fay WP, Sullenger BA. Antidote-Mediated Control of an Anticoagulant Aptamer in Vivo. Nat Biotechnol. 2004;22:1423–1428. doi: 10.1038/nbt1023. [DOI] [PubMed] [Google Scholar]
- 233.Dyke CK, Steinhubl SR, Kleiman NS, Cannon RO, Aberle LG, Lin M, Myles SK, Melloni C, Harrington RA, Alexander JH. First-in-Human Experience of an Antidote-Controlled Anticoagulant Using Rna Aptamer Technology. Circulation. 2006;114:2490–2497. doi: 10.1161/CIRCULATIONAHA.106.668434. [DOI] [PubMed] [Google Scholar]
- 234.Chan MY, Cohen MG, Dyke CK, Myles SK, Aberle LG, Lin M, Walder J, Steinhubl SR, Gilchrist IC, Kleiman NS. Phase 1b Randomized Study of Antidote-Controlled Modulation of Factor Ixa Activity in Patients with Stable Coronary Artery Disease. Circulation. 2008;117:2865–2874. doi: 10.1161/CIRCULATIONAHA.107.745687. [DOI] [PubMed] [Google Scholar]
- 235.Dahlbäck B, Carlsson M, Svensson PJ. Familial Thrombophilia Due to a Previously Unrecognized Mechanism Characterized by Poor Anticoagulant Response to Activated Protein C: Prediction of a Cofactor to Activated Protein C. Proc Natl Acad Sci U S A. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Povsic TJ, Vavalle JP, Aberle LH, Kasprzak JD, Cohen MG, Mehran R, Bode C, Buller CE, Montalescot G, Cornel JH. A Phase 2, Randomized, Partially Blinded, Active-Controlled Study Assessing the Efficacy and Safety of Variable Anticoagulation Reversal Using the Reg1 System in Patients with Acute Coronary Syndromes: Results of the Radar Trial. Eur Heart J. 2013;34:2481–2489. doi: 10.1093/eurheartj/ehs232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cohen MG, Purdy DA, Rossi JS, Grinfeld LR, Myles SK, Aberle LH, Greenbaum AB, Fry E, Chan MY, Tonkens RM. First Clinical Application of an Actively Reversible Direct Factor Ixa Inhibitor as an Anticoagulation Strategy in Patients Undergoing Percutaneous Coronary Intervention. Circulation. 2010;122:614–622. doi: 10.1161/CIRCULATIONAHA.109.927756. [DOI] [PubMed] [Google Scholar]
- 238.Lincoff AM, Mehran R, Povsic TJ, Zelenkofske SL, Huang Z, Armstrong PW, Steg PG, Bode C, Cohen MG, Buller C. Effect of the Reg1 Anticoagulation System Versus Bivalirudin on Outcomes after Percutaneous Coronary Intervention (Regulate-Pci): A Randomised Clinical Trial. Lancet. 2016;387:349–356. doi: 10.1016/S0140-6736(15)00515-2. [DOI] [PubMed] [Google Scholar]
- 239.Ganson NJ, Povsic TJ, Sullenger BA, Alexander JH, Zelenkofske SL, Sailstad JM, Rusconi CP, Hershfield MS. Pre-Existing Anti–Polyethylene Glycol Antibody Linked to First-Exposure Allergic Reactions to Pegnivacogin, a Pegylated Rna Aptamer. J Allergy Clin Immunol. 2016;137:1610–1613. doi: 10.1016/j.jaci.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Keefe AD, Pai S, Ellington A. Aptamers as Therapeutics. Nat Rev Drug Discovery. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Spiel AO, Gilbert JC, Jilma B. Von Willebrand Factor in Cardiovascular Disease. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- 242.Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, Gilbert J, Wagner DD, Schaub R. Inhibition of Von Willebrand Factor-Mediated Platelet Activation and Thrombosis by the Anti-Von Willebrand Factor A1-Domain Aptamer Arc1779. J Thromb Haemostasis. 2009;7:1155–1162. doi: 10.1111/j.1538-7836.2009.03459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Huang R-H, Fremont DH, Diener JL, Schaub RG, Sadler JE. A Structural Explanation for the Antithrombotic Activity of Arc1172, a DNA Aptamer That Binds Von Willebrand Factor Domain A1. Structure. 2009;17:1476. doi: 10.1016/j.str.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, BouFakhreddine S, Holohan TV, Schaub RG. First-in-Human Evaluation of Anti-Von Willebrand Factor Therapeutic Aptamer Arc1779 in Healthy Volunteers. Circulation. 2007;116:2678–2686. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 245.Arzamendi D, Dandachli F, Théorêt J-F, Ducrocq G, Chan M, Mourad W, Gilbert JC, Schaub RG, Tanguay J-F, Merhi Y. An Anti-Von Willebrand Factor Aptamer Reduces Platelet Adhesion among Patients Receiving Aspirin and Clopidogrel in an Ex Vivo Shear-Induced Arterial Thrombosis. Clin Appl Thromb/Hemostasis. 2011;17:E70–E78. doi: 10.1177/1076029610384114. [DOI] [PubMed] [Google Scholar]
- 246.Jilma-Stohlawetz P, Knöbl P, Gilbert JC, Jilma B. The Anti-Von Willebrand Factor Aptamer Arc1779 Increases Von Willebrand Factor Levels and Platelet Counts in Patients with Type 2b Von Willebrand Disease. Thromb Haemostasis. 2012;108:284–290. doi: 10.1160/TH11-12-0889. [DOI] [PubMed] [Google Scholar]
- 247.Jilma-Stohlawetz P, Gorczyca ME, Jilma B, Siller-Matula J, Gilbert JC, Knöbl P. Inhibition of Von Willebrand Factor by Arc1779 in Patients with Acute Thrombotic Thrombocytopenic Purpura. Thromb Haemostasis. 2011;105:545–552. doi: 10.1160/TH10-08-0520. [DOI] [PubMed] [Google Scholar]
- 248.Burger JA, Kipps TJ. Cxcr4: A Key Receptor in the Crosstalk between Tumor Cells and Their Microenvironment. Blood. 2006;107:1761. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 249.Chatterjee S, Behnam Azad B, Nimmagadda S. The Intricate Role of CXCR4 in Cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kluβmann S, Nolte A, Bald R, Erdmann VA, Fürste JP. Mirror-Image Rna That Binds D-Adenosine. Nat Biotechnol. 1996;14:1112–1115. doi: 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- 251.Hoellenriegel J, Zboralski D, Maasch C, Rosin NY, Wierda WG, Keating MJ, Kruschinski A, Burger JA. The Spiegelmer Nox-A12, a Novel Cxcl12 Inhibitor, Interferes with Chronic Lymphocytic Leukemia Cell Motility and Causes Chemosensitization. Blood. 2014;123:1032–1039. doi: 10.1182/blood-2013-03-493924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ludwig H, Weisel K, Petrucci MT, Leleu X, Cafro AM, Garderet L, Leitgeb C, Foa R, Greil R, Yakoub-Agha I. Olaptesed Pegol, an Anti-Cxcl12/Sdf-1 Spiegelmer, Alone and with Bortezomib-Dexamethasone in Relapsed/Refractory Multiple Myeloma: A Phase Iia Study. Leukemia. 2017;31:997–1000. doi: 10.1038/leu.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang Z, Yuan W, Sun L, Szeto FL, Wong KE, Li X, Kong J, Li YC. 1,25-Dihydroxyvitamin D3 Targeting of Nf-Kb Suppresses High Glucose-Induced Mcp-1 Expression in Mesangial Cells. Kidney Int. 2007;72:193–201. doi: 10.1038/sj.ki.5002296. [DOI] [PubMed] [Google Scholar]
- 254.Tesch GH. Mcp-1/Ccl2: A New Diagnostic Marker and Therapeutic Target for Progressive Renal Injury in Diabetic Nephropathy. Am J physiol Renal Physiol. 2008;294:F697–F701. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 255.Oberthür D, Achenbach J, Gabdulkhakov A, Buchner K, Maasch C, Falke S, Rehders D, Klussmann S, Betzel C. Crystal Structure of a Mirror-Image L-Rna Aptamer (Spiegelmer) in Complex with the Natural L-Protein Target Ccl2. Nat Commun. 2015;6:6923. doi: 10.1038/ncomms7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, Więcek A, Haller H. C-C Motif-Ligand 2 Inhibition with Emapticap Pegol (Nox-E36) in Type 2 Diabetic Patients with Albuminuria. Nephrol, Dial, Transplant. 2016;32:307–315. doi: 10.1093/ndt/gfv459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Birn H, Christensen EI. Renal Albumin Absorption in Physiology and Pathology. Kidney Int. 2006;69:440–449. doi: 10.1038/sj.ki.5000141. [DOI] [PubMed] [Google Scholar]
- 258.Doherty EA, Doudna JA. Ribozyme Structures and Mechanisms. Annu Rev Biophys Biomol Struct. 2001;30:457–475. doi: 10.1146/annurev.biophys.30.1.457. [DOI] [PubMed] [Google Scholar]
- 259.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-Splicing Rna: Autoexcision and Autocyclization of the Ribosomal Rna Intervening Sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 260.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The Rna Moiety of Ribonuclease P Is the Catalytic Subunit of the Enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 261.Zhang YC, Taylor MM, Samson WK, Phillips MI. Antisense Therapeutics: Methods in Molecular Medicine. Humana Press; Totowa, NJ: 2005. Antisense Therapeutics; pp. 3–10. [PubMed] [Google Scholar]
- 262.Tanner NK. Ribozymes: The Characteristics and Properties of Catalytic Rnas. FEMS Microbiol Rev. 1999;23:257–275. doi: 10.1111/j.1574-6976.1999.tb00399.x. [DOI] [PubMed] [Google Scholar]
- 263.James HA, Gibson I. The Therapeutic Potential of Ribozymes. Blood. 1998;91:371–382. [PubMed] [Google Scholar]
- 264.Carbonell A, Flores R, Gago S. From Nucleic Acids Sequences to Molecular Medicine. Springer-Verlag; Berlin: 2012. p. 11. [Google Scholar]
- 265.Walter F, Westhof E. eLS. John Wiley & Sons, Ltd; 2001. Catalytic RNA. [Google Scholar]
- 266.Burnett JC, Rossi JJ. Rna-Based Therapeutics: Current Progress and Future Prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Beaudry AA, Joyce GF. Directed Evolution of an Rna Enzyme. Science. 1992;257:635–641. doi: 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- 268.Beaudry A, DeFoe J, Zinnen S, Burgin A, Beigelman L. In Vitro Selection of a Novel Nuclease-Resistant Rna Phosphodiesterase. Chem Biol. 2000;7:323–334. doi: 10.1016/S1074-5521(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 269.Yu Q, Pecchia DB, Kingsley SL, Heckman JE, Burke JM. Cleavage of Highly Structured Viral Rna Molecules by Combinatorial Lbraries of Hairpin Ribozymes: The Most Effective Ribozymes Are Not Predicted by Substrate Selection Rules. J Biol Chem. 1998;273:23524–23533. doi: 10.1074/jbc.273.36.23524. [DOI] [PubMed] [Google Scholar]
- 270.Scanlon KJ. Anti-Genes: Sirna, Ribozymes and Antisense. Curr Pharm Biotechnol. 2004;5:415–420. doi: 10.2174/1389201043376689. [DOI] [PubMed] [Google Scholar]
- 271.Usman N, Blatt LM. Nuclease-Resistant Synthetic Ribozymes: Developing a New Class of Therapeutics. J Clin Invest. 2000;106:1197–1202. doi: 10.1172/JCI11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Sun LQ, Wang L, Gerlach WL, Symonds G. Target Sequence-Specific Inhibition of Hiv-1 Replication by Ribozymes Directed to Tat Rna. Nucleic Acids Res. 1995;23:2909–2913. doi: 10.1093/nar/23.15.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pavco P, McSwiggen J, Stinchcomb D, Escobedo J. United States of America: 2006. [Google Scholar]
- 274.Zinnen SP, Domenico K, Wilson M, Dickinson BA, Beaudry A, Mokler V, Daniher AT, Burgin A, Beigelman L. Selection, Design, and Characterization of a New Potentially Therapeutic Ribozyme. RNA. 2002;8:214–228. doi: 10.1017/S1355838202014723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Shippy R, Lockner R, Farnsworth M, Hampel A. The Hairpin Ribozyme. Mol Biotechnol. 1999;12:117–129. doi: 10.1385/MB:12:1:117. [DOI] [PubMed] [Google Scholar]
- 276.Wang L, Witherington C, King A, Gerlach WL, Carr A, Penny R, Cooper D, Symonds G, Sun L-Q. Preclinical Characterization of an Anti-Tat Ribozyme for Therapeutic Application. Hum Gene Ther. 1998;9:1283–1291. doi: 10.1089/hum.1998.9.9-1283. [DOI] [PubMed] [Google Scholar]
- 277.Frankel AD, Young JA. Hiv-1: Fifteen Proteins and an Rna. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 278.Wong-Staal F, Poeschla EM, Looney DJ. A Controlled, Phase 1 Clinical Trial to Evaluate the Safety and Effects in Hiv-1 Infected Humans of Autologous Lymphocytes Transduced with a Ribozyme That Cleaves Hiv-1 Rna. University of California San Diego, La Jolla, California. Hum Gene Ther. 1998;9:2407–2425. doi: 10.1089/hum.1998.9.16-2407. [DOI] [PubMed] [Google Scholar]
- 279.Krüger M, Beger C, Wong-Staal F. Use of Ribozymes to Inhibit Gene Expression. Methods Enzymol. 1999;306:207–225. doi: 10.1016/S0076-6879(99)06014-0. [DOI] [PubMed] [Google Scholar]
- 280.Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, Workman C, Bloch M, Lalezari J, Becker; S, Thornton L. Phase 2 Gene Therapy Trial of an Anti-Hiv Ribozyme in Autologous Cd34+ Cells. Nat Med. 2009;15:285–292. doi: 10.1038/nm.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Amado RG, Mitsuyasu RT, Rosenblatt JD, Ngok FK, Bakker A, Cole S, Chorn N, Lin L-S, Bristol G, Boyd MP. Anti-Human Immunodeficiency Virus Hematopoietic Progenitor Cell-Delivered Ribozyme in a Phase I Study: Myeloid and Lymphoid Reconstitution in Human Immunodeficiency Virus Type-1-Infected Patients. Hum Gene Ther. 2004;15:251–262. doi: 10.1089/104303404322886101. [DOI] [PubMed] [Google Scholar]
- 282.Symonds GP, Amado RG, Sun L-Q, MacPherson JL, Fanning GC, Gerlach W. Methods for genetic modification of hematopoietic progenitor cells and uses of the modified cells. 11,506,722. US Patent. 2010
- 283.Ngok FK, Mitsuyasu RT, Macpherson JL, Boyd MP, Symonds GP, Amado RG. Ribozymes and siRNA Protocols. Humana Press; Totowa, NJ: 2004. Clinical Gene Therapy Research Utilizing Ribozymes; pp. 581–598. [DOI] [PubMed] [Google Scholar]
- 284.Hicklin DJ, Ellis LM. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 285.Weng DE, Masci PA, Radka SF, Jackson TE, Weiss PA, Ganapathi R, Elson PJ, Capra WB, Parker VP, Lockridge JA. A Phase I Clinical Trial of a Ribozyme-Based Angiogenesis Inhibitor Targeting Vascular Endothelial Growth Factor Receptor-1 for Patients with Refractory Solid Tumors. Mol Cancer Ther. 2005;4:948. doi: 10.1158/1535-7163.MCT-04-0210. [DOI] [PubMed] [Google Scholar]
- 286.Kobayashi H, Gail Eckhardt S, Lockridge JA, Rothenberg ML, Sandler AB, O’Bryant CL, Cooper W, Holden SN, Aitchison RD, Usman N. Safety and Pharmacokinetic Study of Rpi.4610 (Angiozyme), an Anti-Vegfr-1 Ribozyme, in Combination with Carboplatin and Paclitaxel in Patients with Advanced Solid Tumors. Cancer Chemother Pharmacol. 2005;56:329–336. doi: 10.1007/s00280-004-0968-x. [DOI] [PubMed] [Google Scholar]
- 287.Morrow PK, Murthy RK, Ensor JD, Gordon GS, Margolin KA, Elias AD, Urba WJ, Weng DE, Rugo HS, Hortobagyi GN. An Open-Label, Phase 2 Trial of Rpi.4610 (Angiozyme) in the Treatment of Metastatic Breast Cancer. Cancer. 2012;118:4098–4104. doi: 10.1002/cncr.26730. [DOI] [PubMed] [Google Scholar]
- 288.Chow CS, Bogdan FM. A Structural Basis for Rna–Ligand Interactions. Chem Rev. 1997;97:1489–1514. doi: 10.1021/cr960415w. [DOI] [PubMed] [Google Scholar]
- 289.Dervan PB. Molecular Recognition of DNA by Small Molecules. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/S0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 290.Disney MD, Yildirim I, Childs-Disney JL. Methods to Enable the Design of Bioactive Small Molecules Targeting Rna. Org Biomol Chem. 2014;12:1029–1039. doi: 10.1039/C3OB42023J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Guan L, Disney MD. Recent Advances in Developing Small Molecules Targeting Rna. ACS Chem Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 292.Thomas JR, Hergenrother PJ. Targeting Rna with Small Molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 293.Finlay AC, Hochstein FA, Sobin BA, Murphy FX. Netropsin, a New Antibiotic Produced by a Streptomyces. J Am Chem Soc. 1951;73:341–343. doi: 10.1021/ja01145a113. [DOI] [Google Scholar]
- 294.Dimarco A, Gaetani M, Orezzi P, Scottit, Arcamone F. Experimental Studies on Distamycin a New Antibiotic with Cytotoxic Activity. Cancer Chemother Rep. 1962;18:15–19. [PubMed] [Google Scholar]
- 295.Berman HM, Neidle S, Zimmer C, Thrum H. Netropsin, a DNA-Binding Oligopeptide Structural and Binding Studies. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1979;561:124–131. doi: 10.1016/0005-2787(79)90496-9. [DOI] [PubMed] [Google Scholar]
- 296.Dickerson RE, Kopka ML. Nuclear Overhauser Data and Stereochemical Considerations Suggest That Netropsin Binds Symmetrically within the Minor Groove of Poly(Da).Poly(Dt), Forming Hydrogen Bonds with Both Strands of the Double Helix. J Biomol Struct Dyn. 1985;3:423–431. doi: 10.1080/07391102.1985.10508431. [DOI] [PubMed] [Google Scholar]
- 297.Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. Binding of an Antitumor Drug to DNA, Netropsin and C-G-C-G-A-A-T-T-BrC-G-C-G. J Mol Biol. 1985;183:553–563. doi: 10.1016/0022-2836(85)90171-8. [DOI] [PubMed] [Google Scholar]
- 298.Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. The Molecular Origin of DNA-Drug Specificity in Netropsin and Distamycin. Proc Natl Acad Sci U S A. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wartell RM, Larson JE, Wells RD. Netropsin. A Specific Probe for a-T Regions of Duplex Deoxyribonucleic Acid. J Biol Chem. 1974;249:6719–6731. [PubMed] [Google Scholar]
- 300.Pelton JG, Wemmer DE. Structural Characterization of a 2:1 Distamycin A.D(Cgcaaattggc) Complex by Two-Dimensional Nmr. Proc Natl Acad Sci U S A. 1989;86:5723–5727. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Dervan PB. Design of Sequence-Specific DNA-Binding Molecules. Science. 1986;232:464–471. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- 302.Leimgruber W, Batcho AD, Schenker F. The Structure of Anthramycin. J Am Chem Soc. 1965;87:5793–5795. doi: 10.1021/ja00952a051. [DOI] [PubMed] [Google Scholar]
- 303.Leimgruber W, Stefanovic V, Schenker F, Karr A, Berger J. Isolation and Characterization of Anthramycin, a New Antitumor Antibiotic. J Am Chem Soc. 1965;87:5791–5793. doi: 10.1021/ja00952a050. [DOI] [PubMed] [Google Scholar]
- 304.Kohn KW, Bono VH, Jr, Kann HE., Jr Anthramycin, a New Type of DNA-Inhibiting Antibiotic: Reaction with DNA and Effect on Nucleic Acid Synthesis in Mouse Leukemia Cells. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1968;155:121–129. doi: 10.1016/0005-2787(68)90342-0. [DOI] [PubMed] [Google Scholar]
- 305.Kohn KW, Spears CL. Reaction of Anthramycin with Deoxyribonucleic Acid. J Mol Biol. 1970;51:551–572. doi: 10.1016/0022-2836(70)90008-2. [DOI] [PubMed] [Google Scholar]
- 306.Kohn KW, Glaubiger D, Spears CL. The Reaction of Anthramycin with DNA. Ii. Studies of Kinetics and Mechanism. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1974;361:288–302. doi: 10.1016/0005-2787(74)90372-4. [DOI] [PubMed] [Google Scholar]
- 307.Wright WB, Jr, Brabander HJ, Greenblatt EN, Day IP, Hardy RA., Jr Derivatives of 1,2,3,11a-Tetrahydro-5h-Pyrrolo[2,1-C][1,4]Benzodiazepine-5,11(10h)-Dione as Anxiolytic Agents. J Med Chem. 1978;21:1087–1089. doi: 10.1021/jm00208a017. [DOI] [PubMed] [Google Scholar]
- 308.Wright WB, Jr, Greenblatt EN, Day IP, Quinones NQ, Hardy RA., Jr Derivatives of 11-(1-Piperazinyl)-5h-Pyrrolo[2,1-C][1,4]Benzodiazepine as Central Nervous System Agents. J Med Chem. 1980;23:462–465. doi: 10.1021/jm00178a020. [DOI] [PubMed] [Google Scholar]
- 309.Hurley LH, Thurston DE. Pyrrolo(L, 4)Benzodiazepine Antitumor Antibiotics: Chemistry, Interaction with DNA, and Biological Implications. Pharm Res. 1984;01:52–59. doi: 10.1023/A:1016395113085. [DOI] [PubMed] [Google Scholar]
- 310.Hurley LH, Reck T, Thurston DE, Langley DR, Holden KG, Hertzberg RP, Hoover JR, Gallagher G, Jr, Faucette LF, Mong SM. Pyrrolo[1,4]Benzodiazepine Antitumor Antibiotics: Relationship of DNA Alkylation and Sequence Specificity to the Biological Activity of Natural and Synthetic Compounds. Chem Res Toxicol. 1988;1:258–268. doi: 10.1021/tx00005a002. [DOI] [PubMed] [Google Scholar]
- 311.Puvvada MS, Forrow SA, Hartley JA, Stephenson P, Gibson I, Jenkins TC, Thurston DE. Inhibition of Bacteriophage T7 Rna Polymerase in Vitro Transcription by DNA-Binding Pyrrolo[2,1-C][1,4]Benzodiazepines. Biochemistry. 1997;36:2478–2484. doi: 10.1021/bi952490r. [DOI] [PubMed] [Google Scholar]
- 312.Hochhauser D, Meyer T, Spanswick VJ, Wu J, Clingen PH, Loadman P, Cobb M, Gumbrell L, Begent RH, Hartley JA. Phase I Study of Sequence-Selective Minor Groove DNA Binding Agent Sjg-136 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2009;15:2140–2147. doi: 10.1158/1078-0432.CCR-08-1315. [DOI] [PubMed] [Google Scholar]
- 313.Janjigian YY, Lee W, Kris MG, Miller VA, Krug LM, Azzoli CG, Senturk E, Calcutt MW, Rizvi NA. A Phase I Trial of Sjg-136 (Nsc#694501) in Advanced Solid Tumors. Cancer Chemother Pharmacol. 2010;65:833–838. doi: 10.1007/s00280-009-1088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zammarchi F, Williams DG, Adams L, Havenith K, Chivers S, D’Hooge F, Howard PW, Hartley JA, van Berkel PH. Pre-Clinical Development of Adct-402, a Novel Pyrrolobenzodiazepine (Pbd)-Based Antibody Drug Conjugate (Adc) Targeting Cd19-Expressing B-Cell Malignancies. Blood. 2015;126:1564–1564. [Google Scholar]
- 315. [accessed December 6, 2017];Study of ADCT-402 in Patients With Relapsed or Refractory B-cell Lineage Acute Lymphoblastic Leukemia (B-ALL) https://clinicaltrials.gov/ct2/show/NCT02669264.
- 316.Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A. A Dll3-Targeted Antibody-Drug Conjugate Eradicates High-Grade Pulmonary Neuroendocrine Tumor-Initiating Cells in Vivo. Sci Transl Med. 2015;7:302ra136. doi: 10.1126/scitranslmed.aac9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Stein EM, Stein A, Walter RB, Fathi AT, Lancet JE, Kovacsovics TJ, Advani AS, DeAngelo DJ, O’Meara MM, Zaho B. Proceedings of the 56th ASH Annual Meeting and Exposition; San Francisco, CA. Dec 6–9, 2014. [Google Scholar]
- 318. [accessed December 7, 2017];Study of ADCT-402 in Patients With Relapsed or Refractory B-cell Lineage Non Hodgkin Lymphoma (B-NHL) https://clinicaltrials.gov/ct2/show/NCT02669017.
- 319.Tse WC, Boger DL. Sequence-Selective DNA Recognition: Natural Products and Nature’s Lessons. Chem Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 320.Hurley LH. DNA and Its Associated Processes as Targets for Cancer Therapy. Nat Rev Cancer. 2002;2:188. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 321.Boger DL, Johnson DS. Cc-1065 and the Duocarmycins: Understanding Their Biological Function through Mechanistic Studies. Angew Chem, Int Ed Engl. 1996;35:1438–1474. doi: 10.1002/anie.199614381. [DOI] [Google Scholar]
- 322.Boger DL, Garbaccio RM. Catalysis of the Cc-1065 and Duocarmycin DNA Alkylation Reaction: DNA Binding Induced Conformational Change in the Agent Results in Activation. Bioorg Med Chem. 1997;5:263–276. doi: 10.1016/S0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 323.Boger DL, Garbaccio RM. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the Cc-1065 and Duocarmycin DNA Alkylation Reaction. Acc Chem Res. 1999;32:1043–1052. doi: 10.1021/ar9800946. [DOI] [Google Scholar]
- 324.Boger DL, Cai H. Bleomycin: Synthetic and Mechanistic Studies. Angew Chem, Int Ed. 1999;38:448–476. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 325.Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Bleomycins: A Structural Model for Specificity, Binding, and Double Strand Cleavage. Acc Chem Res. 1996;29:322–330. doi: 10.1021/ar9501333. [DOI] [Google Scholar]
- 326.Wade WS, Mrksich M, Dervan PB. Design of Peptides That Bind in the Minor Groove of DNA at 5′-(a, T) G (a, T) C (a, T)-3′sequences by a Dimeric Side-by-Side Motif. J Am Chem Soc. 1992;114:8783–8794. doi: 10.1021/ja00049a006. [DOI] [Google Scholar]
- 327.White S, Baird EE, Dervan PB. On the Pairing Rules for Recognition in the Minor Groove of DNA by Pyrrole-Imidazole Polyamides. Chem Biol. 1997;4:569–578. doi: 10.1016/S1074-5521(97)90243-X. [DOI] [PubMed] [Google Scholar]
- 328.Trauger JW, Baird EE, Dervan PB. Recognition of DNA by Designed Ligands at Subnanomolar Concentrations. Nature. 1996;382:559. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]
- 329.Marques MA, Doss RM, Foister S, Dervan PB. Expanding the Repertoire of Heterocycle Ring Pairs for Programmable Minor Groove DNA Recognition. J Am Chem Soc. 2004;126:10339–10349. doi: 10.1021/ja0486465. [DOI] [PubMed] [Google Scholar]
- 330.Wang CC, Ellervik U, Dervan PB. Expanding the Recognition of the Minor Groove of DNA by Incorporation of B-Alanine in Hairpin Polyamides. Bioorg Med Chem. 2001;9:653–657. doi: 10.1016/S0968-0896(00)00282-0. [DOI] [PubMed] [Google Scholar]
- 331.Bando T, Sugiyama H. Synthesis and Biological Properties of Sequence-Specific DNA-Alkylating Pyrrole–Imidazole Polyamides. Acc Chem Res. 2006;39:935–944. doi: 10.1021/ar030287f. [DOI] [PubMed] [Google Scholar]
- 332.Sugiyama H, Lian C, Isomura M, Saito I, Wang AH-J. Distamycin a Modulates the Sequence Specificity of DNA Alkylation by Duocarmycin A. Proc Natl Acad Sci U S A. 1996;93:14405–14410. doi: 10.1073/pnas.93.25.14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Tao ZF, Fujiwara T, Saito I, Sugiyama H. Sequence-Specific DNA Alkylation by Hybrid Molecules between Segment a of Duocarmycin a and Pyrrole/Imidazole Diamide. Angew Chem, Int Ed. 1999;38:650–653. doi: 10.1002/(SICI)1521-3773(19990301)38:5<650::AID-ANIE650>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 334.Fujiwara T, Tao Z-F, Ozeki Y, Saito I, Wang H-J, Lee M, Sugiyama H. Modulation of Sequence Specificity of Duocarmycin-Dependent DNA Alkylation by Pyrrole–Imidazole Triamides. J Am Chem Soc. 1999;121:7706–7707. doi: 10.1021/ja991331i. [DOI] [Google Scholar]
- 335.Bando T, Iida H, Saito I, Sugiyama H. Sequence-Specific DNA Interstrand Cross-Linking by Imidazole–Pyrrole Cpi Conjugate. J Am Chem Soc. 2001;123:5158–5159. doi: 10.1021/ja003660c. [DOI] [PubMed] [Google Scholar]
- 336.Bando T, Narita A, Saito I, Sugiyama H. Highly Efficient Sequence-Specific DNA Interstrand Cross-Linking by Pyrrole/Imidazole Cpi Conjugates. J Am Chem Soc. 2003;125:3471–3485. doi: 10.1021/ja028459b. [DOI] [PubMed] [Google Scholar]
- 337.Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB. Regulation of Gene Expression by Small Molecules. Nature. 1997;387:202. doi: 10.1038/387202a0. [DOI] [PubMed] [Google Scholar]
- 338.Dickinson LA, Gulizia RJ, Trauger JW, Baird EE, Mosier DE, Gottesfeld JM, Dervan PB. Inhibition of Rna Polymerase Ii Transcription in Human Cells by Synthetic DNA-Binding Ligands. Proc Natl Acad Sci U S A. 1998;95:12890–12895. doi: 10.1073/pnas.95.22.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Mapp AK, Ansari AZ, Ptashne M, Dervan PB. Activation of Gene Expression by Small Molecule Transcription Factors. Proc Natl Acad Sci U S A. 2000;97:3930–3935. doi: 10.1073/pnas.97.8.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Arndt H-D, Hauschild KE, Sullivan DP, Lake K, Dervan PB, Ansari AZ. Toward Artificial Developmental Regulators. J Am Chem Soc. 2003;125:13322–13323. doi: 10.1021/ja0371395. [DOI] [PubMed] [Google Scholar]
- 341.Olenyuk BZ, Zhang G-J, Klco JM, Nickols NG, Kaelin WG, Dervan PB. Inhibition of Vascular Endothelial Growth Factor with a Sequence-Specific Hypoxia Response Element Antagonist. Proc Natl Acad Sci U S A. 2004;101:16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Campuzano V, Montermini L, Moltò MD, Pianese L. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic Gaa Triplet Repeat Expansion. Science. 1996;271:1423. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 343.Burnett R, Melander C, Puckett JW, Son LS, Wells RD, Dervan PB, Gottesfeld JM. DNA Sequence-Specific Polyamides Alleviate Transcription Inhibition Associated with Long Gaa Ttc Repeats in Friedreich’s Ataxia. Proc Natl Acad Sci U S A. 2006;103:11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Nickols NG, Dervan PB. Suppression of Androgen Receptor-Mediated Gene Expression by a Sequence-Specific DNA-Binding Polyamide. Proc Natl Acad Sci U S A. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Yang F, Nickols NG, Li BC, Marinov GK, Said JW, Dervan PB. Antitumor Activity of a Pyrrole-Imidazole Polyamide. Proc Natl Acad Sci U S A. 2013;110:1863–1868. doi: 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Raskatov JA, Meier JL, Puckett JW, Yang F, Ramakrishnan P, Dervan PB. Modulation of Nf-Kb-Dependent Gene Transcription Using Programmable DNA Minor Groove Binders. Proc Natl Acad Sci U S A. 2012;109:1023–1028. doi: 10.1073/pnas.1118506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Raskatov JA, Nickols NG, Hargrove AE, Marinov GK, Wold B, Dervan PB. Gene Expression Changes in a Tumor Xenograft by a Pyrrole-Imidazole Polyamide. Proc Natl Acad Sci U S A. 2012;109:16041–16045. doi: 10.1073/pnas.1214267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Hurley LH, Gairola C, Zmijewski M. Pyrrolo(1,4)Benzodiazepine Antitumor Antibiotics. In Vitro Interaction of Anthramycin, Sibiromycin and Tomaymycin with DNA Using Specifically Radiolabelled Molecules. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1977;475:521–535. doi: 10.1016/0005-2787(77)90067-3. [DOI] [PubMed] [Google Scholar]
- 349.Jones GB, Davey CL, Jenkins TC, Kamal A, Kneale GG, Neidle S, Webster GD, Thurston DE. The Non-Covalent Interaction of Pyrrolo[2, 1-C] [1, 4]Benzodiazepine-5, 11-Diones with DNA. Anti Cancer Drug Des. 1990;5:249–264. [PubMed] [Google Scholar]
- 350.Mantaj J, Jackson PJ, Rahman KM, Thurston DE. From Anthramycin to Pyrrolobenzodiazepine (Pbd)-Containing Antibody-Drug Conjugates (Adcs) Angew Chem, Int Ed. 2017;56:462–488. doi: 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Farmer JD, Rudnicki SM, Suggs JW. Synthesis and Dna Crosslinking Ability of a Dimeric Anthramycin Analog. Tetrahedron Lett. 1988;29:5105–5108. doi: 10.1016/S0040-4039(00)80691-7. [DOI] [Google Scholar]
- 352.Bose DS, Thompson AS, Ching J, Hartley JA, Berardini MD, Jenkins TC, Neidle S, Hurley LH, Thurston DE. Rational Design of a Highly Efficient Irreversible DNA Interstrand Cross-Linking Agent Based on the Pyrrolobenzodiazepine Ring System. J Am Chem Soc. 1992;114:4939–4941. doi: 10.1021/ja00038a089. [DOI] [Google Scholar]
- 353.Jenkins TC, Hurley LH, Neidle S, Thurston DE. Structure of a Covalent DNA Minor Groove Adduct with a Pyrrolobenzodiazepine Dimer: Evidence for Sequence-Specific Interstrand Crosslinking. J Med Chem. 1994;37:4529–4537. doi: 10.1021/jm00052a012. [DOI] [PubMed] [Google Scholar]
- 354.Walton MI, Goddard P, Kelland LR, Thurston DE, Harrap KR. Preclinical Pharmacology and Antitumour Activity of the Novel Sequence-Selective DNA Minor-Groove Cross-Linking Agent Dsb-120. Cancer Chemother Pharmacol. 1996;38:431–438. doi: 10.1007/s002800050507. [DOI] [PubMed] [Google Scholar]
- 355.Gregson SJ, Howard PW, Hartley JA, Brooks NA, Adams LJ, Jenkins TC, Kelland LR, Thurston DE. Design, Synthesis, and Evaluation of a Novel Pyrrolobenzodiazepine DNA-Interactive Agent with Highly Efficient Cross-Linking Ability and Potent Cytotoxicity. J Med Chem. 2001;44:737–748. doi: 10.1021/jm001064n. [DOI] [PubMed] [Google Scholar]
- 356.Alley MC, Hollingshead MG, Pacula-Cox CM, Waud WR, Hartley JA, Howard PW, Gregson SJ, Thurston DE, Sausville EA. Sjg-136 (Nsc 694501), a Novel Rationally Designed DNA Minor Groove Interstrand Cross-Linking Agent with Potent and Broad Spectrum Antitumor Activity: Part 2: Efficacy Evaluations. Cancer Res. 2004;64:6700–6706. doi: 10.1158/0008-5472.CAN-03-2942. [DOI] [PubMed] [Google Scholar]
- 357.Hartley JA, Spanswick VJ, Brooks N, Clingen PH, McHugh PJ, Hochhauser D, Pedley RB, Kelland LR, Alley MC, Schultz R. Sjg-136 (Nsc 694501), a Novel Rationally Designed DNA Minor Groove Interstrand Cross-Linking Agent with Potent and Broad Spectrum Antitumor Activity: Part 1: Cellular Pharmacology, in Vitro and Initial in Vivo Antitumor Activity. Cancer Res. 2004;64:6693–6699. doi: 10.1158/0008-5472.CAN-03-2941. [DOI] [PubMed] [Google Scholar]
- 358.Martin C, Ellis T, McGurk CJ, Jenkins TC, Hartley JA, Waring MJ, Thurston DE. Sequence-Selective Interaction of the Minor-Groove Interstrand Cross-Linking Agent Sjg-136 with Naked and Cellular DNA: Footprinting and Enzyme Inhibition Studies. Biochemistry. 2005;44:4135–4147. doi: 10.1021/bi0479813. [DOI] [PubMed] [Google Scholar]
- 359.Rahman KM, Thompson AS, James CH, Narayanaswamy M, Thurston DE. The Pyrrolobenzodiazepine Dimer Sjg-136 Forms Sequence-Dependent Intrastrand DNA Cross-Links and Monoalkylated Adducts in Addition to Interstrand Cross-Links. J Am Chem Soc. 2009;131:13756–13766. doi: 10.1021/ja902986x. [DOI] [PubMed] [Google Scholar]
- 360.Rahman KM, James CH, Thurston DE. Effect of Base Sequence on the DNA Cross-Linking Properties of Pyrrolobenzodiazepine (Pbd) Dimers. Nucleic Acids Res. 2011;39:5800–5812. doi: 10.1093/nar/gkr122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Snapper I. Stilbamidine and Pentamidine in Multiple Myeloma. JAMA. 1947;133:157–161. doi: 10.1001/jama.1947.02880030011002. [DOI] [PubMed] [Google Scholar]
- 362.Seager LD. Some Pharmacological Actions of the Diamidines. Fed Proc. 1947;6:370. [PubMed] [Google Scholar]
- 363.Rosenberg EF. The Diamidines in Chemotherapy; a Survey of Recent Developments with a Note Regarding Therapeutic Trials in Patients with Rheumatoid Arthritis. Ann Intern Med. 1946;25:832–844. doi: 10.7326/0003-4819-25-5-832. [DOI] [PubMed] [Google Scholar]
- 364.Heathcote RS. The Diamidines; Their Pharmacological Actions and Their Therapeutic Uses in Some Tropical Diseases. J Trop Med Hyg. 1946;49:1. [PubMed] [Google Scholar]
- 365.Collier HO, Lourie EM. The Action in Vitro of Diamidines and Other Compounds on Leishmania Donovani. Ann Trop Med Parasitol. 1946;40:88–100. doi: 10.1080/00034983.1946.11685265. [DOI] [PubMed] [Google Scholar]
- 366.Das BP, Wallace RA, Boykin DW., Jr Synthesis and Antitrypanosomal Activity of Some Bis(4-Guanylphenyl) Five- and Six-Membered Ring Heterocycles. J Med Chem. 1980;23:578–581. doi: 10.1021/jm00179a022. [DOI] [PubMed] [Google Scholar]
- 367.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Antiparasitic Compounds That Target DNA. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Wilson WD, Tanious FA, Buczak H, Venkatramanan MK, Das BP, Boykin DW. The Effects of Ligand Structure on Binding Mode and Specificity in the Interaction of Unfused Aromatic Cations with DNA. Molecular Basis of Specificity in Nucleic Acid-Drug Interactions: Proceedings of the Twenty-Third Jerusalem Symposium on Quantum Chemistry and Biochemistry; Jerusalem, Israel. May 14–17, 1970; The Netherlands: Springer; 1970. p. 331. [DOI] [Google Scholar]
- 369.Wilson WD, Ratmeyer L, Zhao M, Strekowski L, Boykin D. The Search for Structure-Specific Nucleic Acid-Interactive Drugs: Effects of Compound Structure on Rna Versus DNA Interaction Strength. Biochemistry. 1993;32:4098–4104. doi: 10.1021/bi00066a035. [DOI] [PubMed] [Google Scholar]
- 370.Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, Boykin DW. Dications That Target the DNA Minor Groove: Compound Design and Preparation, DNA Interactions, Cellular Distribution and Biological Activity. Curr Med Chem: Anti-Cancer Agents. 2005;5:389–408. doi: 10.2174/1568011054222319. [DOI] [PubMed] [Google Scholar]
- 371.Tidwell RR, Boykin DW. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes. Wiley-VCH; Weinheim, Germany: 2002. Dicationic DNA Minor Groove Binders as Antimicrobial Agents. [Google Scholar]
- 372.Tanious FA, Spychala J, Kumar A, Greene K, Boykin DW, Wilson WD. Different Binding Mode in at and Gc Sequences for Unfused-Aromatic Dications. J Biomol Struct Dyn. 1994;11:1063–1083. doi: 10.1080/07391102.1994.10508053. [DOI] [PubMed] [Google Scholar]
- 373.Nguyen B, Neidle S, Wilson WD. A Role for Water Molecules in DNA-Ligand Minor Groove Recognition. Acc Chem Res. 2009;42:11–21. doi: 10.1021/ar800016q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Nguyen B, Boykin DW, Wilson WD. Synthetic and Biophysical Studies of DNA Binding Compounds. Research Signpost; Kerala, India: 2007. DNA Minor Groove Interactions of Antiparasitic Diamidines: Re-evaluation of the Crescent-Shape Concept in Groove-Binding; pp. 39–66. [Google Scholar]
- 375.Farahat AA, Kumar A, Wenzler T, Brun R, Paul A, Wilson WD, Boykin DW, Ismail MA. Indole and Benzimidazole Bichalcophenes: Synthesis, DNA Binding and Antiparasitic Activity. Eur J Med Chem. 2017;5234:30858–30859. doi: 10.1016/j.ejmech.2017.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Neidle S. DNA Minor-Groove Recognition by Small Molecules. Nat Prod Rep. 2001;18:291–309. doi: 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]
- 377.Laughton CA, Tanious F, Nunn CM, Boykin DW, Wilson WD, Neidle S. A Crystallographic and Spectroscopic Study of the Complex between D(Cgcgaattcgcg)2 and 2,5-Bis(4-Guanylphenyl)Furan, an Analogue of Berenil. Structural Origins of Enhanced DNA-Binding Affinity. Biochemistry. 1996;35:5655–5661. doi: 10.1021/bi952162r. [DOI] [PubMed] [Google Scholar]
- 378.Boykin DW, Kumar A, Spychala J, Zhou M, Lombardy RJ, Wilson WD, Dykstra CC, Jones SK, Hall JE. Dicationic Diarylfurans as Anti-Pneumocystis Carinii Agents. J Med Chem. 1995;38:912–916. doi: 10.1021/jm00006a009. [DOI] [PubMed] [Google Scholar]
- 379.Wang L, Carrasco C, Kumar A, Stephens CE, Bailly C, Boykin DW, Wilson WD. Evaluation of the Influence of Compound Structure on Stacked-Dimer Formation in the DNA Minor Groove. Biochemistry. 2001;40:2511–2521. doi: 10.1021/bi002301r. [DOI] [PubMed] [Google Scholar]
- 380.Wang L, Bailly C, Kumar A, Ding D, Bajic M, Boykin DW, Wilson WD. Specific Molecular Recognition of Mixed Nucleic Acid Sequences: An Aromatic Dication That Binds in the DNA Minor Groove as a Dimer. Proc Natl Acad Sci U S A. 2000;97:12–16. doi: 10.1073/pnas.97.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Munde M, Ismail MA, Arafa R, Peixoto P, Collar CJ, Liu Y, Hu L, David-Cordonnier MH, Lansiaux A, Bailly C. Design of DNA Minor Groove Binding Diamidines That Recognize Gc Base Pair Sequences: A Dimeric-Hinge Interaction Motif. J Am Chem Soc. 2007;129:13732–13743. doi: 10.1021/ja074560a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Guo P, Paul A, Kumar A, Farahat AA, Kumar D, Wang S, Boykin DW, Wilson WD. The Thiophene “Sigma-Lole” as a Concept for Preorganized, Specific Recognition of Gc Base Pairs in the DNA Minor Groove. Chem - Eur J. 2016;22:15404–15412. doi: 10.1002/chem.201603422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Paul A, Kumar A, Nanjunda R, Farahat AA, Boykin DW, Wilson WD. Systematic Synthetic and Biophysical Development of Mixed Sequence DNA Binding Agents. Org Biomol Chem. 2017;15:827–835. doi: 10.1039/C6OB02390H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Liu Y, Chai Y, Kumar A, Tidwell RR, Boykin DW, Wilson WD. Designed Compounds for Recognition of 10 Base Pairs of DNA with Two at Binding Sites. J Am Chem Soc. 2012;134:5290–5299. doi: 10.1021/ja211628j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Laughlin-Toth S, Carter EK, Ivanov I, Wilson WD. DNA Microstructure Influences Selective Binding of Small Molecules Designed to Target Mixed-Site DNA Sequences. Nucleic Acids Res. 2017;45:1297–1306. doi: 10.1093/nar/gkw1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Harika NK, Germann MW, Wilson WD. First Structure of a Designed Minor Groove Binding Heterocyclic Cation That Specifically Recognizes Mixed DNA Base Pair Sequences. Chem - Eur J. 2017;23:17612. doi: 10.1002/chem.201704563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Wells RD. Non-B DNA Conformations, Mutagenesis and Disease. Trends Biochem Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 388.Choi J, Majima T. Conformational Changes of Non-B DNA. Chem Soc Rev. 2011;40:5893–5909. doi: 10.1039/c1cs15153c. [DOI] [PubMed] [Google Scholar]
- 389.Balasubramanian S, Neidle S. G-Quadruplex Nucleic Acids as Therapeutic Targets. Curr Opin Chem Biol. 2009;13:345–353. doi: 10.1016/j.cbpa.2009.04.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Balasubramanian S, Hurley LH, Neidle S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat Rev Drug Discovery. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Maizels N, Gray LT. The G4 Genome. PLoS Genet. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Bochman ML, Paeschke K, Zakian VA. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat Rev Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Williamson JR, Raghuraman MK, Cech TR. Monovalent Cation-Induced Structure of Telomeric DNA: The G-Quartet Model. Cell. 1989;59:871–880. doi: 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- 394.Sundquist WI, Klug A. Telomeric DNA Dimerizes by Formation of Guanine Tetrads between Hairpin Loops. Nature. 1989;342:825–829. doi: 10.1038/342825a0. [DOI] [PubMed] [Google Scholar]
- 395.Davis JT. G-Quartets 40 Years Later: From 5′-Gmp to Molecular Biology and Supramolecular Chemistry. Angew Chem, Int Ed. 2004;43:668–698. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- 396.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Miyoshi D, Sugimoto N. Molecular Crowding Effects on Structure and Stability of DNA. Biochimie. 2008;90:1040–1051. doi: 10.1016/j.biochi.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 398.Phan AT. Human Telomeric G-Quadruplex: Structures of DNA and Rna Sequences. FEBS J. 2010;277:1107–1117. doi: 10.1111/j.1742-4658.2009.07464.x. [DOI] [PubMed] [Google Scholar]
- 399.Patel DJ, Phan AT, Kuryavyi V. Human Telomere, Oncogenic Promoter and 5′-Utr G-Quadruplexes: Diverse Higher Order DNA and Rna Targets for Cancer Therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Zahler AM, Williamson JR, Cech TR, Prescott DM. Inhibition of Telomerase by G-Quartet DNA Structures. Nature. 1991;350:718–720. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 401.Neidle S, Parkinson G. Telomere Maintenance as a Target for Anticancer Drug Discovery. Nat Rev Drug Discovery. 2002;1:383–393. doi: 10.1038/nrd793. [DOI] [PubMed] [Google Scholar]
- 402.Rezler EM, Bearss DJ, Hurley LH. Telomere Inhibition and Telomere Disruption as Processes for Drug Targeting. Annu Rev Pharmacol Toxicol. 2003;43:359–379. doi: 10.1146/annurev.pharmtox.43.100901.135733. [DOI] [PubMed] [Google Scholar]
- 403.Mergny JL, Helene C. G-Quadruplex DNA: A Target for Drug Design. Nat Med. 1998;4:1366–1367. doi: 10.1038/3949. [DOI] [PubMed] [Google Scholar]
- 404.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound. J Med Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 405.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress C-Myc Transcription. Proc Natl Acad Sci U S A. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.De Cian A, Cristofari G, Reichenbach P, De Lemos E, Monchaud D, Teulade-Fichou MP, Shin-Ya K, Lacroix L, Lingner J, Mergny JL. Reevaluation of Telomerase Inhibition by Quadruplex Ligands and Their Mechanisms of Action. Proc Natl Acad Sci U S A. 2007;104:17347–17352. doi: 10.1073/pnas.0707365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Gowan SM, Harrison JR, Patterson L, Valenti M, Read MA, Neidle S, Kelland LR. A G-Quadruplex-Interactive Potent Small-Molecule Inhibitor of Telomerase Exhibiting in Vitro and in Vivo Antitumor Activity. Mol Pharmacol. 2002;61:1154–1162. doi: 10.1124/mol.61.5.1154. [DOI] [PubMed] [Google Scholar]
- 408.Read M. Structure-Based Design of Selective and Potent G Quadruplex-Mediated Telomerase Inhibitors. Proc Natl Acad Sci U S A. 2001;98:4844–4849. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Gunaratnam M, Swank S, Haider SM, Galesa K, Reszka AP, Beltran M, Cuenca F, Fletcher JA, Neidle S. Targeting Human Gastrointestinal Stromal Tumor Cells with a Quadruplex-Binding Small Molecule. J Med Chem. 2009;52:3774–3783. doi: 10.1021/jm900424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Huang FC, Chang CC, Lou PJ, Kuo IC, Chien CW, Chen CT, Shieh FY, Chang TC, Lin JJ. G-Quadruplex Stabilizer 3,6-Bis(1-Methyl-4-Vinylpyridinium)Carbazole Diiodide Induces Accelerated Senescence and Inhibits Tumorigenic Properties in Cancer Cells. Mol Cancer Res. 2008;6:955–964. doi: 10.1158/1541-7786.MCR-07-0260. [DOI] [PubMed] [Google Scholar]
- 411.Monchaud D, Teulade-Fichou M-P. A Hitchhiker’s Guide to G-Quadruplex Ligands. Org Biomol Chem. 2008;6:627. doi: 10.1039/B714772B. [DOI] [PubMed] [Google Scholar]
- 412.Rodriguez Rl, Müller S, Yeoman JA, Trentesaux C, Riou J-Fo, Balasubramanian S. A Novel Small Molecule That Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J Am Chem Soc. 2008;130:15758–15759. doi: 10.1021/ja805615w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Muller S, Kumari S, Rodriguez R, Balasubramanian S. Small-Molecule-Mediated G-Quadruplex Isolation from Human Cells. Nat Chem. 2010;2:1095–1098. doi: 10.1038/nchem.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP. Small-Molecule-Induced DNA Damage Identifies Alternative DNA Structures in Human Genes. Nat Chem Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP. Anticancer Activity of Cx-3543: A Direct Inhibitor of Rrna Biogenesis. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 416.Duan WH, Rangan A, Vankayalapati H, Kim MY, Zeng QP, Sun DK, Han HY, Fedoroff OY, Nishioka D, Rha SY. Design and Synthesis of Fluoroquinophenoxazines That Interact with Human Telomeric G-Quadruplexes and Their Biological Effects. Mol Cancer Ther. 2001;1:103–120. [PubMed] [Google Scholar]
- 417.Todd AK, Johnston M, Neidle S. Highly Prevalent Putative Quadruplex Sequence Motifs in Human DNA. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Huppert JL, Balasubramanian S. G-Quadruplexes in Promoters Throughout the Human Genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Huppert JL, Bugaut A, Kumari S, Balasubramanian S. G-Quadruplexes: The Beginning and End of Utrs. Nucleic Acids Res. 2008;36:6260–6268. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. High-Throughput Sequencing of DNA G-Quadruplex Structures in the Human Genome. Nat Biotechnol. 2015;33:877–881. doi: 10.1038/nbt.3295. [DOI] [PubMed] [Google Scholar]
- 421.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. Putative DNA Quadruplex Formation within the Human C-Kit Oncogene. J Am Chem Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Cogoi S, Xodo LE. G-Quadruplex Formation within the Promoter of the Kras Proto-Oncogene and Its Effect on Transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Dai J, Chen D, Jones RA, Hurley LH, Yang D. Nmr Solution Structure of the Major G-Quadruplex Structure Formed in the Human Bcl2 Promoter Region. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Guo K, Gokhale V, Hurley LH, Sun D. Intramolecularly Folded G-Quadruplex and I-Motif Structures in the Proximal Promoter of the Vascular Endothelial Growth Factor Gene. Nucleic Acids Res. 2008;36:4598–4608. doi: 10.1093/nar/gkn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Lim KW, Lacroix L, Yue DJ, Lim JK, Lim JM, Phan AT. Coexistence of Two Distinct G-Quadruplex Conformations in the Htert Promoter. J Am Chem Soc. 2010;132:12331–12342. doi: 10.1021/ja101252n. [DOI] [PubMed] [Google Scholar]
- 426.Wang JM, Huang FC, Kuo MH, Wang ZF, Tseng TY, Chang LC, Yen SJ, Chang TC, Lin JJ. Inhibition of Cancer Cell Migration and Invasion through Suppressing the Wnt1-Mediating Signal Pathway by G-Quadruplex Structure Stabilizers. J Biol Chem. 2014;289:14612–14623. doi: 10.1074/jbc.M114.548230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Métifiot M, Amrane S, Litvak S, Andreola ML. G-Quadruplexes in Viruses: Function and Potential Therapeutic Applications. Nucleic Acids Res. 2014;42:12352. doi: 10.1093/nar/gku999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Cahoon LA, Seifert HS. An Alternative DNA Structure Is Necessary for Pilin Antigenic Variation in Neisseria Gonorrhoeae. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Amrane S, Kerkour A, Bedrat A, Vialet B, Andreola ML, Mergny JL. Topology of a DNA G-Quadruplex Structure Formed in the Hiv-1 Promoter: A Potential Target for Anti-Hiv Drug Development. J Am Chem Soc. 2014;136:5249–5252. doi: 10.1021/ja501500c. [DOI] [PubMed] [Google Scholar]
- 430.Gude L, Berkovitch SS, Santos WL, Kutchukian PS, Pawloski AR, Kuimelis R, McGall G, Verdine GL. Mapping Targetable Sites on Human Telomerase Rna Pseudoknot/Template Domain Using 2′-Ome Rna-Interacting Polynucleotide (Riptide) Microarrays. J Biol Chem. 2012;287:18843–18853. doi: 10.1074/jbc.M111.316596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Poehlsgaard J, Douthwaite S. The Bacterial Ribosome as a Target for Antibiotics. Nat Rev Microbiol. 2005;3:870–881. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- 432.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural Basis for the Interaction of Antibiotics with the Peptidyl Transferase Centre in Eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 433.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Structural Basis for Aminoglycoside Inhibition of Bacterial Ribosome Recycling. Nat Struct Mol Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 434.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic Control by a Metabolite Binding Mrna. Chem Biol. 2002;9:1043–1049. doi: 10.1016/S1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 435.Johnson LF, Abelson HT, Penman S, Green H. The Relative Amounts of the Cytoplasmic Rna Species in Normal, Transformed and Senescent Cultured Cell Lines. J Cell Physiol. 1977;90:465–470. doi: 10.1002/jcp.1040900310. [DOI] [PubMed] [Google Scholar]
- 436.Zuker M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating Chemical Modification Constraints into a Dynamic Programming Algorithm for Prediction of Rna Secondary Structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Tinoco I, Jr, Borer PN, Dengler B, Levine MD, Uhlenbeck OC, Crothers DM, Gralla J. Improved Estimation of Secondary Structure in Ribonucleic Acids. Nat New Biol. 1973;246:40–41. doi: 10.1038/newbio246040a0. [DOI] [PubMed] [Google Scholar]
- 439.Tinoco I, Jr, Uhlenbeck OC, Levine MD. Estimation of Secondary Structure in Ribonucleic Acids. Nature. 1971;230:362–367. doi: 10.1038/230362a0. [DOI] [PubMed] [Google Scholar]
- 440.Uhlenbeck OC, Baller J, Doty P. Complementary Oligonucleotide Binding to the Anticodon Loop of Fmet-Transfer Rna. Nature. 1970;225:508–510. doi: 10.1038/225508a0. [DOI] [PubMed] [Google Scholar]
- 441.Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH. Improved Free-Energy Parameters for Predictions of Rna Duplex Stability. Proc Natl Acad Sci U S A. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Xia T, SantaLucia J, Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of Rna Duplexes with Watson-Crick Base Pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 443.Zuker M. On Finding All Suboptimal Foldings of an Rna Molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 444.Jaeger JA, Turner DH, Zuker M. Improved Predictions of Secondary Structures for Rna. Proc Natl Acad Sci U S A. 1989;86:7706–7710. doi: 10.1073/pnas.86.20.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Jaeger JA, Turner DH, Zuker M. Predicting Optimal and Suboptimal Secondary Structure for Rna. Methods Enzymol. 1990;183:281–306. doi: 10.1016/0076-6879(90)83019-6. [DOI] [PubMed] [Google Scholar]
- 446.Mathews DH. Using the RNAstructure Software Package to Predict Conserved Rna Structures. Current Protocols in Bioinformatics. 2014;46:11–22. doi: 10.1002/0471250953.bi1204s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Peattie DA. Direct Chemical Method for Sequencing Rna. Proc Natl Acad Sci U S A. 1979;76:1760–1764. doi: 10.1073/pnas.76.4.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the Structure of Rnas in Solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate Shape-Directed Rna Structure Determination. Proc Natl Acad Sci U S A. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Kwok CK, Ding Y, Tang Y, Assmann SM, Bevilacqua PC. Determination of in Vivo Rna Structure in Low-Abundance Transcripts. Nat Commun. 2013;4:2971. doi: 10.1038/ncomms3971. [DOI] [PubMed] [Google Scholar]
- 451.Wells SE, Hughes JM, Haller Igel A, Ares M., Jr Use of Dimethyl Sulfate to Probe Rna Structure in Vivo. Methods Enzymol. 2000;318:479–493. doi: 10.1016/S0076-6879(00)18071-1. [DOI] [PubMed] [Google Scholar]
- 452.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In Vivo Genome-Wide Profiling of Rna Secondary Structure Reveals Novel Regulatory Features. Nature. 2013;505:696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 453.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-Wide Probing of Rna Structure Reveals Active Unfolding of Mrna Structures in Vivo. Nature. 2013;505:701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E. Landscape and Variation of Rna Secondary Structure across the Human Transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. Rna Shape Analysis in Living Cells. Nat Chem Biol. 2012;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Spitale RC, Flynn RA, Torre EA, Kool ET, Chang HY. Rna Structural Analysis by Evolving Shape Chemistry. Wiley Interdisp Rev RNA. 2014;5:867–881. doi: 10.1002/wrna.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG. Motor Neuron Disease. Smn2 Splicing Modifiers Improve Motor Function and Longevity in Mice with Spinal Muscular Atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 458.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG. Smn2 Splice Modulators Enhance U1-Pre-Mrna Association and Rescue Sma Mice. Nat Chem Biol. 2015;11:511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- 459.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S. Ptc124 Targets Genetic Disorders Caused by Nonsense Mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 460.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL. Two-Dimensional Combinatorial Screening Identifies Specific Aminoglycoside-Rna Internal Loop Partners. J Am Chem Soc. 2008;130:11185–11194. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 461.Tran T, Disney MD. Identifying the Preferred Rna Motifs and Chemotypes That Interact by Probing Millions of Combinations. Nat Commun. 2012;3:1125. doi: 10.1038/ncomms2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Velagapudi SP, Luo Y, Tran T, Haniff HS, Nakai Y, Fallahi M, Martinez GJ, Childs-Disney JL, Disney MD. Defining Rna–Small Molecule Affinity Landscapes Enables Design of a Small Molecule Inhibitor of an Oncogenic Noncoding Rna. ACS Cent Sci. 2017;3:205–216. doi: 10.1021/acscentsci.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. A Small Molecule Microarray Platform to Select Rna Internal Loop-Ligand Interactions. ACS Chem Biol. 2007;2:745–754. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 464.Llano-Sotelo B, Azucena EF, Jr, Kotra LP, Mobashery S, Chow CS. Aminoglycosides Modified by Resistance Enzymes Display Diminished Binding to the Bacterial Ribosomal Aminoacyl-Trna Site. Chem Biol. 2002;9:455–463. doi: 10.1016/S1074-5521(02)00125-4. [DOI] [PubMed] [Google Scholar]
- 465.Tran T, Disney MD. Molecular Recognition of 6′-N-5-Hexynoate Kanamycin a and Rna 1 × 1 Internal Loops Containing Ca Mismatches. Biochemistry. 2011;50:962–969. doi: 10.1021/bi101724h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Velagapudi SP, Seedhouse SJ, Disney MD. Structure-Activity Relationships through Sequencing (Starts) Defines Optimal and Suboptimal Rna Motif Targets for Small Molecules. Angew Chem, Int Ed. 2010;49:3816–3818. doi: 10.1002/anie.200907257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Velagapudi SP, Seedhouse SJ, French J, Disney MD. Defining the Rna Internal Loops Preferred by Benzimidazole Derivatives Via 2d Combinatorial Screening and Computational Analysis. J Am Chem Soc. 2011;133:10111–10118. doi: 10.1021/ja200212b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Velagapudi SP, Pushechnikov A, Labuda LP, French JM, Disney MD. Probing a 2-Aminobenzimidazole Library for Binding to Rna Internal Loops Via Two-Dimensional Combinatorial Screening. ACS Chem Biol. 2012;7:1902–1909. doi: 10.1021/cb300213g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Velagapudi SP, Gallo SM, Disney MD. Sequence-Based Design of Bioactive Small Molecules That Target Precursor Micrornas. Nat Chem Biol. 2014;10:291–297. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, Childs-Disney JL. Inforna 2.0: A Platform for the Sequence-Based Design of Small Molecules Targeting Structured Rnas. ACS Chem Biol. 2016;11:1720–1728. doi: 10.1021/acschembio.6b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. Mirbase: Microrna Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Guttilla IK, White BA. Coordinate Regulation of Foxo1 by Mir-27a, Mir-96, and Mir-182 in Breast Cancer Cells. J Biol Chem. 2009;284:23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Velagapudi SP, Cameron MD, Haga CL, Rosenberg LH, Lafitte M, Duckett DR, Phinney DG, Disney MD. Design of a Small Molecule against an Oncogenic Noncoding Rna. Proc Natl Acad Sci U S A. 2016;113:5898–5903. doi: 10.1073/pnas.1523975113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Childs-Disney JL, Tsitovich PB, Disney MD. Using Modularly Assembled Ligands to Bind Rna Internal Loops Separated by Different Distances. ChemBioChem. 2011;12:2143–2146. doi: 10.1002/cbic.201100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Velagapudi SP, Disney MD. Two-Dimensional Combinatorial Screening Enables the Bottom-up Design of a Microrna-10b Inhibitor. Chem Commun. 2014;50:3027–3029. doi: 10.1039/c3cc00173c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Pang F, Zha R, Zhao Y, Wang Q, Chen D, Zhang Z, Chen T, Yao M, Gu J, He X. Mir-525–3p Enhances the Migration and Invasion of Liver Cancer Cells by Downregulating Znf395. PLoS One. 2014;9:e90867. doi: 10.1371/journal.pone.0090867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Childs-Disney JL, Disney MD. Small Molecule Targeting of a Microrna Associated with Hepatocellular Carcinoma. ACS Chem Biol. 2016;11:375–380. doi: 10.1021/acschembio.5b00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Al Sibae MR, McGuire BM. Current Trends in the Treatment of Hepatic Encephalopathy. Ther Clin Risk Manage. 2009;5:617–626. doi: 10.2147/TCRM.S4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Haga CL, Velagapudi SP, Strivelli JR, Yang W-Y, Disney MD, Phinney DG. Small Molecule Inhibition of Mir-544 Biogenesis Disrupts Adaptive Responses to Hypoxia by Modulating Atm-Mtor Signaling. ACS Chem Biol. 2015;10:2267–2276. doi: 10.1021/acschembio.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, Disney MD. Small Molecule Inhibition of Microrna-210 Reprograms an Oncogenic Hypoxic Circuit. J Am Chem Soc. 2017;139:3446–3455. doi: 10.1021/jacs.6b11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Haga CL, Phinney DG. Micrornas in the Imprinted Dlk1-Dio3 Region Repress the Epithelial-to-Mesenchymal Transition by Targeting the Twist1 Protein Signaling Network. J Biol Chem. 2012;287:42695–42707. doi: 10.1074/jbc.M112.387761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Kelly TJ, Souza AL, Clish CB, Puigserver P. A Hypoxia-Induced Positive Feedback Loop Promotes Hypoxia-Inducible Factor 1alpha Stability through Mir-210 Suppression of Glycerol-3-Phosphate Dehydrogenase 1-Like. Mol Cell Biol. 2011;31:2696–2706. doi: 10.1128/MCB.01242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The Tumour Suppressor Protein Vhl Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 484.Grosso S, Doyen J, Parks SK, Bertero T, Paye A, Cardinaud B, Gounon P, Lacas-Gervais S, Noel A, Pouyssegur J. Mir-210 Promotes a Hypoxic Phenotype and Increases Radioresistance in Human Lung Cancer Cell Lines. Cell Death Dis. 2013;4:e544. doi: 10.1038/cddis.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ. A Microrna Polycistron as a Potential Human Oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Hsu TI, Hsu CH, Lee KH, Lin JT, Chen CS, Chang KC, Su CY, Hsiao M, Lu PJ. Microrna-18a Is Elevated in Prostate Cancer and Promotes Tumorigenesis through Suppressing Stk4 in Vitro and in Vivo. Oncogenesis. 2014;3:e99. doi: 10.1038/oncsis.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic Dystrophy Type 2 Caused by a Cctg Expansion in Intron 1 of Znf9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 488.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic Dystrophy in Transgenic Mice Expressing an Expanded Cug Repeat. Science. 2000;289:1769–1772. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 489.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J. Expanded Ggggcc Hexanucleotide Repeat in Noncoding Region of C9orf72 Causes Chromosome 9p-Linked Ftd and Als. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L. A Hexanucleotide Repeat Expansion in C9orf72 Is the Cause of Chromosome 9p21-Linked Als-Ftd. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ. Sam68 Sequestration and Partial Loss of Function Are Associated with Splicing Alterations in Fxtas Patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L. Large Expansion of the Attct Pentanucleotide Repeat in Spinocerebellar Ataxia Type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 493.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang W-Y, Fostvedt E, Jansen-West K, Belzil VV, Desaro P. Discovery of a Biomarker and Lead Small Molecules to Target R(Ggggcc)-Associated Defects in C9ftd/Als. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Yang W-Y, He F, Strack RL, Oh SY, Frazer M, Jaffrey SR, Todd PK, Disney MD. Small Molecule Recognition and Tools to Study Modulation of R(Cgg)Exp in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem Biol. 2016;11:2456–2465. doi: 10.1021/acschembio.6b00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, Disney MD. Structure of the Myotonic Dystrophy Type 2 Rna and Designed Small Molecules That Reduce Toxicity. ACS Chem Biol. 2014;9:538–550. doi: 10.1021/cb4007387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Disney MD, Liu B, Yang W-Y, Sellier C, Tran T, Charlet-Berguerand N, Childs-Disney JL. A Small Molecule That Targets R(Cgg)Exp and Improves Defects in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem Biol. 2012;7:1711–1718. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, Disney MD. Precise Small-Molecule Recognition of a Toxic Cug Rna Repeat Expansion. Nat Chem Biol. 2016;13:188–193. doi: 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Rzuczek SG, Gao Y, Tang ZZ, Thornton CA, Kodadek T, Disney MD. Features of Modularly Assembled Compounds That Impart Bioactivity against an Rna Target. ACS Chem Biol. 2013;8:2312–2321. doi: 10.1021/cb400265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Yang W-Y, Gao R, Southern M, Sarkar PS, Disney MD. Design of a Bioactive Small Molecule That Targets R(Auucu) Repeats in Spinocerebellar Ataxia 10. Nat Commun. 2016;7:11647. doi: 10.1038/ncomms11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Rzuczek SG, Park H, Disney MD. A Toxic Rna Catalyzes the in Cellulo Synthesis of Its Own Inhibitor. Angew Chem, Int Ed. 2014;53:10956–10959. doi: 10.1002/anie.201406465. [DOI] [PubMed] [Google Scholar]
- 501.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V. Cgg Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC. Non-Atg–Initiated Translation Directed by Microsatellite Expansions. Proc Natl Acad Sci U S A. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD. Targeting the R(Cgg) Repeats That Cause Fxtas with Modularly Assembled Small Molecules and Oligonucleotides. ACS Chem Biol. 2014;9:904–912. doi: 10.1021/cb400875u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P. Distinct Brain Transcriptome Profiles in C9orf72-Associated and Sporadic Als. Nat Neurosci. 2015;18:1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC. Ggggcc Repeat Expansion in C9orf72 Compromises Nucleocytoplasmic Transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M. C9orf72 Poly(Ga) Aggregates Sequester and Impair Hr23 and Nucleocytoplasmic Transport Proteins. Nat Neurosci. 2016;19:668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C. The C9orf72 Ggggcc Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in Ftld/Als. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 508.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R. Unconventional Translation of C9orf72 Ggggcc Expansion Generates Insoluble Polypeptides Specific to C9ftd/Als. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Park H, Tran T, Lee JH, Park H, Disney MD. Controlled Dehydration Improves the Diffraction Quality of Two Rna Crystals. BMC Struct Biol. 2016;16:19. doi: 10.1186/s12900-016-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic Dystrophy Type 1 Is Associated with Nuclear Foci of Mutant Rna, Sequestration of Muscleblind Proteins and Deregulated Alternative Splicing in Neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 511.Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N. Misregulated Alternative Splicing of Bin1 Is Associated with T Tubule Alterations and Muscle Weakness in Myotonic Dystrophy. Nat Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 512.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of Trinucleotide Repeat Transcripts in Nuclei of Myotonic Dystrophy Cells and Tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Furling D, Lemieux D, Taneja K, Puymirat J. Decreased Levels of Myotonic Dystrophy Protein Kinase (Dmpk) and Delayed Differentiation in Human Myotonic Dystrophy Myoblasts. Neuromuscul Disord. 2001;11:728–735. doi: 10.1016/S0960-8966(01)00226-7. [DOI] [PubMed] [Google Scholar]
- 514.Hamshere MG, Newman EE, Alwazzan M, Athwal BS, Brook JD. Transcriptional Abnormality in Myotonic Dystrophy Affects Dmpk but Not Neighboring Genes. Proc Natl Acad Sci U S A. 1997;94:7394–7399. doi: 10.1073/pnas.94.14.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Angelbello AJ, Gonzalez AL, Rzuczek SG, Disney MD. Development of Pharmacophore Models for Small Molecules Targeting Rna: Application to the Rna Repeat Expansion in Myotonic Dystrophy Type 1. Bioorg Med Chem Lett. 2016;26:5792–5796. doi: 10.1016/j.bmcl.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Childs-Disney JL, Stepniak-Konieczna E, Tran T, Yildirim I, Park H, Chen CZ, Hoskins J, Southall N, Marugan JJ, Patnaik S. Induction and Reversal of Myotonic Dystrophy Type 1 Pre-Mrna Splicing Defects by Small Molecules. Nat Commun. 2013;4:2044. doi: 10.1038/ncomms3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA. Design of a Bioactive Small Molecule That Targets the Myotonic Dystrophy Type 1 Rna Via an Rna Motif–Ligand Database and Chemical Similarity Searching. J Am Chem Soc. 2012;134:4731–4742. doi: 10.1021/ja210088v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Click Chemistry in Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks. Angew Chem, Int Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::AID-ANIE1053>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 519.Krasinski A, Radic Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC. In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J Am Chem Soc. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 520.Poulin-Kerstien AT, Dervan PB. DNA-Templated Dimerization of Hairpin Polyamides. J Am Chem Soc. 2003;125:15811–15821. doi: 10.1021/ja030494a. [DOI] [PubMed] [Google Scholar]
- 521.Morgan BS, Forte JE, Culver RN, Zhang Y, Hargrove AE. Discovery of Key Physicochemical, Structural, and Spatial Properties of Rna-Targeted Bioactive Ligands. Angew Chem, Int Ed. 2017;56:13498–13502. doi: 10.1002/anie.201707641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Conformation of the Tar Rna-Arginine Complex by Nmr Spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 523.Athanassiou Z, Patora K, Dias RL, Moehle K, Robinson JA, Varani G. Structure-Guided Peptidomimetic Design Leads to Nanomolar Beta-Hairpin Inhibitors of the Tat-Tar Interaction of Bovine Immunodeficiency Virus. Biochemistry. 2007;46:741–751. doi: 10.1021/bi0619371. [DOI] [PubMed] [Google Scholar]
- 524.Davidson A, Patora-Komisarska K, Robinson JA, Varani G. Essential Structural Requirements for Specific Recognition of Hiv Tar Rna by Peptide Mimetics of Tat Protein. Nucleic Acids Res. 2011;39:248–256. doi: 10.1093/nar/gkq713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Hamy F, Felder ER, Heizmann G, Lazdins J, Aboul-ela F, Varani G, Karn J, Klimkait T. An Inhibitor of the Tat/Tar Rna Interaction That Effectively Suppresses Hiv-1 Replication. Proc Natl Acad Sci U S A. 1997;94:3548–3553. doi: 10.1073/pnas.94.8.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of Selective Bioactive Small Molecules by Targeting an Rna Dynamic Ensemble. Nat Chem Biol. 2011;7:553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Filikov AV, Mohan V, Vickers TA, Griffey RH, Cook PD, Abagyan RA, James TL. Identification of Ligands for Rna Targets Via Structure-Based Virtual Screening: Hiv-1 Tar. J Comput-Aided Mol Des. 2000;14:593–610. doi: 10.1023/A:1008121029716. [DOI] [PubMed] [Google Scholar]
- 528.Lind KE, Du Z, Fujinaga K, Peterlin BM, James TL. Structure-Based Computational Database Screening, in Vitro Assay, and Nmr Assessment of Compounds That Target Tar Rna. Chem Biol. 2002;9:185–193. doi: 10.1016/S1074-5521(02)00106-0. [DOI] [PubMed] [Google Scholar]
- 529.Bose D, Jayaraj G, Suryawanshi H, Agarwala P, Pore SK, Banerjee R, Maiti S. The Tuberculosis Drug Streptomycin as a Potential Cancer Therapeutic: Inhibition of Mir-21 Function by Directly Targeting Its Precursor. Angew Chem, Int Ed. 2012;51:1019–1023. doi: 10.1002/anie.201106455. [DOI] [PubMed] [Google Scholar]
- 530.Shortridge MD, Walker MJ, Pavelitz T, Chen Y, Yang W, Varani G. A Macrocyclic Peptide Ligand Binds the Oncogenic Microrna-21 Precursor and Suppresses Dicer Processing. ACS Chem Biol. 2017;12:1611–1620. doi: 10.1021/acschembio.7b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A Simple Ligand That Selectively Targets Cug Trinucleotide Repeats and Inhibits Mbnl Protein Binding. Proc Natl Acad Sci U S A. 2009;106:16068–16073. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Wong C-H, Nguyen L, Peh J, Luu LM, Sanchez JS, Richardson SL, Tuccinardi T, Tsoi H, Chan WY, Chan HYE. Targeting Toxic Rnas That Cause Myotonic Dystrophy Type 1 (Dm1) with a Bisamidinium Inhibitor. J Am Chem Soc. 2014;136:6355–6361. doi: 10.1021/ja5012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Ofori LO, Hoskins J, Nakamori M, Thornton CA, Miller BL. From Dynamic Combinatorial ‘Hit’ to Lead: In Vitro and in Vivo Activity of Compounds Targeting the Pathogenic Rnas That Cause Myotonic Dystrophy. Nucleic Acids Res. 2012;40:6380–6390. doi: 10.1093/nar/gks298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Nakamori M, Taylor K, Mochizuki H, Sobczak K, Takahashi MP. Oral Administration of Erythromycin Decreases Rna Toxicity in Myotonic Dystrophy. Ann Clin Transl Neurol. 2016;3:42–54. doi: 10.1002/acn3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.García-López A, Llamusí B, Orzáez M, Pérez-Payá E, Artero RD. In Vivo Discovery of a Peptide That Prevents Cug–Rna Hairpin Formation and Reverses Rna Toxicity in Myotonic Dystrophy Models. Proc Natl Acad Sci U S A. 2011;108:11866–11871. doi: 10.1073/pnas.1018213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Nguyen L, Lee J, Zimmerman SC, Wong C-H. Small Molecules That Target the Toxic RNA in Myotonic Dystrophy Type 2. ChemMedChem. 2014;9:2455–2462. doi: 10.1002/cmdc.201402095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Zamiri B, Reddy K, Macgregor RB, Pearson CE. Tmpyp4 Distorts Rna G-Quadruplex Structures of the Disease-Associated R(Ggggcc)N Repeat of the C9orf72 Gene and Blocks Interaction of Rna-Binding Proteins. J Biol Chem. 2014;289:4653. doi: 10.1074/jbc.C113.502336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S. The C9orf72 Repeat Expansion Disrupts Nucleocytoplasmic Transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Winkler W, Nahvi A, Breaker RR. Thiamine Derivatives Bind Messenger Rnas Directly to Regulate Bacterial Gene Expression. Nature. 2002;419:952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 540.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. Sensing Small Molecules by Nascent Rna: A Mechanism to Control Transcription in Bacteria. Cell. 2002;111:747–756. doi: 10.1016/S0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 541.Blount KF, Wang JX, Lim J, Sudarsan N, Breaker RR. Antibacterial Lysine Analogs That Target Lysine Riboswitches. Nat Chem Biol. 2007;3:44–49. doi: 10.1038/nchembio842. [DOI] [PubMed] [Google Scholar]
- 542.Furukawa K, Gu H, Sudarsan N, Hayakawa Y, Hyodo M, Breaker RR. Identification of Ligand Analogues That Control C-Di-Gmp Riboswitches. ACS Chem Biol. 2012;7:1436–1443. doi: 10.1021/cb300138n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Kim JN, Blount KF, Puskarz I, Lim J, Link KH, Breaker RR. Design and Antimicrobial Action of Purine Analogs That Bind Guanine Riboswitches. ACS Chem Biol. 2009;4:915–927. doi: 10.1021/cb900146k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Gilbert SD, Reyes FE, Edwards AL, Batey RT. Adaptive Ligand Binding by the Purine Riboswitch in the Recognition of Guanine and Adenine Analogs. Structure. 2009;17:857–868. doi: 10.1016/j.str.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Lunse CE, Schmidt MS, Wittmann V, Mayer G. Carba-Sugars Activate the Glms-Riboswitch of Staphylococcus Aureus. ACS Chem Biol. 2011;6:675–678. doi: 10.1021/cb200016d. [DOI] [PubMed] [Google Scholar]
- 546.Daldrop P, Reyes FE, Robinson DA, Hammond CM, Lilley DM, Batey RT, Brenk R. Novel Ligands for a Purine Riboswitch Discovered by Rna-Ligand Docking. Chem Biol. 2011;18:324–335. doi: 10.1016/j.chembiol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Blount KF, Breaker RR. Riboswitches as Antibacterial Drug Targets. Nat Biotechnol. 2006;24:1558–1564. doi: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- 548.Park SJ, Kim YG, Park HJ. Identification of Rna Pseudoknot-Binding Ligand That Inhibits the −1 Ribosomal Frameshifting of Sars-Coronavirus by Structure-Based Virtual Screening. J Am Chem Soc. 2011;133:10094–10100. doi: 10.1021/ja1098325. [DOI] [PubMed] [Google Scholar]
- 549.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Conformational Inhibition of the Hcv Ires Rna. Nat Chem Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Bugaut A, Balasubramanian S. 5′-Utr Rna G-Quadruplexes: Translation Regulation and Targeting. Nucleic Acids Res. 2012;40:4727–4741. doi: 10.1093/nar/gks068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Hermann T. Small Molecules Targeting Viral Rna. Wiley Interdiscip Rev RNA. 2016;7:726–743. doi: 10.1002/wrna.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Connelly CM, Moon MH, Schneekloth JS., Jr The Emerging Role of Rna as a Therapeutic Target for Small Molecules. Cell Chem Biol. 2016;23:1077–1090. doi: 10.1016/j.chembiol.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Tor Y. Targeting Rna with Small Molecules. ChemBioChem. 2003;4:998–1007. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 554.Stern S, Moazed D, Noller HF. Structural Analysis of Rna Using Chemical and Enzymatic Probing Monitored by Primer Extension. Methods Enzymol. 1988;164:481–489. doi: 10.1016/S0076-6879(88)64064-X. [DOI] [PubMed] [Google Scholar]
- 555.Moazed D, Noller HF. Interaction of Antibiotics with Functional Sites in 16s Ribosomal Rna. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 556.Woese CR, Magrum LJ, Gupta R, Siegel RB, Stahl DA, Kop J, Crawford N, Brosius J, Gutell R, Hogan JJ. Secondary Structure Model for Bacterial 16s Ribosomal Rna: Phylogenetic, Enzymatic and Chemical Evidence. Nucleic Acids Res. 1980;8:2275–2293. doi: 10.1093/nar/8.10.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Regulski EE, Breaker RR. In-Line Probing Analysis of Riboswitches. Methods Mol Biol. 2008;419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 558.Lomenick B, Olsen RW, Huang J. Identification of Direct Protein Targets of Small Molecules. ACS Chem Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Guan L, Disney MD. Covalent Small-Molecule-Rna Complex Formation Enables Cellular Profiling of Small-Molecule-Rna Interactions. Angew Chem, Int Ed. 2013;52:10010. doi: 10.1002/anie.201301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Yang W-Y, Wilson HD, Velagapudi SP, Disney MD. Inhibition of Non-Atg Translational Events in Cells Via Covalent Small Molecules Targeting Rna. J Am Chem Soc. 2015;137:5336–5345. doi: 10.1021/ja507448y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Hostetter AA, Osborn MF, DeRose VJ. Rna-Pt Adducts Following Cisplatin Treatment of Saccharomyces Cerevisiae. ACS Chem Biol. 2012;7:218–225. doi: 10.1021/cb200279p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Osborn MF, White JD, Haley MM, DeRose VJ. Platinum-Rna Modifications Following Drug Treatment in S. Cerevisiae Identified by Click Chemistry and Enzymatic Mapping. ACS Chem Biol. 2014;9:2404–2411. doi: 10.1021/cb500395z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Rijal K, Chow; CS. A New Role for Cisplatin: Probing Ribosomal Rna Structure. Chem Commun. 2008:107–109. doi: 10.1039/B816633A. [DOI] [PubMed] [Google Scholar]
- 564.Guan L, Disney MD. Small Molecule-Mediated Cleavage of Rna in Living Cells. Angew Chem, Int Ed. 2013;52:1462–1465. doi: 10.1002/anie.201206888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Carter BJ, de Vroom E, Long EC, van der Marel GA, van Boom JH, Hecht SM. Site-Specific Cleavage of Rna by Fe(Ii).Bleomycin. Proc Natl Acad Sci U S A. 1990;87:9373–9377. doi: 10.1073/pnas.87.23.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Xu H, Xu H, Lin M, Wang W, Li Z, Huang J, Chen Y, Chen X. Learning the Drug Target-Likeness of a Protein. Proteomics. 2007;7:4255–4263. doi: 10.1002/pmic.200700062. [DOI] [PubMed] [Google Scholar]
- 567.Schork NJ. Personalized Medicine: Time for One-Person Trials. Nature. 2015;520:609–611. doi: 10.1038/520609a. [DOI] [PubMed] [Google Scholar]
- 568.Sawyers CL. Chronic Myeloid Leukemia. N Engl J Med. 1999;340:1330–1340. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- 569.Cox MC, Maffei L, Buffolino S, Del Poeta G, Venditti A, Cantonetti M, Aronica G, Aquilina P, Masi M, Amadori S. A Comparative Analysis of Fish, Rt-Pcr, and Cytogenetics for the Diagnosis of Bcr-Abl-Positive Leukemias. Am J Clin Pathol. 1998;109:24–31. doi: 10.1093/ajcp/109.1.24. [DOI] [PubMed] [Google Scholar]
- 570.Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl Oncoprotein Oligomerization Domain. Nat Struct Biol. 2002;9:117–120. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 571.Salesse S, Verfaillie CM. Bcr/Abl: From Molecular Mechanisms of Leukemia Induction to Treatment of Chronic Myelogenous Leukemia. Oncogene. 2002;21:8547–8559. doi: 10.1038/sj.onc.1206082. [DOI] [PubMed] [Google Scholar]
- 572.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (Sti571, Imatinib), a Rationally Developed, Targeted Anticancer Drug. Nat Rev Drug Discovery. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 573.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal Structures of the Kinase Domain of C-Abl in Complex with the Small Molecule Inhibitors Pd173955 and Imatinib (Sti-571) Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 574.Milojkovic D, Apperley J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin Cancer Res. 2009;15:7519–7527. doi: 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- 575.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW. Imatinib Induces Hematologic and Cytogenetic Responses in Patients with Chronic Myelogenous Leukemia in Myeloid Blast Crisis: Results of a Phase Ii Study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.V99.10.3530. [DOI] [PubMed] [Google Scholar]
- 576.O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 577.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple Bcr-Abl Kinase Domain Mutations Confer Polyclonal Resistance to the Tyrosine Kinase Inhibitor Imatinib (Sti571) in Chronic Phase and Blast Crisis Chronic Myeloid Leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/S1535-6108(02)00096-X. [DOI] [PubMed] [Google Scholar]
- 578.Reddy EP, Aggarwal AK. The Ins and Outs of Bcr-Abl Inhibition. Genes Cancer. 2012;3:447–454. doi: 10.1177/1947601912462126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Valent P. Imatinib-Resistant Chronic Myeloid Leukemia (Cml): Current Concepts on Pathogenesis and New Emerging Pharmacologic Approaches. Biol: Targets Ther. 2007;1:433–448. [PMC free article] [PubMed] [Google Scholar]
- 580.Pelz-Ackermann O, Cross M, Pfeifer H, Deininger M, Wang SY, Al-Ali HK, Niederwieser D, Lange T. Highly Sensitive and Quantitative Detection of Bcr-Abl Kinase Domain Mutations by Ligation Pcr. Leukemia. 2008;22:2288–2291. doi: 10.1038/leu.2008.180. [DOI] [PubMed] [Google Scholar]
- 581.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of Tyrosine Kinases by Mutation of the Gatekeeper Threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, O’Hare T, Hu S, Narasimhan NI, Rivera VM. Ponatinib in Refractory Philadelphia Chromosome-Positive Leukemias. N Engl J Med. 2012;367:2075–2088. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q. Ap24534, a Pan-Bcr-Abl Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315i Mutant and Overcomes Mutation-Based Resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MO, Dror RO, Kuriyan J, Shaw DE. A Conserved Protonation-Dependent Switch Controls Drug Binding in the Abl Kinase. Proc Natl Acad Sci U S A. 2009;106:139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Schilsky RL. Personalized Medicine in Oncology: The Future Is Now. Nat Rev Drug Discovery. 2010;9:363–366. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 586.Shim JS, Liu JO. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int J Biol Sci. 2014;10:654–663. doi: 10.7150/ijbs.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn J, Lazar V, Kurzrock R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase Ii Clinical Trials. J Clin Oncol. 2015;33:3817–3825. doi: 10.1200/JCO.2015.61.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Gonzalez de Castro D, Clarke PA, Al-Lazikani B, Workman P. Personalized Cancer Medicine: Molecular Diagnostics, Predictive Biomarkers, and Drug Resistance. Clin Pharmacol Ther. 2013;93:252–259. doi: 10.1038/clpt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Scheerens H, Malong A, Bassett K, Boyd Z, Gupta V, Harris J, Mesick C, Simnett S, Stevens H, Gilbert H. Current Status of Companion and Complementary Diagnostics: Strategic Considerations for Development and Launch. Clin Transl Sci. 2017;10:84–92. doi: 10.1111/cts.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Roskoski R., Jr The Erbb/Her Family of Protein-Tyrosine Kinases and Cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 591.Gazdar AF. Activating and Resistance Mutations of Egfr in Non-Small-Cell Lung Cancer: Role in Clinical Response to Egfr Tyrosine Kinase Inhibitors. Oncogene. 2009;28:S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J Clin Oncol. 2008;26:1742–1751. doi: 10.1200/JCO.2007.12.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of Lung Cancer-Derived Egfr Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural Basis for Inhibition of the Epidermal Growth Factor Receptor by Cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 595. [accessed July 25, 2017];List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools) https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm.
- 596.Wong SF. Cetuximab: An Epidermal Growth Factor Receptor Monoclonal Antibody for the Treatment of Colorectal Cancer. Clin Ther. 2005;27:684–694. doi: 10.1016/j.clinthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 597.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 598.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Kohne CH. Addition of Cetuximab to Chemotherapy as First-Line Treatment for Kras Wild-Type Metastatic Colorectal Cancer: Pooled Analysis of the Crystal and Opus Randomised Clinical Trials. Eur J Cancer. 2012;48:1466–1475. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 599.Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ. Clinical Relevance of Kras Mutation Detection in Metastatic Colorectal Cancer Treated by Cetuximab Plus Chemotherapy. Br J Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology Provisional Clinical Opinion: Testing for Kras Gene Mutations in Patients with Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 601.Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. Fda Drug Approval Summary: Panitumumab (Vectibix) Oncologist. 2007;12:577–583. doi: 10.1634/theoncologist.12-5-577. [DOI] [PubMed] [Google Scholar]
- 602.Sickmier EA, Kurzeja RJM, Michelsen K, Vazir M, Yang E, Tasker AS. The Panitumumab Egfr Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS One. 2016;11:e0163366. doi: 10.1371/journal.pone.0163366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J. Randomized, Phase Iii Trial of Panitumumab with Infusional Fluorouracil, Leucovorin, and Oxaliplatin (Folfox4) Versus Folfox4 Alone as First-Line Treatment in Patients with Previously Untreated Metastatic Colorectal Cancer: The Prime Study. J Clin Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 604.Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Zhang K. Panitumumab Versus Cetuximab in Patients with Chemotherapy-Refractory Wild-Type Kras Exon 2 Metastatic Colorectal Cancer (Aspecct): A Randomised, Multicentre, Open-Label, Non-Inferiority Phase 3 Study. Lancet Oncol. 2014;15:569–579. doi: 10.1016/S1470-2045(14)70118-4. [DOI] [PubMed] [Google Scholar]
- 605.Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L. Studies Leading to the Identification of Zd1839 (Iressa): An Orally Active, Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Targeted to the Treatment of Cancer. Bioorg Med Chem Lett. 2001;11:1911–1914. doi: 10.1016/S0960-894X(01)00344-4. [DOI] [PubMed] [Google Scholar]
- 606.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 607.Yun C-H, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of Lung Cancer-Derived Egfr Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. Zd1839 (Iressa): An Orally Active Inhibitor of Epidermal Growth Factor Signaling with Potential for Cancer Therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 609.Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V. Gefitinib Plus Best Supportive Care in Previously Treated Patients with Refractory Advanced Non-Small-Cell Lung Cancer: Results from a Randomised, Placebo-Controlled, Multicentre Study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 610.Kazandjian D, Blumenthal GM, Yuan W, He K, Keegan P, Pazdur R. Fda Approval of Gefitinib for the Treatment of Patients with Metastatic Egfr Mutation-Positive Non-Small Cell Lung Cancer. Clin Cancer Res. 2016;22:1307–1312. doi: 10.1158/1078-0432.CCR-15-2266. [DOI] [PubMed] [Google Scholar]
- 611.Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, DiOrio C, Doty J, Morin MJ, Moyer MP. Induction of Apoptosis and Cell Cycle Arrest by Cp-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 1997;57:4838–4848. [PubMed] [Google Scholar]
- 612.Johnson JR, Cohen M, Sridhara R, Chen YF, Williams GM, Duan J, Gobburu J, Booth B, Benson K, Leighton J. Approval Summary for Erlotinib for Treatment of Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer after Failure of at Least One Prior Chemotherapy Regimen. Clin Cancer Res. 2005;11:6414–6421. doi: 10.1158/1078-0432.CCR-05-0790. [DOI] [PubMed] [Google Scholar]
- 613.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S. Erlotinib Versus Chemotherapy as First-Line Treatment for Patients with Advanced Egfr Mutation-Positive Non-Small-Cell Lung Cancer (Optimal, Ctong-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 614.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target Binding Properties and Cellular Activity of Afatinib (Bibw 2992), an Irreversible Erbb Family Blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 615.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M. Phase Iii Study of Afatinib or Cisplatin Plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with Egfr Mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 616.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY. Afatinib Versus Cisplatin Plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring Egfr Mutations (Lux-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 617.Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y. Afatinib Versus Placebo for Patients with Advanced, Metastatic Non-Small-Cell Lung Cancer after Failure of Erlotinib, Gefitinib, or Both, and One or Two Lines of Chemotherapy (Lux-Lung 1): A Phase 2b/3 Randomised Trial. Lancet Oncol. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 618.Wu SG, Liu YN, Tsai MF, Chang YL, Yu CJ, Yang PC, Yang JC, Wen YF, Shih JY. The Mechanism of Acquired Resistance to Irreversible Egfr Tyrosine Kinase Inhibitor-Afatinib in Lung Adenocarcinoma Patients. Oncotarget. 2016;7:12404–12413. doi: 10.18632/oncotarget.7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ. Azd9291, an Irreversible Egfr Tki, Overcomes T790m-Mediated Resistance to Egfr Inhibitors in Lung Cancer. Cancer Discovery. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS. Osimertinib or Platinum-Pemetrexed in Egfr T790m-Positive Lung Cancer. N Engl J Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Gutierrez C, Schiff R. Her 2: Biology, Fetection, and Clinical Implications. Arch Pathol Lab Med. 2011;135:55–62. doi: 10.1043/2010-0454-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Fink MY, Chipuk JE. Survival of Her2-Positive Breast Cancer Cells: Receptor Signaling to Apoptotic Control Centers. Genes Cancer. 2013;4:187–195. doi: 10.1177/1947601913488598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M. Use of Chemotherapy Plus a Monoclonal Antibody against Her2 for Metastatic Breast Cancer That Overexpresses Her2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 624.Cho H-S, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr, Leahy DJ. Structure of the Extracellular Region of Her2 Alone and in Complex with the Herceptin Fab. Nature. 2003;421:756. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 625.Vu T, Claret FX. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front Oncol. 2012;2:62. doi: 10.3389/fonc.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Gennari R, Menard S, Fagnoni F, Ponchio L, Scelsi M, Tagliabue E, Castiglioni F, Villani L, Magalotti C, Gibelli N. Pilot Study of the Mechanism of Action of Preoperative Trastuzumab in Patients with Primary Operable Breast Tumors Overexpressing Her2. Clin Cancer Res. 2004;10:5650–5655. doi: 10.1158/1078-0432.CCR-04-0225. [DOI] [PubMed] [Google Scholar]
- 627.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA. Trastuzumab Plus Adjuvant Chemotherapy for Operable Her2-Positive Breast Cancer. N Engl J Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 628.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K. A Unique Structure for Epidermal Growth Factor Receptor Bound to Gw572016 (Lapatinib): Relationships among Protein Conformation, Inhibitor Off-Rate, and Receptor Activity in Tumor Cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 629.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-Tumor Activity of Gw572016: A Dual Tyrosine Kinase Inhibitor Blocks Egf Activation of Egfr/Erbb2 and Downstream Erk1/2 and Akt Pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 630.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S, Arribas J. Lapatinib, a Her2 Tyrosine Kinase Inhibitor, Induces Stabilization and Accumulation of Her2 and Potentiates Trastuzumab-Dependent Cell Cytotoxicity. Oncogene. 2009;28:803–814. doi: 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 631.Segovia-Mendoza M, González-González ME, Barrera D, Díaz L, García-Becerra R. Efficacy and Mechanism of Action of the Tyrosine Kinase Inhibitors Gefitinib, Lapatinib and Neratinib in the Treatment of Her2-Positive Breast Cancer: Preclinical and Clinical Evidence. Am J Cancer Res. 2015;5:2531–2561. [PMC free article] [PubMed] [Google Scholar]
- 632.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B. Lapatinib Plus Capecitabine for Her2-Positive Advanced Breast Cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 633.Johnston S, Pippen J, Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ. Lapatinib Combined with Letrozole Versus Letrozole and Placebo as First-Line Therapy for Postmenopausal Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol. 2009;27:5538–5546. doi: 10.1200/JCO.2009.23.3734. [DOI] [PubMed] [Google Scholar]
- 634.Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff J. Randomized Study of Lapatinib Alone or in Combination with Trastuzumab in Women with Erbb2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J Clin Oncol. 2010;28:1124–1130. doi: 10.1200/JCO.2008.21.4437. [DOI] [PubMed] [Google Scholar]
- 635.Adams CW, Allison DE, Flagella K, Presta L, Clarke J, Dybdal N, McKeever K, Sliwkowski MX. Humanization of a Recombinant Monoclonal Antibody to Produce a Therapeutic Her Dimerization Inhibitor, Pertuzumab. Cancer Immunol Immunother. 2006;55:717–727. doi: 10.1007/s00262-005-0058-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into Erbb Signaling from the Structure of the Erbb2-Pertuzumab Complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/S1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 637.Citri A, Skaria KB, Yarden Y. The Deaf and the Dumb: The Biology of Erbb-2 and Erbb-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/S0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 638.Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly Enhanced Antitumor Activity of Trastuzumab and Pertuzumab Combination Treatment on Her2-Positive Human Xenograft Tumor Models. Cancer Res. 2009;69:9330. doi: 10.1158/0008-5472.CAN-08-4597. [DOI] [PubMed] [Google Scholar]
- 639.Swain SM, Kim SB, Cortes J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Knott A. Pertuzumab, Trastuzumab, and Docetaxel for Her2-Positive Metastatic Breast Cancer (Cleopatra Study): Overall Survival Results from a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2013;14:461–471. doi: 10.1016/S1470-2045(13)70130-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-Independent Her2/Her3/Pi3k Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the Pi3k Inhibitor Gdc-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 641.Lord CJ, Ashworth A. Targeted Therapy for Cancer Using Parp Inhibitors. Curr Opin Pharmacol. 2008;8:363–369. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 642.Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L, Moche M, Cohen MS, Schüler H. Structural Basis for Potency and Promiscuity in Poly(Adp-Ribose) Polymerase (Parp) and Tankyrase Inhibitors. J Med Chem. 2017;60:1262–1271. doi: 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Livraghi L, Garber JE. Parp Inhibitors in the Management of Breast Cancer: Current Data and Future Prospects. BMC Med. 2015;13:188. doi: 10.1186/s12916-015-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M. New Response Evaluation Criteria in Solid Tumours: Revised Recist Guideline (Version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 645.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline Brca1/2 Mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A. Olaparib for Metastatic Breast Cancer in Patients with a Germline Brca Mutation. N Engl J Med. 2017;377:523. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 647.Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ. Preclinical Selection of a Novel Poly(Adp-Ribose) Polymerase Inhibitor for Clinical Trial. Mol Cancer Ther. 2007;6:945–956. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 648.Dockery LE, Gunderson CC, Moore KN. Rucaparib: The Past, Present, and Future of a Newly Approved Parp Inhibitor for Ovarian Cancer. OncoTargets Ther. 2017;10:3029–3037. doi: 10.2147/OTT.S114714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (Ariel2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 650.Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D. Phase 2 Multicentre Trial Investigating Intermittent and Continuous Dosing Schedules of the Poly(Adp-Ribose) Polymerase Inhibitor Rucaparib in Germline Brca Mutation Carriers with Advanced Ovarian and Breast Cancer. Br J Cancer. 2016;114:723–730. doi: 10.1038/bjc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Friday BB, Adjei AA. Advances in Targeting the Ras/Raf/Mek/Erk Mitogen-Activated Protein Kinase Cascade with Mek Inhibitors for Cancer Therapy. Clin Cancer Res. 2008;14:342–346. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 652.Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M, Palmieri G, Testori A, Marincola FM, Mozzillo N. The Role of Braf V600 Mutation in Melanoma. J Transl Med. 2012;10:85. doi: 10.1186/1479-5876-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D. Mechanism of Activation of the Raf-Erk Signaling Pathway by Oncogenic Mutations of B-Raf. Cell. 2004;116:855–867. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 654.Cantwell-Dorris ER, O’Leary JJ, Sheils OM. Brafv600e: Implications for Carcinogenesis and Mol. Ther. Mol Cancer Ther. 2011;10:385–394. doi: 10.1158/1535-7163.MCT-10-0799. [DOI] [PubMed] [Google Scholar]
- 655.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK. Discovery of a Selective Inhibitor of Oncogenic B-Raf Kinase with Potent Antimelanoma Activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Rustad EH, Dai HY, Hov H, Coward E, Beisvag V, Myklebost O, Hovig E, Nakken S, Vodak D, Meza-Zepeda LA. Braf V600e Mutation in Early-Stage Multiple Myeloma: Good Response to Broad Acting Drugs and No Relation to Prognosis. Blood Cancer J. 2015;5:e299. doi: 10.1038/bcj.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M. Improved Survival with Vemurafenib in Melanoma with Braf V600e Mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: The First Drug Approved for Braf-Mutant Cancer. Nat Rev Drug Discovery. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- 659.Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: Targeted Inhibition of Mutated Braf for Treatment of Advanced Melanoma and Its Potential in Other Malignancies. Drugs. 2012;72:2207–2222. doi: 10.2165/11640870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 660.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L. Combined Vemurafenib and Cobimetinib in Braf-Mutated Melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 661.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C. Mechanism of Mek Inhibition Determines Efficacy in Mutant Kras- Versus Braf-Driven Cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 662.Rheault TR, Stellwagen JC, Adjabeng GM, Hornberger KR, Petrov KG, Waterson AG, Dickerson SH, Mook RA, Laquerre SG, King AJ. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med Chem Lett. 2013;4:358–362. doi: 10.1021/ml4000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH. The B-Raf(V600e) Inhibitor Dabrafenib Selectively Inhibits Rip3 and Alleviates Acetaminophen-Induced Liver Injury. Cell Death Dis. 2014;5:e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, Smitheman KN, Erhardt JA, Hughes-Earle A, Kane-Carson LS. Dabrafenib; Preclinical Characterization, Increased Efficacy When Combined with Trametinib, While Braf/Mek Tool Combination Reduced Skin Lesions. PLoS One. 2013;8:e67583. doi: 10.1371/journal.pone.0067583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr, Kaempgen E. Dabrafenib in Braf-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 666.Abe H, Kikuchi S, Hayakawa K, Iida T, Nagahashi N, Maeda K, Sakamoto J, Matsumoto N, Miura T, Matsumura K. Discovery of a Highly Potent and Selective Mek Inhibitor: Gsk1120212 (Jtp-74057 Dmso Solvate) ACS Med Chem Lett. 2011;2:320–324. doi: 10.1021/ml200004g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P. Improved Survival with Mek Inhibition in Braf-Mutated Melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 668.Cooper WA, Lam DCL, O’Toole SA, Minna JD. Molecular Biology of Lung Cancer. J Thorac Dis. 2013;5:S479–S490. doi: 10.3978/j.issn.2072-1439.2013.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H. Identification of the Transforming Eml4-Alk Fusion Gene in Non-Small-Cell Lung Cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 670.Chin LP, Soo RA, Soong R, Ou SH. Targeting Ros1 with Anaplastic Lymphoma Kinase Inhibitors: A Promising Therapeutic Strategy for a Newly Defined Molecular Subset of Non-Small-Cell Lung Cancer. J Thorac Oncol. 2012;7:1625–1630. doi: 10.1097/JTO.0b013e31826baf83. [DOI] [PubMed] [Google Scholar]
- 671.Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine Kinase Gene Rearrangements in Epithelial Malignancies. Nat Rev Cancer. 2013;13:772–787. doi: 10.1038/nrc3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Ou SH. Crizotinib: A Novel and First-in-Class Multitargeted Tyrosine Kinase Inhibitor for the Treatment of Anaplastic Lymphoma Kinase Rearranged Non-Small Cell Lung Cancer and Beyond. Drug Des, Dev Ther. 2011;5:471–485. doi: 10.2147/DDDT.S19045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I. Structure Based Drug Design of Crizotinib (Pf-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (C-Met) Kinase and Anaplastic Lymphoma Kinase (Alk) J Med Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 674.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, Pao W. Insights into Alk-Driven Cancers Revealed through Development of Novel Alk Tyrosine Kinase Inhibitors. Cancer Res. 2011;71:4920–4931. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.Okamoto W, Okamoto I, Arao T, Kuwata K, Hatashita E, Yamaguchi H, Sakai K, Yanagihara K, Nishio K, Nakagawa K. Antitumor Action of the Met Tyrosine Kinase Inhibitor Crizotinib (Pf-02341066) in Gastric Cancer Positive for Met Amplification. Mol Cancer Ther. 2012;11:1557–1564. doi: 10.1158/1535-7163.MCT-11-0934. [DOI] [PubMed] [Google Scholar]
- 676.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T. First-Line Crizotinib Versus Chemotherapy in Alk-Positive Lung Cancer. N Engl J Med. 2014;371:2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 677.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F. Crizotinib Versus Chemotherapy in Advanced Alk-Positive Lung Cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 678.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB. Crizotinib in Ros1-Rearranged Non-Small-Cell Lung Cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Mazieres J, Zalcman G, Crino L, Biondani P, Barlesi F, Filleron T, Dingemans AM, Lena H, Monnet I, Rothschild SI. Crizotinib Therapy for Advanced Lung Adenocarcinoma and a Ros1 Rearrangement: Results from the Euros1 Cohort. J Clin Oncol. 2015;33:992–999. doi: 10.1200/JCO.2014.58.3302. [DOI] [PubMed] [Google Scholar]
- 680.Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]