

Abstract
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, encounters variable and hostile environments within the host. A major component of these hostile conditions is reductive and oxidative stresses induced by factors modified by the host immune response, such as oxygen tension, NO or CO gases, reactive oxygen and nitrogen intermediates, the availability of different carbon sources and changes in pH. It is therefore essential for Mtb to continuously monitor and appropriately respond to the microenvironment. To this end, Mtb has developed various redox-sensitive systems capable of monitoring its intracellular redox environment and coordinating a response essential for virulence. Various aspects of Mtb physiology are regulated by these systems, including drug susceptibility, secretion systems, energy metabolism and dormancy. While great progress has been made in understanding the mechanisms and pathways that govern the response of Mtb to the host's redox environment, many questions in this area remain unanswered. The answers to these questions are promising avenues for addressing the tuberculosis crisis.
Keywords: redox, dormancy, Dos, WhiB, tuberculosis, virulence
Mycobacterium tuberculosis senses and responds to redox stresses within the host to survive and cause disease.
INTRODUCTION
Tuberculosis (TB) afflicted more than 10 million people and resulted in 1.7 million deaths in 2016 alone (World Health Organization, 2017). Recent estimates suggest that currently 1.7 billion people, ∼1/4 of the global population, are latently infected with Mycobacterium tuberculosis (Mtb), the causative agent of TB (Houben and Dodd 2016). Mtb is transmitted to new hosts via the respiratory route where it exists as a facultative intracellular pathogen that causes disease in approximately 10% of immunocompetent hosts. A hallmark of the host's immune response to Mtb is the formation of caseous granulomas—organized collections of immune cells, classically considered necrotic and hypoxic. There is growing appreciation, however, for the heterogeneity of granulomas, both within and between hosts (Barry et al.2009; Lenaerts, Barry and Dartois 2015; Hunter 2016). More specifically, granulomas manifest as lesions with differing degrees of cellularity, hypoxia, acidosis and necrosis; the success of Mtb as a pathogen, therefore, can be credited in part to its ability to monitor, respond to, and resist wide-ranging microenvironments within the host.
Interestingly, the stresses encountered by Mtb within the host can be considered within the framework of oxido-reductive stress. Hypoxia, acidity, carbon source availability, the respiratory burst of phagocytes and host-derived gasotransmitters like nitric oxide (NO) and carbon monoxide (CO) are all environmental forces that Mtb may encounter throughout the course of infection. These factors have the potential to impact major reduction-oxidation (redox) couples within Mtb, the transport of electrons or directly damage cellular components (Kumar et al.2011; Cumming et al.2014; Chinta et al.2016). In fact, host susceptibility to Mtb infection is in part determined by the host's ability to produce redox active compounds known as reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI) (Lamichhane 2011; Voskuil et al.2011). Additionally, the redox environment of the granuloma as well as the ability of Mtb to maintain redox homeostasis have been recognized as contributing factors in prolonged therapy and drug resistance in Mtb infection (Via et al.2008; Lanoix, Lenaerts and Nuermberger 2015; Saini et al.2016).
Thus, it is necessary for Mtb to monitor and respond to numerous environmental factors to maintain redox homeostasis. This process requires Mtb to modify its activity to suit the dynamic demands of its microenvironment, ultimately achieving its pathogenic potential. By understanding the host's response to Mtb as well as Mtb’s response to the host, we may identify potential targets for host-directed and anti-bacterial therapies in TB.
SCOPE
This review will focus on the molecular machinery that Mtb employs to sense and respond to various reductive and oxidative stresses encountered within the host. We will begin by discussing fundamental principles of redox reactions and how they are affected by environmental conditions. Next, we will review the systems within Mtb that are essential for maintaining redox homeostasis and responding to environmental redox stresses. Lastly, we will integrate these systems and describe their ability to coordinate an effective response to redox stresses. In sum, we propose a conceptual model of Mtb pathogenesis that features interactions between the host environment and Mtb.
FROM REDOX CHEMISTRY TO REDOX BIOLOGY
To fully understand how Mtb senses and responds to shifting redox environments, an understanding of the fundamental concepts in redox chemistry is essential. We direct the reader to these excellent resources for additional reading on redox chemistry and biology (Schafer and Buettner 2001; Halliwell and Gutteridge 2008). Redox chemistry encompasses chemical reactions in which the oxidation state of participating molecules is changed through the gain or loss of electrons. A molecule is oxidized if it loses electrons, and it is reduced if it gains electrons. Redox biology can often be distilled into the activities of biologically relevant redox couples (Table 1), each composed of the two states of a redox-active molecule: the oxidized state and the reduced state (e.g. NAD+/NADH).
Table 1.
Redox potentials of biologically relevant redox couples.

|
Redox couples are ordered by increasing reduction potential (ΔEo΄). ΔEo΄ values were adjusted from the reduction potential under standard conditions to reflect the potential at a pH = 7 using the Nernst equation, as H+ is an important constituent of many of the couples in question. It is important to recognize that these values do not directly reflect ΔE values in vivo, as various conditions, including temperature and concentration of reactants can vary dramatically. ΔEo΄ values shown were originally reported by Schafer and Buettner (2001) and Halliwell and Gutteridge (2008).
The properties of redox-active molecules, such as the presence of polar groups or the ability to undergo a conformational change, contribute to their tendency to be in an oxidized or reduced state. This tendency can be described by the half-cell reduction potential, ΔE, which is determined experimentally under standard conditions (ΔEo) that are 1.0 M for solutes and ions or 1 atm for gases at 25°C and pH = 0. The reduction potential for an alternative set of fixed conditions is denoted by ΔEo΄. For example, it is useful to establish ΔEo΄ as the standard conditions above with the exception of pH = 7 for biologically relevant redox couples, as shown in Table 1. The direction of a redox reaction under constant conditions can be determined by the relationship between the ΔE of each couple: the reduced member of a given redox couple will reduce the oxidized constituent of any other redox couple with a more positive ΔEo.
Major redox couples serve varied functions, including carrying electrons (e.g. NAD+/NADH, NADP+/NADPH) and maintaining redox homeostasis as a ‘buffer’ (e.g. glutathione/glutathione disulfide GSSG/GSH). Other couples composed of ROI and RNI, however, may threaten redox homeostasis. As their names suggest, ROI and RNI contain molecules with at least one redox-active oxygen or nitrogen atom, respectively. A number of important members of the ROI and RNI groups are also considered free radicals. The name indicates that these molecules are stable, or free, despite containing at least one unpaired electron, or radical. Key examples of ROI include the radicals O2•− (superoxide), XO2• (peroxyl radical) and XO• (alkoxyl radical) and the non-radicals hydrogen peroxide (H2O2), ONOO− (peroxynitrite, anion) and HOCl (hypochlorous acid). Examples of RNI include the radicals •NO (nitric oxide, radical) and •NO2, (nitrogen dioxide) and the non-radicals HNO2 (nitrous acid), NO+ (nitrosonium), NO¯ (nitroxyl anion), and others (Halliwell and Gutteridge 2008).
ROI and RNI are feared mediators of oxidative stress and oxidative damage. Oxidative stress refers to an imbalance between ROI and the capacity of the cell to detoxify the species or repair cellular damage. Under these circumstances, the cell has a relatively higher ΔE and a decreased reductive capacity, as the abundance of oxidized species tends to oxidize other species in the cell. This can lead to oxidative damage if cellular components such as DNA or membrane lipids are damaged. The reductive capacity of a cell can often be defined by the central redox buffer in a cell, such as GSH/GSSG in most cells. Similarly, a cell can also experience reductive stress, a condition opposite to oxidative stress. Reductive stress can be conceptualized by an excess of electrons in a cell and exemplified by the loss of a terminal electron acceptor in or disruption of the central electron transport chain in a respiring cell. The threat of increased oxidative or reductive damage, therefore, underscores the importance of maintaining redox homeostasis within a cell.
One way cells maintain homeostasis is by coupling some reactions through the use of a common redox couple (e.g. NAD+/NADH, fatty acid catabolism) and separating these reactions from others by using a separate redox couple (e.g. NADP+/NADPH, fatty acid synthesis). This prevents certain reactions from reaching equilibrium, which generates redox potentials different from those measured under standard conditions and allows cells to manipulate their internal redox environment to their benefit. The impact various conditions have on the reduction potential of a redox reaction, including the concentration of the reactants, can be calculated with the Nernst equation:
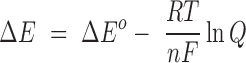 |
(1) |
where R is the gas constant, T is the temperature in Kelvin, n is the number of electrons being transferred in the given reaction, F is the Faraday constant, and Q is the equilibrium constant calculated for the given reaction using the concentrations of the reactants and stoichiometry indicated by the balanced chemical equation. If the reaction is at equilibrium and Q = 1, then ΔE = ΔEo; it is therefore the concentration of reactants and the temperature that shift the favorability of reactions, processes, and the overall redox environment within a cell. H+ and O2, for example, are important environmental factors and participants in numerous redox reactions; the Nernst equation, then, provides information on how their availability influences redox homeostasis within a cell. Other factors, such as the availability of various carbon sources, their concentrations, or host-derived gasotransmitters capable of inhibiting metabolism can shape the overall redox environment within a cell secondary to affecting cell metabolism. These factors in particular bear relevance to the following discussion due to their variable presence within the tuberculous granuloma (Lenaerts, Barry and Dartois 2015; Chinta et al.2016).
Gases, lipids, and other factors external to Mtb follow a heterogeneous distribution between and within granulomas due to variation in gross TB pathology, which ranges from early, non-necrotizing granulomas to necrotic and cavitating lesions to confluent collections of immune cells filling the available space within the lung (Lenaerts, Barry and Dartois 2015). Features of a specific lesion type can generate inter- and intra-lesion heterogeneity; for example, a fibrotic rim present in some forms of TB pathology limits the oxygen within a necrotic granuloma and generates an oxygen gradient flowing from normoxia to extreme hypoxia toward the center of the granuloma (Datta et al.2016). Lesions can be dynamic, too. Progressive necrosis in the center of a lesion can shift the carbon source available to Mtb to lipid-rich caseum, which in some cases is thought to drain into an open airway to form a cavity (Lenaerts, Barry and Dartois 2015). In this instance, oxygen is then able to reach the interior of the formerly hypoxic lesion. Through all of these conditions, Mtb exposure to host gases, such as NO and CO, as well as various oxidative species produced by the phagocyte oxidative burst, will vary, resulting in fluctuating, dynamic stresses on the pathogen.
MYCOBACTERIAL REDOX BUFFERS: ERGOTHIONEINE AND MYCOTHIOL
One strategy that Mtb employs to survive in hostile oxidizing environments is to synthesize low molecular weight thiols to help it maintain a reducing environment and redox homeostasis (Fig. 1) (Kumar et al.2011). In many cell types, including human cells, this role falls to glutathione (GSH) (Newton et al.1996; Venketaraman et al.2003). Interestingly, Mycobacteria do not produce GSH; rather, they synthesize two main types of thiols, ergothioneine (EGT) and mycothiol (MSH), which maintain intracellular redox balance during the course of infection (Kumar et al.2011; Saini et al.2016). Both are synthesized in mycobacteria via a multistep process involving multiple gene products encoded by the gene clusters egtA-E (EGT) and mshA-D (MSH) (Newton et al.2006; Seebeck 2010). EGT contains a histidine as its core amino acid, and, at physiological pH, it exists as a thione that can be reduced to a disulfide form (ΔEo΄ = − 60 mV) (Jocelyn 1972; Cumming et al.2018). MSH has cysteine as its core amino acid and, under the same conditions, exists as a thiol that can also be reduced to a disulfide (ΔEo΄ = − 230 mV) (Sharma et al.2016; Cumming et al.2018). The antioxidant properties of MSH result from the sulfur atom in the cysteine residue, which is active only in the reduced form (Rawat and Av-Gay 2007). Reduced MSH can be oxidized to MSH disulfide (MSSM) in the context of oxidative stress, and MSSM can be converted back to the reduced state, MSH, by MSSM reductase in the presence of NADPH (Loi, Rossius and Antelmann 2015). MSH serves as a cofactor for enzymes involved in the detoxification of peroxides, electrophiles and NO, and it is required for Mtb survival in the host (Sareen et al.2003; Buchmeier and Fahey 2006; Rawat and Av-Gay 2007; Newton, Buchmeier and Fahey 2008).
Figure 1.

EGT and MSH Counter Oxidative Stress. Various exogenous factors can induce oxidative or nitrosative stress (yellow) in Mtb, leading to the generation of ROI and RNI. Major cytosolic redox buffers (orange), EGT and MSH, detoxify ROI/RNI, protect the cell from oxidative damage and contribute to virulence (red) by resisting anti-TB drugs and promoting survival.
EGT and MSH appear to have overlapping as well as distinct roles within Mtb. Significantly increased levels of EGT were observed in a MtbΔmshA deletion mutant; however, lack of EGT in egtA:Tn, a transposon mutant of Mtb deficient in EGT production, did not lead to a corresponding increase in MSH (Saini et al.2016). Instead, the egtA:Tn mutant had a 25-fold increase in ROI-producing cells relative to wild-type Mtb, whereas the ΔmshA mutant had only an 8-fold increase in ROI producing cells relative to wild-type Mtb. This observation indicates that increased EGT production may compensate for a lack of MSH, but not vice versa. Additionally, transcriptome analysis of EGT and MSH mutants in Mtb revealed that while they serve similar functions, they also have distinct roles. These distinct roles may abrogate competition between the EGT and MSH biosynthetic pathways based on the redox status of the cell, allowing Mtb to synthesize the most appropriate buffer (Saini et al.2016).
Nonpathogenic, fast-growing Mycobacterium smegmatis (Ms) has also been used to study fundamental mechanisms of redox homeostasis in mycobacteria, as it is an appealing alternative to logistical challenges of Mtb research. Ms ΔegtA is highly sensitive to oxidative stress caused by cumene hydroperoxide (CHP), whereas ΔegtD is sensitive to lipid peroxides, tert-butyl hydroperoxide and CHP (Sao Emani et al.2013; Singh et al.2016). To this point MsΔmshA significantly increased levels of EGT by 26-fold in the exponential phase and 5-fold in the stationary phase (Ta et al.2011). Similar results were also observed in Mtb ΔmshA (Saini et al.2016). That being said, a Ms double mutant lacking both EGT and MSH is highly sensitive to peroxide stress compared to single mutants, which suggests that the functions of MSH and EGT might partly compensate for the function of other (Sao Emani et al.2013).
Additionally, EGT and MSH alter peroxide and drug susceptibilities in Mtb. Saini et al. (2016) showed that egtA and egtD transposon mutants of Mtb are significantly more sensitive to H2O2, CHP, paraquat and menadione, suggesting a role for EGT in protecting Mtb against oxidative stress. Egt mutants of Mtb are highly susceptible to anti-TB drugs such as rifampicin, isoniazid, clofazimine and bedaquiline. MtbΔmshA, was shown to be more susceptible to clofazimine and bedaquiline than wild-type and egt mutants. Mtb lacking MSH exhibited increased sensitivity towards rifampicin and streptomycin and decreased sensitivity towards isoniazid and ethionamide, suggesting that MSH may be required for the activation of some drugs while contributing to tolerance against others (Hernick 2013). Overall, the shared and unique aspects of anti-TB drug susceptibilities in EGT and MSH mutants appear consistent with their shared and unique roles in maintaining redox homeostasis. This highlights an important relationship between maintaining redox homeostasis and survival.
In addition to its role in drug susceptibility, EGT also appears to be important to Mtb proliferation within murine cell lines and in vivo. Intracellular growth of MtbΔegtA and ΔegtD in murine RAW264.7 macrophages was reduced three-fold at 120 h post-infection, and the growth of MtbΔegtD mutant was reduced five-fold relative to wild-type Mtb at 120 h post-infection of murine J774A.1 macrophages (Richard-Greenblatt et al.2015; Saini et al.2016). These studies demonstrate that EGT is important for Mtb survival in macrophages. In addition, Saini et al. (2016) observed significantly reduced bacillary burden in the lungs of mice infected with MtbΔegtA and ΔegtD relative to wild type, establishing a role for EGT in Mtb pathogenesis. These findings complement the increased drug susceptibility in EGT mutants and suggest that EGT biosynthesis might be an attractive target for the development of new anti-TB therapeutic molecules intended to enhance existing therapies or provide a direct anti-mycobacterial effect.
MSH is also important to the virulence and survival of Mtb, as the loss of MSH biosynthesis and MSH-dependent detoxification inhibit the growth of mycobacteria (Sareen et al.2003; Hernick 2013). In Mtb Erdman, the mshA and mshC genes are essential (Sareen et al.2003; Buchmeier and Fahey 2006); interestingly, however, one group identified a spontaneous mshA mutant of Mtb H37RV that is resistant to ethionamide and that grows in immunodeficient mice but requires catalase for growth in vitro (Vilchèze et al.2008). This suggests important differences in the regulation of the redox environment may exist between different strains of Mtb. Survival of a mshB transposon mutant with only 20% wild-type MSH levels was markedly reduced in the presence of CHP; this mutant was resistant to H2O2, however, suggesting that 20% of MSH production is enough for Mtb to withstand oxidative stress caused by H2O2, in conjunction with other mechanisms for managing this stress (Buchmeier et al.2003). Enhanced killing of the mshB mutant under oxidative stress was observed during mid-exponential phase of growth, and the mutant also showed increased sensitivity to rifampicin, implicating MSH as an important part of Mtb’s survival strategy (Buchmeier et al.2003). Lastly, an earlier study showed that MtbΔmshD was sensitive to oxidative and acidic stress and showed reduced survival in macrophages (Buchmeier, Newton and Fahey 2006). In this study, MtbΔmshD had higher levels of the MshD substrate, Cys-GlcN-Inositol, and produced a novel thiol, N-formyl-Cys-GlcN-Inositol; however, these molecules could not protect Mtb from the oxidative stress. The cumulative research on MSH biosynthesis demonstrates the importance of MSH in Mtb growth and withstanding oxidative stress, both endogenous and exogenous. Importantly, several studies have demonstrated that Ms msh mutants are sensitive to oxidative stress, antibiotic stresses, alkylating stress and low pH, strengthening the claim that MSH protects mycobacteria against various stresses and toxic agents (Newton et al.1999; Rawat et al. 2002, 2003, 2007; Xu et al.2011).
The relationship between drug susceptibility and redox homeostasis was further established by a study from Bhaskar and colleagues (2014) that measured the changes in intra-mycobacterial MSH redox potential by coupling the MSH-dependent oxidoreductase, mycoredoxin 1, to redox sensitive GFP. Treatment of Mtb infected macrophages with anti-TB drugs led to an oxidative shift in the MSH redox potential, disrupting MSH homeostasis and leading to efficient killing of Mtb. Additionally, it was observed that mycobacteria with a higher MSH redox potential were more susceptible to antibiotics (Bhaskar et al.2014). In a follow up study, Mehta, Rajmani and Singh (2016) showed that Mtb WhiB3 responds to the acidic pH in macrophages during infection by modulating the MSH redox potential, which plays an important role in phagosomal maturation and virulence. This study showed that the redox sensitive WhiB3 and MSH are regulatory components mediating gene expression and are required to resist the acidic stress caused during Mtb infection (Mehta, Rajmani and Singh 2016). The findings discussed in this section as well as the specificity of MSH expression in mycobacteria make MSH an attractive drug target for the treatment of TB with the added advantage that genes involved in biosynthesis of MSH are absent in humans (Rawat et al.2007; Newton, Buchmeier and Fahey 2008; Gutierrez-Lugo et al.2009; Hernick 2013).
THE MULTIFUNCTIONAL WHIB FAMILY
Mtb WhiB proteins are redox-sensing transcription factors of the WhiB-like (Wbl) family characterized by conserved Cys residues (Cys-Xn-Cys-X2-Cys-X5-Cys; where X is an amino acid and n is a variable number) that coordinate an Fe–S cluster. There are seven members of the WhiB family in Mtb, namely WhiB1 (Rv3219), WhiB2 (Rv3260c), WhiB3 (Rv3416), WhiB4 (Rv3681c), WhiB5 (Rv0022c), WhiB6 (Rv3862c) and WhiB7 (Rv3197A) (Saini, Farhana and Steyn 2012). Expression of these independent genes requires different stimuli, and their mRNA levels vary at mid-logarithmic growth phase. During the mid-logarithmic growth phase, whiB1 was highly expressed and whiB5 was poorly expressed relative to the other whiB genes that exhibited a similar number of mRNA copies as sigA (Larsson et al.2012). Likewise, these proteins share little sequence homology, and the microenvironment around their Fe-S clusters may vary considerably despite belonging to the same family. For example, the Fe–S cluster in WhiB3 contains multiple arginine (Arg) residues adjacent to the cluster-coordinating Cys residues (Cys23-Arg24-Cys53-Arg54-Arg55-Cys56-Cys62-Arg63). At neutral pH, these Arg residues may significantly lower the pKa of Cys, resulting in the formation of Cys thiolates. These thiolates are highly susceptible to oxidation that can lead to the loss of the Fe–S cluster (Saini et al.2012).
Owing to these differences, individual WhiB proteins also respond to different physiological stimuli such as hypoxia, NO and cyclic AMP, thereby augmenting their redox-sensitive function as transcription factors. Central to this role is the presence of an AT hook-like DNA binding motif that facilitates DNA binding. Hence, it can be argued that the unique physiochemical attributes of Mtb WhiB proteins facilitate a broad range of redox functionality that may explain their key roles in diverse aspects of Mtb biology. The reported roles of WhiB proteins in mycobacteria include their function as transcriptional regulators (all WhiB proteins), maintenance of redox homeostasis (WhiB3, WhiB4 and WhiB7), regulating protein secretion systems (WhiB1, WhiB5 and WhiB6), virulence (WhiB3, WhiB4, WhiB5 and WhiB6), antibiotic resistance (WhiB7) and reactivation from latency (WhiB1 and WhiB5) (Steyn et al.2002; Singh et al.2007; Smith et al.2010; Burian et al. 2012, 2013; Chawla et al.2012; Solans et al.2014) (Fig. 2).
Figure 2.

WhiB proteins regulate Mtb virulence. Mtb can encounter environments with various concentrations of O2 and NO within the host, leading to the development of intracellular redox stresses (yellow). (A) In the context of reductive stress, WhiB3 and WhiB6 (orange) adopt a conformation that promotes the transcription of genes involved in lipid synthesis while inhibiting the expression of ESX-1. These responses enhance Mtb’s ability to evade the immune system. (B) In the context of oxidative stress, WhiB3, Whib4 and WhiB5 (orange) contribute to increased virulence (red) by upregulating key virulence genes, including secretion systems ESX-2 and ESX-4. (C) Under nitrosative stresses, a number of WhiB proteins (orange) contribute to the transcription of genes involved in lipid synthesis while the inhibiting the expression of ESX-1 genes, and other processes involved in evasion of the immune system (red). While WhiB1 and WhiB4 are sensitive to NO, their involvement in this context remains unclear.
WhiB3 is probably the most widely studied member of the WhiB family in Mtb. WhiB3 was shown to interact with the principal sigma factor of Mtb, SigA, and to be involved in Mtb virulence in mice (Steyn et al.2002). Subsequent studies have shown that WhiB3 modulates different aspects of Mtb pathogenesis. For example, as O2 can oxidize and NO can nitrosylate the WhiB3 Fe-S cluster, WhiB3 can respond to dormancy signals, including hypoxia and NO (Singh et al.2009). Furthermore, WhiB3 controls redox homeostasis in Mtb in part by modulating the production of redox buffer, EGT (Saini et al.2016). WhiB3 also facilitates maintenance of intracellular redox homeostasis by channeling reducing equivalents into lipid anabolism to produce polyketides such as phthiocerol dimycocerosate, triacylglycerol, and lipid cyclomodulins, which in turn impact the host cell-cycle during Mtb infection (Singh et al.2009). More recently, it was shown that Mtb infection blocks the G1/S transition in macrophages, and polyketides produced under WhiB3 control arrests the infected macrophages in the G0/G1 phase (Cumming et al.2017). Thus, WhiB3 manipulates the immune system and promotes long-term persistence by altering the host cell cycle of infected macrophages. Taken together, these studies firmly established that the redox-sensitive WhiB3 regulates aspects of Mtb virulence.
WhiB proteins have been implicated in modulating Mtb virulence by regulating the expression of 6 kDa early secretory antigenic target (ESAT-6) protein family secretion (ESX) systems also known as type VII secretion systems. ESX systems represent sophisticated secretory apparatuses that export a set of effector proteins, such as ESAT-6, which enables Mtb to evade the host immune response. WhiB6 and WhiB3 appear to regulate the expression of the ESX-1 (Bosserman et al.2017; Cumming et al.2017). WhiB3 specifically regulates genes that contribute to the ESX-1 system, including esxA, esxB, espA, espC and espD (Cumming et al.2017). Similarly, microarray analysis has shown that WhiB5 controls the expression of members of two other type VII secretion systems, namely ESX-2 and ESX-4 (Casonato et al.2012). This leads to speculation that Mtb modulates ESX expression through WhiB3, WhiB5 and WhiB6 to evade or suppress the immune system. Supporting this claim, both whiB3 and whiB5 mutants have both shown attenuation in murine models of TB (Steyn et al.2002; Casonato et al.2012).
Solans et al. (2014) observed that clinical TB strains secrete more ESAT-6 compared to lab-adapted Mtb strains. This phenomenon was ascribed to a genetic polymorphism in WhiB6 unique to the clinical strains. The reintroduction of a copy of whiB6 from clinical strains to H37Rv increased ESAT-6 production and secretion to the level of clinical strains. Additionally, chromatin immunoprecipitation sequencing (ChIP-seq) and high-resolution transcriptomic studies showed that PhoP controls ESX-1 via EspR and ESX-1 expression is regulated by WhiB6 as part of the PhoP regulon (Solans et al.2014; Broset, Martín and Gonzalo-Asensio 2015). As stated earlier, the ESX-1 secreted factor ESAT-6 is a major Mtb virulence factor; it can therefore be argued that alterations in whiB6 impact virulence and pathogenicity of TB strains. A recent study by Chen et al. (2016) further advanced our understanding of WhiB6-mediated regulation of the ESX-1 cluster by showing WhiB6 differentially regulates both the ESX-1 and DosR regulons in Mycobacterium marinum (Mm), a species used to model Mtb infection in zebrafish, through environmental manipulation of the WhiB6 Fe-S cluster. The Fe-S cluster in WhiB6 is sensitive to oxidative and nitrosative stresses and plays an important role in regulating ESX-1 secretion as disruption of the intact Fe-S clusters in WhiB6 following exposure to O2 abolished the secretion of ESX-1 substrates (Chen et al.2016). The lack of ESX-1 substrates in the culture media of MmΔwhib6 is seemingly at odds with findings from Bosserman et al. (2017) who showed residual secretion of some ESX-1 components in MmΔwhiB6. Though the Mm Δwhib6 strains in both reports were virulent and induced hemolysis, these findings may be reconciled through the observation that low levels of ESX-1 activity are sufficient to translocate ESX-1 substrates to the cell surface of Mm and produce a virulent phenotype (Kennedy et al.2014). However, these levels of ESX-1 activity may or may not be high enough to detect these substrates in the culture media. Regardless, a clear role has emerged for WhiB6 in the regulation of ESX-1 and, subsequently, Mtb pathogenicity.
WhiB4 has been shown to regulate the PE/PPE gene family of mycobacteria. This gene family is unique to mycobacteria and contributes to the antigenic variation observed in Mtb (Gey van Pittius et al.2006). Mtb alone has 168 members in this gene family, and their secretion is reported to be regulated in part by the ESX-5 system, ESX-1 system, and several other regulators (Voskuil et al.2004; Houben et al.2009; Sani et al.2010; Fishbein et al.2015). Thus, the relationship of WhiB4, the PE/PPE gene family, and ESX expression may also impact the virulence of mycobacterial species. For example, Wu et al. (2017) showed WhiB4 is important for virulence of Mm. MmΔwhiB4 exhibited impaired replication in macrophages and diminished virulence in zebrafish. In contrast, MtbΔwhiB4 exhibited enhanced replication in macrophages and increased virulence in lungs of guinea pigs, although its dissemination to spleens was reduced (Chawla et al.2012). These seemingly contrasting roles for WhiB4 in Mm and Mtb were observed despite the high level of similarity in the WhiB4 proteins of the two species—83% identity in the amino acid sequence and 100% identity in the Cys-X14–22-Cys-X2-Cys-X5-Cys motif. Closer examination showed that, in Mm, a large number of pe/ppe genes (98) were regulated by WhiB4 compared to only three pe/ppe genes (pe35, ppe19, ppe68) in Mtb (Wu et al.2017). Considering the emerging evidence about a link between PE/PPE gene family members and regulation of ESX secretion systems, it is possible that differences in the expression of genes regulated by these proteins across different organisms may have uncovered an additional layer of complexity to WhiB functioning.
The complex nature of WhiB proteins is emphasized by an emerging, large body of research that fails to address more fundamental aspects of WhiB proteins, such as the redox potentials each of their Fe–S clusters. A major step forward in understanding WhiB biochemistry, however, came from the determination of the 3D structure of WhiB1, determined by nuclear magnetic resonance (Kudhair et al.2017). WhiB1 was shown to be a four-helix bundle with a core of three α-helices held together by an [4Fe-4S]2+ cluster. It also contains a cAMP receptor protein binding site. Its [4Fe-4S]2+ cluster is stable in the presence of O2 but is highly reactive to NO, as it reacts with eight equivalents of NO and yields two dinuclear dinitrosyl-iron thiol complexes. The Fe–S cluster, which is disassembled by nitrosylation, is required for WhiB1 to interact with the major sigma factor, SigA. On the other hand, NO mediated loss of the Fe–S cluster facilitates DNA binding to regulate the gene expression of different pathways, including repression of the espA operon that codes for proteins that are essential for the function of the major virulence factor ESX-1.
In sum, the sensitivity of WhiB proteins to environmental gases allows Mtb to differentially respond to stresses during infection by modulating the expression of various virulence factors (Fig. 2).
THE DOS SYSTEM AND THE DORMANCY REGULON
Perhaps one of the most important aspects of Mtb infection is the ability of Mtb to persist within the host for decades in a condition known as latent TB infection (LTBI). While patients with LTBI are asymptomatic, Mtb can ‘reactivate’ and cause disease after years or decades. The factors that contribute to latent and reactivation TB, however, remain poorly defined. One explanation is the ability of Mtb to enter and exit a state of extremely low metabolic activity under unfavorable or favorable conditions, respectively. This state, termed dormancy, is conceptually, but not yet empirically associated with the clinical condition LTBI, making it a topic of great interest. A collection of 48 genes induced as Mtb enters dormancy has been identified and is referred to as the Dos dormancy regulon (Voskuil et al.2003). Expression of the dormancy regulon is sensitive to O2, NO and CO and ultimately controlled by the Dos three-component system (Fig. 3) (Voskuil et al.2003; Kumar et al.2008).
Figure 3.

DosR/S/T respond to reductive stress. Environmental factors such as hypoxia; the host-derived gasotransmitters, NO and CO; and a lipid-rich environment can induce reductive stress in Mtb. DosS and DosT are heme-kinase sensors that sense reductive stress or hypoxia, respectively. Both sensors can directly bind NO and CO to maintain signaling through the regulatory element, DosR, which induces the Dos dormancy regulon. The dormancy regulon is a collection of 48 genes theoretically involved in the induction of dormancy and the rewiring of metabolism. Fatty acid synthesis in particular is thought to be served as an electron sink to combat the reductive environment.
DosS and DosT
Initially described as the DevR/S dormancy system (Dasgupta et al.2000; Saini et al.2004), the Dos system is composed of two histidine sensor-kinases, DosS and DosT, and a regulatory element, DosR, which acts as a transcription factor when phosphorylated by the upstream sensors (Roberts et al.2004). DosS and DosT are membrane-tethered proteins that contain a heme group and a histidine kinase essential for autophosphorylation and subsequent signaling (Chim, Johnson and Goulding 2014). DosT is inactive when its heme is in the O2-bound state; as environmental O2 decreases (i.e. hypoxia), O2 dissociates from the heme group causing DosT to undergo autophosphorylation (Kumar et al.2007). Signaling through DosS, on the other hand, is dependent on the oxidation state of the iron in its prosthetic heme group; DosS is inactive when its heme iron is in the ferric (Fe3+) state, whereas it undergoes autophosphorylation when its heme iron is reduced to the ferrous (Fe2+) state (Kumar et al.2007). Initial experiments conducted in vitro also observed O2 binding directly to the DosS heme iron; these studies, however, were conducted at a partial pressure of O2 exceeding what is likely encountered within the host (Ioanoviciu et al. 2007, 2009). Thus, the more plausible mechanism of in vivo signaling is that exposure to O2 induces autooxidation of the iron to the Fe3+ state, preventing signaling in an aerobic environment, while a reductant could reduce the heme-iron to the Fe2+ state and induce expression of the dormancy regulon (Kumar et al.2007; Honaker et al.2010). Furthermore, the responsiveness of these sensors to host gasotransmitters capable of inhibiting cellular metabolism, such as NO and CO, is thought to maintain or ‘lock’ the activated heme group in both DosS and DosT in a stable conformation, allowing both sensors to maintain active signaling (Kumar et al. 2007, 2008). This coordinated response to various gases appears to be further augmented by the NO-sensitive serine/threonine protein kinase, PknH, which phosphorylates DosR at additional sites and appears to be required for the full induction of the Dos regulon (Chao et al.2010).
Hypoxia
A progressively hypoxic environment induces the expression of genes and proteins within the dormancy regulon in Mtb (Gopinath et al.2015; Schubert et al.2015; Iona et al.2016; Kumari et al.2017). In the Wayne model of dormancy, the expression of these genes as well as the protein expression of DosR peaked during the induction of dormancy and decreased relative to peak expression during the remainder of dormancy, measured at day 40 at the latest (Gopinath et al.2015; Iona et al.2016). In a standing model of dormancy, however, transcripts of important genes were upregulated throughout dormancy with a peak expression between 60 and 90 days (Kumari et al.2017). In this study, the expression of the dosR and tgs1 genes appears to follow an expression pattern similar to that observed in the study by Iona et al. (2016) through day 30; however, peak expression was observed beyond the latest time points in previous experiments. Though the difference in findings may be due to the use of different models, it is possible the dormancy regulon may be expressed at the highest levels well after the initial exposure to hypoxia at a time point not previously observed. Lastly, additional proteomic analysis estimated that proteins encoded in the dormancy regulon make up 20% of the proteome of Mtb throughout much of hypoxia-induced dormancy (Schubert et al.2015). In sum, it appears that genes and proteins of the dormancy regulon are expressed to varying extents throughout hypoxia-induced dormancy, and they may play roles in inducing, maintaining and emerging from dormancy.
Lipids
The source of carbon for Mtb has similarly been shown to impact the expression of genes within the dormancy regulon in vitro. Culturing Mtb in equal-length long chain fatty acids (LC-FAs) induces expression of more than half of the genes in the dormancy regulon (Rodríguez et al.2014). Interestingly, genes within this group, including the gene encoding triacylglycerol synthase, tgs1, are associated with Mtb lipid synthesis in lipid-rich, hypoxic environments (Daniel et al.2011). This suggests that reductive stress may be sufficient to induce the dormancy regulon. This is further supported by work from Honaker and colleagues who demonstrated a possible DosS-dependent signaling mechanism through which the excess electrons within a cell may be sensed (Honaker et al.2010). Sensing may occur through a direct interaction between the electron transport chain of the cell or perhaps through monitoring the balance of intracellular redox couples, such as NAD+/NADH. Recently, more nuanced studies have investigated the contribution different lipids make to gene expression in Mtb. The addition of cholesterol to media containing LC-FAs led to earlier induction of DosR-dependent genes thought to be essential to the reductive stress response, such as tgs1 (Soto-Ramirez et al.2017). This was supported in a follow-up proteomic analysis of Mtb cultured in LC-FA media supplemented with cholesterol conducted by the same group (Garcia-Morales et al.2017). Together, these studies suggest that the dormancy regulon plays a role in responding to the availability of various carbon sources and the associated intracellular redox environment.
Vitamin C
It appears that vitamin C may also induce the dormancy regulon. Taneja et al. (2010) found that physiological levels of vitamin C depleted O2 from Mtb cultures, induced the dormancy regulon, and produced a dormancy phenotype. A follow-up study revealed that in addition to the induction of the dormancy regulon, the overall transcriptional response to vitamin C treatment mimicked a number of other intracellular stresses, including oxidative and nitrosative stresses (Kumari et al.2015). This was supported by comprehensive study detailing the response of Mtb to vitamin C treatment that involved transcriptome remodeling of 67% of Mtb genes and the induction of a viable but non-culturable state in Mtb. Vitamin-C-treated Mtb in this state were resistant to rifampicin and isoniazid; however, killing of Mtb by pyrazinamide was potentiated to an extent that negated the effects of the aforementioned tolerance (Sikri et al.2018).
In contrast, another group determined that vitamin C kills Mtb by inducing high levels of ferrous iron in the cell and oxidative stress via the Fenton reaction, rather than inducing the dormancy regulon (Vilchèze et al.2013). The authors point to the repression of the mbtD gene as evidence for increased intracellular iron in Mtb following treatment with vitamin C; repression of mbtD in the context of high levels of iron, however, is dependent on the activity of ideR (Rodriguez et al.2002). Surprisingly, though, ideR was also downregulated in the gene expression data (Vilchèze et al.2013). Furthermore, the gene expression data did not reveal any change in the expression of genes encoding for proteins essential for the Mtb response to increased iron concentrations (Vilchèze et al.2013), such as bacterioferritin A and B (Rodriguez and Smith 2003). Likewise, the gene for a key oxidative response regulator, ahpD, was also down regulated. Contrary to their in vitro findings, vitamin C treatment alone has no effect in vivo, though it appears to potentiate the effects of rifampicin and isoniazid combination therapy (Vilchèze, Kim and Jacobs 2018). Notably, this stands in contrast with the tolerance of these drugs in vitro, reported by Sikri et al. (2018). A possible explanation is that the upregulation of sodC, ahpC, katG and MSH and EGT biosynthesis genes observed by Sikri et al. (2018) may counter the ROI-dependent actions of rifampin and isoniazid, thereby conferring resistance to these drugs (Wang, Burger and Drlica 1998; Dwyer et al.2014; Piccaro et al.2014). While vitamin C would certainly be an appealing adjunct to TB therapies for logistical reasons alone, the discrepancies of its impact on Mtb and its effects in vitro highlight the need for further investigation.
The Dos system in Mtb pathogenesis
The importance of DosR/S/T and the dormancy regulon to pathogenicity have been investigated in various animal models of TB, as well. Infection studies in C57BL/6J and SCID mice using Mtb lacking dosR showed no attenuation and, in one study, led to increased virulence compared to wild type (Parish et al.2003a; Rustad et al.2008). The inability of most mouse strains to reproduce major aspects of human TB pathology, including hypoxic granulomas, has since been appreciated (Aly et al.2006; Ernst 2012). Subsequent studies in C3HeB/FeJ mice that do form hypoxic, necrotic, and even cavitating lesions have demonstrated roles in Mtb virulence for specific components of the Dos system, though surprisingly not dosR (Gautam et al.2015; Gautam, Mehra and Kaushal 2015). Compared to the H37Rv strain, Converse and colleagues (2009) observed a decreased bacterial burden in C57BL/6J mice, rabbits and guinea pigs infected with a dosR/dosS double mutant (Converse et al.2009). These results suggest that the Dos system may play a role in the survival and persistence of Mtb in potentially hypoxic environments, in vivo; however, the failure of any of these models to fully capture the environment of the human lungs must be noted (Ernst 2012; Lenaerts, Barry and Dartois 2015).
This issue was recently addressed using the non-human primate (NHP) model of TB, which most closely resembles human TB. Mehra et al. (2015) infected NHPs with Mtb lacking either dosR, dosS or dosT. These mutants displayed no defect in replication relative to WT and complemented Mtb strains; however, all three single mutants were compromised in their ability to cause disease and persist within the host. Not surprisingly, this phenotype was strongly associated with hypoxia described within the lesions of host NHPs and a more robust T-cell response to infection, suggesting that components of the Dos system or the downstream regulon may play a role in suppressing or evading the host immune response. The relationship between hypoxia and the expression of the dormancy regulon was further validated in the second study from this group, which found that the dormancy regulon was induced in Mtb in association with lesion hypoxia and latency in infected NHPs (Hudock et al.2017). It is interesting to note that the most hypoxic lesions also contained central necrosis and the subsequent accumulation of a lipid-rich caseum within the granuloma. Thus, these studies do not preclude a contribution of lipid metabolism to Dos system-mediated virulence or the expression of genes within the dormancy regulon in vivo. Ultimately, the major contribution of these two studies is identifying a direct link between hypoxia and the expression of genes involved in the dormancy regulon in vivo and demonstrating an essential role for the components of the Dos system in Mtb virulence in the model that most closely approximates human TB.
Lastly, the clinical relevance of the Dos system and dormancy regulon was recently investigated in HIV-positive and HIV-negative TB patients. The authors observed increased expression of genes within the dormancy regulon in the cells collected by bronchoalveolar lavage (BAL) from HIV uninfected patients (Walter et al.2016). The authors propose two possible explanations for this observation. The first is that impaired host immunity in HIV patients leads to disordered granuloma formation and a potentially increased oxygen tension in granulomas (Diedrich, O’Hern and Wilkinson 2016). An alternative explanation stems from the analysis of host gene expression in the same BAL samples. Increased expression of the arginase gene and a decreased expression of the gene for inducible nitric oxide synthase in HIV infected patients suggest that macrophages in HIV infected patients were potentially less capable of producing NO, stressing Mtb, and inducing the dormancy regulon in Mtb (Walter et al.2016). Regardless, this finding underscores the importance of the host response on cellular activity of Mtb in human TB.
OTHER SYSTEMS FOR MAINTAINING REDOX HOMEOSTASIS
SenX3-RegX3
Mtb has a number of other systems important for responding to threats to redox homeostasis. Similar to DosR/S/T, the SenX3-RegX3 system is a two-component system that is sensitive to O2, NO and CO through the heme sensor kinase, SenX3 (Singh and Kumar 2015). Available evidence suggests that SenX3 may operate as a redox sensor that signals under the opposite conditions as DosS and DosT; hypoxia, NO and CO increase the activity of DosS and DosT, whereas these factors decrease SenX3-dependent signaling (Singh and Kumar 2015). It follows, then, that RegX3 would be active in the presence of O2 and the absence of NO and CO, though this has not been shown. Microarray data from a SenX3-RegX3 deletion mutant of Mtb demonstrate direct or indirect regulation of genes involved in the synthesis of DNA, RNA, lipids and cell wall components, as well as genes involved in lipid metabolism, suggesting that the SenX3-RegX3 system may serve to sense the suitability of the external environment for Mtb replication (Parish et al.2003b). This is supported by evidence that SenX3-RegX3 mutants exhibit impaired growth after infecting macrophages and are attenuated in mice and guinea pigs (Parish et al.2003b; Rickman et al.2004; Rifat, Bishai and Karakousis 2009). More specifically, RegX3 appears to play a direct role in the regulation of cydB, gltA and ald—three genes important for oxygen-dependent metabolism in Mtb (Roberts et al.2011).
Sigma factors
Specialized sigma factors also provide Mtb with transcriptional control in the context of redox stress. Specifically, SigH and SigE are important transcriptional regulators in the response to oxidative stress that share a number of striking similarities (Wu et al.1997; Raman et al.2001). Both recognize nearly identical promoter sequences with the exception of a discriminating base in the sixth position of the -35 hexamer (Song et al.2008; Sharp et al.2016), both are important to the pathogenesis of TB (Kaushal et al.2002; Ando et al.2003; Mehra et al.2012), and, importantly, both SigH and SigE are bound by redox-sensitive anti-sigma factors in a reducing environment and are released under oxidizing conditions (Song et al.2003; Barik et al.2010). Despite these similarities, SigH and SigE maintain functionally distinct roles.
Recently, Sharp et al. (2016) identified new members of the direct SigH regulon and expanded our understanding of its role in the response to oxidative stress. ChIP-seq analysis confirmed the regulation of genes for the antioxidant proteins thioredoxin and thioredoxin reductase, as well as various oxidoreductases, but it also identified genes important in mitigating oxidative damage. These include genes important in the synthesis and salvage of cysteine-containing proteins (mec, cysO, cysM, moeZ and moeB1), the repair of oxidized methionine residues (msrB), DNA repair (udgB) and ribosome replenishment (rpmE). In contrast, SigE appears to govern the transcription of genes involved in a general stress response, such as those encoding heat shock proteins (hsp and htpX) and enzymes involved in fatty acid oxidation (aceA, fadB2, fadE24, and fadE23; Manganelli et al.2001). However, the SigE regulon has not been as rigorously defined as the SigH regulon. Lastly, it appears that SigE may help Mtb survive oxidative stress by moderating the potentially harmful aspects of the WhiB4 response to such stress, such as nucleoid condensation (Chawla et al.2017). Thus, there is an appreciation for greater diversity in the activity of the redox-responsive transcriptional elements, SigH and SigE.
Serine/Threonine protein kinases
PknH (mentioned above) and PknG are two serine/threonine protein kinases that have emerged as important players in the maintenance of redox homeostasis in Mtb. PknG contains a rubredoxin domain with two thioredoxin motifs that regulates its kinase activity (Scherr et al.2007). Initial studies identified important roles for PknG in Mtb survival in macrophages (Walburger et al.2004), antibiotic resistance (Wolff et al.2009) and virulence in vivo (Cowley et al.2004). More recent studies show that these phenotypes are dependent, at least in part, on the maintenance of redox homeostasis by PknG through the regulation of the Nudix hydrolase, RenU (Wolff et al.2015), and the metabolic regulator, GarA (Khan et al.2017). Mtb mutants lacking either PknG or RenU exhibited decreased survival relative to WT Mtb when exposed to H2O2 and diamide (Wolff et al.2015). Additionally, M. smegmatis mutants lacking PknG or RenU accumulated the RenU substrates NADH and FAD following exposure to H2O2. Ultimately, it was shown that a ribosomal protein, L13, was capable of enhancing RenU activity following phosphorylation by PknG, establishing a redox homeostatic system under the control of PknG that is important for Mtb survival within macrophages. In addition, PknG may regulate redox homeostasis via phosphorylation of GarA. An Mtb mutant lacking PknG exhibited a wider range of redox environments between stressed and unstressed conditions, which was associated with decreased viability in the Wayne model of hypoxia-induced dormancy and when grown on nutrient-limited media (Khan et al.2017). Under hypoxic conditions, PknG promoted survival of M. smegmatis by reprogramming metabolism via phosphorylation of GarA, a regulator of key steps in the TCA cycle. Taken together, this work expands the role of PknG from withstanding oxidative stress within macrophages to a role as a central redox regulator capable of managing instances of reductive and oxidative stress during infection and dormancy-inducing conditions, ultimately making it a more attractive therapeutic target.
Enzymatic detoxification of ROI
Other enzymes in Mtb exist to counteract specific ROI or RNI; for example, superoxide dismutases (SODs) and catalases directly counter O2•− and H2O2, respectively (Ehrt and Schnappinger 2009). SODs are metalloproteinases responsible for the conversion of O2•− to H2O2 that can then be converted into OH− by catalase. Mtb catalase KatG also has the ability to detoxify other peroxides as well as peroxynitrite (ONOO−) (Wengenack et al.1999). These enzymes are considered virulence factors for Mtb and aid in countering oxidative stress from exogenous sources, including the phagocyte respiratory burst (Li et al.1998; Edwards et al.2001). Several other enzymes are important for the detoxification of specific ROI and RNI, and a full discussion of these would exceed the scope of this review. For an in depth discussion on Mtb strategies to directly counter such stresses, we direct the reader to the following review (Ehrt and Schnappinger 2009). These particular examples, however, illustrate the specificity of the Mtb armament against ROI and RNI.
CONCLUSIONS AND DIRECTIONS
We have described numerous factors encountered by Mtb during infection, as well as the major cellular machinery coordinate an output that allows it to adapt to the presence, absence or a mix of these factors (Fig. 4). The variable presence of these factors contributes to the dynamic, heterogeneous environment within the host, comprising an important aspect of Mtb pathogenesis. While great strides have been made in the last few years, many fundamental aspects of Mtb redox physiology still elude us.
Figure 4.

Mtb senses and responds to its redox environment. Hypoxia, gasotransmitters and lipid metabolism saturate the electron transport chain and electron carriers within the cell, inducing reductive stress (left). Elements responsive to reductive stress (left) execute a coordinated output, involving fatty acids synthesis to counter reductive stress and inhibit aspects of the immune response, expression of proteins important in the induction of dormancy, and decreasing the expression of highly antigenic secretion systems. On the other end of the spectrum, increased oxygen, the presence of numerous anti-TB drugs, and the respiratory burst of host phagocytes (right) can induce intracellular oxidative stress and ROI capable of damaging DNA, lipids and proteins. EGT and MSH can protect the cell by detoxifying ROI, promoting survival and resistance to drugs. WhiB proteins respond by upregulating the expression of secretion systems ESX-2 and ESX-4 (right). Ultimately, the ability of Mtb to sense and respond to a dynamic redox environment during infection is essential for Mtb virulence and pathogenesis.
For example, fundamental facts regarding redox buffers, such as the reduction potentials of MSH and EGT in vivo remain unknown. We partially understand the impact of these redox buffers on drug susceptibility and resisting exogenous ROI; however, the significance of Mtb utilizing two major redox buffers as opposed to one is unclear. The ability of MSH or EGT to compensate for the other may provide an obvious advantage for a bacterium that must withstand significant oxidative stress prior to escaping the phagosome. However, it is possible that the existence of two major redox buffers may provide other advantages for Mtb. This remains an area of open speculation and investigation.
Similarly, most fundamental aspects of WhiB protein physiology are unknown. The redox potentials for the iron-sulfur clusters common to this family have not been determined, and the 3D structures of WhiB proteins remain unsolved, with the notable exception of WhiB1 (Kudhair et al.2017). The relationship between these proteins and Mtb virulence is an exciting area of investigation, given the ability of these proteins to positively and negatively regulate the expression of numerous genes in diverse redox environments. Special attention should be paid to the relationship of these proteins and the expression of secretion systems central to the virulence of Mtb.
In contrast, the fundamental aspects of the components of the DosR/S/T system are relatively well characterized in terms of their structure, responsive elements and signaling pathways. Recent work by Mehra et al. (2015) and Hudock et al. (2017) established the Dos system as an essential part of Mtb virulence in vivo and that the expression of genes within the Dos dormancy regulon is associated with local hypoxia. A link between induction of the dormancy regulon and dormant Mtb, however, has yet to be conclusively established in vivo. A mechanistic explanation for Mtb persistence within the host would address a crucial question in the TB field and would truly represent a substantial advance in our understanding of Mtb pathogenesis.
Regarding redox physiology, many of the redox-sensitive systems in Mtb play a role in drug resistance and susceptibility. This includes both major redox buffers, members of the WhiB family, and components of the DosR/S/T system. Notably these systems and their activity do not always convey resistance to drugs; for example, Mtb lacking MSH showed increased susceptibility to some drugs (e.g. rifampicin and streptomycin) while resistance to others (e.g. ethionamide and isoniazid). Such insights highlight the intricate interactions of anti-TB drugs and the redox machinery of Mtb as well as the potential of these cellular components as targets for adjunct therapies to existing anti-TB drugs. Their unique expression within the pathogen alone makes them excellent targets for novel anti-TB drugs.
Ultimately, great strides have been made in the past decade in understanding how Mtb senses and responds to our best efforts to eradicate it from our bodies. The essential nature of many of the components Mtb uses to this end speak to the significance of sensing and reacting to the dynamic redox environments within the host. The remaining gaps in our understanding of these cellular components and their place in the pathogenesis of TB, therefore, hold promise in addressing the TB crisis.
FUNDING
This work was supported by NIH grants R01Al111940 and R21A127182, a Bill and Melinda Gates Foundation Award (OPP1130017) and by pilot funds from the UAB CFAR, CFRB, and Infectious Diseases and Global Health and Vaccines Initiative to A.J.C.S. The research from which this publication emanated was cofounded by the South African Medical Research Council. H.T.P. was supported by National Institute of General Medical Sciences (NIGMS) Medical Scientist Training Program (MSTP) T32GM008361. V.S. was supported by a UAB CFAR development grant derived from NIH P30 AI027767.
Conflict of Interest. None declared.
REFERENCES
- Aly S, Wagner K, Keller C et al. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol 2006;210:298–305. [DOI] [PubMed] [Google Scholar]
- Ando M, Yoshimatsu T, Ko C et al. Deletion of Mycobacterium tuberculosis sigma factor E results in delayed time to death with bacterial persistence in the lungs of aerosol-infected mice. Infect Immun 2003;71:7170–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik S, Sureka K, Mukherjee P et al. RseA, the SigE specific anti-sigma factor of Mycobacterium tuberculosis, is inactivated by phosphorylation-dependent ClpC1P2 proteolysis. Mol Microbiol 2010;75:592–606. [DOI] [PubMed] [Google Scholar]
- Barry CE, Boshoff HI, Dartois V et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Micro 2009;7:845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar A, Chawla M, Mehta M et al. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog 2014;10:e1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosserman RE, Nguyen TT, Sanchez KG et al. WhiB6 regulation of ESX-1 gene expression is controlled by a negative feedback loop in Mycobacterium marinum. Proc Natl Acad Sci USA 2017;114:E10772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broset E, Martín C, Gonzalo-Asensio J. Evolutionary landscape of the Mycobacterium tuberculosis complex from the viewpoint of PhoPR: implications for virulence regulation and application to vaccine development. mBio 2015;6:e01289–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmeier NA, Newton GL, Fahey RC. A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J Bacteriol 2006;188:6245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmeier NA, Newton GL, Koledin T et al. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol 2003;47:1723–32. [DOI] [PubMed] [Google Scholar]
- Buchmeier N, Fahey RC. The mshA gene encoding the glycosyltransferase of mycothiol biosynthesis is essential in Mycobacterium tuberculosis Erdman. FEMS Microbiol Lett 2006;264:74–9. [DOI] [PubMed] [Google Scholar]
- Burian J, Ramón-García S, Sweet G et al. The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J Biol Chem 2012;287:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian J, Yim G, Hsing M et al. The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res 2013;41:10062–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casonato S, Cervantes Sánchez A, Haruki H et al. WhiB5, a transcriptional regulator that contributes to Mycobacterium tuberculosis virulence and reactivation. Infect Immun 2012;80:3132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JD, Papavinasasundaram KG, Zheng X et al. Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J Biol Chem 2010;285:29239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla M, Mehta M, Parikh P et al. Redox-dependent condensation of mycobacterial genome by WhiB4. bioRxiv 2017;133181, DOI: 10.1101/133181. [DOI] [PMC free article] [PubMed]
- Chawla M, Parikh P, Saxena A et al. Mycobacterium tuberculosis WhiB4 regulates oxidative stress response to modulate survival and dissemination in vivo. Mol Microbiol 2012;85:1148–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hu Y, Cumming BM et al. Mycobacterial WhiB6 differentially regulates ESX-1 and the Dos regulon to modulate granuloma formation and virulence in zebrafish. Cell Reports 2016;16:2512–24. [DOI] [PubMed] [Google Scholar]
- Chim N, Johnson PM, Goulding CW. Insights into redox sensing metalloproteins in Mycobacterium tuberculosis. J Inorg Biochem 2014;133:118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta KC, Saini V, Glasgow JN et al. The emerging role of gasotransmitters in the pathogenesis of tuberculosis. Nitric Oxide 2016;59:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Converse PJ, Karakousis PC, Klinkenberg LG et al. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect Immun 2009;77:1230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S, Ko M, Pick N et al. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol 2004;52:1691–702. [DOI] [PubMed] [Google Scholar]
- Cumming BM, Chinta KC, Reddy VP et al. Role of ergothioneine in microbial physiology and pathogenesis. Antioxid Redox Signal 2018;28:431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Mazorodze JH, Steyn AJC et al. The physiology and genetics of oxidative stress in mycobacteria. In: Hatful GF, Jacobs WR Jr (eds.). Molecular Genetics of Mycobacteria. Second Ed Washington, DC: ASM Press, 2014, 299–322. [Google Scholar]
- Cumming BM, Rahman MA, Lamprecht DA et al. Mycobacterium tuberculosis arrests host cycle at the G1/S transition to establish long term infection. PLoS Pathog 2017;13:e1006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J, Maamar H, Deb C et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog 2011;7:e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N, Kapur V, Singh KK et al. Characterization of a two-component system, devR-devS, of Mycobacterium tuberculosis. Tuber Lung Dis 2000;80:141–59. [DOI] [PubMed] [Google Scholar]
- Datta M, Via LE, Chen W et al. Mathematical model of oxygen transport in tuberculosis granulomas. Ann Biomed Eng 2016;44:863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich CR, O’Hern J, Wilkinson RJ. HIV-1 and the Mycobacterium tuberculosis granuloma: a systematic review and meta-analysis. Tuberculosis 2016;98:62–76. [DOI] [PubMed] [Google Scholar]
- Dwyer DJ, Belenky PA, Yang JH et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci 2014;111:E2100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards KM, Cynamon MH, Voladri RKR et al. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med 2001;164:2213–9. [DOI] [PubMed] [Google Scholar]
- Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol 2009;11:1170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol 2012;12:581–91. [DOI] [PubMed] [Google Scholar]
- Fishbein S, van Wyk N, Warren RM et al. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol 2015;96:901–16. [DOI] [PubMed] [Google Scholar]
- Garcia-Morales L, Leon-Solis L, Monroy-Muñoz IE et al. Comparative proteomic profiles reveal characteristic Mycobacterium tuberculosis proteins induced by cholesterol during dormancy conditions. Microbiology 2017;163:1237–47. [DOI] [PubMed] [Google Scholar]
- Gautam US, McGillivray A, Mehra S et al. DosS is required for the complete virulence of Mycobacterium tuberculosis in mice with classical granulomatous lesions. Am J Respir Cell Mol Biol 2015;52:708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam US, Mehra S, Kaushal D. In-vivo gene signatures of Mycobacterium tuberculosis in C3HeB/FeJ Mice. PLoS One 2015;10:e0135208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gey van Pittius NC, Sampson SL, Lee H et al. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 2006;6:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath V, Raghunandanan S, Gomez RL et al. Profiling the proteome of Mycobacterium tuberculosis during dormancy and reactivation. Mol Cell Proteomics 2015;14:2160–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Lugo M-T, Baker H, Shiloach J et al. Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. J Biomol Screen 2009;14:643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC (eds.) Chemistry of free radicals and related “reactive species.” In: Free Radicals in Biology and Medicine. 4th ed New York, NY: Oxford University Press, 2008, 30–78. [Google Scholar]
- Hernick M. Mycothiol: a target for potentiation of rifampin and other antibiotics against Mycobacterium tuberculosis. Expert Rev Anti Infect Ther 2013;11:49–67. [DOI] [PubMed] [Google Scholar]
- Honaker RW, Dhiman RK, Narayanasamy P et al. DosS responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. J Bacteriol 2010;192:6447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben ENG, Walburger A, Ferrari G et al. Differential expression of a virulence factor in pathogenic and non-pathogenic mycobacteria. Mol Microbiol 2009;72:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben RMGJ, Dodd PJ. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med 2016;13:e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudock TA, Foreman TW, Bandyopadhyay N et al. Hypoxia sensing and persistence genes are expressed during the intragranulomatous survival of Mycobacterium tuberculosis. Am J Respir Cell Mol Biol 2017;56:637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RL. Tuberculosis as a three-act play: a new paradigm for the pathogenesis of pulmonary tuberculosis. Tuberculosis 2016;97:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioanoviciu A, Meharenna YT, Poulos TL et al. DevS oxy complex stability identifies this heme protein as a gas sensor in Mycobacterium tuberculosis dormancy. Biochemistry 2009;48:5839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioanoviciu A, Yukl ET, Moënne-Loccoz P et al. DevS, a heme-containing two-component oxygen sensor of Mycobacterium tuberculosis. Biochemistry 2007;46:4250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iona E, Pardini M, Mustazzolu A et al. Mycobacterium tuberculosis gene expression at different stages of hypoxia-induced dormancy and upon resuscitation. J Microbiol 2016;54:565–72. [DOI] [PubMed] [Google Scholar]
- Jocelyn PC. Biochemistry of the SH Group: The Occurence, Chemical Properties, Metabolism and Biological Function of Thiols and Disulphides. London: Academic Press, 1972. [Google Scholar]
- Kaushal D, Schroeder BG, Tyagi S et al. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative factor, SigH. Proc Natl Acad Sci 2002;99:8330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GM, Hooley GC, Champion MM et al. A novel ESX-1 locus reveals that surface-associated ESX-1 substrates mediate virulence in Mycobacterium marinum. J Bacteriol 2014;196:1877–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Bhaskar A, Upadhyay S et al. Protein kinase G confers survival advantage to Mycobacterium tuberculosis during latency-like conditions. J Biol Chem 2017;292:16093–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudhair BK, Hounslow AM, Rolfe MD et al. Structure of a Wbl protein and implications for NO sensing by M. tuberculosis. Nat Commun 2017;8:2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Deshane JS, Crossman DK et al. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J Biol Chem 2008;283:18032–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Farhana A, Guidry L et al. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med 2011;13:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Toledo JC, Patel RP et al. Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc Natl Acad Sci 2007;104:11568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P, Sikri K, Kaur K et al. Sustained expression of DevR/DosR during long-term hypoxic culture of Mycobacterium tuberculosis. Tuberculosis 2017;106:33–7. [DOI] [PubMed] [Google Scholar]
- Kumari P, Sikri K, Tyagi JS et al. The pleiotropic transcriptional response of Mycobacterium tuberculosis to vitamin C is robust and overlaps with the bacterial response to multiple intracellular stresses. Microbiology 2015;161:739–53. [DOI] [PubMed] [Google Scholar]
- Lamichhane G. Mycobacterium tuberculosis response to stress from reactive oxygen and nitrogen species. Front Microbio 2011;2:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix J-P, Lenaerts AJ, Nuermberger EL. Heterogeneous disease progression and treatment response in a C3HeB/FeJ mouse model of tuberculosis. Dis Model Mech 2015;8:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson C, Luna B, Ammerman NC et al. Gene expression of Mycobacterium tuberculosis putative transcription factors whiB1-7 in redox environments. PLoS One 2012;7:e37516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts A, Barry CE, Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev 2015;264:288–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Kelley C, Collins F et al. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J Infect Dis 1998;177:1030–5. [DOI] [PubMed] [Google Scholar]
- Loi V Van, Rossius M, Antelmann H. Redox regulation by reversible protein S-thiolation in bacteria. Front Microbiol 2015;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Voskuil MI, Schoolnik GK et al. The Mycobacterium tuberculosis ECF sigma factor σE: role in global gene expression and survival in macrophages†. Mol Microbiol 2001;41:423–37. [DOI] [PubMed] [Google Scholar]
- Mehra S, Foreman TW, Didier PJ et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am J Respir Crit Care Med 2015;191:1185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Golden NA, Stuckey K et al. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis 2012;205:1203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta M, Rajmani RS, Singh A. Mycobacterium tuberculosis WhiB3 responds to vacuolar pH-induced changes in mycothiol redox potential to modulate phagosomal maturation and virulence. J Biol Chem 2016;291:2888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton GL, Arnold K, Price MS et al. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol 1996;178:1990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev 2008;72:471–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton GL, Ta P, Bzymek KP et al. Biochemistry of the initial steps of mycothiol biosynthesis. J Biol Chem 2006;281:33910–20. [DOI] [PubMed] [Google Scholar]
- Newton GL, Unson MD, Anderberg SJ et al. Characterization of Mycobacterium smegmatis mutants defective in 1-d-myo-Inosityl-2-amino-2-deoxy-?-d-glucopyranoside and mycothiol biosynthesis. Biochem Biophys Res Commun 1999;255:239–44. [DOI] [PubMed] [Google Scholar]
- Parish T, Smith DA, Kendall S et al. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect Immun 2003;71:1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T, Smith DA, Roberts G et al. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 2003;149:1423–35. [DOI] [PubMed] [Google Scholar]
- Piccaro G, Pietraforte D, Giannoni F et al. Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2014;58:7527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S, Song T, Puyang X et al. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J Bacteriol 2001;183:6119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat M, Av-Gay Y. Mycothiol-dependent proteins in actinomycetes. FEMS Microbiol Rev 2007;31:278–92. [DOI] [PubMed] [Google Scholar]
- Rawat M, Johnson C, Cadiz V et al. Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem Biophys Res Commun 2007;363:71–6. [DOI] [PubMed] [Google Scholar]
- Rawat M, Kovacevic S, Billman-Jacobe H et al. Inactivation of mshB, a key gene in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Microbiology 2003;149:1341–9. [DOI] [PubMed] [Google Scholar]
- Rawat M, Newton GL, Ko M et al. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 2002;46:3348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard-Greenblatt M, Bach H, Adamson J et al. Regulation of ergothioneine biosynthesis and its effect on Mycobacterium tuberculosis growth and infectivity. J Biol Chem 2015;290:23064–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman L, Saldanha JW, Hunt DM et al. A two-component signal transduction system with a PAS domain-containing sensor is required for virulence of Mycobacterium tuberculosis in mice. Biochem Biophys Res Commun 2004;314:259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifat D, Bishai WR, Karakousis PC. Phosphate depletion: a novel trigger for Mycobacterium tuberculosis persistence. J Infect Dis 2009;200:1126–35. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Liao RP, Wisedchaisri G et al. Two sensor kinases contribute to the hypoxic response of Mycobacterium tuberculosis. J Biol Chem 2004;279:23082–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts G, Vadrevu IS, Madiraju MV et al. Control of CydB and GltA1 expression by the SenX3 RegX3 two component regulatory system of Mycobacterium tuberculosis. PLoS One 2011;6:e21090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Smith I. Mechanisms of iron regulation in mycobacteria: role in physiology and virulence. Mol Microbiol 2003;47:1485–94. [DOI] [PubMed] [Google Scholar]
- Rodriguez GM, Voskuil MI, Gold B et al. ideR, An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun 2002;70:3371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez JG, Hernández AC, Helguera-Repetto C et al. Global adaptation to a lipid environment triggers the dormancy-related phenotype of Mycobacterium tuberculosis. mBio 2014;5:e01125–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad TR, Harrell MI, Liao R et al. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One 2008;3:e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini DK, Malhotra V, Dey D et al. DevR-DevS is a bona fide two-component system of Mycobacterium tuberculosis that is hypoxia-responsive in the absence of the DNA-binding domain of DevR. Microbiology 2004;150:865–75. [DOI] [PubMed] [Google Scholar]
- Saini V, Cumming BM, Guidry L et al. Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of Mycobacterium tuberculosis. Cell Reports 2016;14:572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini V, Farhana A, Glasgow JN et al. Iron sulfur cluster proteins and microbial regulation: implications for understanding tuberculosis. Curr Opin Chem Biol 2012;16:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini V, Farhana A, Steyn AJC. Mycobacterium tuberculosis WhiB3: a novel iron–sulfur cluster protein that regulates redox homeostasis and virulence. Antioxidants Redox Signal 2012;16:687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sani M, Houben ENG, Geurtsen J et al. Direct visualization by Cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog 2010;6:e1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sao Emani C, Williams MJ, Wiid IJ et al. Ergothioneine is a secreted antioxidant in Mycobacterium smegmatis. Antimicrob Agents Chemother 2013;57:3202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D, Newton GL, Fahey RC et al. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J Bacteriol 2003;185:6736–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 2001;30:1191–212. [DOI] [PubMed] [Google Scholar]
- Scherr N, Honnappa S, Kunz G et al. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci 2007;104:12151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert OT, Ludwig C, Kogadeeva M et al. Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 2015;18:96–108. [DOI] [PubMed] [Google Scholar]
- Seebeck FP. In vitro reconstitution of Mycobacterial ergothioneine biosynthesis. J Am Chem Soc 2010;132:6632–3. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Van Laer K, Messens J et al. Thiol redox and p Ka properties of mycothiol, the predominant low-molecular-weight thiol cofactor in the actinomycetes. ChemBioChem 2016;17:1689–92. [DOI] [PubMed] [Google Scholar]
- Sharp JD, Singh AK, Park ST et al. Comprehensive definition of the SigH regulon of Mycobacterium tuberculosis reveals transcriptional control of diverse stress responses. PLoS One 2016;11:e0152145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikri K, Duggal P, Kumar C et al. Multifaceted remodeling by vitamin C boosts sensitivity of Mycobacterium tuberculosis subpopulations to combination treatment by anti-tubercular drugs. Redox Biol 2018;15:452–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Crossman DK, Mai D et al. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 2009;5:e1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Guidry L, Narasimhulu KV et al. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci 2007;104:11562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AR, Strankman A, Orkusyan R et al. Lack of mycothiol and ergothioneine induces different protective mechanisms in Mycobacterium smegmatis. Biochem Biophys Reports 2016;8:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Kumar A. Virulence factor SenX3 is the oxygen-controlled replication switch of Mycobacterium tuberculosis. Antioxid Redox Signal 2015;22:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LJ, Stapleton MR, Fullstone GJM et al. Mycobacterium tuberculosis WhiB1 is an essential DNA-binding protein with a nitric oxide-sensitive iron–sulfur cluster. Biochem J 2010;432:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solans L, Aguiló N, Samper S et al. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies WhiB6 as a novel ESX-1 component. Infect Immun 2014;82:3446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, Dove SL, Lee KH et al. RshA, an anti-sigma factor that regulates the activity of the mycobacterial stress response sigma factor SigH. Mol Microbiol 2003;50:949–59. [DOI] [PubMed] [Google Scholar]
- Song T, Song S-E, Raman S et al. Critical role of a single position in the -35 element for promoter recognition by Mycobacterium tuberculosis SigE and SigH. J Bacteriol 2008;190:2227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Ramirez MD, Aguilar-Ayala DA, Garcia-Morales L et al. Cholesterol plays a larger role during Mycobacterium tuberculosis in vitro dormancy and reactivation than previously suspected. Tuberculosis 2017;103:1–9. [DOI] [PubMed] [Google Scholar]
- Steyn AJC, Collins DM, Hondalus MK et al. Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc Natl Acad Sci 2002;99:3147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta P, Buchmeier N, Newton GL et al. Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J Bacteriol 2011;193:1981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja NK, Dhingra S, Mittal A et al. Mycobacterium tuberculosis transcriptional adaptation, growth arrest and dormancy phenotype development is triggered by vitamin C. PLoS One 2010;5:e10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venketaraman V, Dayaram YK, Amin AG et al. Role of glutathione in macrophage control of mycobacteria. Infect Immun 2003;71:1864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via LE, Lin PL, Ray SM et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 2008;76:2333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Av-Gay Y, Attarian R et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol 2008;69:1316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Hartman T, Weinrick B et al. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat Comms 2013;4:1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C, Kim J, Jacobs WR. Vitamin C potentiates the killing of Mycobacterium tuberculosis by the first-line tuberculosis drugs isoniazid and rifampin in mice. Antimicrob Agents Chemother 2018;62:e02165–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Bartek IL, Visconti K et al. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbio 2011;2:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Schnappinger D, Rutherford R et al. Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis 2004;84:256–62. [DOI] [PubMed] [Google Scholar]
- Voskuil MI, Schnappinger D, Visconti KC et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 2003;198:705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walburger A, Koul A, Ferrari G et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 2004;304:1800–4. [DOI] [PubMed] [Google Scholar]
- Walter ND, de Jong BC, Garcia BJ et al. Adaptation of Mycobacterium tuberculosis to impaired host immunity in HIV-infected patients. J Infect Dis 2016;214:1205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Burger RM, Drlica K. Role of superoxide in catalase-peroxidase-mediated isoniazid action against mycobacteria. Antimicrob Agents Chemother 1998;42:709–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengenack NL, Jensen MP, Rusnak F et al. Mycobacterium tuberculosis KatG is a peroxynitritase. Biochem Biophys Res Commun 1999;256:485–7. [DOI] [PubMed] [Google Scholar]
- WHO Global tuberculosis report 2017. World Health Organization, 2017. [Google Scholar]
- Wolff KA, de la Peña AH, Nguyen HT et al. A redox regulatory system critical for Mycobacterial survival in macrophages and biofilm development. PLoS Pathog 2015;11:e1004839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff KA, Nguyen HT, Cartabuke RH et al. Protein kinase G is required for intrinsic antibiotic resistance in mycobacteria. Antimicrob Agents Chemother 2009;53:3515–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ru H-W, Xiang Z-H et al. WhiB4 regulates the PE/PPE gene family and is essential for virulence of Mycobacterium marinum. Sci Rep 2017;7:3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu QL, Kong D, Lam K et al. A mycobacterial extracytoplasmic function sigma factor involved in survival following stress. J Bacteriol 1997;179:2922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Vilchèze C, Av-Gay Y et al. Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 2011;55:3133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]