

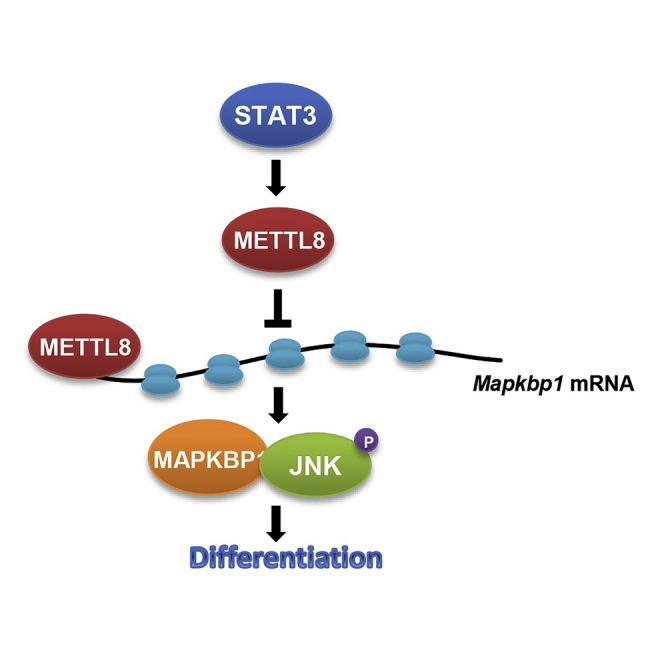
Summary
The capacity of embryonic stem cells (ESCs) to differentiate into all lineages of mature organism is precisely regulated by cellular signaling factors. STAT3 is a crucial transcription factor that plays a central role in maintaining ESC identity. However, the underlying mechanism by which STAT3 directs differentiation is still not completely understood. Here, we show that STAT3 positively regulates gene expression of methyltransferase-like protein 8 (Mettl8) in mouse ESCs. We found that METTL8 is dispensable for pluripotency but affects ESC differentiation. Subsequently, we discovered that METTL8 interacts with Mapkbp1's mRNA, which is an intermediate factor in c-Jun N-terminal kinase (JNK) signaling, and inhibits the translation of the mRNA. Thereby, METTL8 prohibits the activation of JNK signaling and enhances the differentiation of mouse ESCs. Collectively, our study uncovers a STAT3 target, Mettl8, which regulates mouse ESC differentiation via JNK signaling.
Keywords: embryonic stem cell, STAT3, Mettl8, JNK, differentiation
Graphical Abstract
Highlights
-
•
Mettl8 is transcriptionally regulated by STAT3
-
•
METTL8 inhibits mouse ESC differentiation
-
•
METTL8 binds to Mapkbp1 mRNA and inhibits translation
-
•
METTL8 inhibits JNK pathway during differentiation
In this article, Fu and colleagues show that STAT3 positively regulates gene expression of Mettl8 in mouse ESCs. METTL8 is dispensable for pluripotency but affects ESC differentiation by interacting with Mapkbp1 mRNA and inhibiting its translation. MAPKBP1 mediates the effect of METTL8 to JNK signaling during mouse ESC differentiation.
Introduction
Embryonic stem cells (ESCs) are derived from the inner cell mass of the blastocyst, which comprises pluripotent cells with the potential to differentiate into all cell types of the body (Williams et al., 1988b). This capacity enables ESCs a wide utilization in regenerative medicine and cell-based therapies. Therefore, a better understanding of the underlying mechanisms that modulate the differentiation of ESCs is essential for their clinical application in the future (Chen et al., 2008).
Leukemia inhibitory factor (LIF) was found to be the key growth factor for the culture and maintenance of mouse ESCs (mESCs) in vitro (Smith et al., 1988, Williams et al., 1988a). LIF, a member of the interleukin-6 (IL-6) family of cytokines, binds to gp130/LIFR and results in the phosphorylation on tyrosine 705 residues of STAT3, a member of the STAT gene family identified in the interferon-induced regulatory pathways (Darnell et al., 1994, Fu et al., 1990, Fu et al., 1992, Schindler et al., 1992). STAT3, first identified as a transcription factor (TF) for the IL-6 family of cytokines (Akira et al., 1994, Zhong et al., 1994), was subsequently found to be crucial for ESC pluripotency (Boeuf et al., 1997, Boyer et al., 2005, Niwa et al., 1998, Raz et al., 1999, Ying et al., 2003). Conventional knockout of Stat3 in mice results in embryonic lethality at embryonic day 6.5 (E6.5) (Takeda et al., 1997). By eliminating Stat3 in the mouse oocytes and embryos we found that STAT3 has an essential role in inner cell mass lineage specification and maintenance, and in pluripotent stem cell identity through the OCT4-NANOG circuit (Do et al., 2013).
The c-Jun NH2-teminal kinase (JNK) belongs to the mitogen-activated protein (MAP) kinase family, which were initially identified as ultraviolet-responsive protein kinases that activated c-Jun by phosphorylating its NH2-terminal serine/threonine residues (Dérijard et al., 1994, Hibi et al., 1993). In response to growth factors, cytokines, and a number of environmental stresses, JNK is activated through a well-orchestrated cascade of MAP kinase activation (Jaeschke et al., 2006, Sabapathy et al., 2004). In particular, mitogen-activated kinase kinase 4 and 7, isoforms of MAP2K, directly phosphorylate and activate JNK, which in turn leads to the phosphorylation of (TF) c-Jun and switching on of transcriptional regulation exclusively through formation of complex with other TFs, such as c-fos, in the activator protein-1 complex (Davis, 2000, Weston and Davis, 2007). JNK is encoded by two ubiquitously expressed genes (JNK1 and JNK2) and by a third gene (JNK3) that is selectively expressed in neurons. MAPKBP1 is a JNK binding protein and enhances the activation of JNK (Koyano et al., 1999, Lecat et al., 2012). Recent studies indicated that JNK signaling is required for lineage-specific differentiation but not stem cell self-renewal. ESCs lacking JNK1 show transcriptional deregulation of several lineage-commitment genes and fail to undergo neuronal differentiation, as do ESCs lacking JNK pathway scaffold proteins (Xu and Davis, 2010). Studies also found that JNK binds to a large set of active promoters during the differentiation of stem cells and results in histone 3 phosphorylation on chromatin (Tiwari et al., 2011). It is also reported that JNK regulates STAT3 activity via its Ser-727 phosphorylation, showing the crosstalk between STAT3 and JNK pathways (Lim and Cao, 1999).
In this study, we further investigate how STAT3 integrate to the core regulatory circuit in ESC pluripotency and differentiation, and identify Mettl8 as a downstream target of STAT3 in mESCs. We discover the role of METTL8 as a negative regulator of JNK signaling in stem cells. Our results provide insights into the crosstalk between STAT3 and JNK signaling during stem cell differentiation.
Results
Mettl8 Is a Direct Target of STAT3 in mESCs
In this study, we further investigated how STAT3 crosstalk with other potential pathways in ESC pluripotency. Therefore, we screened for unknown factors that were regulated by STAT3 using ESCs treated with STAT3 inhibitors STA-21 and STATTIC (Schust et al., 2006, Song et al., 2005). Real-time PCR results obtained from screening for a library of 200 epigenetic candidates led us to identify Mettl8 (Figure 1A). We found that the mRNA levels of Mettl8 were downregulated after the two-inhibitor treatment (Figure 1B). Meanwhile, we checked Mettl8's protein level, which was also inhibited by STA-21 and STATTIC (Figures 1C and S1).
Figure 1.

Mettl8 Is Transcriptionally Regulated by STAT3
(A) Real-time PCR was performed to screen for changes when ESCs were treated with STA-21 and STATTIC for 1 hr.
(B and C) E14 cells were treated with STA-21 and STATTIC for 6 hr and harvested. (B) Total RNAs were extracted and followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05. (C) Cell lysates were analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(D) Knockdown Stat3 in E14 cells resulted in downregulation of Mettl8 mRNA. Data are shown as the mean ± SD from three independent experiments.
(E) Knockdown Stat3 in E14 cells resulted in downregulation of METTL8 protein. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(F and G) E14 cells were transfected with Flag-vector or Flag-tagged STAT3 at increasing concentrations. (F) Total RNAs were extracted followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05. (G) Cell lysates were analyzed by western blot.
(H) Bioinformatic analysis identifies three possible STAT3 binding sites on Mettl8 gene labeled as P1, P2, and P3. Data are shown as the mean ± SD from three independent experiments.
(I) Inducible Flag-METTL8 overexpression E14 cells were treated with or without doxycycline and culture in mediums with or without LIF for 6 days. Then colonies were stained for AP activity and cell lysates were analyzed by western blot. Scale bars, 0.5 cm.
To further confirm that Mettl8 is a target of STAT3, we reduced Stat3 transcript levels by employing Stat3 small hairpin RNAs (shRNAs), and found that both Mettl8 mRNA and protein levels in ESCs were reduced (Figures 1D and 1E). Next we validated this result with STAT3 overexpression in mESCs. When STAT3 was overexpressed, both Mettl8 mRNA and protein levels were upregulated (Figures 1F and 1G). To examine whether STAT3 regulates Mettl8 directly or indirectly, we used bioinformatics, and identified three possible STAT3 binding sites on Mettl8 gene at −2,672, −83, and 1,020 bp (PROMO software). We then performed chromatin immunoprecipitation (ChIP) assay using the pY705-STAT3-specific antibody. We found the highest enrichment in a promoter region at −83 bp and a proximal enhancer at 1,020 bp of Mettl8 gene (Figure 1H). These results show that STAT3 directly interacts with the regulatory regions of the Mettl8. Taken together, our evidence demonstrate STAT3 directly and positively regulates Mettl8 expression in pluripotent cells.
To further figure out whether METTL8 exerts a key function downstream of STAT3 in ESCs, we created inducible METTL8 overexpression mESCs. After LIF withdrawal ESCs became differentiated, but METTL8 overexpression rescued this phenomenon and showed more ESC colonies stained positive for alkaline phosphatase (AP) (Figure 1I), suggesting that METTL8 mediates the role of STAT3 in pluripotent cells.
METTL8 Is Dispensable for the Maintenance of Pluripotency in mESCs
To examine the function of METTL8 in mESCs, we first induced mESCs to differentiation by retinoic acid (RA) treatment and found both mRNA and protein levels of Mettl8 were reduced (Figures 2A and 2B). Similarly, during embryoid body (EB) formation, Mettl8 was also significantly downregulated from day 6 onward and barely detectible at days 9–15 (Figures 2C and 2D). These data imply that expression of Mettl8 is correlated with the pluripotent state of ESCs, thus we next evaluated the direct effect of METTL8 on ESC pluripotency. However, when Mettl8 was knocked out, the morphology of ESCs showed no difference from the wild-type cells, which were positive in AP staining (Figure 2E). Moreover, mRNA and protein expressions of key pluripotent genes were not changed in Mettl8 deficient or overexpressed mESCs (Figures 2F, 2G, 2H, and S2A), and Mettl8 overexpression or knockout had little impact on the clonogenicity of mESCs (Figures 2I and 2J). However, when cultured in sub-optimal conditions, self-renewal of Mettl8-null ESCs is weakened (Figure 2J). These results suggested METTL8 is dispensable for ESC pluripotency when Mettl8-null ESCs are cultivated in normal conditions but might play a role in ESC differentiation.
Figure 2.

METTL8 Affects ESCs Differentiation Rather Than Pluripotency
(A) Total RNAs were extracted from E14 cells treated with RA for 0 or 4 days, followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments. ∗∗∗p < 0.001.
(B) E14 cells were treated with RA for the indicated times and cell lysates were analyzed by western blot.
(C and D) E14 cells were subjected to EB formation and cells were harvested at the indicated times. (C) Total RNAs were extracted followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments.∗p < 0.05, ∗∗∗p < 0.001. (D) Cell lysates were analyzed by western blot.
(E) Mettl8 KO and control E14 cells were stained for AP activity and pictures were taken. Scale bars, 100 μm.
(F) Total RNAs were extracted from Mettl8 KO and control E14 cells followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments.
(G) Cell lysates of Mettl8 KO and control E14 cells were analyzed by western blot.
(H and I) E14 cells were transfected with Flag-vector or Flag-tagged METTL8 and (H) 48 hr later cell lysates were analyzed by western blot. (I) Colony-forming assays were performed and the number of AP-positive colonies was counted.
(J) Mettl8 KO and control cells were cultured under normal condition or LIF deprivation for 4 days. Then colony-forming assays were performed and the number of AP-positive colonies was counted. ∗∗p < 0.01.
(K) Mettl8 KO and control E14 cells were subjected to EB formation for 7 days. EBs were trypsinized and re-plated into gelatin-coated dished. After 3 days culture in ESC medium, colonies were stained for AP activity and AP-positive colonies were counted. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05.
(L) Inducible METTL8 overexpression E14 cells were treated with or without doxycycline and subjected to EB formation for 7 days, or treated with doxycycline for the first 3 days of EB formation. Then EBs were trypsinized and re-plated into gelatin-coated dishes. After 3 days culture in ESC medium, colonies were stained for AP activity and AP-positive colonies were counted. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
METTL8 Inhibits mESC Differentiation
We next examined the function of METTL8 in differentiation assay of ESCs. Interestingly, after 10 days of EB formation, the numbers of AP-positive ESCs decreased in the Mettl8 knockout (KO) group (Figures 2K and S2B). Consistently, more ectopic expression of METTL8 led to higher AP-positive numbers from EBs (Figure 2L), which suggests that METTL8 impedes mESC differentiation. Then global gene transcriptome profiling was performed with whole-genome cDNA microarray to find out the genes with significant changes between Mettl8 or scramble small interfering RNA (siRNA) mESCs (Table S1). We found that 353 genes were upregulated (>2-fold) and 89 transcripts were downregulated (>2-fold) after Mettl8 depletion. To define discreet functional clusters of common gene regulation, we performed a hierarchical clustering of our microarray data. This analysis yielded four discernable clusters of commonly regulated genes with two each associated with up- and downregulation (Figure S2C). Overall upregulated gene clusters were associated with developmental capacity. When we performed gene ontology we found that these upregulated genes are enriched for gene ontology terms such as developmental process and cellular differentiation. Particularly, we found some key upregulated lineage genes such as Pax6, Gata6, T, and Hand1, which have been reported to be upregulated during mESC differentiation (Figure S2C). We also examined the global gene expression changes using RNA sequencing (RNA-seq) comparing the transcriptome from ESCs with either Mettl8 or scramble siRNA. We found that 198 genes were downregulated (>2-fold) and 1,353 transcripts were upregulated (>2-fold) after Mettl8 depletion (Figure S2D). Consistent with microarray data, we also found key upregulated developmental genes such as Pax6, Gata6, T, and Hand1, and then used real-time PCR to validate the increase in mRNA expression of these genes (Figure S2E). Taken together, these results suggested that METTL8 indeed inhibits ESC differentiation, possibly by suppressing expression of some key developmental genes.
METTL8 Functions in Somatic Reprogramming
Given the role of METTL8 in the regulation of ESC differentiation, we sought to further explore its functions in induced pluripotent stem cells (iPSCs) and determine whether METTL8 also regulates somatic reprogramming. Firstly, using the well-established iPSC mouse model (Carey et al., 2010), we found both mRNA and protein levels of Mettl8 were increased during cellular reprogramming after inducing OSKM (OCT4, SOX2, KLF4, and MYC) in MEF cells (Figures 3A and 3B), indicating that METTL8 may facilitate reprogramming. To test this hypothesis, we crossed Mettl8 KO with inducible OSKM strain to introduce Mettl8 KO MEF cells along with OSKM induction. Mettl8 deficiency indeed resulted in a decreased iPSC colony formation (Figure 3C). These Mettl8 KO iPSC colonies shared a similar morphology with the wild-type ones (Figure 3D). In addition, RT-PCR analysis revealed that all of the randomly picked Mettl8 KO MEF-derived colonies expressed endogenous ESC marker genes, including Oct4, Sox2, Nanog, Rex1, Fbx15, and Esg1 (Figure 3E). Immunofluorescence analysis also showed that these colonies were positive for pluripotent markers OCT4 and SOX2 (Figure 3F). Together our results suggest that METTL8 play its role in somatic reprogramming.
Figure 3.

METTL8 Functions in Somatic Reprogramming
(A) Total RNA was extracted from OSKM-inducible MEF cells treated with doxycycline on days 0, 5, and 10, and iPSCs followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05.
(B) Cell lysis from OSKM-inducible MEF cells treated with doxycycline on days 0, 5, and 10, and iPSCs and analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(C) Wild-type and Mettl8 KO OSKM-inducible MEF cells were treated with doxycycline and induced for iPSCs. After 20 days, numbers of iPSC colonies were counted. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05.
(D) Wild-type and Mettl8 KO iPSCs were stained for AP activity and pictures were taken. Scale bars, 100 μm.
(E) Total RNA from MEF cells, E14 cells, and the indicated Mettl8 KO iPSC colonies were subjected to RT-PCR analysis for detection of expression of endogenous Oct4, Sox2, Nanog, Rex1, Fbx15, and Esg1. Actin was used as an internal control.
(F) Mettl8 KO iPSC colonies were subjected to immunofluorescence for analysis of endogenous OCT4 and SOX2 expression. Scale bars, 50 μm.
METTL8 Interacts with Mapkbp1 mRNA
As we discovered simultaneously that METTL8 interacts with mRNAs (Xu et al., 2017), to further understand the mechanism of METTL8 in pluripotent cells, we sought to identify METTL8-interacting RNAs by photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) assay (Hafner et al., 2010) (Figure 4A). The subsequent analysis indeed revealed that mRNAs are potential METTL8 protein interacting partners, which account for a large proportion of the binding peaks (Table S2). This result was matched with the subcellular distribution of METTL8 protein that it is mainly located at the cytoplasm where most mature mRNA resides (Figure 4B). To elucidate the mechanism by which METTL8 works through its mRNA partners, we chose Mapkbp1 mRNA from the candidates for detailed investigation, and RNA immunoprecipitation (RIP) assay was performed. We discovered that Mapkbp1 mRNAs were enriched in Flag-tagged METTL8 expression sample, which confirmed METTL8 protein's ability to bind Mapkbp1 mRNA (Figure 4C). The endogenous complex between METTL8 protein and Mapkap1 mRNA was also detected by RIP assay using METTL8 antibody (Figure 4D). In addition, the interaction of METTL8 with Mapkbp1 mRNA was further verified by affinity pull-down of endogenous Mapkbp1 mRNA using a biotinylated RNA antisense probe to Mapkbp1 mRNA (Figure 4E).
Figure 4.

METTL8 Binds to Mapkbp1 mRNA
(A) Schematic illustration of PAR-CLIP assay.
(B) E14 cells were harvested and subjected to cell fractionation. Each fraction was collected and analyzed by western blot.
(C) HEK293T cells were transfected by the indicated plasmids. After 48 hr cells were harvested and subjected to RIP with flag antibody. Total RNAs were extracted followed by real-time PCR analysis. Actin was used as an internal control. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05.
(D) E14 cells were harvested and subjected to RIP with METTL8 antibody or isotype-matched immunoglobulin G (IgG). Total RNAs were extracted and relative level of Mapkbp1 mRNA was analyzed by quantitative real-time PCR. Actin was used as an internal control. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05.
(E) E14 cells were harvested and cell lysates were then incubated with in-vitro-synthesized biotin-labeled sense or antisense Mapkbp1 mRNA probes for biotin pull-down assay, followed by western blot analysis.
METTL8 Inhibits Mapkbp1 mRNA Translation
To clarify the influence of METTL8 protein on its interacting partner, we next tested the expression level of Mapkbp1 mRNA after knocking out Mettl8 in ESCs. The results showed no significant change between Mettl8 KO and control cells (Figure 5A). Similar results were obtained when METTL8 was overexpressed in HEK293T cells (Figure 5B), demonstrating that METTL8 does not affect mRNA levels of Mapkbp1. Thus, we then checked the protein level of Mapkbp1, and found a higher expression of MAPKBP1 protein in Mettl8 KO cells (Figures 5C and S3A). Based on this data, we speculated that METTL8 decreases the protein stability of MAPKBP1. However, when ESCs treated with cycloheximide (CHX) and protein synthesis was blocked, the degradation rate of MAPKBP1 protein showed no significant change between Mettl8 KO and control cells (Figure 5D), indicating that protein stability of MAPKBP1 was not affected. Then we hypothesized that METTL8 may influence translation, and to directly test the possibility, Mettl8 KO and control ESC lysates were subjected to polysome profiling by fractionation through sucrose gradients. The lightest components sedimented at the top (fractions 1 and 2), small (40S) and large (60S) ribosomal subunits and monosomes (80S) located in fractions 3 to 8, and larger polysomes in fractions 9 to 15 (Figure 5E). Mettl8 KO did not change global polysome distribution profiles, indicating that it did not affect global translation (Figure 5E). After isolating RNA from each fraction, real-time PCR analysis indicated that Mapkbp1 mRNA in Mettl8 KO cells was abundant in fractions 12 to 15, while in control cells it was abundant in fractions 4 to 6 and 11 to 13 (Figure 5F). The result indicates that silencing Mettl8 leads to Mapkbp1 mRNA distribution shifting toward larger polysomes and thus promotes translation. As a negative control, actin mRNA did not show that shift (Figure 5F). Taken together METTL8 affects Mapkbp1 expression through binding to Mapkap1 mRNA and inhibiting its translation.
Figure 5.

Mettl8 Deficiency Enhances Mapkbp1 mRNA Translation
(A) Total RNAs were extracted from Mettl8 KO and control E14 cells followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments.
(B) HEK293T cells were transfected by indicated plasmids. After 48 hr total RNAs were extracted followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments.
(C) Cell lysates of Mettl8 KO and control E14 cells were analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(D) Mettl8 KO and control E14 cells were treated with CHX for the indicated times and cell lysates were analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(E) Global polysome profiles of Mettl8 KO and control E14 cells were analyzed by sucrose gradient sedimentation.
(F) Each fraction from sucrose gradient sedimentation was collected and total RNA was extracted. Levels of Mapkbp1 mRNA were analyzed by quantitative real-time PCR.
METTL8 Inhibits JNK Signaling in mESCs
To further understand the consequences of the interaction between METTL8 and Mapkbp1 mRNA, we next investigated the role of MAPKBP1 in ESCs. We induced differentiation of ESCs by RA treatment and found mRNA levels of Mapkbp1 were not changed during the process (Figure 6A). This result was expected as we postulate that mRNA expression of Mapkbp1 is not affected by METTL8. However, an increase of MAPKBP1 protein level was observed after RA treatment, accompanying the emergence of phosphorylated JNK, which was not detectable in that pluripotent state (Figures 6B and S3B). Moreover, the expression levels of both MAPKBP1 and phosphorylated JNK were higher in Mettl8 KO cells than control cells (Figures 6B and S3B). These results suggest that Mettl8 KO enhances JNK signaling during differentiation. To further confirm the hypothesis, we employed another model of ESC differentiation, EB formation, and also found an enhanced JNK signal in Mettl8 KO EBs (Figure 6C). By contrast, although level of phosphorylated JNK protein was quite low in MEFs, during somatic reprogramming MAPKBP1 and phosphorylation of JNK showed a decreasing tendency and still appeared at higher levels in Mettl8 KO cells (Figure 6D). Finally we performed the JNK assay and confirmed that the enzyme activity of JNK was indeed enhanced in Mettl8-deficient cells (Figure 6E). Taken together, METTL8 inhibits JNK signaling through MAPKBP1 in mESCs.
Figure 6.

METTL8 Inhibits JNK Signaling in ESCs
(A) Total RNAs were extracted from E14 cells treated with RA for 0 or 4 days, followed by real-time PCR analysis. Data are shown as the mean ± SD from three independent experiments.
(B) Mettl8 KO and control E14 cells were treated with RA for the indicated times and cell lysates were analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(C) Mettl8 KO and control E14 cells were subjected to EB formation and 21 days later cell lysates were analyzed by western blot. The value of each band was calculated from three independent replicates and indicates the relative expression level after normalizing to the loading control actin.
(D) Mettl8 KO and control MEF cells were induced to iPSCs and collected at indicated times. Cell lysates were analyzed by western blot.
(E) Mettl8 KO and control E14 cells were treated with RA for 0 or 4 days were harvested and cell lysates were subjected to JNK assay, followed by western blot analysis.
The METTL8-MAPKBP1-JNK Pathway Suppresses ESC Differentiation
To further validate that METTL8 regulates the JNK pathway during differentiation, we next performed in vitro differentiation assays in ESCs. According to a previous study, JNK proteins are mostly enriched for regulating genes involved in nervous and cardiovascular system development (Tiwari et al., 2011). Therefore we next subjected mESCs to neural and cardiac differentiation in vitro. When Mettl8 was silenced, neuronal differentiation was enhanced, which, however, showed no significant change in Mettl8 and Mapkbp1 double KO (DKO) cells (Figures 7A and S4A). Similarly, cardiac differentiation of Mettl8 KO cells followed the same pattern in Mettl8 and Mapkbp1 DKO cells (Figures 7B and S4B). Moreover, we also examined markers of other germline layers, some of which were also upregulated in Mettl8 KO cells and rescued in Mettl8 and Mapkbp1 DKO cells (Figures S4C and S4D). These results indicate that MAPKBP1 mediates the function of METTL8 during differentiation. Meanwhile we performed the same assays in Mettl8 KO cells treated with JNK inhibitor SP600125, and the number of cells with Nestin and Gata4 expression was consistently reduced compared with those without treatment (Figures 7C, 7D, S4E, and S4F). The expression of other lineage markers was also lower in SP600125-treated Mettl8 KO cells (Figures S4G and S4H), indicating that METTL8-MAPKBP1 regulates ESC differentiation by the JNK pathway.
Figure 7.

The METTL8-JNK Pathway in ESC Differentiation
(A) Mettl8 KO, Mapkbp1 KO, Mettl8, and Mapkbp1 DKO, and control E14 cells, were subjected to neuron progenitor cells differentiation. Cells were stained for expression of Nestin and positive cells were counted by cytometer. Data are shown as the mean ± SD from three independent experiments. ∗∗∗p < 0.001. Scale bars, 100 μm.
(B) Mettl8 KO, Mapkbp1 KO, Mettl8, and Mapkbp1 DKO, and control E14 cells, were subjected to cardiac differentiation. Cells were stained for expression of Gata4 and positive cells were counted under fluorescence microscopy. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05. Scale bars, 100 μm.
(C) Mettl8 KO and control E14 cells were treated with or without SP600125 and subjected to neuron progenitor cells differentiation. Cells were stained for expression of Nestin and positive cells were counted by cytometer. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05. Scale bars, 100 μm.
(D) Mettl8 KO and control E14 cells were treated with or without SP600125 and subjected to cardiac differentiation. Cells were stained for expression of Gata4 and positive cells were counted under fluorescence microscopy. Data are shown as the mean ± SD from three independent experiments. ∗p < 0.05. Scale bars, 100 μm.
Taken together, as depicted in Figure S5A, our results unravel a pathway whereby STAT3 regulates mESC differentiation by transcriptionally activating Mettl8, which, in turn, inhibits JNK signaling through interaction with a key JNK modulator, Mapkbp1 mRNA and attenuating its translation.
Discussion
In this study, we identified Mettl8 as a transcriptional target of STAT3 that mediates STAT3’s functions in mESCs. According to previous studies, STAT3 responds to external cytokine LIF and then transcribes critical pluripotent factors in the nucleus, such as Oct4 and Nanog. Nevertheless, it remains undefined if STAT 3 is able to regulate stem cell pluripotency or differentiation by other mechanisms. We report here that METTL8 mediates a role of STAT3 during ESC differentiation. Our results indicate that STAT3, besides modulating Oct4 and Nanog expression, plays diverse roles in maintenance of ESC identity.
We found that METTL8 impedes differentiation of mESCs rather than pluripotency. Through RNA-seq and microarray analysis, it is discovered that lineage markers increased in Mettl8-deficient ESCs, while pluripotency markers showed no change. However, we think those elevated lineage genes are not a strong enough driving force for ESC differentiation. As a result, Mettl8 KO cells maintain a pluripotent state. Nevertheless, once those Mettl8-deficient cells were deprived of pluripotent conditions they would be more vulnerable to differentiate. However, different to embryo lethality caused by Stat3 deficiency, no abnormal phenotype was observed in Mettl8 KO mice. So it needs further investigation if there is redundancy between METTL family members. One of the METTL family genes, Mettl3, is also reported to bind mRNA transcripts, and inhibits mRNA stability (Geula et al., 2015).
We also used an affinity purification approach and subsequent mass spectrometry analysis to explore the interacting proteins with METTL8, and found a number of RNA binding proteins in the candidates list (Table S3). This result suggests that the binding between METTL8 and Mapkbp1 mRNA may be not direct, and that some RNA binding proteins may mediate the interaction, which needs further investigation. In addition, mRNA of Gata6, which is a lineage marker, was also proved to bind to METTL8 by subsequent experiments (data not shown). Coincidently Gata6 mRNA also showed an upregulation in Mettl8 knockdown cells in both microarray and RNA-seq analysis, which suggests that METTL8 may affect differentiation through alternative factors and mechanisms.
It is reported that METTL8 causes 3-methylcytidine (m3C) modification on human mRNAs (Xu et al., 2017). METTL8 contains a conserved methytransferase domain and is predicted to have methytransferase activity. Consistently, from the PAR-CLIP data in our study, most candidate METTL8 binding partners are mRNAs. We should further investigate if METTL8 modifies m3C on Mapkbp1 mRNA and then causes translation inhibition. In addition, other METTL family proteins also reported to modify mRNAs. For example, METTL3 and METTL14 catalyze m(6)A RNA methylation (Liu et al., 2014), which is critical for glioblastoma stem cell self-renewal (Cui et al., 2017).
MAPKBP1 enhances JNK signaling activated by mitogen-activated protein kinase kinase 1 (MEK1) and transforming growth factor β-activated kinase 1 (Koyano et al., 1999). Given that extracellular signal-regulated kinase (ERK) signaling is a major driving force for mESC differentiation and that MEK 1 also activates ERK signaling, the crosstalk between JNK and ERK signaling is worth being further studied. Moreover, studies showed that JNK signaling is required for lineage-specific differentiation but not stem cell self-renewal (Xu and Davis, 2010), which is consistent with our finding that METTL8 affects differentiation but not the pluripotent state. It is reported that JNK signaling can induce STAT3-Ser727 phosphorylation, which is important for the transactivation potency of STAT3 (Lim and Cao, 1999). Here, we also found that, when the STAT3 pathway was inhibited, JNK phosphorylation showed a higher level. However, this phenotype became subtle in Mettl8 KO cells, indicating that STAT3 has a feedback on JNK signaling in mESCs mediated by METTL8 (Figure S5B).
Taken together, our results identified a signaling axis STAT3-METTL8-MAPKBP1-JNK that is important for mESC differentiation and further broadens the landscape of regulatory networks in mESCs.
Experimental Procedures
Cell Culture and RNA Interference
mESCs were maintained in Glasgow minimum essential medium supplemented with 15% fetal bovine serum (FBS), 2 mM L-glutamine, 100 mM non-essential amino acids, 0.1 mM β-mercaptoethanol, 1 mM sodium pyruvate (Invitrogen) and 1,000 U/mL LIF (Millipore) on gelatin-coated plates. MEF and HEK293T cells were cultured in DMEM supplemented with 10% FBS (Invitrogen).
Stat3 shRNA was cloned into pLVX vector with target sequence of GATCCCCGGGCCATCCTAAGCACAAAttcaagagaTTTGTGCTTAGGATGGCCCTTTTTA and co-transfected into HEK293T cells with pVSVG and gag/pol plasmids. Twelve hours after transfection, cells were cultured with DMEM medium containing 20% FBS for an additional 24 hr. The culture medium containing lentivirus particles was filtered through a 0.45-μm filter (Millipore) and incubated with mESCs supplemented with 4 mg/mL polybrene (Sigma) for 12 hr, followed by selection with 5 mg/mL puromycin for another 24 hr.
AP Staining
AP staining was detected using the Alkaline Phosphatase Detection Kit (Millipore) according to the manufacturer's recommendations.
RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
RNA was extracted using TRIzol reagent (Invitrogen) and treated with DNaseI (Amicon). cDNA synthesis was performed with 1 μg of total RNA and the Superscript III kit (Invitrogen) according to the manufacturer's instructions. Endogenous mRNA levels were measured by real-time PCR analysis based on SYBR Green detection with an ABI real-time PCR machine. The sequence of real-time PCR primers is provided in Table S4. Samples were assayed in duplicate and normalized to endogenous actin.
Microarray Analysis
mESCs were transfected with siRNA targeted against either Mettl8 or a scrambled siRNA control. After 72 hr growth in selection media, total RNA was isolated by an RNAeasy purification kit (QIAGEN). Microarray analysis was performed using an Agilent whole-mouse gene expression microarray according to the manufacturer's instructions. Duplicate samples were used to compare transcript expression differences between Mettl8-siRNA and scrambled-siRNA ESCs.
Protein Extraction and Western Blot
Protein extracts were obtained using cell lysis buffer (20 mM HEPES, 400 nM NaCl, 0.5% NP-40, 10% glycerol, 1 mM DTT) with protease inhibitor (Roche) and phosphatase inhibitor. Proteins were separated by SDS-PAGE, transferred to polyvinylidene fluoride membrane (Bio-Rad), and probed with specific primary antibody and appropriate HRP-conjugated secondary antibody. Signals were detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce). The following antibodies were used for western blot: α-STAT3 (1:1,000, Santa Cruz, sc-482), p-STAT3Y705 (1:1,000, Cell Signaling Technology, no. 9131), METTL8 polyclonal (1:2,000, produced by our lab), METTL8 monoclonal (1:1,000, produced by our lab), α-tubulin (1:2,500, Santa Cruz, sc-53646), β-actin (1:2,500, Santa Cruz, sc-8432), SOCS3 (1:1,000, Cell Signaling, no. 2923), OCT4 (1:1,000, Cell Signaling, no. 2840), SOX2 (1:200, Santa Cruz, sc-365964), MAPKBP1 (1:200, Santa Cruz, sc-514754), and Flag (1:1,000, Sigma, F1804).
ChIP
ChIP assays with mESCs were carried out as described previously (Loh et al., 2006). In brief, cells were crosslinked with 1% formaldehyde for 10 min at room temperature and formaldehyde was neutralized by addition of 0.2 M glycine. Chromatin extracts containing DNA fragments with an average size of 500 bp were immunoprecipitated using α-pY705-STAT3 antibody. For all ChIP experiments, quantitative real-time PCR analyses were performed using SYBR-Green Master Mix.
RIP
RIP was performed as described previously (McHugh et al., 2015). In brief, 1–3 × 107 cells were lysed in hypotonic buffer supplemented with RNase A inhibitor and DNase I before centrifugation. Cell lysates were precleared with protein A/G beads (Pierce) before they were incubated with protein A/G beads coated with the indicated antibodies at 4°C for 3 hr. After extensive washing, the bead-bound immunocomplexes were eluted using elution buffer (50 mM Tris [pH 8.0], 1% SDS, and 10 mM EDTA) at 65°C for 10 min. To isolate protein-associated RNAs from the eluted immunocomplexes, samples were treated with proteinase K, and RNAs were extracted by phenol/chloroform. Purified RNAs were then subjected to real-time PCR analysis.
Biotin Pull-Down Assay
All processes were performed in the RNase-free conditions. For antisense oligomer affinity pull-down assay, sense or antisense biotin-labeled DNA oligomers corresponding to Mapkbp1 mRNA (3 mg) were incubated with lysates from 2 to 3 × 107 × 1014 cells. One hour after incubation, streptavidin-coupled Dynabeads (Invitrogen) were added to isolate the RNA-protein complex. For in vitro RNA pull-down assay, 3 mg in-vitro-synthesized biotin-labeled Mapkbp1 mRNA was incubated with purified flag or flag-METTL8 protein for 3 hr. Streptavidin-coupled Dynabeads (Invitrogen) were then added to the reaction mix to isolate the RNA-protein complex.
Polysome Analysis
E14 cells were preincubated with CHX (100 μg/mL, Sigma) for 15 min, and cytoplasmic lysates were prepared and fractionated by ultracentrifugation through 15%–50% linear sucrose gradients; 15 fractions were collected, and RNA extracted from each fraction was used for quantitative real-time PCR analysis.
CRISPR KO in E14 Cells
E14 cells were transfected with pX459 plasmids containing gRNAs for Mettl8 or Mapkbp1. Forty-eight hours later cells were treated with puromycin (5 μg/mL) for 48 hr. Then the live cells were collected, and each single cell was re-plated into 96-well plates and cultured. The knockout efficiency was confirmed by western blot.
Colony-Forming Assay
Colony-forming assay was performed as described previously (Gu et al., 2015). In brief, ESCs were trypsinized to obtain a single-cell suspension, and 600 cells were plated per 10-cm2 well. After 6 days, colonies were stained for AP activity and divided into two categories: differentiated and undifferentiated.
JNK Assay
Cell lysates were incubated with c-Jun-conjugated beads and JNK activity was analyzed using a JNK activity assay kit (New England Bio Labs) according to manufacturer's instructions.
Neuron Progenitor Cell Differentiation
Neuron progenitor cell differentiation assay was performed as described previously (Lau et al., 2006). To produce EBs, 200 × 1014 cells were cultured in hanging drops for 3 days. Ten EBs were plated on gelatin-coated dishes in knockout DMEM containing epidermal growth factor, basic fibroblast growth factor, and L-ascorbic acid. Eight to 24 hr later, the medium was replaced by proliferation medium: DMEM/F12 supplemented with 10% FBS, 2 mM L-glutamine, 20 nM progesterone, 100 mM putrescine, 25 mg/mL insulin, 50 mg/mL transferrin, and 30 nM sodium selenite.
Cardiac Differentiation
Cardiac differentiation assay was performed as described previously (Boheler, 2002). E14 cells were culture in hanging drops with differentiation media (DMEM supplemented with 10% FBS, L-glutamine, non-essential amino acids, sodium pyruvate, penicillin-streptomycin, β-mercaptoethanol, and ascorbic acid) (50 mg/mL), and diluted to 1,000 cells per 25 μL for 3 days. At day 4, EBs were transferred into gelatin-coated dishes, and the beating cardiomyocytes could be observed as early as day 8.
Induced Pluripotent Stem Cells
Animals were maintained in a certified facility, and all protocols were approved by IACUC of National University of Singapore. All procedures related to animals were performed in accordance with guidelines of AAALAC. iPSCs were induced as described previously (Carey et al., 2010). MEF cells were isolated from E13.5 embryos of Gt(ROSA)26Sortm1(rtTA∗M2)Jae Col1a1tm3(tetO-Pou5f1,-Sox2,-Klf4,-Myc)Jae/J mice. Cells were treated with doxycycline and 20 days later iPSC colonies were counted.
Author Contributions
H.G. and X.-Y.F. conceived and designed the project. H.G., D.V.D., and X.L. performed most of the experiments and analyzed the data. L.X. helped on Mettl8 KO ESC, RIP, and translational study. Y.S. performed FACS analysis. Y.W. helped with cardiomyocytes differentiation. N.S. helped to create the Mettl8 KO mice under J.Y.’s and X.L.’s guidance. Y.L. and H.Y. analyzed the RNA-seq data. G.A.T. provided technical assistance. J.M.N. and H.G. performed the sucrose gradient sedimentation. H.G., X.L., and X.F. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
This work is supported by grants from the Singapore National Medical Research Council (R-713-000-181-511) and the Ministry of Education (R-713-000-169-112) and by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative. The authors declare no competing financial interests.
Published: April 26, 2018
Footnotes
Supplemental Information includes seven figures and four tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.03.022.
Contributor Information
Hao Gu, Email: csiguh@nus.edu.sg.
Xin-Yuan Fu, Email: csifxy@nus.edu.sg.
Accession Numbers
The accession number for the RNA-seq reported in this paper is GEO: GSE112360.
Supplemental Information
References
- Akira S., Nishio Y., Inoue M., Wang X.J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Boeuf H., Hauss C., Graeve F.D., Baran N., Kedinger C. Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J. Cell Biol. 1997;138:1207–1217. doi: 10.1083/jcb.138.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boheler K.R. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ. Res. 2002;91:189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey B.W., Markoulaki S., Beard C., Hanna J., Jaenisch R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods. 2010;7:56–59. doi: 10.1038/nmeth.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.-G. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.E., Jr., Kerr I.M., Stark G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Dérijard B., Hibi M., Wu I.-H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Do D.V., Ueda J., Messerschmidt D.M., Lorthongpanich C., Zhou Y., Feng B., Guo G., Lin P.J., Hossain M.Z., Zhang W. A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev. 2013;27:1378–1390. doi: 10.1101/gad.221176.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.Y., Kessler D.S., Veals S.A., Levy D.E., Darnell J.E., Jr. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA. 1990;87:8555–8559. doi: 10.1073/pnas.87.21.8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.Y., Schindler C., Improta T., Aebersold R., Darnell J.E., Jr. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl. Acad. Sci. USA. 1992;89:7840–7843. doi: 10.1073/pnas.89.16.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gu H., Li Q., Huang S., Lu W., Cheng F., Gao P., Wang C., Miao L., Mei Y., Wu M. Mitochondrial E3 ligase March5 maintains stemness of mouse ESCs via suppression of ERK signalling. Nat. Commun. 2015;6:7112. doi: 10.1038/ncomms8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr., Jungkamp A.C., Munschauer M. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M., Lin A., Smeal T., Minden A., Karin M. Identification of an oncoprotein-and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Jaeschke A., Karasarides M., Ventura J.J., Ehrhardt A., Zhang C., Flavell R.A., Shokat K.M., Davis R.J. JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Koyano S., Ito M., Takamatsu N., Shiba T., Yamamoto K.-i., Yoshioka K. A novel Jun N-terminal kinase (JNK)-binding protein that enhances the activation of JNK by MEK kinase 1 and TGF-β-activated kinase 1. FEBS Lett. 1999;457:385–388. doi: 10.1016/s0014-5793(99)01084-4. [DOI] [PubMed] [Google Scholar]
- Lau T., Adam S., Schloss P. Rapid and efficient differentiation of dopaminergic neurons from mouse embryonic stem cells. Neuroreport. 2006;17:975–979. doi: 10.1097/01.wnr.0000223395.66951.01. [DOI] [PubMed] [Google Scholar]
- Lecat A., Di Valentin E., Somja J., Jourdan S., Fillet M., Kufer T.A., Habraken Y., Sadzot C., Louis E., Delvenne P. The c-Jun N-terminal kinase (JNK)-binding protein (JNKBP1) acts as a negative regulator of NOD2 protein signaling by inhibiting its oligomerization process. J. Biol. Chem. 2012;287:29213–29226. doi: 10.1074/jbc.M112.355545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C.P., Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J. Biol. Chem. 1999;274:31055–31061. doi: 10.1074/jbc.274.43.31055. [DOI] [PubMed] [Google Scholar]
- Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- McHugh C.A., Chen C.-K., Chow A., Surka C.F., Tran C., McDonel P., Pandya-Jones A., Blanco M., Burghard C., Moradian A. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236. doi: 10.1038/nature14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H., Burdon T., Chambers I., Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz R., Lee C.K., Cannizzaro L.A., d'Eustachio P., Levy D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA. 1999;96:2846–2851. doi: 10.1073/pnas.96.6.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., Hochedlinger K., Nam S.Y., Bauer A., Karin M., Wagner E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Schindler C., Shuai K., Prezioso V.R., Darnell J.E., Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992;257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- Schust J., Sperl B., Hollis A., Mayer T.U., Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Smith A.G., Heath J.K., Donaldson D.D., Wong G.G., Moreau J., Stahl M., Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988;336:688–690. doi: 10.1038/336688a0. [DOI] [PubMed] [Google Scholar]
- Song H., Wang R., Wang S., Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Noguchi K., Shi W., Tanaka T., Matsumoto M., Yoshida N., Kishimoto T., Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V.K., Stadler M.B., Wirbelauer C., Paro R., Schubeler D., Beisel C. A chromatin-modifying function of JNK during stem cell differentiation. Nat. Genet. 2011;44:94–100. doi: 10.1038/ng.1036. [DOI] [PubMed] [Google Scholar]
- Weston C.R., Davis R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Williams R.L., Hilton D.J., Pease S., Willson T.A., Stewart C.L., Gearing D.P., Wagner E.F., Metcalf D., Nicola N.A., Gough N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Williams R.L., Hiltonl D.J., Nicolai N.A. Potential of embryonic stem cells. Nature. 1988;336:15. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Xu L., Liu X., Sheng N., Oo K.S., Liang J., Chionh Y.H., Xu J., Ye F., Gao Y.-G., Dedon P.C. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J. Biol. Chem. 2017;292:14695–14703. doi: 10.1074/jbc.M117.798298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Davis R.J. c-Jun NH2-terminal kinase is required for lineage-specific differentiation but not stem cell self-renewal. Mol. Cell Biol. 2010;30:1329–1340. doi: 10.1128/MCB.00795-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q.L., Nichols J., Chambers I., Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- Zhong Z., Wen Z., Darnell J.E., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.