

 Catalytic chemo- and enantioselective generation of tertiary benzylic copper complexes from Cu–B(pin) (pin = pinacolato) additions to 1,1-disubstituted alkenes followed by in situ reactions with ketones and carboxylic acid phenol esters to construct multifunctional alkylboron compounds that contain quaternary stereogenic centers is presented.
Catalytic chemo- and enantioselective generation of tertiary benzylic copper complexes from Cu–B(pin) (pin = pinacolato) additions to 1,1-disubstituted alkenes followed by in situ reactions with ketones and carboxylic acid phenol esters to construct multifunctional alkylboron compounds that contain quaternary stereogenic centers is presented.
Abstract
Catalytic chemo- and enantioselective generation of tertiary benzylic copper complexes from Cu–B(pin) (pin = pinacolato) additions to 1,1-disubstituted alkenes followed by in situ reactions with ketones and carboxylic acid phenol esters to construct multifunctional alkylboron compounds that contain quaternary stereogenic centers is presented. The method is distinguished by the unprecedented reaction mode of tertiary benzylic Cu complexes, allowing reaction with a wide range of carbonyl electrophiles in good yields and with high chemo-, site-, diastereo- and enantioselectivity. The catalytic protocol was performed with easily accessible chiral ligands and copper salts at ambient temperature. Functionalization of multifunctional alkylboron products provides useful building blocks that are otherwise difficult to access.
Introduction
Enantioselective construction of all-carbon quaternary stereogenic centers remains both an important and challenging area in organic synthesis.1 Conversion of the carbon–metal bond of tertiary alkyl–metal complexes to the carbon–carbon bond provides a direct approach. However, it is non-trivial to prepare enantiomerically enriched alkyl–metal nucleophiles (e.g. Grignard, organolithium and organoaluminum reagents), which are generally air- and moisture-sensitive reagents that have limited functional group tolerance.2 Moreover, alkyl–metal complexes are not configurationally stable and racemization of the carbon–metal bond might even occur under cryogenic conditions.3,4 Catalytic enantioselective generation of chiral alkyl–metal complexes from easily accessible alkenes constitutes a unique approach to access such types of alkyl nucleophiles. Particularly, impressive developments of the synthesis of enantioenriched secondary alkyl–Cu complexes bearing a C–Cu bond at the stereogenic center through Cu–B(pin) or Cu–H additions to alkenes and the in situ transformation of their stereogenic C–Cu bond to C–C bonds have been reported,5,6 although significant limitations and challenges remain. For example, alkyl–Cu complexes that can be accessed are limited to secondary alkyl nucleophiles, probably due to the larger steric hindrance of tert-alkyl–Cu complexes that might result in lower reactivity and more significant racemization. In addition, enantioselective coupling of 1,1-disubstituted alkenes, B2(pin)2, and ketones or carboxylic acid derivatives is unprecedented.7
In 2014, Buchwald and co-workers reported a method of cyanation of vinylarene through a benzylic Cu intermediate generated from Cu–B(pin) addition to terminal alkenes (Scheme 1a).8a In 2015, Montgomery and co-workers disclosed an NHC–Cu catalyzed approach (Scheme 1a).8b In both protocols, the secondary benzylic Cu complex underwent dearomative addition to the electrophilic cyanation reagent through a six-membered transition state followed by rearomatization, resulting in functionalization at the aryl ring.8c However, there is no report on the reaction mode of tertiary benzylic Cu complexes. Herein we described unprecedented reactions of enantioenriched tertiary benzylic Cu complexes generated from Cu–B(pin) addition to 1,1-disubstituted aryl alkenes with carbonyl electrophiles at the benzylic position, providing a wide range of multifunctional alkylboron building blocks.
Scheme 1. Reaction design and proof of concept.

The challenges for such transformations include: (1) the nucleophilic Cu–B(pin) complex has to react with 1,1-disubstituted alkenes chemo- and regioselectively; (2) the chiral Cu complex has to induce high enantioselectivity of Cu–B(pin) addition even at ambient temperature, as 1,1-disubstituted alkenes represent one of the most challenging classes of substrates for enantioselective catalysis.9 Moreover, the chiral catalyst has to control the diastereoselectivity of addition of the tertiary benzylic Cu intermediate to ketone; (3) the low reactivity of the sterically hindered tertiary benzylic Cu complex has to be enhanced by the ligand to enhance the rate of carbonyl addition and reduce racemization of the C–Cu bond. We hypothesized that a Cu complex derived from an electron-rich ligand might overcome these challenges.
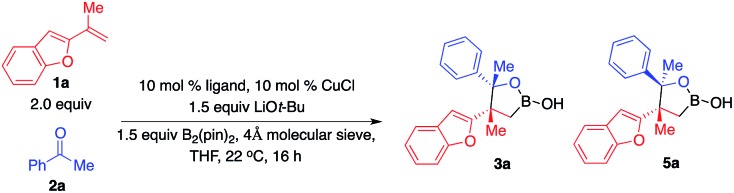
As shown in Scheme 1b, we envisioned that the nucleophilic Cu–B(pin) intermediate in situ generated from ligand–Cu–Ot-Bu and B2(pin)2 reacts with 1,1-disubstituted aryl alkene 1a to afford benzyl–Cu complex I, which might undergo addition with carbonyl compounds to provide alkylboron 3a or 4a with high stereoselectivity.
Results and discussion
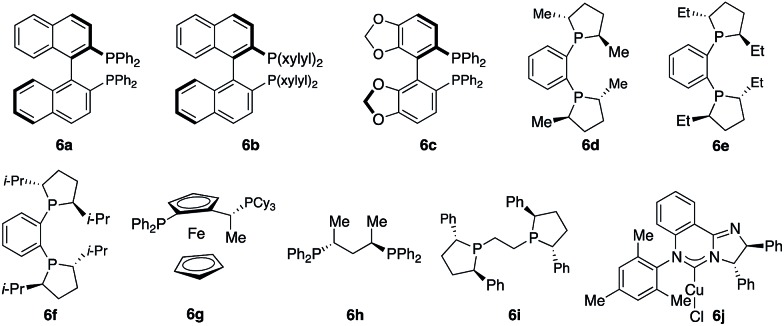
We started exploring the concept by evaluating the reaction of aryl alkene 1a with acetophenone 2a in the presence of the Cy3P–Cu complex. Desired alkylboron products 3a and 5a were afforded in 85% yield and 59 : 41 diastereomeric ratio (d.r.) (Scheme 1c). Intramolecular chelation of the free hydroxyl group to the B(pin) resulted in isolation of the cyclic boronic acid products after work-up with silica gel. In contrast to Buchwald and Montgomery's studies, the addition occurred exclusively at the benzylic site rather than the aryl ring, indicating a completely different reaction mechanism of such transformations. We next investigated a variety of chiral phosphine ligands (Table 1). Reactions of 1a with acetophenone 2a promoted by Cu complexes derived from aryl phosphines (6a–c) delivered a mixture of diastereomers in a 50 : 50–58 : 42 ratio (entries 1–3). The enantiomeric ratio (e.r.) of each diastereomer was not high. Cu complexes generated from phosphines that contain stereogenic centers at phosphorus (6d–f) provided higher d.r. without improvement of e.r. (entries 4–6). Transformation promoted by the phosphine–Cu complex bearing a ferrocene skeleton afforded alkylboron products in a 57 : 43 d.r., 30 : 70 e.r. for 3a and 80 : 20 e.r. for 5a (entry 7). The phosphine–Cu complex that has been previously reported to be able to induce high enantioselectivity in Cu–B(pin) addition to 1,1-disubstituted aryl alkenes gave 75 : 25 d.r. and 92 : 8 e.r. for the major diastereomer 3a (entry 8).9b Further investigation revealed that the reaction catalyzed by the phosphine–Cu complex formed from Ph–BPE (6i) delivered 3a in a 93 : 7 d.r. and 92% yield and >99 : 1 e.r. for 3a (entry 9), indicating that a single catalyst can control the stereoselectivity for Cu–B(pin) addition and ketone addition. Cu complex 6j derived from N-heterocyclic carbene did not provide high stereoselectivity.
Table 1. Ligand screen a .
| ||||||
| Entry | Ligand | Yield of 3a b (%) | Yield of 5a b (%) | d.r. c | e.r. of 3a d | e.r. of 5a d |
| 1 | 6a | 31 | 31 | 50 : 50 | 83 : 17 | 58 : 42 |
| 2 | 6b | 52 | 37 | 58 : 42 | 64 : 36 | 57 : 43 |
| 3 | 6c | 52 | 41 | 56 : 44 | 92 : 8 | 88 : 12 |
| 4 | 6d | 55 | 22 | 71 : 29 | 36 : 64 | 69 : 31 |
| 5 | 6e | 71 | 24 | 75 : 25 | 30 : 70 | 83 : 17 |
| 6 | 6f | 72 | 27 | 73 : 27 | 64 : 36 | 70 : 30 |
| 7 | 6g | 57 | 43 | 57 : 43 | 30 : 70 | 80 : 20 |
| 8 | 6h | 67 | 22 | 75 : 25 | 92 : 8 | 56 : 44 |
| 9 | 6i | 92 | 7 | 93 : 7 | >99 : 1 | nd e |
| 10 | 6j | 51 | 34 | 60 : 40 | 96 : 4 | 65 : 35 |
| ||||||
aReactions were performed under a N2 atmosphere, see the ESI details.
bYields of purified products.
cd.r. was determined by 400 MHz 1HNMR analysis of unpurified mixtures.
de.r was determined by HPLC analysis of the corresponding diol after oxidation of the organoboron product with NaBO3·4H2O.
eNot determined.
With the optimal conditions in hand, we set out to explore the substrate scope of ketones. As shown in Scheme 2, a wide range of ketones are well tolerated. Reactions of aryl methyl ketones that contain electron-withdrawing groups afforded alkylboron compounds in 56–79% yield, 82 : 18–92 : 8 d.r. and >99 : 1 e.r. (3b–g). Ketones bearing halogen units and carbonyl groups are good substrates (3c–g). Transformations of ketones containing electron-donating groups delivered the desired products in 84–89% yield, 86 : 14–92 : 8 d.r. and >99 : 1 e.r. (3h–k). Ketones bearing heterocycles can be tolerated (3l–n), although 2-acetyl furan gave lower diastereoselectivity (3l). The reaction of ketones with alkyl groups larger than methyl provided 3p in 82% yield, 84 : 16 d.r. and >99 : 1 e.r. Less electrophilic dialkyl ketones were transformed with high efficiency and stereoselectivity (3q–r). Competitive enolization of dialkyl ketones was not observed. Transformation of α,β-unsaturated ketones delivered 3s in 65% yield, 83 : 17 d.r. and >99 : 1 e.r. without competitive boron 1,4-conjugate addition.
Scheme 2. Scope of ketones. aThe same conditions as in Table 1; yield refers to the yield of the major diastereomer, see the ESI† for details. bCombined yield of two diastereomers.

The scope of 1,1-disubstituted aryl alkenes was investigated. As indicated in Scheme 3, alkenes containing an alkyl substituent other than methyl were converted in 96% yield, 95 : 5 d.r. and >99 : 1 e.r. (7a). Reactions of alkenes substituted with furan, benzofuran, thiophene and benzothiophene afforded alkylboron products in 72–98% yield, 75 : 25–93 : 7 d.r. and >99 : 1 e.r. (7b–g). Alkenes bearing indole (7h–i), dibenzofuran (7j), and carbazole (7k–l) moieties that commonly exist in pharmaceutically important molecules were transformed in high yield and enantioselectivity, albeit lower diastereoselectivity. Transformations of alkenes that contain naphthalene substituted with a range of functional groups generated desired products in 74–99% yield, 86 : 14–91 : 9 d.r. and 98:2–>99 : 1 e.r. (7m–r). The limitation of this method is that no reaction occurred with alkenes containing a simple phenyl ring.
Scheme 3. Scope of 1,1-disubstituted aryl alkenes for ketone addition.a aThe same conditions as in Table 1; yield refers to the combined yield of two diastereomers, see the ESI† for details. bOne equivalent of alkene and two equivalents of ketone were used. cYield of the major diastereomer after oxidation with NaBO3·4H2O. dYield of the major diastereomer. e6h was used as the ligand.

To further expand the scope of electrophiles for reactions with tertiary benzyl–Cu complexes, we tested other types of carbonyl compounds. Acylation of an enantioenriched organometallic reagent that contains a carbon–metal bond represents a direct approach to access ketones with a quaternary stereogenic center. Although pioneering studies for catalytic enantioselective allylic alkylation of acyl nucleophiles and ketone enolates to generate tertiary alkyl aryl ketones have been disclosed,10–12 direct catalytic enantioselective nucleophilic addition of an enantioenriched tertiary alkyl–Cu complex to carboxylic acid derivatives remains unprecedented.13
We began our studies by treatment of the enantioenriched tertiary benzyl–Cu intermediate in situ generated from Cu–B(pin) addition to 1,1-disubstituted aryl alkene 1a promoted by the Cu complex derived from 6i with a variety of easily accessible benzoic acid p-Cl-phenol esters, delivering a range of tertiary alkyl aryl ketones in high yields and enantioselectivity (Scheme 4). Esters bearing electron-withdrawing groups (4b–d, 4f), halogens (4c–e), carboxylic ester (4f), and electron-donating groups (4g–h, 4i–j) and heterocycles (4k–n) are well tolerated. It is worth mentioning that reactions of esters with electron-rich aryl groups resulted in lower enantioselectivity (cf.4g, 4k–l), illustrating that the tertiary benzyl–Cu intermediate is not configurationally stable. If the rate of the acylation was not fast enough, competitive racemization of the organocopper complex occurred. Alkyl carboxylic acid derivatives cannot provide desired products due to competitive enolization, indicating that the reaction of ketones that overcame competitive enolization might proceed faster than phenol esters.
Scheme 4. Scope of carboxylic acid derivatives for multicomponent acylation.

We next explored the scope of 1,1-disubstituted aryl alkenes (Scheme 5). However, the optimal phosphine–Cu complex for 1a proved to be not effective for alkenes substituted with other types of aryl groups. Reinvestigation of chiral ligands revealed that NHC–Cu complex 6j promoted the transformations with improved efficiency and enantioselectivity.14 The reaction of alkenes that contain benzothiophene and thiophene in the presence of 6j afforded alkylboron compounds (9a–b) in 62–75% yield and 86 : 14–89 : 11 e.r. Alkenes bearing substituted furan moieties were transformed in 66–67% yield and 87 : 13–98 : 2 e.r. (9c–d). Naphthyl alkenes that contain a variety of functional groups are well tolerated (9f–k).
Scheme 5. Scope of 1,1-disubstituted alkenes for multicomponent acylation. a10 mol% 6i and 10 mol% CuCl were used.

The multicomponent reactions can be easily performed on a gram scale with easily accessible starting materials and readily available catalysts. As shown in Scheme 6a, the reaction of 1b (2.52 g) with 2a (1.20 g) in the presence of the 3.0 mol% phosphine–Cu complex derived from 6i afforded 7m (2.64 g) in 85% yield, 91 : 9 d.r. and 99 : 1 e.r. Transformation of 1b (1.34 g) with 8a (2.78 g) promoted by NHC–Cu complex 6j delivered 9f (1.95 g) in 67% yield and 98 : 2 e.r. The cyclic boronic acid moiety exists in a range of biologically active molecules.15 Further studies on the biological activity of such molecules will be conducted. Moreover, the C–B bond can be transformed into other bonds. Conversion of the sterically hindered C–B bond to the C–C bond was not trivial. Screening of reaction conditions revealed that treatment of boronic acid 7m and 4-bromoanisole with the RuPhos–Pd complex and Cs2CO3 provided alcohol 10 in 64% yield as a single enantiomer (Scheme 6b). Conversion of alkyl–B(pin) 9f to potassium trifluoroborate followed by introduction of the RuPhos–Pd complex, K2CO3 and 4-bromoanisole afforded ketone 11 in 86% yield.16 Chromane derivatives are important motifs in pharmaceutically and biologically active molecules. Oxidation of cyclic boronic acid 3t with NaBO3·4H2O generated diol 12 in 92% yield. Chemoselective intramolecular coupling of the primary C–O bond with the C–Br bond led to the formation of chromane derivative 14 that contains two quaternary stereogenic centers in 98% yield and >99 : 1 e.r., providing a new approach for such motifs.17 Oxidation of 3a followed by direct reduction with Me4NBH(OAc)3 afforded diol 15 in a 4 : 1 d.r., 66% yield of the major diastereomer and 94 : 6 e.r.18
Scheme 6. Gram scale reactions and functionalization.

The reaction mode of tertiary benzyl–Cu complexes deserves further discussion. It has been recently reported that the C–Cu bond of an enantioenriched secondary benzyl–Cu complex can undergo facile racemization, when the aryl group of the benzyl–Cu complex is sterically hindered or contains an electron-withdrawing substituent, leading to lower reactivity of the benzyl–Cu complex.19 Therefore, it is unusual that reactions of even more sterically congested tertiary benzyl–Cu provided high enantioselectivity. Considering the previous dearomative allylation mechanism of cyanation of styrene, we hypothesized that it might be possible that dearomative isomerization of tertiary benzyl–Cu complex I to reduce steric repulsion between the congested alkyl group and ligand occurred to generate a new allyl–Cu species IV. Coordination of the carbonyl electrophile followed by addition through a six-membered transition state delivered two continuous quaternary stereogenic centers. Facile intramolecular C–C bond formation might account for minimal racemization. The next question is whether isomerization of I altered the configuration of the stereogenic center. Quenching I with MeOH afforded hydroboration product 16 in 98% yield and 98 : 2 e.r.9c The stereochemistry of 16 is the same as the benzylic stereogenic center of 3a. So the dearomative isomer is suprafacial. The chirality of tertiary benzyl–Cu complexes could be efficiently transferred in the isomerization process. Moreover, the reason why alkenes with simple phenyl groups didn't work might be that the barrier for such dearomative isomerization is too high (Scheme 7).
Scheme 7. Proposed catalytic cycle.

To further expand this unique reaction pathway of tertiary benzyl–Cu complexes, we demonstrated that a range of electrophiles could be employed in this mode of reaction in a highly enantioselective fashion. In our preliminary studies, as shown in Scheme 8, the reaction of 1,1-disubstituted alkene 1a with electrophilic cyanation reagent 17 promoted by the phosphine–Cu complex derived from 6i afforded 18 in 45% yield and 98 : 2 e.r. associated with 11% yield of aryl cyanation product 19, supporting our hypothesis that tertiary benzyl–Cu complexes might undergo facile dearomative isomerization followed by reaction with electrophiles through a six-membered transition state. In contrast to previous studies, the dearomative isomerization process of the tertiary benzyl–Cu intermediate (I to IV) was of lower energy compared with direct addition of Cu complex I. Furthermore, aldehyde 20 was transformed to provide multifunctional alkylboron 21a in 38% yield, 94 : 6 d.r. and 97 : 3 e.r. for the major diastereomer in the presence of the Cu complex generated from 6i. The same catalyst also promoted the reaction of aldimine 22 to afford 23a in 59% yield of both diastereomers, 62 : 38 d.r. and 97.5 : 2.5 e.r. for both diastereomers. In all cases, reactions occurred at the benzylic site preferentially.
Scheme 8. Expanding scope of electrophiles for reactions with tertiary benzyl–Cu complexes.

Conclusions
In conclusion, we have disclosed an unprecedented reaction mode of tertiary benzyl–Cu complexes in situ generated from enantioselective Cu–B(pin) addition to 1,1-disubstituted aryl alkenes. A wide range of electrophiles that can react through a six-membered transition state were successfully applied to such transformations with high efficiency and stereoselectivity, delivering multifunctional alkylboron compounds that are otherwise difficult to access. Our strategy represents a unique departure from the traditional reactivity of secondary benzyl–Cu complexes, delivering a powerful tool for the enantioselective addition of olefin-derived nucleophiles to carbonyl electrophiles that will be of broad synthetic utility. Further mechanistic studies and expanding scope of alkene nucleophiles are underway.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This work was financially supported by the State Key Laboratory of Organometallic Chemistry, the Shanghai Institute of Organic Chemistry, the “Hundred Talents Plan” of Chinese Academy of Sciences, the “Thousand Youth Talents Plan”, the Shanghai Sailing Program (Grant No. 17YF1424100), the National Natural Science Foundation of China (Grant No. 21702222), The Science and Technology Commission of Shanghai Municipality (Grant No. 17JC1401200) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000).
Footnotes
†Electronic supplementary information (ESI) available. CCDC 1579294 and 1579300. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00827b
References
- For recent reviews, see ; (a) Corey E. J., Guzman-Perez A. Angew. Chem., Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Christoffers J., Mann A. Angew. Chem., Int. Ed. 2001;40:4591. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (c) Denissova I., Barriault L. Tetrahedron. 2003;59:10105. [Google Scholar]; (d) Trost B. M., Jiang C. Synthesis. 2006;3:369. [Google Scholar]; (e) Das J. P., Marek I. Chem. Commun. 2011;47:4593. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; (f) Quasdorf K. W., Overman L. E. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of synthesis and reaction of chiral alkyl lithium reagents, see: ; (a) Hoppe D., Hense T. Angew. Chem., Int. Ed. Engl. 1997;36:2282. [Google Scholar]; (b) Beak P., Basu A., Gallagher D. J., Park Y. S., Thayumanavan S. Acc. Chem. Res. 1996;29:552. [Google Scholar]; (c) Basu A., Thayumanavan S. Angew. Chem., Int. Ed. 2002;41:716. doi: 10.1002/1521-3773(20020301)41:5<716::aid-anie716>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- For a recent review on the configurational stability of enantioenriched alkyl nucleophiles, see: ; for examples of configurational stability of Grignard reagents, see: ; (a) Swift E. C., Jarvo E. R. Tetrahedron. 2013;69:5799. doi: 10.1016/j.tet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whitesides G. M., Witanowski M., Roberts J. D. J. Am. Chem. Soc. 1965;87:2854. [Google Scholar]; (c) Whitesides G. M., Roberts J. D. J. Am. Chem. Soc. 1965;87:4878. [Google Scholar]; (d) Hoffmann R. W. Chem. Soc. Rev. 2003;32:225. doi: 10.1039/b300840c. [DOI] [PubMed] [Google Scholar]
- For examples of enantiospecific reactions of nonracemic alkylcuprates, see: ; (a) Dieter R. K., Topping C. M., Chandupatla K. R., Lu K. J. Am. Chem. Soc. 2001;123:5132. doi: 10.1021/ja0156587. [DOI] [PubMed] [Google Scholar]; (b) Dieter R. K., Oba G., Chandupatla K. R., Topping C. M., Lu K., Watson R. T. J. Org. Chem. 2004;69:3076. doi: 10.1021/jo035845i. [DOI] [PubMed] [Google Scholar]
- For recent examples of C–C bond forming reactions of chiral alkyl–Cu complexes in situ generated from Cu–B(pin) addition to alkenes, see: ; (a) Ito H., Kosaka Y., Nonoyama K., Sasaki Y., Sawamura M. Angew. Chem., Int. Ed. 2008;47:7424. doi: 10.1002/anie.200802342. [DOI] [PubMed] [Google Scholar]; (b) Yamamoto E., Kojima R., Kubota K., Ito H. Synlett. 2016;27:272. [Google Scholar]; (c) Jia T., Cao P., Wang B., Lou Y., Yin X., Wang M., Liao J. J. Am. Chem. Soc. 2015;137:13760. doi: 10.1021/jacs.5b09146. [DOI] [PubMed] [Google Scholar]; (d) Chen B., Cao P., Yin X., Liao Y., Jiang L., Ye J., Wang M., Liao J. ACS Catal. 2017;7:2425. [Google Scholar]; (e) Logan K. M., Brown M. K. Angew. Chem., Int. Ed. 2017;56:851. doi: 10.1002/anie.201609844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Green J. C., Joannou M. V., Murray S. A., Zanghi J. M., Meek S. J. ACS Catal. 2017;7:4441. doi: 10.1021/acscatal.7b01123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent examples of C–C bond forming reactions of chiral alkyl–Cu complexes in situ generated from Cu–H addition to alkenes, see: ; (a) Ascic E., Buchwald S. L. J. Am. Chem. Soc. 2015;137:4666. doi: 10.1021/jacs.5b02316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y.-M., Bruno N. C., Placeres Á. L., Zhu S., Buchwald S. L. J. Am. Chem. Soc. 2015;137:10524. doi: 10.1021/jacs.5b07061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang Y.-M., Buchwald S. L. J. Am. Chem. Soc. 2016;138:5024. doi: 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bandar J. S., Ascic E., Buchwald S. L. J. Am. Chem. Soc. 2016;138:5821. doi: 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Friis S. D., Pirnot M. T., Buchwald S. L. J. Am. Chem. Soc. 2016;138:8372. doi: 10.1021/jacs.6b04566. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yang Y., Perry I. B., Buchwald S. L. J. Am. Chem. Soc. 2016;138:9787. doi: 10.1021/jacs.6b06299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhou Y., Bandar J. S., Buchwald S. L. J. Am. Chem. Soc. 2017;139:8126. doi: 10.1021/jacs.7b04937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yang Y., Perry I. B., Lu G., Liu P., Buchwald S. L. Science. 2016;353:144. doi: 10.1126/science.aaf7720. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Lee J., Torker S., Hoveyda A. H. Angew. Chem., Int. Ed. 2017;56:821. doi: 10.1002/anie.201611444. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Han J. T., Jang W. J., Kim N., Yun J. J. Am. Chem. Soc. 2016;138:15146. doi: 10.1021/jacs.6b11229. [DOI] [PubMed] [Google Scholar]
- . For coupling of 1,1-disubstituted allene, imine and B2(pin)2, see: ; (a) Fujihara T., Sawada A., Yamaguchi T., Tani Y., Terao J., Tsuji Y. Angew. Chem., Int. Ed. 2017;56:1539. doi: 10.1002/anie.201611314. [DOI] [PubMed] [Google Scholar]; (b) Boreux A., Indukuri K., Gagosz F., Riant O. ACS Catal. 2017;7:8200. [Google Scholar]; (c) Butcher T. W., McClain E. J., Hamilton T. G., Perrone T. M., Kroner K. M., Donohoe G. C., Akhmedov N. G., Petersen J. L., Popp B. V. Org. Lett. 2016;18:6428. doi: 10.1021/acs.orglett.6b03326. [DOI] [PubMed] [Google Scholar]; (d) Yeung K., Ruscoe R. E., Rae J., Pulis A. P., Procter D. J. Angew. Chem., Int. Ed. 2016;55:11912. doi: 10.1002/anie.201606710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Yang Y., Buchwald S. L. Angew. Chem., Int. Ed. 2014;53:8677. doi: 10.1002/anie.201402449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao W., Montgomery J. Angew. Chem., Int. Ed. 2015;54:12683. doi: 10.1002/anie.201507303. [DOI] [PubMed] [Google Scholar]; (c) Yang Y., Liu P. ACS Catal. 2015;5:2944. [Google Scholar]
- For Cu-catalyzed enantioselective hydroboration of 1,1-disubstituted alkenes, see: ; (a) Corberá R., Mszar N. W., Hoveyda A. H. Angew. Chem., Int. Ed. 2011;50:7079. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]; (b) Wang Z., He X., Zhang R., Zhang G., Xu G., Zhang Q., Xiong T., Zhang Q. Org. Lett. 2017;19:3067. doi: 10.1021/acs.orglett.7b01135. [DOI] [PubMed] [Google Scholar]; (c) Wen L., Cheng F., Li H., Zhang S., Hong X., Meng F. Asian J. Org. Chem. 2018;7:103. [Google Scholar]; (d) Jang W. J., Song S. M., Moon J. H., Lee J. Y., Yun J. J. Am. Chem. Soc. 2017;139:13660. doi: 10.1021/jacs.7b08379. [DOI] [PubMed] [Google Scholar]
- For catalytic enantioselective allylic alkylation of acyl nucleophiles, see: . For catalytic stereospecific allylic substitution with acyl nucleophiles, see: ; (a) Breitler S., Carreira E. M. J. Am. Chem. Soc. 2015;137:5296. doi: 10.1021/jacs.5b01951. [DOI] [PubMed] [Google Scholar]; (b) Hojoh K., Ohmiya H., Sawamura M. J. Am. Chem. Soc. 2017;139:2184. doi: 10.1021/jacs.6b12881. [DOI] [PubMed] [Google Scholar]; (c) Evans P. A., Oliver S., Chae J. J. Am. Chem. Soc. 2012;134:19314. doi: 10.1021/ja306602g. [DOI] [PubMed] [Google Scholar]; (d) Evans P. A., Oliver S. Org. Lett. 2013;15:5626. doi: 10.1021/ol402336u. [DOI] [PubMed] [Google Scholar]; (e) Turnbull B. W. H., Oliver S., Evans P. A. J. Am. Chem. Soc. 2015;137:15374. doi: 10.1021/jacs.5b10270. [DOI] [PubMed] [Google Scholar]
- For catalytic enantioselective allylation of ketone enolates, see: ; (a) Behenna D. C., Stoltz B. M. J. Am. Chem. Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Mohr J. T., Behenna D. C., Harned A. M., Stoltz B. M. Angew. Chem., Int. Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; (c) Mohr J. T., Stoltz B. M. Chem.–Asian J. 2007;2:1476. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost B. M., Xu J., Schmidt T. J. Am. Chem. Soc. 2009;131:18343. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reeves C. M., Eidamshaus C., Kim J., Stoltz B. M. Angew. Chem., Int. Ed. 2013;52:6718. doi: 10.1002/anie.201301815. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vita M. V., Mieville P., Waser J. Org. Lett. 2014;16:5768. doi: 10.1021/ol5028333. [DOI] [PubMed] [Google Scholar]; (g) Craig R. A., Loskot S. A., Mohr J. T., Behenna D. C., Harned A. M., Stoltz B. M. Org. Lett. 2015;17:5160. doi: 10.1021/acs.orglett.5b02376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Akula R., Guiry P. J. Org. Lett. 2016;18:5472. doi: 10.1021/acs.orglett.6b02584. [DOI] [PubMed] [Google Scholar]; (i) de Oliveira M. N., Fournier J., Arseniyadis S., Cossy J. Org. Lett. 2017;19:14. doi: 10.1021/acs.orglett.6b02971. [DOI] [PubMed] [Google Scholar]; (j) James J., Guiry P. J. ACS Catal. 2017;7:1397. [Google Scholar]; (k) Chen W., Chen W., Hartwig J. F. J. Am. Chem. Soc. 2014;136:15825. doi: 10.1021/ja506500u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Liu W.-B., Reeves C. M., Virgil S. C., Stoltz B. M. J. Am. Chem. Soc. 2013;135:10626. doi: 10.1021/ja4052075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Liu W.-B., Reeves C. M., Stoltz B. M. J. Am. Chem. Soc. 2013;135:17298. doi: 10.1021/ja4097829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of catalytic enantioselective α-arylation of enolate, see: ; (a) Åhman J., Wolfe J. P., Troutman M. V., Palucki M., Buchwald S. L. J. Am. Chem. Soc. 1998;120:1918. [Google Scholar]; (b) Hamada T., Chieffi A., Åhman J., Buchwald S. L. J. Am. Chem. Soc. 2002;124:1261. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]; (c) Spielvogel D. J., Buchwald S. L. J. Am. Chem. Soc. 2002;124:3500. doi: 10.1021/ja017545t. [DOI] [PubMed] [Google Scholar]; (d) Liu X., Hartwig J. F. J. Am. Chem. Soc. 2004;126:5182. doi: 10.1021/ja031544e. [DOI] [PubMed] [Google Scholar]; (e) Chen G., Kwong F. Y., Chan H. O., Yu W.-Y., Chan A. S. C. Chem. Commun. 2006:1413. doi: 10.1039/b601691j. [DOI] [PubMed] [Google Scholar]; (f) Liao X., Weng Z., Hartwig J. F. J. Am. Chem. Soc. 2008;130:195. doi: 10.1021/ja074453g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Taylor A. M., Altman R. A., Buchwald S. L. J. Am. Chem. Soc. 2009;131:9900. doi: 10.1021/ja903880q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Ge S., Hartwig J. F. J. Am. Chem. Soc. 2011;133:16330. doi: 10.1021/ja2082087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Guo J., Dong S., Zhang Y., Kuang Y., Liu X., Lin L., Feng X. Angew. Chem., Int. Ed. 2013;52:10245. doi: 10.1002/anie.201303602. [DOI] [PubMed] [Google Scholar]; (j) Ghosh A., Walker J. A., Ellern A., Stanley L. M. ACS Catal. 2016;6:2673. [Google Scholar]; (k) Huang Z., Liu Z., Zhou J. J. Am. Chem. Soc. 2011;133:15882. doi: 10.1021/ja2066829. [DOI] [PubMed] [Google Scholar]; (l) Huang Z., Chen Z., Lim L. H., Quang G. C. P., Hirao H., Zhou J. Angew. Chem., Int. Ed. 2013;52:5807. doi: 10.1002/anie.201300481. [DOI] [PubMed] [Google Scholar]; (m) Huang Z., Lim L. H., Chen Z., Li Y., Zhou F., Su H., Zhou J. Angew. Chem., Int. Ed. 2013;52:4906. doi: 10.1002/anie.201300621. [DOI] [PubMed] [Google Scholar]; (n) Yang J., Zhou J. Org. Chem. Front. 2014;1:365. [Google Scholar]
- For acylation of secondary alkyl–Cu complexes, see: ; for acylation of silyl–Cu complexes, see: ; (a) Huang Y., Smith K. B., Brown K. M. Angew. Chem., Int. Ed. 2017;56:13314. doi: 10.1002/anie.201707323. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cirriez V., Rasson C., Riant O., Adv. Synth. Catal., 2013, 355 , 3137 , ; for acylation of aryl–Cu complexes, see: . [Google Scholar]; (c) Herron J. R., Ball Z. T. J. Am. Chem. Soc. 2008;130:16486. doi: 10.1021/ja8070804. [DOI] [PubMed] [Google Scholar]
- (a) Park J. K., Lackey H. H., Rexford M. D., Kovnir K., Shatruk M., McQuade D. T. Org. Lett. 2010;12:5008. doi: 10.1021/ol1021756. [DOI] [PubMed] [Google Scholar]; (b) Park J. K., McQuade D. T. Angew. Chem., Int. Ed. 2012;51:2717. doi: 10.1002/anie.201107874. [DOI] [PubMed] [Google Scholar]; (c) Park J., McQuade D. T. Synthesis. 2012;44:1485. [Google Scholar]; (d) Delvos L., Hensel A., Oestreich M. Synthesis. 2014;46:2957. [Google Scholar]
- (a) Li X., Zhang Y.-K., Liu Y., Ding C. Z., Li Q., Zhou Y., Plattner J. J., Baker S. J., Qian X., Fan D., Liao L., Ni Z.-J., White G. V., Mordaunt J. E., Lazarides L. X., Slater M. J., Jarvest R. L., Thommes P., Ellis M., Edge C. M., Hubbard J. A., Somers D., Rowland P., Nassau P., McDowell B., Skarzynski T. J., Kazmierski W. M., Grimes R. M., Wright L. L., Smith G. K., Zou W., Wright J., Pennicott L. E. Bioorg. Med. Chem. Lett. 2010;20:3550. doi: 10.1016/j.bmcl.2010.04.129. [DOI] [PubMed] [Google Scholar]; (b) Johns B. A., Shotwell J. B. and Haigh D., US Pat., WO 2012/067663 A1, 2012.; (c) Chong P. Y., Miller J. F., Peat A. J. and Shotwell J. B., US Pat., WO 2013/028371 A1, 2013.
- Molander G. A., Jean-Gérard L. J. Org. Chem. 2009;74:5446. doi: 10.1021/jo900968h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Altman R. A., Shafir A., Choi A., Lichtor P. A., Buchwald S. L. J. Org. Chem. 2008;73:284. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]; (b) Yang W., Liu Y., Zhang S., Cai Q. Angew. Chem., Int. Ed. 2015;54:8805. doi: 10.1002/anie.201503882. [DOI] [PubMed] [Google Scholar]
- Evans D. A., Chapman K. T., Carreira E. M. J. Am. Chem. Soc. 1988;110:3560. [Google Scholar]
- Lee J., Radomkit S., Torker S., del Pozo J., Hoveyda A. H. Nat. Chem. 2018;10:99. doi: 10.1038/nchem.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.