

Abstract
Nutrient and energy recovery is becoming more important for a sustainable future. Recently, we developed a hydrogen gas recycling electrochemical system (HRES) which combines a cation exchange membrane (CEM) and a gas-permeable hydrophobic membrane for ammonia recovery. This allowed for energy-efficient ammonia recovery, since hydrogen gas produced at the cathode was oxidized at the anode. Here, we successfully up-scaled and optimized this HRES for ammonia recovery. The electrode surface area was increased to 0.04 m2 to treat up to 11.5 L/day (∼46 gN/day) of synthetic urine. The system was operated stably for 108 days at current densities of 20, 50, and 100 A/m2. Compared to our previous prototype, this new cell design reduced the anode overpotential and ionic losses, while the use of an additional membrane reduced the ion transport losses. Overall, this reduced the required energy input from 56.3 kJ/gN (15.6 kW h/kgN) at 50 A/m2 (prototype) to 23.4 kJ/gN (6.5 kW h/kgN) at 100 A/m2 (this work). At 100 A/m2, an average recovery of 58% and a TAN (total ammonia nitrogen) removal rate of 598 gN/(m2 day) were obtained across the CEM. The TAN recovery was limited by TAN transport from the feed to concentrate compartment.
Keywords: Ammonia recovery, Up-scaling, Electrochemical system, Hydrogen recycling
Short abstract
The improved design of an electrochemical system allows for energy-efficient ammonia recovery despite an increased reactor size.
Introduction
The recovery of energy and nutrients is gaining an increasing interest from the scientific community. This interest is fueled by an increasing world population, which demands higher energy and resource efficiencies for a sustainable future. Interesting and yet often unexploited sources for both energy and nutrients (i.e., P and N) are wastewater and urine. Urine has the advantage that it can be collected without dilution, and thus, these nutrients are present in high concentrations.1,2 In the past decade, the treatment of urine in bioelectrochemical systems (BESs) and electrochemical systems (ESs) has seen a rise in attention, due to the possibility to recover ammonia at the cost of only little energy input or even with simultaneous production of energy.3−6
In both BESs and ESs, electric current derived from the electrode reactions can be used to recover total ammonia nitrogen (TAN, i.e., ammonium and ammonia) from wastewater by driving it through a cation exchange membrane (CEM) toward the cathode.7−9 In BESs, this electrical current is produced from the organic matter in wastewater by microorganisms that inhabit the anode surface,7 while in an ES externally added electric energy is used to drive electrolysis of water.8 TAN can be recovered from the cathode using an NH3 stripping process,8 a hydrophobic gas-permeable membrane,10,11 or crystallization.12
Recently, we managed to lower the required energy input for TAN recovery in ESs through recycling of the produced hydrogen gas (H2) from the cathode to the anode.13 Limited additional energy input is required in such a system, since the product of the cathodic reduction (H2) is used in the anodic oxidation. In practice, some energy input is needed because of the internal resistance of these systems caused by ion transport through ion-selective membranes, electrolyte resistance, and the overpotentials of the electrochemical reaction.14−16 Compared to the oxidation of water, the oxidation of H2 has an additional advantage since this reaction requires a much lower anode potential and overpotential to occur.17 Therefore, the risk of chloride oxidation to chlorine gas (>1.1 V versus Ag/AgCl) and the formation of adsorbable organic halides (AOX) is minimized.18
Until now, the practical energy input for TAN recovery using a hydrogen gas recycling electrochemical system (HRES) was as low as 26.1 kJ/gN (7.3 kW h/kgN) recovered at a current density of 20 A/m2.13 This is already significantly lower compared to the combined 51 kJ/gN required to produce pure and concentrated ammonia from N2 with the Haber–Bosch process (37 kJ/gN natural gas) and to remove ammonia from wastewater streams by nitrification/denitrification (14 kJ/gN electricity).19 At higher current density of 50 A/m2, however, the required input increased to around 56.3 kJ/gN (15.6 kW h/kgN).13
In addition to reducing the energy demand, an increase of treatment capacity is required to make this technology practically applicable. Therefore, in this study we improved the cell configuration for recovery of TAN from synthetic urine, reducing the internal resistance of the system. At the same time, the size (surface area) of the electrodes was increased 4-fold, from 0.01 m2 (0.1 m × 0.1 m) to 0.04 m2 (0.2 m × 0.2 m) to increase the treatment capacity. The new cell configuration was envisioned to recover 46 gN/day at a current density of 100 A/m2, which corresponds to the daily collected volume of urine from about 10 persons.20
Overall, we successfully optimized and operated an up-scaled electrochemical cell for the TAN recovery from synthetic urine. We show a stable performance over 108 days and a maximum TAN recovery efficiency of 74%. These improvements bring energy-efficient ammonium recovery closer to practice.
Experimental Section
Setup
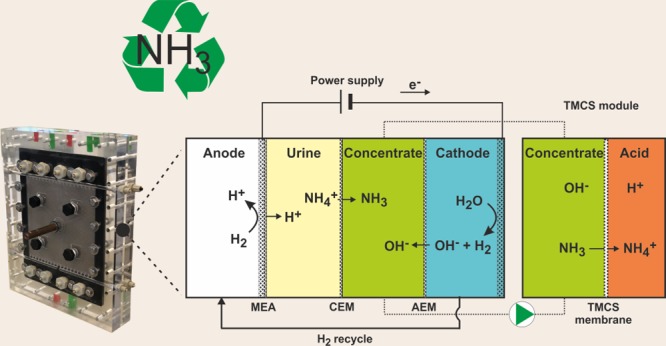
The optimized hydrogen gas recycling electrochemical system (HRES) consisted of four compartments (Figure 1). Compared to the previous cell configuration, an additional compartment was introduced to concentrate the ammonia. This concentrate compartment was placed between the feed compartment and the cathode compartment. The main advantage of this additional compartment is that hydrogen gas is not produced in the same compartment from which the ammonia is recovered. Therefore, the ammonia and hydrogen are not mixed which is advantageous for both the recycling of pure hydrogen to the anode and the recovery of pure ammonia in the sulfuric acid solution. Table 1 summarizes the modification to the configuration of our up-scaled HRES compared to our prototype HRES.
Figure 1.

Scheme of the up-scaled electrochemical system for TAN recovery. H2 produced at the cathode is transported to the anode, using N2 as carrier gas, where it is oxidized. From left to right the anode, feed, concentrate, and cathode compartments are shown. On the far right, the TMCS module is shown which employs a gas-permeable hydrophobic membrane for ammonia extraction. The produced protons in the oxidation reaction move through a CEM to the feed compartment. The electric current produced is used to transport ammonium (NH4+) and other cations from the feed to the concentrate compartment. Finally, the cathodically generated hydrogen gas (H2) is fed to the anode, and hydroxide ions (OH–) move from the cathode to the concentrate compartment. The liquid from the concentrate compartment is continuously circulated over the TMCS module, where the ammonia is extracted into a sulfuric acid solution producing a concentrated ammonium sulfate solution.
Table 1. Modification of the Optimized and Up-Scaled HRES and Its Operation Compared to the Prototype HRES13.
| unit | prototype HRES13 | optimized and up-scaled HRES (this study) | |
|---|---|---|---|
| electrode/IEM surface area | m2 | 0.01 | 0.04 |
| no. and types of IEM used | 1 MEA, 1 CEM | 1 MEA, 1 CEM, 1 AEM | |
| applied current densities | A/m2 | 10, 20, and 50 | 20, 50, and 100 |
| TAN recovery from | cathode compartment | concentrate compartment | |
| TAN loading | gN/d | 1.2, 2.3, and 5.8 | 9.2, 23, and 46 |
| load ratio (LN) | 1.3 | 1.3 |
Figure 1 shows a schematic overview of the HRES. From left to right the compartments were as follows: (i) anode, (ii) feed, (iii) concentrate, and (iv) cathode. The hydrogen gas produced at the cathode was recycled to the anode compartment where it was oxidized at a gas diffusion electrode.
The anode compartment was separated from the feed compartment by a (Nafion-based) membrane electrode assembly (MEA), with the electrode facing the anode compartment. The feed compartment, containing the synthetic urine, was separated from the concentrate compartment by another CEM (CMH-PP Ralex, MEGA a. s., Stráž pod Ralskem, Czech Republic). The TAN, extracted from the feed compartment, is transported through this CEM to the neighboring concentrate compartment which is separated from the cathode compartment by an anion exchange membrane (AEM) (AMH-PP Ralex, MEGA a. s.). TAN was recovered from the concentrate compartment using a gas-permeable hydrophobic membrane module (TMCS, transmembrane chemisorption) as previously described by Kuntke et al. (2016).21
The anode MEA was a 24 cm × 24 cm Nafion N117 CEM coated with a 20 cm × 20 cm platinum Vulcan (carbon) catalyst (0.5 mg Pt/cm2) and an integrated gas diffusion layer (GDL) purchased from FuelCellsETC. The gas diffusion electrode (GDE, catalyst + GDL) side of the MEA was facing the anode compartment, while the side without catalyst and GDL was facing the feed compartment.
The dimensions of the anode and cathode compartment were 20 cm × 20 cm × 0.2 cm. Inside the anode compartment, four Pt-coated titanium mesh electrodes (9.8 cm × 9.8 cm × 0.2 cm, 5 mg Pt/cm2, Magneto Special Anodes BV) acted as current collectors. Inside the cathode compartment, four Pt-coated titanium mesh electrodes (9.8 cm × 9.8 cm × 0.2 cm, 5 mg Pt/cm2 Magneto Special Anodes BV) were used as the cathode. The dimensions of the feed and concentrate compartment were 20 cm × 20 cm × 0.05 cm. Inside the feed, concentrate, and cathode compartments, spacers (SEFAR FLUORTEX, 20 cm × 20 cm × 0.05 cm, 47% open area) were employed to separate the membranes. The housing for both the anode and cathode compartment was made from PMMA (poly methyl methacrylate) with a flow field of 20 cm × 20 cm × 0.2 cm and integrated influent and effluent channels for the four compartments. Custom-made 2 mm thick silicone rubber gaskets were used to seal off the compartments. All ion exchange membranes and electrodes had a projected surface area of 400 cm2.
The produced hydrogen gas was purged from the cathode compartment to the anode with nitrogen gas (15 mL/min). This carrier gas was enriched with approximately 10% of extra H2 from a custom-made electrolyzer (operated at constant current and 10 mM H2SO4 as electrolyte) which was injected into the anode compartment in close vicinity to the MEA.
The TMCS modules were made in-house from a custom-made polypropylene (PP) housing and commercially available membranes (1.5 m, 0.2 μm pore size, V8/2 Type TP, MICRODYN-NADIR GmbH, Wiesbaden, Germany).
Operational Conditions and Electrolyte Composition
The anode compartment contained a gas diffusion electrode and therefore did not contain any liquid. The feed compartment (0.83 L) was fed with synthetic wastewater which consisted of 4.45 g/L NaCl, 1.85 g/L KCl, 1.04 g/L K2SO4, and 13.7 g/L (NH4)2CO3. This synthetic urine was used to mimic the pretreated urine (used for MEC operation) as described by Zamora et al., 2017.22 The system was always operated at a load ratio of approximately 1.3 meaning that the TAN load in the influent was always 23% lower than the maximum amount that could theoretically be removed by the applied current.23
| 1 |
Here, i is the current density (A/m2), Cfeed,TAN is the molar concentration of TAN in the feed inflow (mol/L), Qfeed the feed inflow rate (mL/min), F is the Faraday constant (96 485 C/mol), and Am the surface area of the cation exchange membrane (m2).
The influent into the feed compartment was fed with a rate of 1.6, 4.0, or 8.0 mL/min to maintain a load ratio of 1.3 at the chosen current densities of 20, 50, and 100 A/m2, respectively.
The catholyte (1 L) consisted of a 0.1 M NaOH (pH ∼ 12.8) solution at the start of an experiment. The composition changed during the experiment because of ion transport to and from this compartment.16,24 The concentrate compartment was filled with feed solution at the start of the experiment.
The acid solution used to absorb the ammonia from the concentrate solution was a 1 M H2SO4. All liquids in the compartments were recycled at a rate of 30 mL/min. Ag/AgCl reference electrodes (3 M KCl/saturated AgCl, +0.205 V versus NHE, QM711X, QiS-Prosence BV, Oosterhout, The Netherlands) were placed in the feed, concentrate, and catholyte compartments to measure anode (MEA) potential, cathode potential, and membrane potentials. The pH values [Orbisint CPS11D sensors connected to a Liquiline CM444 transmitter (Endress+Hauser BV, Naarden, The Netherlands)] of the feed, concentrate, and catholyte were continuously measured. The applied current was controlled by a power supply (ES 030-5, Delta Elektronika BV, Zierikzee, The Netherlands). A Memograph M RSG40 data logger (Endress+Hauser BV) was used to record pH, temperature, current density, cell voltage, anode potential, and cathode potential.
Chemical Analysis
Samples were taken daily on weekdays to determine cations, anions, and conductivity. Conductivity was measured using pH/mV conductivity meter (Seven Excellence S470, Mettler Toledo, Tiel, The Netherlands). Cations (Na+, K+, NH4+) and anions (SO42–, Cl–, NO3–, NO2–) in the feed, concentrate, and catholyte were analyzed with a Metrohm Compact IC Flex 930 instrument with a cation column (Metrosep C 4-150/4.0) and a Metrohm Compact IC 761 instrument with an anion column (Metrosep A Supp 5-150/4.0), each equipped with a conductivity detector (Metrohm Nederland BV, Schiedam, The Netherlands). Ammonium-nitrogen in the acid was determined using a cuvette test kit (LCK303; HACH NEDERLAND, Tiel, The Netherlands).
Calculations
The calculations are based on earlier work13 and explained in detail in the Supporting Information (equations S1–S14).
Results and Discussion
The Removal Rate of TAN Increases at Higher Current Densities, While the Recovery Efficiency Decreases
In this Article, we show an optimized and up-scaled electrochemical cell for TAN recovery from synthetic urine. Since the up-scaled HRES had a 4-times-larger CEM surface area than our previous system, we investigated the dependence of TAN recovery on the TMCS membrane surface area. Figure 2A shows the TAN recovery with 1, 2, and 3 TMCS modules connected in series in the recirculation loop of the concentrate compartment at an applied current density of 20 A/m2. Every TMCS module had a membrane surface area of 0.04 m2 giving a total membrane surface area of 0.12 m2 when three modules were used. The average TAN recovery was 74 ± 2%, and no significant difference was found during operation with 1, 2, or 3 modules.
Figure 2.

Box-and-whisker diagram (with outlier indicated as single points) of TAN recovery evaluated at 20 A/m2 with 1, 2, and 3 TMCS modules (A) and TAN recovery evaluated at applied current densities of 20, 50, and 100 A/m2 (B). Increasing the surface area of the gas-permeable hydrophobic membrane (e.g., number of TMCS modules) did not affect TAN recovery. However, increasing the applied current density while maintaining a load ratio of 1.3 resulted in a decreasing TAN recovery.
Afterward, the TAN recovery was evaluated at current densities of 50 and 100 A/m2 (Figure 2b). The TAN recovery decreased from 74 ± 2% at 20 A/m2 to 63 ± 2% at 50 A/m2. The addition of a fourth TMCS module, however, did not increase the TAN recovery indicating that the extraction of TAN from the concentrate compartment was not limiting. At a current density of 100 A/m2, a TAN recovery of 58 ± 2% was found.
TAN recovery is a function of the TAN transport over the CEM from the feed solution into the concentrate solution and TAN transport over the TMCS membrane from the concentrate solution to the acid solution.23 On first sight, the TAN transport rate did not increase linearly with the increase of current density. At a current density of 20 A/m2 the TAN transport rate over the CEM was 141 ± 8 gN/(m2 day); at 50 A/m2, 311 ± 9 gN/(m2 day); and at 100 A/m2, 598 ± 24 gN/(m2 day). As the transport of TAN from the concentrate solution through the TMCS module was not limiting the overall recovery, the TAN transport from the feed solution into the concentrate solution through the CEM must have been limiting the TAN recovery. For quantification of this transport through the CEM between feed and concentrate compartment, the contribution of each cation species to the total charge transport was calculated (Figure 3). The results confirm that the contribution of TAN to the total charge transport through the CEM decreases with increasing current density, and the relative contribution of other cations increases.
Figure 3.
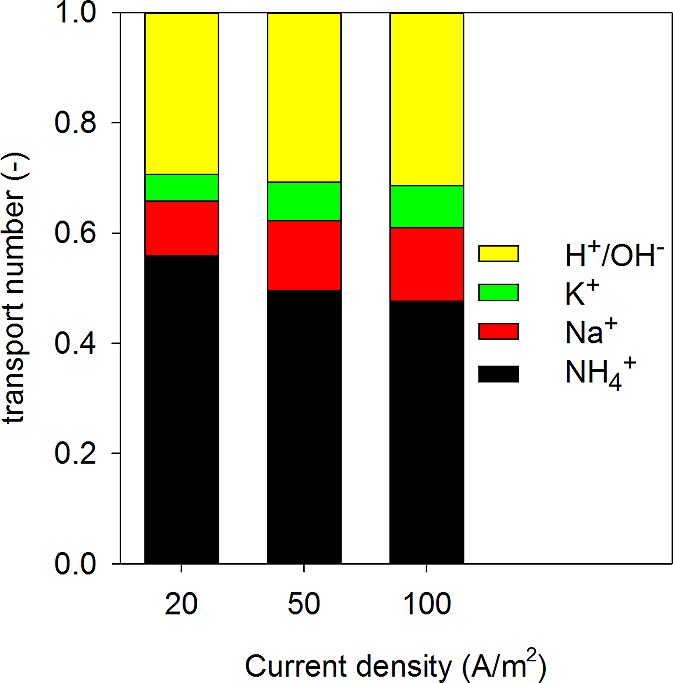
Ion transport numbers across the CEM separating feed from concentrate compartment at the applied current densities of 20, 50, and 100 A/m2. Ammonium is the main ion transported from feed to concentrate compartment through the CEM. At a current density of 20 A/m2 a transport number of 0.56 was determined; at 50 A/m2, 0.50; and at 100 A/m2, 0.48.
This was also confirmed by the continuous increase of the conductivity in both the cathode and concentrate compartment, indicating that cations other than ammonium and protons were transported. At an applied current density of 20 A/m2, the conductivity measured in the concentrate compartment increase by 1.2 mS/(cm day); at 50 A/m2, 2.9 mS/(cm day); and at 100 A/m2, 2.3 mS/(cm day). If all the ion transport was through proton-carrying species (ammonium and protons), the conductivity would remain stable since ammonia is continuously extracted from the concentrate solution in the TMCS module, and protons would react with hydroxide ions to form water. Furthermore, an increase in water level in the concentrate compartment was observed. This water transport from the feed to the concentrate compartment can be explained by electro-osmosis, which has been reported and studied in similar electrochemical systems (e.g., electrodialysis and electroconcentration).25,26 The transport of ions other than proton-carrying species (ammonium and protons) and the transport of water show that the system did not reach steady state yet.
The decrease in transport of ammonium compared to other ions is most likely due to limited mixing of the liquid in the feed recirculation, where new influent was added to the system. Compared to the previous used prototype HRES, the thickness of the feed compartment was reduced from 12 to 0.5 mm in the redesign and up-scaled HRES. Although this reduced the ionic resistance of this compartment, it also limited the possibility to circulate the liquid at high rates over the compartments. It is likely that TAN was depleted at the CEM surface area where the ions are being removed, although sufficient TAN was found in the feed compartment effluent (i.e., 50 A/m2, 1.61 gN/L; and 100 A/m2, 1.99 gN/L).
Overall Energy Demand Improved Compared to the Prototype: Nonetheless, Limitations Remain
Figure 4A shows the energy demand during operation of the up-scaled system as a function of the applied current density. At a current density of 100 A/m2 an energy demand of 23.4 ± 1.0 kJ/gN (6.5 ± 0.3 kW h/kgN) was determined, which is considerably lower than the energy demand reported for our prototype HRES. In those experiments, the energy demand for TAN recovery at 10 A/m2 was 30.5 kJ/gN (8.5 kW h/kgN); at 20 A/m2, 26.1 kJ/gN (7.3 kW h/kgN); and at 50 A/m–2, 56.3 kJ/gN (15.6 kW h/kgN).13
Figure 4.

Energy demand for TAN removal (A) and potential losses (B) at the applied current densities. The dashed lines in both parts show the previous obtained minimum energy demand and potential loss at an applied current density of 20 A/m2 using the smaller prototype HRES.13 These results indicate that the optimized and up-scaled HRES consumes less energy even at a 5-times-higher current density and with 4-times-larger electrode/membrane surface area.
The 10% of additional H2 supplied to the system accounted for approximately 13–15% of the energy demand of the system.
The overall energy demand for TAN recovery in a (bio)electrochemical system is determined by three main factors: cell voltage, applied current density, and the TAN transport rate. Upon comparison of the new system to the prototype HRES at their respective highest applied current densities, the cell voltage was lowered from 4.5 V (prototype) at an applied current density of 50 A/m2 to 1.4 V at an even higher current density of 100 A/m2.13 This cell voltage is affected by different losses (i.e., anode/cathode overpotential, transport losses, ionic losses, and equilibrium losses) inside the system. Table 2 shows the average anode and cathode potential, the conductivities, and pH values measured in the different compartments to calculate the losses within the system.
Table 2. Average Anode Potential, Cathode Potential, pH, and Conductivities Measured at the Applied Current Densities of 20, 50, and 100 A/m2.
| 20 A/m2 | 50 A/m2 | 100 A/m2 | ||
|---|---|---|---|---|
| Ecell | V | 1.19 ± 0.10 | 1.23 ± 0.16 | 1.40 ± 0.01 |
| Eanode | V (vs Ag/AgCl) | –0.02 ± 0.03 | 0.03 ± 0.16 | 0.13 ± 0.01 |
| Ecathode | V (vs Ag/AgCl) | –1.10 ± 0.12 | –1.13 ± 0.01 | –1.18 ± 0.001 |
| pH influent | 9.3 | 9.3 | 9.3 | |
| pH feed effluent | 6.4 ± 0.9 | 6.8 ± 0.3 | 7.3 ± 0.1 | |
| pH concentrate | 12.6 ± 0.8 | 12.8 ± 0.3 | 12.5 ± 0.1 | |
| pH cathode | 13.3 ± 0.1 | 13.7 ± 0.1 | 13.9 ± 0.1 | |
| conductivity influent | mS/cm | 29.7 | 29.7 | 29.7 |
| conductivity feed effluent | mS/cm | 15.3 ± 1.2 | 16.3 ± 0.7 | 17.7 ± 0.4 |
| conductivity concentrate | mS/cm | 55.7 ± 18.3 | 220 ± 32 | 301 ± 16 |
| conductivity cathode | mS/cm | 46.5 ± 15.8 | 236 ± 46 | 351 ± 20 |
A more detailed analysis of the energetic losses was made to identify the main losses contributing to the cell voltage. Figure 4B shows the losses during the operation of the up-scaled system as a function of the applied current density. Overall, the equilibrium losses, anode overpotential, and cathode overpotential are the main contributors to this energy demand, while the ionic and transport losses are marginal. The equilibrium potential is the main contribution to the energy input of the up-scaled HRES. This equilibrium potential is determined by the pH difference that exists between the anode and cathode compartment. Although this pH difference leads to the main energetic input of the system, it is essential to enable the selective removal and recovery of TAN via the membranes employed in the system.
When examining the cell voltage in more detail, the main reasons for the lower cell voltage in these experiments compared to the prototype HRES are the reduced anode overpotential, ionic losses, and transport losses across the IEMs. The anode overpotential in the prototype HRES was 0.93 V at a current density of 50 A/m2, while in the up-scaled system it is reduced to 0.35 V at a current density of 100 A/m2. This improvement is probably a result of the optimized contact between the H2 gas and the GDE, due to the better compression of the GDE. Additionally, adding the concentrate compartment between the feed and cathode compartment allowed for pure hydrogen gas production in the cathode compartment while in the previous design some of the hydrogen was lost across the TMCS module, while also some ammonia was present in the hydrogen gas stream.
The ionic loss in the prototype HRES was 0.4 V at a current density of 50 A/m2, while in the up-scaled system it is reduced to 0.09 V at a current density of 100 A/m2. The main reason was the tighter packing of the electrochemical system resulting in a smaller compartment thickness (1.2 versus 0.05 cm).
The transport loss across the IEMs in the prototype HRES was 2.2 V at a current density of 50 A/m2, while in the up-scaled system it is reduced to 10 mV at a current density of 100 A/m2. This reduction in transport loss, although counterintuitive, was caused by the introduction of the additional concentrate compartment. The dominant ion transports through the system are as follows: (i) proton transport through the CEM (MEA) between the anode and feed compartment, wherein the protons are transported from a high concentration (0.54 M)27 to a low concentration (4.7 × 10–8 M); (ii) ammonium and proton transport through the CEM from the feed to concentrate compartment, from a high concentration in the feed to a lower concentration in the concentrate compartment, wherein the concentration of ammonium in the concentrate compartment is low since it is continuously removed through the TMCS module and because the high pH deprotonation of ammonium to ammonia occurs; and (iii) hydroxide transport from the cathode compartment (0.7 M at 100 A/m2) to the concentrate compartment (0.03 M at 100 A/m2). Overall, the crucial difference between the optimized HRES and prototype was the introduction of the AEM between cathode and concentrate compartment. As a consequence of this modification, the voltage loss caused by cations transported over CEM with a negative membrane potential (separating feed from concentrate compartment) was partially compensated by the voltage gain of anions (hydroxide ions) transported over the AEM with a negative membrane potential (separating cathode from concentrate compartment).16
Hydrogen Gas Recycling Allows Electrochemical Systems To Compete with Bioelectrochemical Systems for Energy-Efficient TAN Recovery
A meaningful comparison of TAN removal/recovery reported for (bio)electrochemical system is challenging, since these reactors are not always operated under identical conditions (i.e., current density, TAN loading rate, reactor size, etc.). While the applied current density influences the TAN transport rate, high TAN recovery/removal efficiencies are only achieved when the applied current density is matched with the TAN loading.8 The load ratio, which relates the applied current density to the TAN loading, can be used to compare different systems. In practice, a load ratio higher than 1 is required to remove/recover all TAN from the feed stream. However, increasing the load ratio to increase the removal/recovery also results in higher energy demand, since the TAN transport number over the CEM decrease with an increasing load ratio. A load ratio below 1 results in relatively high TAN transport rates, while it limits the overall TAN removal/recovery from the feed stream.23 Therefore, we compared our HRES to other current-driven TAN recovery technologies on the basis of their performance at load ratio operated between 0.7 and 1.3.
Our up-scaled HRES performs better than other ESs for TAN recovery; at a load ratio of 1.3, an energy demand as low as 4.2 kW h/kgN (15.1 kJ/gN) was determined for a TAN recovery of 75% at a current density 20 A/m2. Other ESs reported in the literature used even more energy (Table 3); Desloover et al., 2012, reported 16.8 ± 1.4 kW h/kgN (60.5 ± 5.1 kJ/gN) for a TAN removal/recovery of 41% at 30 A/m2 (estimated load ratio of 0.96),8 and Luther et al., 2015, reported 9.5 kW h/kgN (34.2 kJ/gN) for a TAN removal of 53% at 30 A/m2 (estimated load ratio of 0.7).28 Rodriguez-Arredondo et al., 2017, reported 21.5 kJ/gN (about 6 kW h/kgN) for a TAN removal of 83% at 10 A/m2 and a load ratio of 1.3.23
Table 3. Performance of ESs and a BES Reported in the Literature.
| load ratio | current density | energy
input |
recovery | ref | ||
|---|---|---|---|---|---|---|
| (A/m2) | (kW h/kgN) | (kJ/gN) | (%) | |||
| ES | 1.3 | 20 | 4.2 | 15.1 | 75 | this study |
| 1.3 | 50 | 5.5 | 19.8 | 63 | this study | |
| 1.3 | 100 | 6.5 | 23.4 | 58 | this study | |
| 0.96 | 30 | 16.8 | 60.5 | 41 | Desloover et al. 20128 | |
| 0.7 | 30 | 9.5 | 34.2 | 53 | Luther et al. 201528 | |
| 1.3 | 10 | 6 | 21.5 | 83 | Rodriguez-Arredondo et al. 201723 | |
| BES | 0.44 | 29 | 2.4 | 8.6 | 50 | Ledezma et al. 201712 |
BESs for TAN recovery, such as microbial electrolysis cells (MECs), can be more energy-efficient than ESs, as they recover energy stored in organic substrate to lower the energy demand for water splitting.9 Ledezma et al., 2017, reported an energy demand of 2.4 kW h/kgN (8.6 kJ/gN) for a TAN recovery of 49.5 ± 1.8% at a current density of 29.3 A/m2 (estimated load ratio of 0.44).12 These results were obtained in a relatively small MEC (0.01 m2 of CEM), while up-scaling and maintaining high current densities of MECs for TAN recovery is challenging.22 Reaching a higher TAN recovery requires operation at a higher load ratio, which at the same time increases the energy demand.23
Similar to other nonbiological electrochemical systems for TAN recovery, our up-scaled HRES does not remove organic matter contained in the wastewater.29 Therefore, the effluent requires further treatment, which can be done in a BES (i.e., electricity or H2) or in an anaerobic digester (methane production).
Implication
These results show that we successfully scaled up the hydrogen gas recycling electrochemical system for ammonia recovery. At an applied current density of 20 A/m2, the modification to the system reduced the electrical energy demand of the HRES by 33% compared to our prototype. Furthermore, TAN recoveries up to 100 A/m2 were more energy-efficient compared to previous results. Additional optimization of the technology will focus on improving the current efficiency, reducing equilibrium losses, and anode and cathode electrode overpotential. Nevertheless, the technology as developed at our laboratory is deemed for further up-scaling, testing with different wastewater, and piloting.
Acknowledgments
This work was performed in the cooperation framework of Wetsus, European Centre of Excellence for Sustainable Water Technology (www.wetsus.eu). Wetsus is cofunded by the Dutch Ministry of Economic Affairs and Ministry of Infrastructure and Environment, the European Union Regional Development Fund, the Province of Fryslân, and the Northern Netherlands Provinces. The authors would like to thank the participants of the research theme “Resource Recovery” for the fruitful discussions and their financial support. The authors would like to thank Michele Tedesco (Wetsus) for his advice and comments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssuschemeng.8b00457.
Additional calculation details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kirchmann H.; Pettersson S. Human urine - Chemical composition and fertilizer use efficiency. Fert. Res. 1995, 40 (2), 149–154. 10.1007/BF00750100. [DOI] [Google Scholar]
- Larsen T. A.; Gujer W. Separate management of anthropogenic nutrient solutions (human urine). Water Sci. Technol. 1996, 34 (3–4), 87–94. [Google Scholar]
- Ieropoulos I.; Greenman J.; Melhuish C. Urine utilisation by microbial fuel cells; energy fuel for the future. Phys. Chem. Chem. Phys. 2012, 14 (1), 94–98. 10.1039/C1CP23213D. [DOI] [PubMed] [Google Scholar]
- Kelly P. T.; He Z. Nutrients removal and recovery in bioelectrochemical systems: A review. Bioresour. Technol. 2014, 153, 351–360. 10.1016/j.biortech.2013.12.046. [DOI] [PubMed] [Google Scholar]
- Rodríguez Arredondo M.; Kuntke P.; Jeremiasse A. W.; Sleutels T. H. J. A.; Buisman C. J. N.; ter Heijne A. Bioelectrochemical systems for nitrogen removal and recovery from wastewater. Environ. Sci. Water Res. Technol. 2015, 1 (1), 22–33. 10.1039/C4EW00066H. [DOI] [Google Scholar]
- Ledezma P.; Kuntke P.; Buisman C. J. N.; Keller J.; Freguia S. Source-separated urine opens golden opportunities for microbial electrochemical technologies. Trends Biotechnol. 2015, 33 (4), 214–220. 10.1016/j.tibtech.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Kuntke P.; Śmiech K. M.; Bruning H.; Zeeman G.; Saakes M.; Sleutels T. H. J. A.; Hamelers H. V. M.; Buisman C. J. N. Ammonium recovery and energy production from urine by a microbial fuel cell. Water Res. 2012, 46 (8), 2627–2636. 10.1016/j.watres.2012.02.025. [DOI] [PubMed] [Google Scholar]
- Desloover J.; Abate Woldeyohannis A.; Verstraete W.; Boon N.; Rabaey K. Electrochemical resource recovery from digestate to prevent ammonia toxicity during anaerobic digestion. Environ. Sci. Technol. 2012, 46 (21), 12209–12216. 10.1021/es3028154. [DOI] [PubMed] [Google Scholar]
- Kuntke P.; Sleutels T. H. J. A.; Saakes M.; Buisman C. J. N. Hydrogen production and ammonium recovery from urine by a Microbial Electrolysis Cell. Int. J. Hydrogen Energy 2014, 39 (10), 4771–4778. 10.1016/j.ijhydene.2013.10.089. [DOI] [Google Scholar]
- Ulbricht M.; Schneider J.; Stasiak M.; Sengupta A. Ammonia recovery from industrial wastewater by transMembranechemiSorption. Chem. Ing. Tech. 2013, 85 (8), 1259–1262. 10.1002/cite.201200237. [DOI] [Google Scholar]
- Sleutels T. H. J. A.; Hoogland B. J.; Kuntke P.; ter Heijne A.; Buisman C. J. N.; Hamelers H. V. M. Gas-permeable hydrophobic membranes enable transport of CO 2 and NH 3 to improve performance of bioelectrochemical systems. Environ. Sci. Water Res. Technol. 2016, 2 (4), 743–748. 10.1039/C6EW00087H. [DOI] [Google Scholar]
- Ledezma P.; Jermakka J.; Keller J.; Freguia S. Recovering Nitrogen as a Solid without Chemical Dosing: Bio-Electroconcentration for Recovery of Nutrients from Urine. Environ. Sci. Technol. Lett. 2017, 4 (3), 119–124. 10.1021/acs.estlett.7b00024. [DOI] [Google Scholar]
- Kuntke P.; Rodríguez Arredondo M.; Widyakristi L.; Ter Heijne A.; Sleutels T. H. J. A.; Hamelers H. V. M; Buisman C. J. N. Hydrogen Gas Recycling for Energy Efficient Ammonia Recovery in Electrochemical Systems. Environ. Sci. Technol. 2017, 51 (5), 3110–3116. 10.1021/acs.est.6b06097. [DOI] [PubMed] [Google Scholar]
- Liu H.; Cheng S.; Logan B. E. Power generation in fed-batch microbial fuel cells as a function of ionic strength, temperature, and reactor configuration. Environ. Sci. Technol. 2005, 39 (14), 5488–5493. 10.1021/es050316c. [DOI] [PubMed] [Google Scholar]
- Sleutels T. H. J. A.; Hamelers H. V. M; Rozendal R. A.; Buisman C. J. N. Ion transport resistance in Microbial Electrolysis Cells with anion and cation exchange membranes. Int. J. Hydrogen Energy 2009, 34 (9), 3612–3620. 10.1016/j.ijhydene.2009.03.004. [DOI] [Google Scholar]
- Sleutels T. H. J. A.; ter Heijne A.; Kuntke P.; Buisman C. J. N.; Hamelers H. V. M. Membrane Selectivity Determines Energetic Losses for Ion Transport in Bioelectrochemical Systems. ChemistrySelect 2017, 2 (12), 3462–3470. 10.1002/slct.201700064. [DOI] [Google Scholar]
- Sheng W.; Zhuang Z.; Gao M.; Zheng J.; Chen J. G.; Yan Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6 (1), 5848. 10.1038/ncomms6848. [DOI] [PubMed] [Google Scholar]
- Zöllig H.; Remmele A.; Fritzsche C.; Morgenroth E.; Udert K. M. Formation of Chlorination Byproducts and Their Emission Pathways in Chlorine Mediated Electro-Oxidation of Urine on Active and Nonactive Type Anodes. Environ. Sci. Technol. 2015, 49 (18), 11062–11069. 10.1021/acs.est.5b01675. [DOI] [PubMed] [Google Scholar]
- Maurer M.; Schwegler P.; Larsen T. A. Nutrients in urine: energetic aspects of removal and recovery. Water Sci. Technol. 2003, 48 (1), 37–46. [PubMed] [Google Scholar]
- Maurer M.; Pronk W.; Larsen T. A. Treatment processes for source-separated urine. Water Res. 2006, 40 (17), 3151–3166. 10.1016/j.watres.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Kuntke P.; Zamora P.; Saakes M.; Buisman C. J. N.; Hamelers H. V. M. Gas-permeable hydrophobic tubular membranes for ammonia recovery in bio-electrochemical systems. Environ. Sci. Water Res. Technol. 2016, 2 (2), 261–265. 10.1039/C5EW00299K. [DOI] [Google Scholar]
- Zamora P.; Georgieva T.; Ter Heijne A.; Sleutels T. H. J. A.; Jeremiasse A. W.; Saakes M.; Buisman C. J. N.; Kuntke P. Ammonia recovery from urine in a scaled-up Microbial Electrolysis Cell. J. Power Sources 2017, 356, 491–499. 10.1016/j.jpowsour.2017.02.089. [DOI] [Google Scholar]
- Rodríguez Arredondo M.; Kuntke P.; ter Heijne A.; Hamelers H. V. M; Buisman C. J. N. Load ratio determines the ammonia recovery and energy input of an electrochemical system. Water Res. 2017, 111, 330–337. 10.1016/j.watres.2016.12.051. [DOI] [PubMed] [Google Scholar]
- Sleutels T. H. J. A.; Heijne A. T.; Buisman C. J. N.; Hamelers H. V. M. Steady-state performance and chemical efficiency of Microbial Electrolysis Cells. Int. J. Hydrogen Energy 2013, 38 (18), 7201–7208. 10.1016/j.ijhydene.2013.04.067. [DOI] [Google Scholar]
- Pronk W.; Biebow M.; Boller M. Electrodialysis for Recovering Salts from a Urine Solution Containing Micropollutants. Environ. Sci. Technol. 2006, 40 (7), 2414–2420. 10.1021/es051921i. [DOI] [PubMed] [Google Scholar]
- Thompson Brewster E.; Jermakka J.; Freguia S.; Batstone D. J. Modelling recovery of ammonium from urine by electro-concentration in a 3-chamber cell. Water Res. 2017, 124 (7), 210–218. 10.1016/j.watres.2017.07.043. [DOI] [PubMed] [Google Scholar]
- Herrera O. E.; Wilkinson D. P.; Mérida W. Anode and cathode overpotentials and temperature profiles in a PEMFC. J. Power Sources 2012, 198, 132–142. 10.1016/j.jpowsour.2011.09.042. [DOI] [Google Scholar]
- Luther A. K.; Desloover J.; Fennell D. E.; Rabaey K. Electrochemically driven extraction and recovery of ammonia from human urine. Water Res. 2015, 87, 367–377. 10.1016/j.watres.2015.09.041. [DOI] [PubMed] [Google Scholar]
- Kuntke P.; Sleutels T. H. J. A.; Rodríguez Arredondo M.; Georg S.; Barbosa S. G.; Heijne A. (Bio) electrochemical ammonia recovery: progress and perspectives. Appl. Microbiol. Biotechnol. 2018, 102, 3865–3878. 10.1007/s00253-018-8888-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.