

Abstract
This report describes the stereoselective synthesis of 3-azido-tetralins, -chromanes, and -tetrahy-droquinolines via a tandem allylic azide rearrangement/Friedel–Crafts alkylation. Exposure of allylic azides with a pendant trichloroacetimidate to catalytic quantities of AgSbF6 proved optimal for this transformation. This cascade successfully differentiates the equilibrating azide isomers, providing products in excellent yield and selectivity (>25 examples, up to 94% yield and >25:1 dr). In many cases, the reactive isomer is only a trace fraction of the equilibrium mixture, keenly illustrating the dynamic nature of these systems. We demonstrate the utility of this process via a synthesis of hasubanan.
Tetralins, chromanes, and tetrahydroquinolines represent privileged structural motifs present in pharmaceuticals, agrochemicals, and natural products. Many feature an amino substituent at the C-3 position (Figure 1). These molecules display a wide range of activity including treatments or potential treatments for cancer, pain, depression, thrombosis, Parkinson’s disease, and malaria.1–9 Due to this rich history, numerous synthetic methods can generate these systems including cyclization,10–15 annulation,16,17 cycloaddition,18,19 partial reduction,20,21 and others.6,22–24 Presented herein is a distinctive carbon–carbon bond forming reaction that generates these privileged motifs with the required 3-amino-functionality by manipulating the allylic azide rearrangement.
Figure 1.

Representative biologically active compounds.
Allylic azides exist as an equilibrating mixture of isomers. This was first noted by Winstein, who documented the rate of isomerization for prenyl and crotyl azides.25 Since then, chemists have struggled to exploit this rearrangement synthetically because of difficulty differentiating the azide isomers.26–33 Only a few reports accomplish selective elaboration. The Craig lab reported a tandem Claisen rearrangement (Figure 2a).34 Selectivity was achieved by orchestrating a second irreversible sigmatropic process. The Aubé lab reported a tandem Schmidt reaction that attained selectivity through cyclization and achieved diastereocontrol via chairlike intermediates (Figure 2b).35 Recently, we initiated a program to explore the allylic azide rearrangement’s potential.36 We reported the first example of enantioselective resolution of symmetric systems.37 Herein, we report a synergistic approach that combines the use of proximal functionality with a complexity generating cyclization. By merging these tactics (Figure 2c), we obtain selectivity on unsymmetric systems and accomplished a tandem Friedel–Crafts alkylation (Figure 2d). This reaction results in the highest combined yield and diastereoselectivity reported to date for a dynamic allylic azide functionalization. We demonstrate the utility of this chemistry through a succinct synthesis of hasubanan.
Figure 2.

Selectivity for Allylic Azide Functionalization.
Our strategy incorporated a second proximal allylic functional group. When the azide is proximal (Figure 2c, OIM group, isomers i E and iv Z), the second group would be primary. However, when the azide is distal (Figure 2c, ii trans isomer), the second group would be allylic and therefore substantially more reactive. We chose a trichloroacteimidate because it could be readily activated under electrophilic conditions, such as those for glycosylation,38,39 rearrangement,40–45 or substitution.46–50 A pendent arene might intercept the putative electrophile in a Friedel–Crafts alkylation. Ring closure could potentially proceed from either allylic terminus and the tether would enforce regioselectivity. Imposing selectivity by these combined approaches could afford a highly chemo-, site-, regio-, and diastereoselective protocol for the synthesis of 3-azido-tetralins, -chromanes, and -tetrahydroquinolines (X = CH2, O, or N-PG, respectively; Figure 2d).
We began with allylic azide 1a (Table 1). This allylic azide exists as an equilibrium mixture (1.3:1.0:0.7:trace E:trans:Z:cis, representations shown in Figure 2c). The reactive trans isomer is only ∼30% of the mixture. We exposed azide 1a to a number of activators, including Lewis acids (entries 1–3), Brønsted acids (entries 4–7), and transition metal complexes (entries 8–10). Most conditions led to decomposition (entry 1), poor selectivity (entries 2,3), or poor reactivity (entries 3–5, 8, and 9). Control experiments, conducted after an initial hit with cationic gold(I) (entry 10), provided encouraging results with silver salts. Silver salts with noncoordinating counterions were efficacious (entries 12–14) and those with more lipophilic counterions provided superior results. Conditions with catalytic AgSbF6 are mild, high yielding, and highly stereoselective (entry 14). In a control experiment, 2,6-di-tert-buty-4-methyl-pyridine inhibited the reaction (entry 15). The reaction is slow in ethanol stabilized chloroform (entry 16) or in the presence of deliberately added water (entry 17). These observations implicate general acid catalysis.51 The reaction was tolerant to ambient conditions (entry 18).
Table 1.
Optimization of Tandem Rearrangement Friedel-Crafts Alkylation

| |||
|---|---|---|---|
|
| |||
| entrya | catalyst | yield %b | drc |
| 1 | BF3·OEt2 | 35 | 28:1 |
| 2 | Cu(OTf)2 | 75 | 9:1 |
| 3 | Zn(OTf)2 | 58 | 9:1 |
| 4 | TFA | 4 | 4:1 |
| 5 | TsOH | 4 | 7:1 |
| 6 | Tf2NH | 80 | 13:1 |
| 7 | TfOH | 49 | 7:1 |
| 8 | PdCl2 | 0 | nd |
| 9 | [(COP)PdCl]2 | 0 | nd |
| 10 | JohnPhosAuSbF6 | 75 | 21:1 |
| 11 | AgOTs | 0 | nd |
| 12 | AgClO4 | 81 | 7:1 |
| 13 | AgOTf | 80 | 7:1 |
| 14 | AgSbF6 | 92 | 21:1 |
| 15d | AgSbF6 | 3 | 3:1 |
| 16e | AgSbF6 | 55 | 16:1 |
| 17f | AgSbF6 | 71 | 21:1 |
| 18g | AgSbF6 | 77 | 20:1 |
0.10 mmol substrate at 0.1 M in CHCl3 for 24 h.
Determined by GC-FID analysis using naphthalene as an internal standard. Values are the average of duplicate trials.
Determined by GC-FID analysis.
20 mol % 2,6-di-tert-butyl-4-methylpyridine was added. nd = not determined.
The reaction was conducted in CHCl3 stabilized with EtOH.
5 equiv of water were deliberately added.
The reaction was conducted under ambient conditions.
We explored the scope of this cyclization with a series of allylic azides (Scheme 1). Common substituents on the aryl ring were tolerated (H, OMe, Cl, or Br, 2a–2d). Conveniently, derivative 2d provided diffraction quality crystals. Analysis unambiguously demonstrated the relative configuration of 2d. Other compounds were assigned by analogy to compound 2d. Different tethers were tolerated. The 3-methyl group is not required for diastereoselectivity (2e). Azide 2e maps onto biologically active 3-amino-tetralins (Figure 1). Interestingly, imidate 1f exists almost exclusively as the unreactive isomers by 1H NMR analysis (>99% E and Z). The reactive trans isomer is destabilized by syn-pentane interactions with the geminal methyl groups. However, this does not inhibit reactivity and compound 2f was isolated in acceptable yield. This observation supports Curtin–Hammett kinetics with rate limiting aromatic substitution. Moving the methyl group to the center of the allylic system eroded the dr (1.7:1 for compound 2g). We propose a putative stereochemical model based on chair like transition states (Figure 3). The pathways differ in the orientation of the vinyl group. The major diastereomer could arise from the pseudoequatorial orientation. When the hydrogen atom is replaced with a methyl (1g), then the relative energy approaches unity and diastereoselectivity is reduced (2g). Lastly, a group larger than methyl was permitted in the backbone (2h).
Scheme 1.

Formation of Tetralins 2a–h by Tandem Processa
aYields reported for isolated material as the average of duplicate trials. The dr was determined by 1H NMR.
Figure 3.
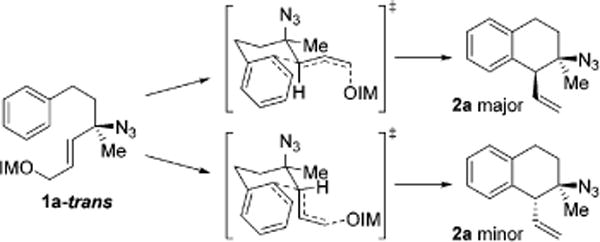
Putative cyclic stereochemical model.
Incorporating a heteroatom into the system would generate valuable heterocycles. A number of ethereal allylic azides were converted into chromanes in high yield and selectivity (Scheme 2, 4a). Activating groups such as methyl (4b) or methoxy (4c) were competent as were compounds with a halogen substituent (entries 4d–4f). Poly substituted aromatics were tolerated (entries 4g–4i). These results are gratifying because the heteroatom is a basic site that could slow catalysis. The proximal oxygen would inductively reduce the stability of a cationic intermediate and could disrupt the azide equilibrium.52 These substrates also indicate limitations in this method. The formation of 4j was slow and imidate decomposition occurred. Chromane 4k was isolated as a mixture of regioisomers (1.4:1).
Scheme 2.

Formation of Chromanes 4a–l by Tandem Processa
aYields reported for isolated material as the average of duplicate trials. The dr was determined by 1H NMR.
We explored aniline derived allylic azides that lead to azido-tetrahydroquinolines (Scheme 3). Several azides were prepared with varying arene substitution and N-protecting group. All imidate precursors in this class contained less than 10% of the reactive trans isomer at equilibrium. Gratifyingly, these azides readily afforded tetrahydroquinolines in good yield with high dr. A number of aryl-substituents were tolerated (entries 6a–6f).
Scheme 3.
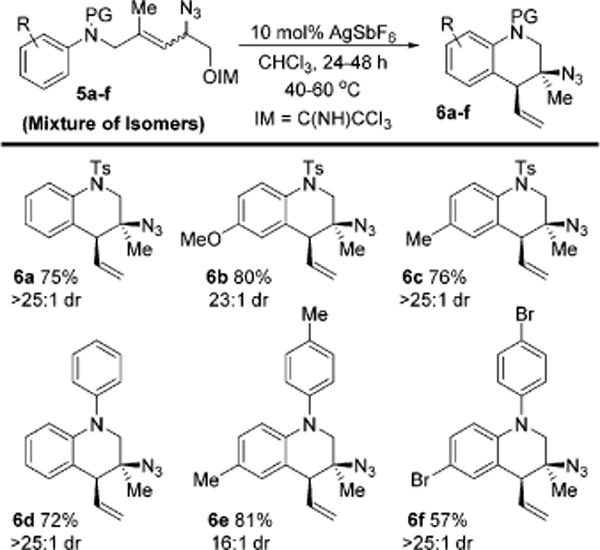
Formation of Tetrahydroquinolines 6a–fa
aYields reported for isolated material as the average of duplicate trials. The dr was determined by 1H NMR.
We conducted the reaction on a gram scale (Scheme 4). Using >1 g of imidate 1a provided tetralin 2a in 82% yield. The product could be oxidized under the Upjohn protocol53 to afford diol 7. Reduction using palladium on carbon provided amine 8. Selective reduction of the azide with LiAlH4 afforded amine 9. The protocol of Evans54 was used to form pyrrolidine 10. Cycloaddition provided triazole 11.
Scheme 4.

Gram Scale Reaction and Diversification of Product
We were emboldened to attempt a synthesis of hasubanan (Scheme 5). Hasubanan is the parent structure of the hasubanan alkaloids, a large family of botanical natural products. Members of this family have been studied synthetically and display diverse biological activities.55–60 The vicinal tetrasubstituted centers in the core provide a significant synthetic challenge. To the best of our knowledge, there are no prior reported syntheses of the parent molecule hasubanan. We began with ester 12, which is directly available from commercial material.61–63 Reduction and partial reoxidation afforded aldehyde 13, which was elaborated by Corey–Chaykovsky epoxidation. Epoxide 14 was opened regioselectively with NaN3 in acetone/water yielding allylic azide 15. Imidate 16 was isolated after activation with trichloroaceto-nitrile. The key cyclization proceeded as expected to establish the necessary stereochemical relationship and the quaternary carbon center. The minor diastereomer was not observed. The final functional group manipulation was accomplished through reduction and ring closure with dicyclohexyl borane.54 This last step clearly illustrates the advantage of using an allylic azide in this synthesis because the final amine functionality was established with concurrent C–N bond formation.
Scheme 5.
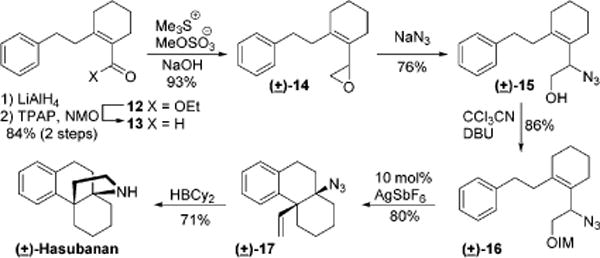
Synthesis of Hasubanan by Tandem Process
In conclusion, we report a tandem allylic azide rearrangement-Friedel–Crafts alkylation. This dynamic cyclization process resolves a mixture of equilibrating allylic azide isomers by exploiting the enhanced reactivity of the allylic electrophile. We demonstrated that this process is general and leads to valuable 3-azido-tetralin, - chromane, and -tetrahydroquinoline hetero-cycles. Furthermore, the products of this reaction are readily diversifiable.
Supplementary Material
Acknowledgments
We thank Ryan Daley for assistance in the analysis of compound 2d. Madeline D. Newcomb for creating the cover art associated with this manuscript. Financial support was provided by the University of Minnesota and The American Chemical Society’s Petroleum Research Fund (PRF # 56505-DNI1). This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM124718. We also acknowledge NIH Shared Instrumentation Grant #S10OD011952.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11299.
Experimental procedures and characterization data (PDF) Crystallographic data for compound 2d (CIF)
ORCID
Joseph J. Topczewski: 0000-0002-9921-5102
Notes
The authors declare no competing financial interest.
References
- 1.Lin YC, Chang JC, Cheng SY, Wang CM, Jhan YL, Lo IW, Hsu YM, Liaw CC, Hwang CC, Chou CH. J Agric Food Chem. 2015;63:2472. doi: 10.1021/jf5056387. [DOI] [PubMed] [Google Scholar]
- 2.Iwata N, Kitanaka S. J Nat Prod. 2010;73:1203. doi: 10.1021/np900543r. [DOI] [PubMed] [Google Scholar]
- 3.Kantarjian HM, O’Brien S, Cortes J. Clin Lymphoma, Myeloma Leuk. 2013;13:530. doi: 10.1016/j.clml.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forster PG, Ghisalberti EL, Jefferies PR, Poletti VM, Whiteside NJ. Phytochemistry. 1986;25:1377. [Google Scholar]
- 5.Hall SR, Raston CL, Skelton BW, White AH. J Chem Soc Perkin Trans. 1981;211:1467. [Google Scholar]
- 6.Sridharan V, Suryavanshi PA, Menéndez JC. Chem Rev. 2011;111:7157. doi: 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]
- 7.Ward RS. Nat Prod Rep. 1999;16:75. [Google Scholar]
- 8.Cragg GM, Kingston DGI, Newman DJ. Anticancer Agents from Natural Products. 2nd. CRC Press; 2011. [Google Scholar]
- 9.Keller-Juslen C, Kuhn M, Von Wartburg A, Staehelin H. J Med Chem. 1971;14:936. doi: 10.1021/jm00292a012. [DOI] [PubMed] [Google Scholar]
- 10.Jefferies LR, Cook SP. Org Lett. 2014;16:2026. doi: 10.1021/ol500606d. [DOI] [PubMed] [Google Scholar]
- 11.Venning ARO, Bohan PT, Alexanian EJ. J Am Chem Soc. 2015;137:3731. doi: 10.1021/jacs.5b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denmark SE, Kornfilt DJP. J Org Chem. 2017;82:3192. doi: 10.1021/acs.joc.7b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimogaki M, Fujita M, Sugimura T. J Org Chem. 2017;82:11836. doi: 10.1021/acs.joc.7b01141. [DOI] [PubMed] [Google Scholar]
- 14.Mahlthau F, Schuster O, Bach T. J Am Chem Soc. 2005;127:9348. doi: 10.1021/ja050626v. [DOI] [PubMed] [Google Scholar]
- 15.Ammann SE, Rice GT, White MC. J Am Chem Soc. 2014;136:10834. doi: 10.1021/ja503322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddel JCT, Wang W, Koukounas K, Thomson RJ. Chem Sci. 2017;8:2156. doi: 10.1039/c6sc04762a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X-Y, An X-T, Zhao X-H, Du J-Y, Deng Y-H, Zhang X-Z, Fan C-A. Org Lett. 2017;19:2965. doi: 10.1021/acs.orglett.7b01202. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Wu F, Chen J, Nicewicz DA, Huang Y. Angew Chem Int Ed. 2017;56:6896. doi: 10.1002/anie.201702940. [DOI] [PubMed] [Google Scholar]
- 19.Kuwano R, Shige T. J Am Chem Soc. 2007;129:3802. doi: 10.1021/ja070012l. [DOI] [PubMed] [Google Scholar]
- 20.Carreño MC, González-López M, Latorre A, Urbano A. J Org Chem. 2006;71:4956. doi: 10.1021/jo060688j. [DOI] [PubMed] [Google Scholar]
- 21.Lim CS, Quach TT, Zhao Y. Angew Chem Int Ed. 2017;56:7176. doi: 10.1002/anie.201703704. [DOI] [PubMed] [Google Scholar]
- 22.Tsoung J, Krämer K, Zajdlik A, Liébert C, Lautens M. J Org Chem. 2011;76:9031. doi: 10.1021/jo201781x. [DOI] [PubMed] [Google Scholar]
- 23.Webster R, Boyer A, Fleming MJ, Lautens M. Org Lett. 2010;12:5418. doi: 10.1021/ol1022239. [DOI] [PubMed] [Google Scholar]
- 24.Ye B, Cramer N. Synlett. 2015;26:1490. [Google Scholar]
- 25.Gagneux A, Winstein S, Young WG. J Am Chem Soc. 1960;82:5956. [Google Scholar]
- 26.Gagnon D, Lauzon S, Godbout C, Spino C. Org Lett. 2005;7:4769. doi: 10.1021/ol052034n. [DOI] [PubMed] [Google Scholar]
- 27.Lauzon S, Tremblay F, Gagnon D, Godbout C, Chabot C, Mercier-Shanks C, Perreault S, DeSeve H, Spino C. J Org Chem. 2008;73:6239. doi: 10.1021/jo800817p. [DOI] [PubMed] [Google Scholar]
- 28.Maag H, Rydzewski RM. J Org Chem. 1992;57:5823. [Google Scholar]
- 29.Vekariya RH, Liu R, Aubé J. Org Lett. 2014;16:1844. doi: 10.1021/ol500011f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman AK, Colasson BB, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:13444. doi: 10.1021/ja050622q. [DOI] [PubMed] [Google Scholar]
- 31.Askin D, Angst C, Danishefsky S. J Org Chem. 1985;50:5005. [Google Scholar]
- 32.Cardillo G, Fabbroni S, Gentilucci L, Perciaccante R, Piccinelli F, Tolomelli A. Org Lett. 2005;7:533. doi: 10.1021/ol047815n. [DOI] [PubMed] [Google Scholar]
- 33.Takasu H, Tsuji Y, Sajiki H, Hirota K. Tetrahedron. 2005;61:11027. [Google Scholar]
- 34.Craig D, Harvey JW, O’Brien AG, White AJP. Org Biomol Chem. 2011;9:7057. doi: 10.1039/c1ob05972f. [DOI] [PubMed] [Google Scholar]
- 35.Liu R, Gutierrez O, Tantillo DJ, Aubé J. J Am Chem Soc. 2012;134:6528. doi: 10.1021/ja300369c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goswami PP, Suding VP, Carlson AS, Topczewski JJ. Eur J Org Chem. 2016;2016:4805. [Google Scholar]
- 37.Ott AA, Goshey CS, Topczewski JJ. J Am Chem Soc. 2017;139:7737. doi: 10.1021/jacs.7b04203. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt RR, Michel J. Angew Chem Int Ed Engl. 1980;19:731. [Google Scholar]
- 39.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc; 2010. [Google Scholar]
- 40.Adhikari AA, Suzuki T, Gilbert RT, Linaburg MR, Chisholm JD. J Org Chem. 2017;82:3982. doi: 10.1021/acs.joc.7b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]
- 42.Overman LE. J Am Chem Soc. 1974;96:597. [Google Scholar]
- 43.Overman LE. J Am Chem Soc. 1976;98:2901. [Google Scholar]
- 44.Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031. doi: 10.1021/ja0676962. [DOI] [PubMed] [Google Scholar]
- 45.Kirsch SF, Overman LE, Watson MP. J Org Chem. 2004;69:8101. doi: 10.1021/jo0487092. [DOI] [PubMed] [Google Scholar]
- 46.Reznichenko AL, Nguyen HN, Hultzsch KC. Angew Chem Int Ed. 2010;49:8984. doi: 10.1002/anie.201004570. [DOI] [PubMed] [Google Scholar]
- 47.Arnold JS, Nguyen HM. J Am Chem Soc. 2012;134:8380. doi: 10.1021/ja302223p. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, Stockdale DP, Mixdorf JC, Topczewski JJ, Nguyen HM. J Am Chem Soc. 2015;137:11912. doi: 10.1021/jacs.5b07492. [DOI] [PubMed] [Google Scholar]
- 49.Topczewski JJ, Quinn DM. Org Lett. 2013;15:1084. doi: 10.1021/ol400054m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topczewski JJ, Tewson TJ, Nguyen HMJ. J Am Chem Soc. 2011;133:19318. doi: 10.1021/ja2087213. [DOI] [PubMed] [Google Scholar]
- 51.Jiang X, Pan Z, Douglas CJ. Tetrahedron Lett. 2015;56:5324. [Google Scholar]
- 52.Packard MH, Cox JH, Suding VP, Topczewski JJ. Eur J Org Chem. 2017;2017:6365. [Google Scholar]
- 53.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973. [Google Scholar]
- 54.Evans DA, Weber AE. J Am Chem Soc. 1987;109:7151. [Google Scholar]
- 55.King SM, Herzon SB. Alkaloids Chem Biol. 2014;73:161. doi: 10.1016/B978-0-12-411565-1.00003-2. [DOI] [PubMed] [Google Scholar]
- 56.Calandra NA, King SM, Herzon SB. J Org Chem. 2013;78:10031. doi: 10.1021/jo401889b. [DOI] [PubMed] [Google Scholar]
- 57.Jones SB, He L, Castle SL. Org Lett. 2006;8:3757. doi: 10.1021/ol0613564. [DOI] [PubMed] [Google Scholar]
- 58.Herzon SB, Calandra NA, King SM. Angew Chem Int Ed. 2011;50:8863. doi: 10.1002/anie.201102226. [DOI] [PubMed] [Google Scholar]
- 59.Chuang KV, Navarro R, Reisman SE. Angew Chem Int Ed. 2011;50:9447. doi: 10.1002/anie.201104487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schultz AG, Wang A. J Am Chem Soc. 1998;120:8259. [Google Scholar]
- 61.Trost BM, Imi K, Davies IW. J Am Chem Soc. 1995;117:5371. [Google Scholar]
- 62.Gärtner D, Stein AL, Grupe S, Arp J, Jacobi von Wangelin A. Angew Chem Int Ed. 2015;54:10545. doi: 10.1002/anie.201504524. [DOI] [PubMed] [Google Scholar]
- 63.Li B-J, Xu L, Wu Z-H, Guan B-T, Sun C-L, Wang B-Q, Shi Z-J. J Am Chem Soc. 2009;131:14656. doi: 10.1021/ja907281f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.