

Abstract
Osteoarthritis (OA) is a disabling degenerative joint disease that prompts pain with limited treatment options. To permit early diagnosis and treatment of OA, a high resolution mechanistic understanding of human chondrocytes in normal and diseased states is necessary. In this study, we assessed the biological effects of OA-related changes in the synovial microenvironment on chondrocytes embedded within anatomically intact cartilage from joints with different pathological grades by next generation RNA-sequencing (RNA-seq). We determined the transcriptome of primary articular chondrocytes derived from pristine knees and ankles, as well as from joints affected by OA. The GALAXY bioinformatics platform was used to facilitate biological interpretations. Comparisons of patient samples by k-means, hierarchical clustering and principal component analysis reveal that primary chondrocytes exhibit OA grade-related differences in gene expression, including genes involved in cell-adhesion, ECM production and immune response. We conclude that diseased synovial microenvironments in joints with different histopathological OA grades directly alter gene expression in chondrocytes. One ramification of this finding is that sampling anatomically intact cartilage from OA joints is not an ideal source of healthy chondrocytes, nor should they be used to generate a normal baseline for the molecular characterization of diseased joints.
Keywords: GALAXY, Collins grade, cartilage, ECM, cell adhesion
INTRODUCTION
Osteoarthritis (OA) is a complex, multifactorial process that variably and asynchronously affects the integrity of the total joint organ (1). This degenerative disorder diminishes the quality of life for millions of people worldwide, and costs billions of dollars in medical care and lost productivity each year (2). Complex interactions between histological, biochemical, microenvironmental and mechanical changes affecting these tissues limit the clinical ability to identify specific treatments based solely on pathologic state. Identifying the common molecular mechanisms among patients and joints that underlie early clinical symptoms of OA is imperative to facilitate a path of intervention before articular cartilage is permanently degraded and lost. Characterization of changes in gene expression during OA progression at the whole genome level offers opportunities for gaining mechanistic insights that could be translated into disease modifying treatments.
The anatomical and histological cascade of events leading to articular cartilage loss in OA has been established, but the underlying molecular mechanisms that dictate its advancement remain elusive. Early disease modifying events include an initial chondrocyte repair response consisting of increased cell proliferation and extracellular protein synthesis (3). This response is followed by chondrocyte hypertrophy and the amplified production of catabolic cytokines and extracellular matrix (ECM) degrading proteases (4, 5). Degradation of collagens and proteoglycans within the ECM is one of the most characteristic features of OA, and reflects a failure of chondrocytes to balance ECM synthesis and degradation. Loss of cartilage is mediated, in part, by the activation of matrix metalloproteinases (MMPs) that are mechanistically controlled by several cytokine-responsive cell signaling pathways linked to OA (6–8).
Although local inflammation is also typical of OA, increasing evidence suggests that systemic inflammation correlates with disease progression (9). Importantly, acute and/or chronic cycles of inflammation both occur in early OA, before extensive loss of articular cartilage and joint destruction (10–12). Our group has studied the molecular biology of human cartilage and chondrocytes under normal and diseased conditions to define pathways that may promote long-term success of OA mitigation strategies (13–16). A number of natural compounds are being explored for possible use as inhibiting mechanisms that mediate OA-related cartilage degradation and/or pain (17–19).
Microarray transcriptome surveys have identified many genes encoding collagenous and non-collagenous ECM-related proteins and enzymes, as well as many distinct inflammatory genes that are differentially up- or down-regulated in damaged OA tissues (20–23). These modulations in cartilage anabolic and catabolic responses largely reflect a perturbation in ECM remodeling. The goal of the present study is to investigate differentially expressed genes in chondrocytes subjected to diseased synovial joint microenvironments with different histopathological grades. To examine grade-specific gene expression patterns and identify new pathways that are amenable to remediation strategies, we used next generation RNA-sequencing (RNA-seq) of primary articular chondrocytes isolated from independent joints with a full range of disease-severity based on clinically-defined OA grades.
MATERIALS AND METHODS
Intact articular cartilage samples were collected from 23 joints of 17 human cadaver donors through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL), with approval by the local ethics committee and informed consent from each family (ORA#: L03090306). To facilitate independent analysis of samples collected within each distinct synovial environment, we treated each joint as a single sample and cells were isolated from intact (apparently healthy) cartilage. Contamination of hyaline cartilage by other tissue types (e.g., subchondral bone, blood) was avoided by careful dissection of cartilage tissue followed by a limited enzyme digestion. Articular cartilage tissues were harvested from either knee or ankle joints (12 ankles, 11 knees). OA disease states were determined by macroscopic histological examination of the articular cartilage within the entire joint and classified using the Collins grading (G) scheme (24). This well-documented grading system was designed for autopsy tissue to assess lesion depth and distribution, and is also more sensitive to changes during early progression of OA than radiological classifications based on joint-space narrowing (24). For this study, joints were specifically classified using the following criteria: G0 joints displayed normal articular cartilage throughout (n=5), G1 joints had some softer than normal articular cartilage (n=7), G2 joints exhibited extensive superficial damage to articular cartilage (n=6) without damage to bone, G3 joints had areas with total articular cartilage loss and observable penetration underlying bone (n=4), and a G4 joint was identified as having full thickness exposure to bone along the articular surface of the joint (n=1) (Fig. 1a). All tissues were stored at 4°C in phosphate buffered solution until chondrocyte isolation.
Figure 1.

Cartilage tissue sampling. (a) Representative arthroscopic images of Collins grades (G0 thru G4 from left to right) provided by one of the co-authors (individuals were not sampled for this study). (b) a cartoon of the synovial joint microenvironment illustrates sampling of articular cartilage, cartilage defects, synovial membrane, and the joint cavity.
Human primary chondrocytes were harvested from identifiable cartilage tissue that was dissected from cadaver joints with different Collins grades. To isolate viable articular chondrocytes, tissues were shaved with a scalpel from the articulating surface and placed in culture medium (DMEM/F12, 10% FBS, 1% penicillin-streptomycin and 50 μg/mL gentamicin), followed by a double enzymatic digestion (8 g of cartilage per 50 mL of media). For the initial digestion, 200 mg of Pronase (25 KU; Calbiochem, San Diego, CA) was added to 100 mL of medium, and incubated at 37°C for one hour. Tissues were rinsed twice with PBS and collagenase-P (1.7 U/mg; Roche, Mannheim, Germany) was added at a concentration of 360 mg/L, followed by an overnight incubation at 37°C on a shaking platform. The resulting solution was filtered using a BD Falcon cell strainer (70 μm; Corning Inc., Corning, NY). Isolated chondrocytes were rinsed three times with PBS before storage at −80°C in Qiazol, and RNA extraction following protocol of the RNeasy Mini kit (Qiagen, Valencia, CA).
RNA-seq data collection was performed by the Gene Expression Core, part of the Advanced Genomics Technology Center at Mayo Clinic (Rochester, MN). Detailed procedures on the creation of RNA libraries, cDNA synthesis, sequencing and base calling have been described in detail elsewhere (25). To avoid biasing insert size calculation and produce unsorted FASTQ files, the BAM files resulting from sequencing (HiSeq 2000, Illumina, San Diego, CA) were shuffled using the HTSlib package in the Genome Analysis Toolkit (26). The resulting FASTQ files were then processed through an analysis workflow in Galaxy v.15.07 (27, 28). Specifically, FASTQ files were post-processed using groomer and trimmer tools and then aligned using Tophat 2.0.6 (29). Transcript assembly was done using Cufflinks and differential gene expression analysis was conducted using Cuffdiff following protocols detailed in (30, 31). The results obtained in the Cuffdiff analysis were visualized and scrutinized using several data exploration tools available in the R package CummeRbund (32). Among these tools, we utilized distance matrices, heat maps and dimensionality reduction (principal component analysis; PCA) to explore relationships between chondrocytes from joints with distinct OA grades using significantly differentially expressed genes (alpha= 0.05). We additionally employed k-means clustering to identify clusters of genes with similar expression profiles across sample groups. In this analysis we experimented with multiple k-values (k=4, 6, 14; defined a priori) to obtain results that allowed for meaningful biological interpretation of clusters. Gene lists obtained in these analyses where further explored using the functional annotation tool within David v6.7 (33, 34). GeneMania (35) was then used to create an integrated network of differentially expressed genes detected by the Cuffdiff analysis (n=533) and David v6.7 gene ontology sub-category for cytokines (n=30). Default analysis parameters were used, and outputs included specific networks of co-localized genes and genes with shared protein domains.
RESULTS
The complex interaction between histological and biochemical changes involved in OA has limited the ability to identify stage-specific treatments based solely on joint pathology. To understand molecular changes during OA progression, we performed RNA-seq analysis on chondrocytes isolated from identifiable cartilage tissues within a cohort of joints with distinct grades of OA (Fig. 1). Primary articular chondrocytes were isolated from knee and ankle joints of elderly male and female donors (19 tissues; 14 donors; age range=60 to 77 years; average age=70.6 years; median=71 years; mode=75 years) with different Collins grades (Fig. 2). The cohort was supplemented with four tissues from three younger donors to improve representation of Collins grades and anatomic location (Fig. 2). No two samples were collected from within the same joint, although some samples were obtained from different joints of the same donor. The majority of samples were from male donors (20 tissues; 14 donors), but chondrocytes from females were also included in the analysis to bolster grade representation (3 tissues; 3 females). In summary, we obtained multiple samples for each disease grade (i.e., n=5, n=7, n=4, n=4 for G0, G1, G2 and G3, respectively), except G4 (n=1) (Fig. 2). Primary chondrocytes were harvested from carefully cleaned cartilage specimens and total poly-adenylated mRNA was isolated for paired end RNA-seq analysis (RNA integrity scores >7). To permit statistical analysis of RNA-seq data from chondrocytes, we grouped multiple donors by joint OA grade regardless of demographics.
Figure 2.
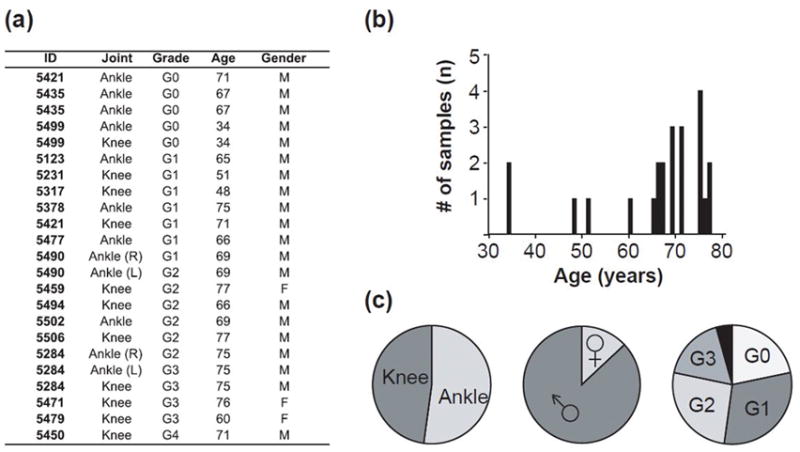
Cartilage donor information. (a) A complete list of samples used to isolate chondrocytes from articular cartilage. Ankle and knee joints were used, and tissues from both the left (L) and right (R) leg were sampled in two individuals. Grades (G) were defined by a clinical pathologist using the Collins grading scale which categorizes G0 as pristine healthy cartilage, and G4 as the most severely diseased; (b) numbers of samples by age; (c) proportional comparisons of samples by anatomic location, gender and grade (a single G4 sample is depicted in black).
RNA isolation from cartilage is challenging because only 2–5% of the tissue is comprised of living chondrocytes (36), but we reliably obtained high-resolution transcriptome data from primary cells derived from articular cartilage. Although previous studies used paired samples from within the same joint (13), our study compares RNA derived from chondrocytes isolated from cartilage specimens that were subjected to independent synovial microenvironments in joints with distinct Collins grades. Our experimental protocol accounts for microenvironmental influences that could be obscured if samples are collected from within the same synovial joint space, and thereby permits a direct focus on disease-related effects of cartilage damage and the surrounding synovial fluid on chondrocytes. RNA-seq data were obtained from chondrocytes in each cartilage specimen and raw reads (~20 to 25 million reads) were initially mapped against the human genome scaffold (hg19 assembly) to yield a matrix of expression values for 23,338 genes in 23 samples.
To specifically test the differences and similarities between expression patterns for chondrocytes from joints with distinct Collins grades, we examined the Jensen-Shannon divergence to measure pairwise similarity between samples based on probability distribution, principal component analysis to assess the inherent variability in expression data, and heat map to identify differentially expressed genes between RNA samples from different Collins grades. For each of these analyses, we remapped reads at a high level of stringency using a TopHat2-Cufflinks-Cuffdiff pipeline (30, 31, 37, 38) and probability of Type I error (alpha=0.05) within the Galaxy platform (Fig. 3). Jensen-Shannon distance estimations revealed that the comparison between samples with joint Collins grades G0 and G1 yields the highest number of differentially expressed genes (n=422). The comparison between primary chondrocytes from G0 and G2 joints uncovered a similar number of genes with different expression values (n=393). Jensen-Shannon distance estimations calculated in a pairwise similarity matrix between conditions also indicated G1 as the most dissimilar sample relative to others (0.139–0.157; Fig. 4a).
Figure 3.
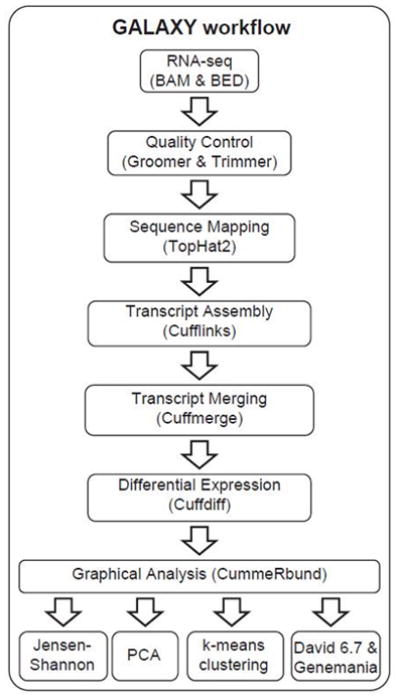
GALAXY bioinformatics workflow showing intermediate steps, software packages and outputs.
Figure 4.
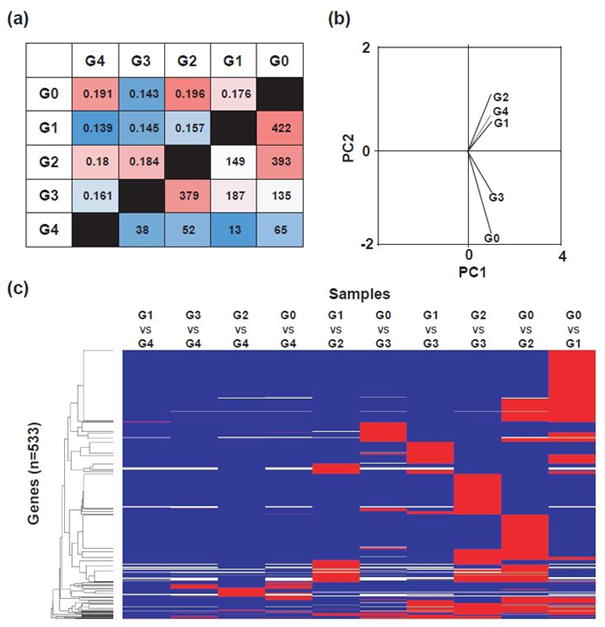
Pairwise comparisons of chondrocyte data by histological grade. (a) Significance matrix highlighting elevated numbers of genes differentially expressed (alpha = 0.05) between groups of chondrocytes from G0 vs G1 & G2 and G2 vs G3 (below diagonal) and J–S distance values (above diagonal). (b) PCA of samples on a set of significant genes. (c) Heat map of 533 significantly different genes among treatment groups (G0–G4). Blue = not significant in this comparison, red = significant in this comparison.
The principal component analysis conducted within CummeRbund provides additional support for these findings, because the gene expression profiles of chondrocyte RNA samples from joints with Collins grades G1 and G2 were highly correlated, while G0 and G3 chondrocytes group together (Fig. 4b). For illustration, we note that the gene expression pattern of a single G4-derived chondrocyte sample appeared to correlate with that of G1 and G2, but this result remains uninformative because of our inability to acquire other G4 samples. More importantly, the principal component analysis result (Fig. 4b) was consistent with the interpretation that G1 and G2 samples have unique expression features that were also detected by analysis of Jensen–Shannon divergence (Fig. 4a). Furthermore, heat-mapping highlighted that the bulk of genes with significant differences (in red) are seen in comparisons between Collins grades G0 versus G1, G0 versus G2, G2 versus G3 and G1 versus G3 (Fig. 4c). The main biological interpretation that can be derived from these global expression analyses is that major differences in gene expression are observed in chondrocytes that have been subjected to a microenvironment influenced by the histopathological damage evident in joints with Collins grades G1 and G2. Collectively, these results are consistent with extensive ECM remodeling and repair in response to anatomical joint damage.
To investigate how gene expression profiles in primary chondrocytes differ between joints with distinct Collins grades, we analyzed gene expression values across our samples by k-means clustering (Fig. 5). Expression patterns were categorized in multiple bins (k=14). Two gene clusters present clear maxima in transcript levels in Collins grade G2 (i.e., cluster 6, n=336 genes; cluster 11, n=62 genes), while one gene cluster had highest expression in Collins grade G1 (cluster 14; n=67 genes). Functional annotation analyses by gene ontology (GO) terms for these clusters revealed over-representation of cell adhesion-related genes in cluster 6 (GO: 0007155) and cell cycle markers in cluster 11 (GO: 007049). Cluster 14 was enriched with immune-response genes (GO: 0006955), cytokine activity (GO: 0005125) and extracellular functions (GO: 0005576). In summary, k-means clustering combined with gene ontology analysis demonstrated that the global gene expression changes we observed for G1- and G2-associated primary chondrocytes are related to specific changes in expression of different classes of genes supporting extracellular matrix synthesis/production and immune-responses.
Figure 5.
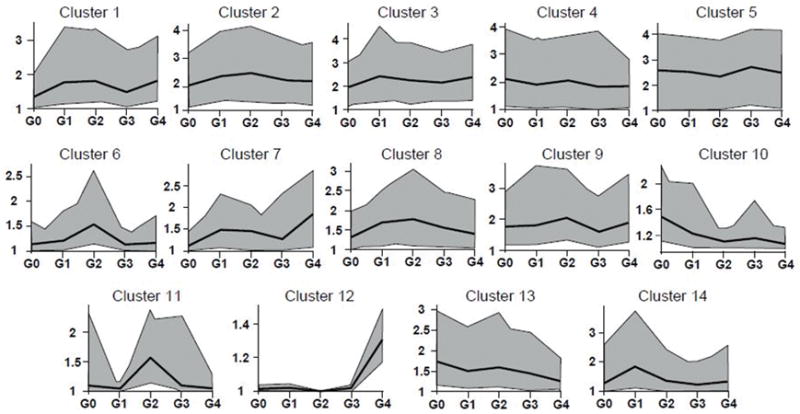
K-means clustering of samples by Cuffdiff analyses yields 14 clusters of genes differentially expressed between different OA grades. To facilitate the presentation, multiple individual lines from numerous individual genes are simplified into a single gray polygon that encompasses these lines. The general shape of these lines follows the expression average for each grade that is represented by the black line within the polygon.
To complement k-means clustering and permit biological interpretations relative to OA disease states, we further interrogated the clustering analysis to assess specific genes with similar functional categories within selected k-means groupings. We focused on genes that increase or decrease expression with increasing Collins grades, or genes that show a biphasic correlation (i.e., increasing and decreasing). Expression of genes related to inflammation, kinase activity, ossification, ECM, and MMP activity decreased in primary chondrocytes with increasing joint OA grade (Fig. 6a). Another category of differentially expressed genes, which were only elevated in joints with intermediate OA Collins grades, are related to cytokine activity, metallopeptidase activity, basement membrane production, phosphorylation, and inflammation (Fig. 6b). Other genes linked to ECM and MMP activity (e.g., ADAMTS15, LAMC2, MMP10), as well as several genes related to phosphorylation increased with increasing OA grade (Fig. 6c). One interpretation we derive from these data is that both structural ECM proteins (i.e., collagens and non-collagenous proteins) and enzymes that support ECM remodeling and repair (i.e., MMP and ADAMTS proteins) are differentially regulated when chondrocytes are exposed to distinct joint conditions (i.e., due to Collins grade). However, cartilage cells respond to their changing environment by switching isoform production with increasing Collins grade as inflammatory pathways and tissue repair processes are initiated, sustained and/or re-initiated. Our findings suggest that there are very dynamic and intricate changes in gene expression with OA progression. Because most prominent changes in gene expression in chondrocytes occur when joints transition from G0 to Collins grades 1 and 2, there is an opportunity for pharmacological inhibition to mitigate the symptoms of OA in patients with limited OA damage.
Figure 6.
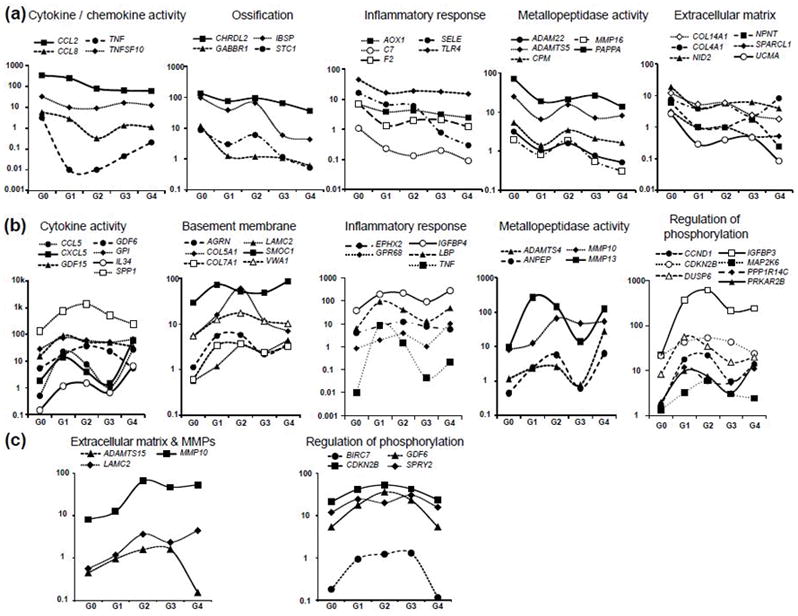
Illustrative line graphs showing the relative expression (Y-axis in arbitrary units) of select differentially expressed genes as defined by K-means clustering. The graphs show (a) decreased expression of cytokine/chemokine activity, ossification, inflammatory response, metallopeptidase activity, and extracellular matrix (ECM) markers (specified in the analysis as G0>G1 and G0>G2); (b) expression of genes related to cytokine activity, basement membrane, inflammatory response, metallopeptidase activity, and regulation of phosphorylation markers that are elevated in one or more grades of OA (specified in the analysis as G0<G1 AND G0<G2 AND G2>G3 OR G0<G1 AND G1>G2 AND G1<G3); (c) genes related to ECM and MMP activity or phosphorylation pathways that are consistently elevated in OA samples (specified in the analysis as G0<G1 and G0<G2 and G1<G3 OR G0<G1 and G0<G2 and G2<G3). These line drawings are presented for illustrative purposes and represent the average RNA-seq values for multiple samples except G4 which is a singular sample. Error bars are omitted for clarity because of excessive overlay with the primary lines within the graphs. We note that expression trends were consistent across the grades.
David and GeneMania were again used to characterize functional pathways involving interrelated genes that are significantly differently expressed (starting with the list of 533 genes generated above). As one striking example, we found that these genes are enriched for many cytokines and chemokines (n=30). The relationships of these differentially expressed genes are further reflected by similarities in co-localization (Fig. 7a) and shared protein domains (Fig. 7b). These findings are consistent with the activation of inflammatory and/or tissue repair pathways in joints with Collins scores greater than zero.
Figure 7.
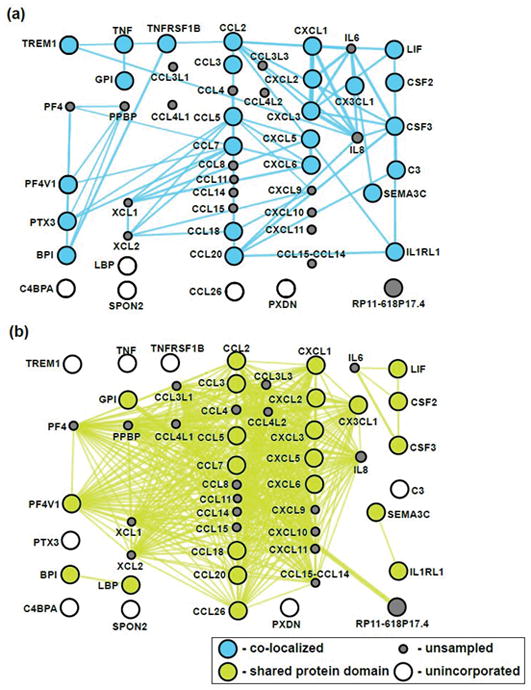
Gene ontology networks of all differentially expressed genes (n=533) across all samples (as defined by Cuffdiff analysis) were visualized by GeneMania (35) to generate integrated networks for cytokines (n=30) as a disease-related category relevant to tissue repair and inflammatory processes operative in synovial joints affected by OA progression. The presentation was customized to emphasize logical gene relationships focuses on (a) proteins known to co-localize and (b) have shared protein domains.
DISCUSSION
Our study examined the molecular signature of chondrocytes isolated from articular cartilage tissues in either healthy or OA-afflicted joints by whole-transcriptome (RNA-seq) analysis. Using bioinformatics, we identified genes and gene ontology categories characteristic of chondrocytes that respond to the microenvironment of joints with different pathological grades. Our results corroborate the concept that OA development is concomitant with whole-joint changes that trigger molecular responses in articular chondrocytes derived from undamaged cartilage within the joint. Because the most prominent changes in gene expression were seen in donors with early stages of OA, this patient population may be most amenable to discovery efforts aimed at defining molecular targets that prevent OA progression. However, patients with G1 joints do not typically seek treatment, so following at-risk populations (e.g., post-traumatic ACL injury) may be most pertinent for testing strategies of early detection and intervention.
Gene ontology categories of genes differentially expressed in donors with early or late OA are related to basic cell functions (e.g., cell cycle, apoptosis, cellular metabolism, RNA binding). Thus, chondrocyte proliferation and/or survival may be compromised. Several of the genes that are modulated in joints with different OA grades have been examined in mouse models of OA (39, 40). For example, mutations in ECM genes (e.g., COL2, MMP13) cause mice to develop OA (41). MMP13 is controlled by other regulatory proteins (e.g., basic fibroblast growth factor) that alter the phenotype of human articular chondrocytes (42). Disease modifying OA-events are co-regulated. The biological pathways linked to modified gene signatures include enzymatic components (e.g., kinases, phosphatases, and acetylases) that are amenable to drug inhibition and may support development of novel therapeutic targets for reducing the severity of OA symptoms, including pain. Our findings are consistent with previous studies showing that genetic determinants, epigenetic chromatin-related mechanisms, transcriptional regulators, microRNAs and long non-coding RNAs, as well as mediators of inflammation, pain, angiogenesis, joint lubrication, cyto-protective responses to oxidative stress and mitochondrial function collectively contribute to OA progression (43–49).
Differences between healthy (G0) and G1 chondrocytes are reflected by reductions in their ability to produce cytokine/chemokine signals, ECM, ossification and metallopeptidase activity. Because cell-matrix contact regulates chondrocyte activity, failure to produce ECM would elicit a negative feedback on chondrocyte growth and maturation. Furthermore, failing to produce appropriate cytokine/chemokine signals would compound the effect of a poor matrix, by either failing to recruit other chondrocytes, or recruiting inappropriate cell types. In addition to the ECM, the pericellular matrix is a critical component of the chondrocyte microenvironment (50). Reductions in metallopeptidase activity, as suggested by the RNA-seq analysis, would be particularly important to consider in patients with a diminished ability to regulate metal ions.
Gene expression patterns underlying OA pathology have been observed in human tissues using microarrays (51) and more recently RNA-seq analysis (52). Typically, transcriptome surveys have been previously conducted on paired, macroscopically damaged and non-damaged tissues sampled from within the same diseased joint (20–23, 53). The limitation of this approach is that any cartilage or chondrocytes sampled will potentially be in contact with synovial fluid of the inflamed degenerating joint. Hence, gene expression in normal appearing tissue within a damaged joint may be affected by gene regulatory cell signaling events that are triggered by inflammatory cytokines. Our cartilage sampling strategy was designed to isolate effects of the synovial microenvironment by comparing cartilage specimens from unaffected limb joints with a cohort of joints with different degrees of cartilage damage. Because expression signatures of chondrocytes from joints with different pathological grades differs, it appears chondrocytes are sensitive to the synovial microenvironments, in particular in cells from G1 joints that initiate expression of inflammatory signals that accompany biological tissue-repair programs.
By examining isolated chondrocytes from the potential contaminants normally found in a synovial joint, we observed OA-related and cell-autonomous transcriptional modulations due to changes in the joint environment. Our analysis involves a large RNA-seq dataset of more than 23 joints that validates, complements and extends previous findings in the field. Yet, we appreciate that our cohort remains limited considering stratification of our results over five different pathological grades (Collins G0 to G4). The resulting small number of specimens per category generates statistical limitations. While our sampling limitations reduce our statistical confidence in categorical changes in gene expression, a strength of our analysis is that we detected gene expression patterns that are linked to disease-related pathways. We also note that in order to obtain a sufficient sample size for meaningful biological interpretations, we combined data from both ankle and knee joints, as well as for different patient demographic parameters. In particular, it was difficult to obtain pristine cartilage (Collins grade G0) because joints without evidence of OA are very rare in deceased donors (i.e., typically of advanced age) and have significant clinical value as allograft tissue for surgical cartilage repair. Future expansion of this cohort with more samples will further balance the data matrix and provide additional statistical power to permit stratification of gene expression patterns by anatomic location, gender, age and ethnicity as discussed previously (54, 55).
Chondrocytes from diseased joints clearly have disease-related gene expression signatures that correlate with different Collins grades. Nevertheless, we cannot fully exclude the possibility that expression patterns seen here are at least in part affected by the biological circumstance of each specimen during RNA isolation. Ideally, RNA should be harvested instantly to ensure that the molecular signature reflects live tissue. However, the biological condition of the patient at time of death, the storage and shipment of the specimen prior to harvest, as well as the tissue digestion procedure to obtain primary chondrocytes are likely to contribute to the expression signatures we observed here. Regardless of these constraints, our samples were collected using standard protocols that permit direct comparisons that revealed OA-related differences.
One finding of our study is that the joint microenvironment clearly affects gene expression in undamaged cartilage segments from joints affected by OA with different Collins grades. In contrast to other approaches that utilize tissue biopsies or samples from within the same microenvironment, our approach allows for increased temporal and molecular resolution in OA joints with low Collins-grades. The observed changes in gene expression may be a direct reflection of alterations in synovial fluid that are known to occur during OA and these changes could be monitored clinically. We are optimistic that further examination of gene expression differences between normal and early stage arthritis will be informative and facilitate identification of specific target genes to prevent disease progression to later stages of OA.
Supplementary Material
Acknowledgments
Ethics approval and consent to participate
All human-derived materials used in this study were obtained and handled in accordance with ethical guidelines and informed consent to participate: Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL) (ORA#: L03090306).
Availability of data and materials
The dataset supporting the conclusions of this article are available upon request until they are made publicly available per the regulations and requirements of NIH funded studies.
Competing interests
David Lewallen reports personal fees and other from Stryker, Pipeline Biomedical, Zimmer, and Ketai Medical Devices. In addition, DGL has patents on selected hip and knee implants with royalties paid by Zimmer, and is employed part time as the Medical Director for The American Joint Replacement registry. None of the other authors have conflicts of interest to disclose.
Funding
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases [R01 AR049069 to AJVW; T32 AR056950 to EAL; R03 AR066342 to ANL; R01 AR068103 to JJW; R01 AR062136 to HJI; R01 DE020194 to JJW and CAB]; VA Merit Award [BX002647 to HJI]; Mayo Clinic Center for Regenerative Medicine [Training and Career Development Award to JS]; and the generous philanthropic support from the William H. and Karen J. Eby Foundation.
Authors’ contributions
Study design: AJVW, HJI, EAL, CAB, AAL
Data collection: XL, EAL, CAB
Interpretation of data: JS, MK, ANL, DGL, SMC, AJK, JJW, AAL, HJI, AJVW
Drafting of article: AJVW, HJI, EAL, CAB, JS, MK, ANL, AJK
Final approval: AJVW, HJI, AAL, EAL, CAB
We thank members of the van Wijnen, Sampen, and Westendorf laboratories, and in particular Scott Riester and Elizabeth Bradley, for stimulating discussions and/or assistance with reagents and procedures. We thank the Gift of Hope Organ Tissue Donor Network as well as Drs. Chubinskaya and Margulis for making human tissues available, and we also extend our appreciation to the family members of the tissue donors who made it possible.
ABBREVIATIONS
- OA
osteoarthritis
- MMP
matrix metalloproteinases
- G
Collins grade
- PCA
principal component analysis
- GO
gene ontology
References
- 1.Burr DB, Gallant MA. Bone remodelling in osteoarthritis. Nat Rev Rheumatol. 2012;8(11):665–73. doi: 10.1038/nrrheum.2012.130. [DOI] [PubMed] [Google Scholar]
- 2.Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. 2014;10(7):437–41. doi: 10.1038/nrrheum.2014.44. [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB. Update on the biology of the chondrocyte and new approaches to treating cartilage diseases. Best Pract Res Clin Rheumatol. 2006;20(5):1003–25. doi: 10.1016/j.berh.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Goldring MB. Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep. 2000;2(6):459–65. doi: 10.1007/s11926-000-0021-y. [DOI] [PubMed] [Google Scholar]
- 5.Appleton CTG, Usmani SE, Pest MA, Pitelka V, Mort JS, Beier F. Reduction in Disease Progression by Inhibition of Transforming Growth Factor α–CCL2 Signaling in Experimental Posttraumatic Osteoarthritis. Arthritis & Rheumatology. 2015;67(10):2691–701. doi: 10.1002/art.39255. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Ellman MB, Kroin JS, Chen D, Yan D, Mikecz K, et al. Species-specific biological effects of FGF-2 in articular cartilage: implication for distinct roles within the FGF receptor family. J Cell Biochem. 2012;113(7):2532–42. doi: 10.1002/jcb.24129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan D, Chen D, Cool SM, van Wijnen AJ, Mikecz K, Murphy G, et al. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res Ther. 2011;13(4):R130. doi: 10.1186/ar3441. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Muddasani P, Norman JC, Ellman M, van Wijnen AJ, Im HJ. Basic fibroblast growth factor activates the MAPK and NFkappaB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J Biol Chem. 2007;282(43):31409–21. doi: 10.1074/jbc.M706508200. [DOI] [PubMed] [Google Scholar]
- 9.Berenbaum F. Osteoarthritis: Physiopathology and Risk Factors for Progression. Osteoporosis Int. 2014;25:S142–S3. [Google Scholar]
- 10.Benito MJ, FitzGerald O, Bresnihan B. Inmunohistologic analysis of synovial tissue from early and late osteoarthritic patients. A potential role for COX-2 and NF-kappa B1 (p50) regulation in early disease. Arthritis Res Ther. 2002:4. [Google Scholar]
- 11.Benito MJ, Veale DJ, Fitzgerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–7. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Y, Tuan RS. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat Rev Rheumatol. 2015;11(4):206–12. doi: 10.1038/nrrheum.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonin CA, Lewallen EA, Baheti S, Bradley EW, Stuart MJ, Berry DJ, et al. Identification of differentially methylated regions in new genes associated with knee osteoarthritis. Gene. 2015 doi: 10.1016/j.gene.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Gibson G, Kim JS, Kroin J, Xu S, van Wijnen AJ, et al. MicroRNA-146a is linked to pain- related pathophysiology of osteoarthritis. Gene. 2011;480(1–2):34–41. doi: 10.1016/j.gene.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Im HJ, Li X, Chen D, Yan D, Kim J, Ellman MB, et al. Biological effects of the plant-derived polyphenol resveratrol in human articular cartilage and chondrosarcoma cells. J Cell Physiol. 2012;227(10):3488–97. doi: 10.1002/jcp.24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Wijnen A, Westendorf J. Osteoporosis and osteoarthritis. Preface. Methods Mol Biol. 2015:1226:v. [PubMed] [Google Scholar]
- 17.Yan D, Chen D, Hawse JR, van Wijnen AJ, Im HJ. Bovine lactoferricin induces TIMP-3 via the ERK1/2-Sp1 axis in human articular chondrocytes. Gene. 2013;517(1):12–8. doi: 10.1016/j.gene.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan D, Chen D, Shen J, Xiao G, van Wijnen AJ, Im HJ. Bovine lactoferricin is anti- inflammatory and anti-catabolic in human articular cartilage and synovium. J Cell Physiol. 2013;228(2):447–56. doi: 10.1002/jcp.24151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Kroin JS, Kc R, Gibson G, Chen D, Corbett GT, et al. Altered spinal microRNA-146a and the microRNA-183 cluster contribute to osteoarthritic pain in knee joints. J Bone Miner Res. 2013;28(12):2512–22. doi: 10.1002/jbmr.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T, Konomi K, Yamasaki S, Aratani S, Tsuchimochi K, Yokouchi M, et al. Comparative analysis of gene, expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006;54(3):808–17. doi: 10.1002/art.21638. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson C, Dehne T, Lindahl A, Brittberg M, Pruss A, Sittinger M, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010;18(4):581–92. doi: 10.1016/j.joca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Barter MJ, Swan DC, Rankin KS, Rowan AD, Santibanez-Koref M, et al. Comparison of Osteoarthritis and Normal Hip Cartilage Transcriptomes Using Rna-Seq Reveals New Candidate Gene Targets and Associated Pathways. Osteoarthr Cartilage. 2012;20:S43-S. doi: 10.1016/j.joca.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Ramos YFM, den Hollander W, Bovee JVMG, Bomer N, van der Breggen R, Lakenberg N, et al. Genes Involved in the Osteoarthritis Process Identified through Genome Wide Expression Analysis in Articular Cartilage; the RAAK Study. Plos One. 2014;9(7) doi: 10.1371/journal.pone.0103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brismar B, Wredmark T, Movin T, Leandersson J, Svensson O. Observer reliability in the arthroscopic classification of osteoarthritis of the knee. Journal of Bone & Joint Surgery, British Volume. 2002;84(1):42–7. doi: 10.1302/0301-620x.84b1.11660. [DOI] [PubMed] [Google Scholar]
- 25.Dudakovic A, Camilleri E, Riester SM, Lewallen EA, Kvasha S, Chen XY, et al. High-Resolution Molecular Validation of Self-Renewal and Spontaneous Differentiation in Clinical-Grade Adipose- Tissue Derived Human Mesenchymal Stem Cells. J Cell Biochem. 2014;115(10):1816–28. doi: 10.1002/jcb.24852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–5. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goecks J, Nekrutenko A, Taylor J, Team G. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8) doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks (vol 7, pg 562, 2012) Nature Protocols. 2014;9(10):2513. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goff LA, Trapnell C, Kelley D. R Package Version 22. 2012. CummeRbund: visualization and exploration of Cufflinks high- throughput sequencing data. [Google Scholar]
- 33.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(suppl 2):W214–W20. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldring MB. Update on the biology of the chondrocyte and new approaches to treating cartilage diseases. Best Practice & Research Clinical Rheumatology. 2006;20(5):1003–25. doi: 10.1016/j.berh.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 37.Kim MK, Oh JY, Il Lee H, Ko JH, Lee HJ, Wee WR, et al. Antigenicity of porcine cornea in xenocorneal transplantation. Cornea. 2008;27(8):S12-S. [Google Scholar]
- 38.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–U174. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon P, Beier F. Novel Insights into Osteoarthritis Joint Pathology from Studies in Mice. Curr Rheumatol Rep. 2015;17(8):1–11. doi: 10.1007/s11926-015-0524-1. [DOI] [PubMed] [Google Scholar]
- 40.Piel MJ, Kroin JS, van Wijnen AJ, Kc R, Im HJ. Pain assessment in animal models of osteoarthritis. Gene. 2014;537(2):184–8. doi: 10.1016/j.gene.2013.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang H, Beier F. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat Rev Rheumatol. 2014;10(7):413–21. doi: 10.1038/nrrheum.2014.46. [DOI] [PubMed] [Google Scholar]
- 42.Im HJ, Muddasani P, Natarajan V, Schmid TM, Block JA, Davis F, et al. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cdelta pathways in human adult articular chondrocytes. J Biol Chem. 2007;282(15):11110–21. doi: 10.1074/jbc.M609040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.den Hollander W, Ramos YFM, Bomer N, Elzinga S, van der Breggen R, Lakenberg N, et al. Transcriptional Associations of Osteoarthritis-Mediated Loss of Epigenetic Control in Articular Cartilage. Arthritis & Rheumatology. 2015;67(8):2108–16. doi: 10.1002/art.39162. [DOI] [PubMed] [Google Scholar]
- 44.Lee AS, Ellman MB, Yan D, Kroin JS, Cole BJ, van Wijnen AJ, et al. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene. 2013;527(2):440–7. doi: 10.1016/j.gene.2013.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller RE, Miller RJ, Malfait AM. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014;70(2):185–93. doi: 10.1016/j.cyto.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lotz M. Osteoarthritis year 2011 in review: biology. Osteoarthritis Cartilage. 2012;20(3):192–6. doi: 10.1016/j.joca.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vo N, Niedernhofer LJ, Nasto LA, Jacobs L, Robbins PD, Kang J, et al. An overview of underlying causes and animal models for the study of age-related degenerative disorders of the spine and synovial joints. J Orthop Res. 2013;31(6):831–7. doi: 10.1002/jor.22204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyaki S, Asahara H. Macro view of microRNA function in osteoarthritis. Nat Rev Rheumatol. 2012;8(9):543–52. doi: 10.1038/nrrheum.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu M, Huang G, Zhang Z, Liu J, Huang Z, Yu B, et al. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthritis and Cartilage. 2015;23(3):423–32. doi: 10.1016/j.joca.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Wilusz RE, Sanchez-Adams J, Guilak F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014;39:25–32. doi: 10.1016/j.matbio.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54(11):3533–44. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 52.Lin Y, Lewallen EA, Camilleri ET, Bonin CA, Jones DL, Dudakovic A, et al. RNA-seq analysis of clinical-grade osteochondral allografts reveals activation of early response genes. Journal of orthopaedic research. 2016 doi: 10.1002/jor.23209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geyer M, Grassel S, Straub RH, Schett G, Dinser R, Grifka J, et al. Differential transcriptome analysis of intraarticular lesional vs intact cartilage reveals new candidate genes in osteoarthritis pathophysiology. Osteoarthritis Cartilage. 2009;17(3):328–35. doi: 10.1016/j.joca.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Boyan BD, Tosi LL, Coutts RD, Enoka RM, Hart DA, Nicolella DP, et al. Addressing the gaps: sex differences in osteoarthritis of the knee. Biol Sex Differ. 2013;4(1):4. doi: 10.1186/2042-6410-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reynard LN, Loughlin J. Insights from human genetic studies into the pathways involved in osteoarthritis. Nat Rev Rheumatol. 2013;9(10):573–83. doi: 10.1038/nrrheum.2013.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.