

Introduction
The major physiological role of platelets is in the formation of hemostatic plugs at sites of penetrating vascular injury that serve to limit blood loss. The aberrant intravascular activation of platelets can, if unchecked, lead to thrombotic events that cause myocardial infarction and stroke. A number of antiplatelet agents are used clinically to limit platelet activation in patients at risk of arterial thrombotic events. However, their use can be associated with a significant risk of bleeding. An improved understanding of platelet signaling mechanisms should identify safer targets for antiplatelet therapy.
Our understanding of the breadth and complexity of signaling pathways that marshal platelet activation has expanded rapidly over the past decade1–4. Recent work published in Arteriosclerosis, Thrombosis, and Vascular Biology (ATVB) and other journals has provided further insight into the regulation of platelet signaling events and identified new targets against which to develop novel antiplatelet agents.
Purinergic Receptors
One of the cornerstones of current antiplatelet therapy targets ADP mediated platelet activation and aggregation via the P2Y12 receptor. However, a major challenge of the thienopyridine-based P2Y12 inhibitors, such as clopidogrel and prasugrel, is the occurrence of high on-treatment platelet reactivity, defined as a higher than expected platelet response to agonist5. Armstrong and colleagues have recently described an important contribution of platelet turnover to high on-treatment platelet reactivity6. In vitro and ex vivo studies demonstrated that the relatively short half-life of clopidogrel and prasugrel was associated with poor inhibition of aggregation of newly formed reticulated platelets on ADP stimulation6. By comparison non-thienopyridine based P2Y12 inhibitors, such as ticagrelor, maintained a good level of platelet inhibition even when untreated platelets were introduced6. However, further studies are required to establish if there is any clinical benefit of non-thienopyridine based P2Y12 inhibitors in the context of high on-treatment platelet reactivity.
In addition to expressing P2Y12 platelets also express P2Y1, thought to regulate initial Ca2+ mobilization and shape change. Currently available therapies, however, only target P2Y12 leaving P2Y1 mediated platelet activation intact. The diadenosine tetraphosphate derivative, GLS-409, has recently been developed as an inhibitor of both P2Y1 and P2Y12 (Figure) (Table)7. GLS-409 potently inhibited platelet aggregation and protected against recurrent coronary thrombosis in a canine model8. Encouragingly, bleeding was only marginally, and non-significantly, prolonged after administration of GLS-409 in a canine model. It remains to be determined if dual P2Y1/P2Y12 antagonists will be used in the clinic in place of the classical unimodal P2Y12 inhibitors9.
Figure. Platelet signaling pathways.
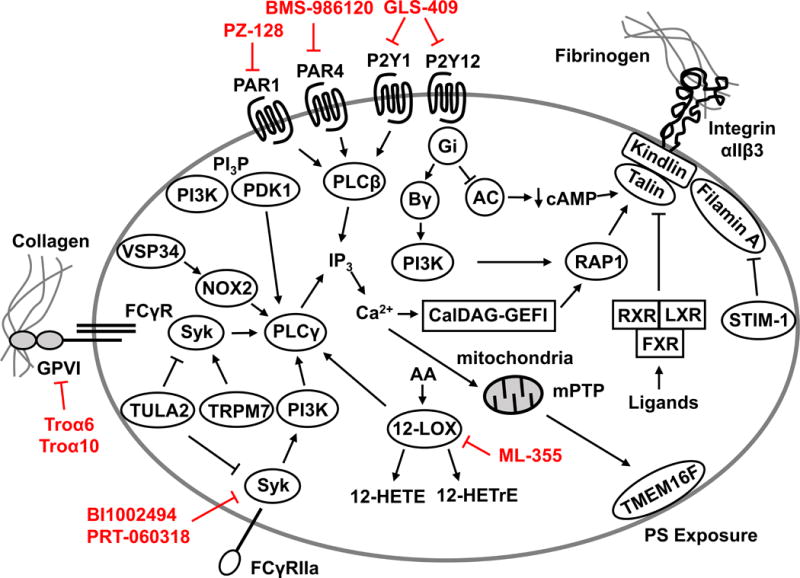
Overview of some of the intracellular platelet signaling pathways required for platelet activation. Novel anti-platelet agents in various stages of preclinical and clinical development are annotated (red).
Table.
Novel antiplatelet agents under development
P2Y12-dependent platelet activation also plays a role in the pathologic response to sepsis. Wild-type mice treated with clopidogrel and P2Y12 deficient mice had reduced platelet aggregation, platelet-leukocyte aggregate formation and lung injury in a sepsis-induced inflammation model10. Interestingly, no protection was observed in P2Y1 deficient mice, which indicates that platelet activation in response to septic challenge is primarily driven by activation of P2Y1210.These findings are consistent with a study showing reduced neutrophil recruitment and lung injury in tricagrelor treated mice subject to abdominal sepsis. Together, these studies suggest that platelets contribute to sepsis-induced lung injury by enhancing the recruitment of neutrophils11.
Protease-activated receptors (PARs)
Thrombin is an extremely potent platelet agonist activating human platelets by proteolytic cleavage of PAR1 and PAR4, which are high and low affinity receptors, respectively (Figure)12. Interestingly, PAR1 activation leads to rapid and transient signaling whereas PAR4 activation leads to prolonged signaling that is required for stable thrombus formation13. Two PAR1-specific antagonists, vorapaxar and atopaxar, have been developed14, 15. Recent studies analyzed the effect of triple antiplatelet therapy on cardiovascular death by adding vorapaxar to standard antiplatelet therapy (aspirin and clopidogrel). Patients receiving triple therapy had reduced cardiovascular death but also had an increase in intracranial hemorrhage14, 16. Nevertheless, vorapaxar has been approved for use in patients with a history of cardiovascular disease with no history of a stroke but it needs to be used with another platelet inhibitor.
Although effective in inhibiting PAR1 activation, a limitation of vorapaxar is its long half-life and slow off rate. This limitation has driven the search for alternative PAR1 antagonists with improved pharmacokinetic profiles. One such agent is the cell-permeable peptide based inhibitor PZ-128, which binds to the intracellular C-terminus of PAR1 and blocks downstream G-protein signaling (Figure) (Table)17. In preclinical studies, PZ-128 inhibited platelet aggregation and thrombus formation17. In a recent phase I study published in ATVB, intravenous administration of PZ-128 resulted in inhibition of platelet activation18. These studies indicate that PZ-128 is a rapid, specific and reversible inhibitor that can be used for short-term inhibition of platelet activation.
To date, the majority of studies have focused on PAR1. Recently, there has been a shift to understanding the contribution of PAR4 to platelet activation. For instance, a recent study identified seven PAR4 variants in a cohort of 236 cardiac patients. One of these PAR4 variants, Tyr157Cys, was predicted to lead to significant structural changes and partial loss of function19. Indeed, platelets isolated from patients with the Tyr157Cys PAR4 variant demonstrated impaired aggregation in response to a PAR4 specific agonist and thrombin. Interestingly, the Tyr157Cys PAR4 variant localized primarily to the perinuclear space likely limiting PAR4 availability at the plasma membrane19. In addition, a racially dimorphic gain-of-function PAR4 variant Ala120Thr has been described20, 21. This PAR4 variant is associated with increased PAR4 reactivity and calcium flux in platelets21. These clinical studies highlight the potential for genetic diversity in PAR4 in the human population.
PAR4 is also being considered as an alternative target for the development of antiplatelet agents. Indeed, a first-in-class small molecule PAR4 inhibitor, BMS-986120, has been developed (Table)22. In a non-human primate model, BMS-986120 potently inhibited arterial thrombosis with only a minor prolongation of bleeding time22. In a recent phase I clinical trial published in ATVB, BMS-986120 was well tolerated and inhibited both platelet aggregation and thrombosis ex vivo23. It will be interesting to see if these PAR1 and PAR4 inhibitors can overcome some of the limitations of current therapies and join our arsenal of antiplatelet agents.
Collagen-GPVI-Fcγ receptor
The platelet-specific collagen receptor glycoprotein (GP)VI, in complex with the Fcγ receptor (FcγR), plays a critical role in both platelet adhesion and activation (Figure)24. A number of GPVI inhibitors have been developed as possible antiplatelet agents, such as inhibitory anti-GPVI antibodies and a soluble GPVI-Fc fusion protein called Revacept25. Additionally, a number of C-type lectin-like proteins have been identified in snake venoms that modulate GPVI activity. This includes the GPVI specific agonist Trowaglerix, which was purified from Tropidolaemus wagleri venom and potently induces platelet aggregation26. In contrast to full-length Trowaglerix protein, however, hexa and decapeptides (Troα6 and Troα10) derived from the C-terminal region of Trowaglerix function as potent inhibitors of collagen-induced platelet aggregation (Figure) (Table)27. Interestingly, docking studies indicate that the decapeptide binds to a cleft between the D1 and D2 domains but does not disrupt GPVI-collagen binding28. Both the hexa and decapeptide also strongly inhibited thrombus formation in mesenteric and carotid ferric chloride models without prolonging bleeding after tail transection27.
GPVI inhibitors are also effective in protecting against cardiac ischemia-reperfusion injury. For instance, both anti-GPVI monoclonal antibody infusion, which induces shedding of GPVI from platelets, and Revacept administration reduced infarct size in a murine cardiac ischemia-reperfusion model29, 30. These studies indicate that GPVI-dependent activation of platelets contributes to cardiac injury and that anti-GPVI therapies may be useful adjuvants in patients with myocardial infarction undergoing revascularization.
Collagen-mediated activation of GPVI can also be negatively regulated by endogenous mechanisms. Leukocyte-associated immunoglobulin-like receptor-1 (LAIR1) is expressed in megakaryocytes and negatively regulates GPVI signaling by binding to collagen (Figure)31, 32. Smith and colleagues generated megakaryocyte-specific LAIR1 knockouts to study the effect of LAIR1 on megakaryocyte and platelet function33. LAIR1 deficiency did not affect megakaryocyte development. Consistent with serving as a negative regulator of GPVI, collagen related peptide stimulation of GPVI on platelets from megakaryocyte specific LAIR1 knockouts caused an enhancement in aggregation that was accompanied by increased phosphorylation of FcγR, Src and spleen tyrosine kinase (Syk)33. It should be noted that LAIR1 expression has not been detected in platelets34. It is proposed that the impaired capacity to activate GPVI in megakaryocytes may, in some way, be transmitted to and persist in formed platelets although the mechanism behind this remains to be elucidated33.
GPVI-mediated platelet activation can also be interrupted at a point further downstream in the intracellular signaling pathway. T-cell ubiquitin ligand-2 (TULA2) is a histidine phosphatase that binds and dephosphorylates Syk (Figure)35. Studies with TULA2 knockout mice showed that TULA2 limits Syk phosphorylation in collagen related peptide stimulated platelets leading to a restriction of GPVI-mediated activation36. In addition, another study found that dephosphorylation of Syk by TULA2 may also serve to limit activation downstream of FcγRIIA37. Mice with reduced TULA2 and expressing human FcγRIIA had enhanced Syk and phospholipase Cγ2 (PLCγ2) phosphorylation after stimulation with an FcγRIIA-specific agonist when compared to wild-type controls37. Finally, in a model of anti-GPIX antibody mediated heparin-induced thrombocytopenia the absence of TULA2 expression markedly worsened thrombocytopenia and shortened bleeding times consistent with increased activation of TULA2-deficient platelets37.
Activation of Syk downstream of the tyrosine kinase coupled receptors GPVI, C-type lectin-like receptor 2 and FcγRIIA is critical for platelet activation. Despite early studies suggesting a central role of Syk in regulating platelet activation, deletion of Syk was not associated with increased bleeding38. Recent work by van Eeuwijk and colleagues revealed a relatively mild hemostatic defect in platelet-specific Syk knockout mice39. However, these mice were strongly protected in a model of arterial thrombosis39. Similar findings were observed when a selective Syk inhibitor, BI1002494, was administered to wild-type mice (Table)39. Syk inhibitors have also been investigated as a possible therapy for heparin-induced thrombocytopenia. For instance, administration of PRT-060318 protected against spontaneous formation of thrombi in the pulmonary vasculature and preserved platelet counts in a humanized FcγRIIA and Platelet Factor 4 mouse model (Table)40.
CalDAG-GEFI
The calcium- and DAG-regulated guanine exchange factor-1 guanine exchange factor (CalDAG-GEFI) is a critical activator of the small GTPases of the Ras-related protein 1 (Rap1) subfamily41. As the major guanine nucleotide exchange factor expressed in platelets the capacity of CalDAG-GEFI to regulate Rap1 mediated platelet activation has been studied extensively. GTP-loading of Rap1 by CalDAG-GEFI induces platelet adhesion, in part, through activation of integrin αIIbβ342, 43. Rap1 activation is also required for a number of other platelet processes, including granule secretion, thromboxane A2 generation, spreading and clot retraction41. Platelets from CalDAG-GEFI knockout mice revealed a blunted aggregatory response after stimulation with a range of physiological agonists44. These results indicated that CalDAG-GEFI-mediated activation of Rap1 provides a common signaling pathway for platelet activation downstream of numerous receptors, including the PARs, P2Y12 and GPVI (Figure)44. In addition, CalDAG-GEFI knockout mice had reduced thrombosis in a variety of experimental models44–46. However, loss of CalDAG-GEFI also resulted in a pronounced bleeding phenotype after tail transection indicating an important function in primary hemostasis. In recent work published in ATVB, Piatt and colleagues generated CalDAG-GEFI transgenic mice that express ~10% of wild-type levels of CalDAG-GEFI47. Platelets from these mice had an impaired response to PAR4, GPVI and P2Y12/P2Y1 specific agonists but it was less severe than platelets from CalDAG-GEFI knockout mice47. Importantly, CalDAG-GEFI low mice had a similar level of protection against thrombosis compared to CalDAG-GEFI knockouts but had a much milder bleeding diathesis47.
Platelets have also been shown to play a role in the development of atherosclerotic plaques in mouse models48–50. These studies highlight the potential of antiplatelet therapies to limit atherogenesis51. Recent studies have shown that P2Y12-deficient, Apolipoprotein E double deficient mice had smaller lesions than controls52. Similarly, low density lipoprotein receptor deficient mice reconstituted with CalDAG-GEFI deficient bone marrow had significantly smaller lesions than controls53. These studies indicate that platelet activation contributes to atherogenesis in mice and an additional benefit of antiplatelet therapy in humans maybe a reduction in lesion progression.
Nuclear receptor subfamily 1 members
A recent study found that two members of the nuclear receptor subfamily 1 of transcription factors, Farnesoid X Receptor (FXR) and Liver X Receptor (LXR), and the associated Retinoid X Receptor (RXR), are expressed in human platelets (Figure)54–56. RXR forms heterodimeric complexes with both FXR and LXR56. Recent studies revealed a surprising non-genomic function for these transcription factors in the regulation of platelet activation. Stimulation of human platelets with FXR and LXR ligands strongly inhibited both platelet aggregation and granule release54, 55. The capacity of FXR ligands to induce accumulation of cGMP likely accounts for the observed impairment in platelet activation55. Interestingly, treatment of platelets with FXR and LXR ligands impaired integrin αIIbβ3 activation and outside-in signaling56. Moreover, infusion of either FXR or LXR ligands into mice inhibited platelet accumulation at sites of vascular injury54, 55. Stimulation of platelets with RXR ligands also results in inhibition of platelet aggregation, granule secretion and integrin αIIbβ3 outside-in signaling56. Further work is required to determine whether FXR, LXR and RXR ligands could serve as viable antiplatelet agents.
Phosphoinositides and platelet kinases
Activation of platelet receptors initiates downstream signaling events that includes the generation of the small molecule signaling intermediate phosphatidylinositol 3-phosphate through the action of phosphatidylinositol 3-kinases (Figure). Phosphoinositide-dependent protein kinase 1 (PDK1) is activated through binding of phosphatidylinositol 3-phosphate enabling phosphorylation of downstream signaling targets that include members of the Akt family of kinases57. Platelet-specific PDK1 deficiency is associated with reduced platelet activation due to impaired integrin αIIbβ3-mediated outside-in signaling58. A small molecule PDK1 inhibitor has also been found to inhibit activation of human platelets59. Another study showed that PDK1 deficient platelets had reduced aggregation and granule release after stimulation with the GPVI specific agonist collagen related peptide60. PDK1 was also found to be essential for the collagen-induced increase in intracellular [Ca2+] in part through a Rac1-PLCγ2 dependent pathway60. As in earlier reports58, platelet specific PDK1 knockout mice exhibit reduced arterial thrombosis and were also protected from ischemic stroke60.
Additional platelet kinases have been found to regulate the metabolism of phosphoinositides in platelets, including the transient receptor potential melastatin-like 7 (TRPM7) channel (Figure). TRMP7 is a constitutively active divalent cation selective channel that regulates intracellular [Ca2+] and [Mg2+] but also functions as a serine/threonine kinase61. Studies with TRPM7 deficient megakaryocytes and megakaryocytes expressing kinase-dead TRPM7 revealed an important role of the cation channel, but not kinase, function in platelet biogenesis62. TRPM7 deficiency in megakaryocytes was associated with impaired proplatelet formation and abnormal megakaryocyte microtubule assembly that led to macrothrombocytopenia62. In a subsequent study, Gotru and colleagues explored the kinase function of TRPM7 in platelets63. While platelet biogenesis was normal in mice expressing a kinase-dead TRPM7, loss of kinase activity markedly reduced arterial thrombosis63. Interrogation of signaling downstream of GPVI and C-type lectin-like receptor-2-mediated receptor activation in platelets containing kinase-dead TRPM7 revealed a common deficit in Syk, linker for activation of T cells and PLCγ2 phosphorylation. Consistent with an impaired activation of PLCγ2, platelets containing a kinase-dead TRPM7 produced significantly less inositol 1,4,5-trisphosphate.
Platelet Oxidases
Lipoxygenases are a family of enzymes that catalyze the oxygenation of polyunsaturated fatty acids that leads to the generation of a variety of active signaling molecules. 12-Lipoxygenase (12-LOX), named for the ability of this family member to oxidize arachidonic acid at carbon 12, is expressed in both megakaryocytes and platelets64. Oxidation of arachidonic acid by 12-LOX results in the formation of 12(S)- hydroperoxyicosa-5,8,10,14-tetraenoic acid that is reduced to 12(S)-hydroxy-5,8,10,14-eicosatetraenoic acid by glutathione peroxidase (Figure). Oxidation of dihomo-γ-linolenic acid by 12-LOX also generates 12(S)-hydroxy-8Z,10E,14Z-eicosatrienoic acid (Figure). Administration of dihomo-γ-linolenic acid or 12(S)-hydroxy-8Z,10E,14Z-eicosatrienoic acid to mice resulted in a 12-LOX-dependent inhibition of platelet activation and thrombosis65, 66. There is a growing body of evidence supporting the involvement of 12-LOX in platelet activation with inhibition or gene deletion effectively abrogating platelet aggregation in response to stimulation through PAR1, PAR4, GPVI and FcγRIIa67–69. A recent study showed that a novel and selective 12-LOX small molecule inhibitor, ML-355 (Table), reduced platelet activation and arterial thrombosis in mice with a minimal effect on hemostasis69.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) is a membrane bound enzyme that catalyzes the generation of superoxide from NADPH. Studies of patients with Chronic Granulomatous Disease, which is caused by a genetic deficiency for NOX2, indicate that NOX2 is the primary producer of superoxide in stimulated platelets70. Reactive oxygen species (ROS) generated from superoxide can markedly increase platelet reactivity71. ROS contributes significantly to thrombosis with deletion of the anti-oxidant enzyme glutathione peroxidase-3 is resulting in increased platelet activation and thrombosis72. Consistent with these findings, NOX2-deficient platelets had a selective defect in response to collagen related peptide stimulation accompanied by a reduced capacity for ROS production and impaired phosphorylation of PLCγ2 and Syk73. In addition, NOX2 deficient mice had reduced thrombosis in the laser-injury model but had no impairment of hemostasis73. These data strongly support a role for NOX2 in GPVI-dependent platelet activation and suggests that selective NOX2 inhibitors could find utility as antiplatelet agents.
A recent study suggested that platelet NOX activity is regulated by the class III phosphatidylinositol 3-kinase vacuolar protein sorting 34 (VPS34)74. VPS34-dependent generation of phosphatidylinositol 3-phosphate has previously been found to regulate NOX-dependent ROS generation in leukocytes by binding to the gp40phox subunit75, 76. Studies with platelets from platelet-specific VPS34 knockout mice demonstrated that VPS34 is also essential for NOX-dependent ROS generation74. Importantly, VPS34 deficient platelets had a reduced capacity to form the active NOX complex at the plasma membrane leading to impaired ROS generation74. This impairment led to reduced platelet aggregation and reduced thrombus formation74. However, an independent study found that VPS34 deficiency in megakaryocytes disrupts platelet biogenesis and resulted in lower platelet counts and abnormal granule formation. These changes may also contribute to the reduced thrombosis77.
Filamin A
The actin-binding protein filamin A functions as an important regulator of platelet activation and shape change. Filamin A mediates platelet activation through interaction with a variety of binding partners, including GPIbα, GPVI and Syk78. Studies have suggested that filamin A-GPIbα binding facilitates activation of integrin αIIbβ379. Structural studies, however, indicate that filamin A may also directly interact with and regulate the activation of integrin αIIbβ3 (Figure)80. Interestingly, platelets isolated from a patient with a filamin A gain-of-function mutation that potentiates its interaction with integrin αIIbβ3 had increased ADP-induced platelet aggregation, dense granule release and integrin αIIbβ3 activation81. Despite enhanced recruitment of talin to integrin αIIbβ3 in ADP-stimulated platelets from the patient, Rap1 activation was unchanged supporting a GPIbα independent mechanism81. The retinue of confirmed filamin A binding partners increased recently with the identification of stromal interaction molecule 1, which is a critical regulator of store operated calcium entry82. In contrast to other filamin A interactions, binding of stromal interaction molecule 1 likely inhibits platelet activation by limiting store operated calcium entry82.
Phosphatidylserine Exposure
Strong agonist-mediated activation of platelets leads to exposure of phosphatidylserine on the outer leaflet of the plasma membrane in a subpopulation of platelets. Exposure of phosphatidylserine confers procoagulant activity on this subpopulation by increasing the assembly of coagulation protease/cofactor complexes, such as prothrombinase complex, which enhances thrombin generation. Agonist-mediated phosphatidylserine exposure requires intracellular and mitochondrial accumulation of calcium, which facilitates cycophilin D mediated formation of the mitochondrial transition pore and the disruption of the inner mitochondrial membrane83, 84. Activation of apoptotic pathways, involving Bax/Bak mediated activation of caspase-9, can also lead to platelet phosphatidylserine exposure85. It was previously unclear if phosphatidylserine exposure driven by apoptosis caused inner mitochondrial membrane disruption. Recent work published in ATVB demonstrated that both agonist and apoptosis-initiated phosphatidylserine exposure involves disruption of the inner, but not the outer mitochondrial membrane86. Furthermore, the authors showed that activation of caspase-9 is required for inner mitochondrial membrane disruption and phosphatidylserine exposure after initiation of apoptosis86.
Exposure of phosphatidylserine also requires activation of phospholipid scramblases that transfer phosphatidylserine from the inner leaflet of the membrane to the outer plasma membrane surface. Scott syndrome is a bleeding disorder that is caused by defective phosphatidylserine exposure on platelets. Scott syndrome patients have a truncated version of the critical calcium-sensitive phospholipid scramblase TMEM16F present in the plasma membrane of platelets and other cells (Figure)87, 88. Platelets from TMEM16F knockout mice showed impaired exposure of phosphatidylserine after stimulation with calcium ionophore or a combination of collagen and thrombin88, 89. This evidence strongly supports the involvement of TMEM16F in phosphatidylserine exposure on platelets. Consistent with the importance of TMEM16F-mediated phosphatidylserine exposure TMEM16F gene specific knockout mice were also protected against arterial thrombosis88. The role of platelet resident TMEM16F has been further explored using platelet specific TMEM16F knockout mice90. The thromboprotection observed in platelet specific TMEM16F largely phenocopies that seen in global TMEM16F knockouts90. Although TMEM16F is also expressed in the endothelium these findings indicate that TMEM16F-mediated phosphatidylserine exposure on the platelet surface is sufficient to support the formation of procoagulant platelets. An additional study showed that phosphatidylserine exposure on platelets stimulated with convulxin/thrombin was dependent on TMEM16F, whereas phosphatidylserine exposure on platelets stimulated with collagen/thrombin was dependent on two pathways one of which involved mitochondrial depolarization mediated by TMEM16F91.
Summary
Recent studies have revealed a series of novel mechanisms that either positively or negatively regulate signaling events downstream of receptor mediated platelet activation. In a number of cases, it appears that disruption of these pathways can selectively inhibit thrombosis while leaving essential hemostatic processes largely intact. These pathways may be of considerable interest as potential targets for development of a new generation of antiplatelet agents. Other work has focused on the development of novel anti-platelet agents that inhibit established targets such as P2Y12, PAR1 and GPVI. These agents may overcome some of the limitations of established therapies. It remains to be determined whether these novel agents will find clinical utility.
Acknowledgments
We would like to acknowledge Dr. Peter Newman for helpful comments.
Sources of Funding
This work was supported by funding from the John C. Parker Professorship (N Mackman) and the National Institute of Health grant number R01 HL130404 (W Bergmeier).
Non-standard abbreviations and acronyms
- 12-LOX
12-Lipoxygenase
- CalDAG-GEFI
calcium- and DAG-regulated guanine exchange factor-1; guanine exchange factor
- FXR
Farnesoid X Receptor
- FcγR
Fcγ receptor
- GP
glycoprotein
- LAIR1
Leukocyte-associated immunoglobulin-like receptor-1
- LXR
Liver X Receptor
- PAR
protease-activated receptor
- NOX2
NADPH oxidase 2
- PDK1
phosphoinositide-dependent protein kinase 1
- PLCγ2
phospholipase Cγ2
- RAP1
Ras-related protein 1
- ROS
reactive oxygen species
- RXR
Retinoid X Receptor
- Syk
spleen tyrosine kinase
- TRPM7
transient receptor potential melastatin-like 7
- TULA2
T-cell ubiquitin ligand-2
- VPS34
vacuolar protein sorting 34
Footnotes
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9(Suppl 1):92–104. doi: 10.1111/j.1538-7836.2011.04361.x. [DOI] [PubMed] [Google Scholar]
- 3.Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: A complex interplay between inhibitory and activatory networks. J Thromb Haemost. 2016;14:918–930. doi: 10.1111/jth.13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiology (Bethesda) 2017;32:162–177. doi: 10.1152/physiol.00020.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tantry US, Bonello L, Aradi D, et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62:2261–2273. doi: 10.1016/j.jacc.2013.07.101. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong PC, Hoefer T, Knowles RB, Tucker AT, Hayman MA, Ferreira PM, Chan MV, Warner TD. Newly formed reticulated platelets undermine pharmacokinetically short-lived antiplatelet therapies. Arterioscler Thromb Vasc Biol. 2017;37:949–956. doi: 10.1161/ATVBAHA.116.308763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanachkov IB, Chang H, Yanachkova MI, Dix EJ, Berny-Lang MA, Gremmel T, Michelson AD, Wright GE, Frelinger AL. New highly active antiplatelet agents with dual specificity for platelet P2Y1 and P2Y12 adenosine diphosphate receptors. Eur J Med Chem. 2016;107:204–218. doi: 10.1016/j.ejmech.2015.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gremmel T, Yanachkov IB, Yanachkova MI, Wright GE, Wider J, Undyala VV, Michelson AD, Frelinger AL, Przyklenk K. Synergistic inhibition of both P2Y1 and P2Y12 adenosine diphosphate receptors as novel approach to rapidly attenuate platelet-mediated thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:501–509. doi: 10.1161/ATVBAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaduganathan M, Bhatt DL. Simultaneous platelet P2Y12 and P2Y1 ADP receptor blockade: Are two better than one? Arterioscler Thromb Vasc Biol. 2016;36:427–428. doi: 10.1161/ATVBAHA.115.307097. [DOI] [PubMed] [Google Scholar]
- 10.Liverani E, Rico MC, Tsygankov AY, Kilpatrick LE, Kunapuli SP. P2Y12 receptor modulates sepsis-induced inflammation. Arterioscler Thromb Vasc Biol. 2016;36:961–971. doi: 10.1161/ATVBAHA.116.307401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahman M, Gustafsson D, Wang Y, Thorlacius H, Braun O. Ticagrelor reduces neutrophil recruitment and lung damage in abdominal sepsis. Platelets. 2014;25:257–263. doi: 10.3109/09537104.2013.809520. [DOI] [PubMed] [Google Scholar]
- 12.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han X, Nieman MT. PAR4 (protease-activated receptor 4): Particularly important 4 antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2018;38:287–289. doi: 10.1161/ATVBAHA.117.310550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–1413. doi: 10.1056/NEJMoa1200933. [DOI] [PubMed] [Google Scholar]
- 15.O’Donoghue ML, Bhatt DL, Wiviott SD, et al. Safety and tolerability of atopaxar in the treatment of patients with acute coronary syndromes: The lessons from antagonizing the cellular effects of thrombin–acute coronary syndromes trial. Circulation. 2011;123:1843–1853. doi: 10.1161/CIRCULATIONAHA.110.000786. [DOI] [PubMed] [Google Scholar]
- 16.Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- 17.Zhang P, Gruber A, Kasuda S, Kimmelstiel C, O’Callaghan K, Cox DH, Bohm A, Baleja JD, Covic L, Kuliopulos A. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation. 2012;126:83–91. doi: 10.1161/CIRCULATIONAHA.112.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurbel PA, Bliden KP, Turner SE, Tantry US, Gesheff MG, Barr TP, Covic L, Kuliopulos A. Cell-penetrating pepducin therapy targeting par1 in subjects with coronary artery disease. Arterioscler Thromb Vasc Biol. 2016;36:189–197. doi: 10.1161/ATVBAHA.115.306777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norman JE, Cunningham MR, Jones ML, Walker ME, Westbury SK, Sessions RB, Mundell SJ, Mumford AD. Protease-activated receptor 4 variant p.Tyr157Cys reduces platelet functional responses and alters receptor trafficking. Arterioscler Thromb Vasc Biol. 2016;36:952–960. doi: 10.1161/ATVBAHA.115.307102. [DOI] [PubMed] [Google Scholar]
- 20.Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, Dong JF, Shaw C, Bray PF. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and mir-376c. Nat Med. 2013;19:1609–1616. doi: 10.1038/nm.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel-Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, Holinstat M, Shaw CA, Bray PF. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124:3450–3458. doi: 10.1182/blood-2014-04-572479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong PC, Seiffert D, Bird JE, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaf5294. [DOI] [PubMed] [Google Scholar]
- 23.Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, Yang J, Newby DE. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38:448–456. doi: 10.1161/ATVBAHA.117.310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dütting S, Bender M, Nieswandt B. Platelet GPVI: A target for antithrombotic therapy? Trends Pharmacol Sci. 2012;33:583–590. doi: 10.1016/j.tips.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Andrews RK, Arthur JF, Gardiner EE. Targeting GPVI as a novel antithrombotic strategy. J Blood Med. 2014;5:59–68. doi: 10.2147/JBM.S39220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CH, Chung CH, Kuo HL, Hsu CC, Huang TF. The highly specific platelet glycoprotein (GP) VI agonist trowaglerix impaired collagen-induced platelet aggregation ex vivo through matrix metalloproteinase-dependent GPVI shedding. J Thromb Haemost. 2008;6:669–676. doi: 10.1111/j.1538-7836.2008.02914.x. [DOI] [PubMed] [Google Scholar]
- 27.Chang CH, Chung CH, Tu YS, Tsai CC, Hsu CC, Peng HC, Tseng YJ, Huang TF. Trowaglerix venom polypeptides as a novel antithrombotic agent by targeting immunoglobulin-like domains of glycoprotein VI in platelet. Arterioscler Thromb Vasc Biol. 2017;37:1307–1314. doi: 10.1161/ATVBAHA.116.308604. [DOI] [PubMed] [Google Scholar]
- 28.Herr AB. Charming the snake: Venom-derived peptides show surprising efficacy as glycoprotein VI-targeting antithrombotic agents. Arterioscler Thromb Vasc Biol. 2017;37:1266–1268. doi: 10.1161/ATVBAHA.117.309627. [DOI] [PubMed] [Google Scholar]
- 29.Pachel C, Mathes D, Arias-Loza AP, Heitzmann W, Nordbeck P, Deppermann C, Lorenz V, Hofmann U, Nieswandt B, Frantz S. Inhibition of platelet GPVI protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2016;36:629–635. doi: 10.1161/ATVBAHA.115.305873. [DOI] [PubMed] [Google Scholar]
- 30.Schönberger T, Ziegler M, Borst O, Konrad I, Nieswandt B, Massberg S, Ochmann C, Jürgens T, Seizer P, Langer H, Münch G, Ungerer M, Preissner KT, Elvers M, Gawaz M. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am J Physiol Cell Physiol. 2012;303:C757–766. doi: 10.1152/ajpcell.00060.2012. [DOI] [PubMed] [Google Scholar]
- 31.Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, Farndale RW, Lisman T, Sonnenberg A, Lenting PJ, Meyaard L. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med. 2006;203:1419–1425. doi: 10.1084/jem.20052554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brondijk TH, de Ruiter T, Ballering J, Wienk H, Lebbink RJ, van Ingen H, Boelens R, Farndale RW, Meyaard L, Huizinga EG. Crystal structure and collagen-binding site of immune inhibitory receptor LAIR-1: Unexpected implications for collagen binding by platelet receptor GPVI. Blood. 2010;115:1364–1373. doi: 10.1182/blood-2009-10-246322. [DOI] [PubMed] [Google Scholar]
- 33.Smith CW, Thomas SG, Raslan Z, Patel P, Byrne M, Lordkipanidzé M, Bem D, Meyaard L, Senis YA, Watson SP, Mazharian A. Mice lacking the inhibitory collagen receptor LAIR-1 exhibit a mild thrombocytosis and hyperactive platelets. Arterioscler Thromb Vasc Biol. 2017;37:823–835. doi: 10.1161/ATVBAHA.117.309253. [DOI] [PubMed] [Google Scholar]
- 34.Steevels TA, Westerlaken GH, Tijssen MR, Coffer PJ, Lenting PJ, Akkerman JW, Meyaard L. Co-expression of the collagen receptors leukocyte-associated immunoglobulin-like receptor-1 and glycoprotein VI on a subset of megakaryoblasts. Haematologica. 2010;95:2005–2012. doi: 10.3324/haematol.2010.026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas DH, Getz TM, Newman TN, Dangelmaier CA, Carpino N, Kunapuli SP, Tsygankov AY, Daniel JL. A novel histidine tyrosine phosphatase, TULA-2, associates with Syk and negatively regulates GPVI signaling in platelets. Blood. 2010;116:2570–2578. doi: 10.1182/blood-2010-02-268136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reppschläger K, Gosselin J, Dangelmaier CA, Thomas DH, Carpino N, McKenzie SE, Kunapuli SP, Tsygankov AY. TULA-2 protein phosphatase suppresses activation of Syk through the gpvi platelet receptor for collagen by dephosphorylating Tyr(p)346, a regulatory site of Syk. J Biol Chem. 2016;291:22427–22441. doi: 10.1074/jbc.M116.743732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Abraham S, Renna S, Edelstein LC, Dangelmaier CA, Tsygankov AY, Kunapuli SP, Bray PF, McKenzie SE. TULA-2 (t-cell ubiquitin ligand-2) inhibits the platelet Fc receptor for IgG IIa (FcγRIIa) signaling pathway and heparin-induced thrombocytopenia in mice. Arterioscler Thromb Vasc Biol. 2016;36:2315–2323. doi: 10.1161/ATVBAHA.116.307979. [DOI] [PubMed] [Google Scholar]
- 38.Law DA, Nannizzi-Alaimo L, Ministri K, Hughes PE, Forsyth J, Turner M, Shattil SJ, Ginsberg MH, Tybulewicz VL, Phillips DR. Genetic and pharmacological analyses of Syk function in alphaIIbbeta3 signaling in platelets. Blood. 1999;93:2645–2652. [PubMed] [Google Scholar]
- 39.van Eeuwijk JM, Stegner D, Lamb DJ, Kraft P, Beck S, Thielmann I, Kiefer F, Walzog B, Stoll G, Nieswandt B. The novel oral Syk inhibitor, Bl1002494, protects mice from arterial thrombosis and thromboinflammatory brain infarction. Arterioscler Thromb Vasc Biol. 2016;36:1247–1253. doi: 10.1161/ATVBAHA.115.306883. [DOI] [PubMed] [Google Scholar]
- 40.Reilly MP, Sinha U, André P, Taylor SM, Pak Y, Deguzman FR, Nanda N, Pandey A, Stolla M, Bergmeier W, McKenzie SE. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. 2011;117:2241–2246. doi: 10.1182/blood-2010-03-274969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stefanini L, Bergmeier W. Rap1-GTPase signaling and platelet function. J Mol Med (Berl) 2016;94:13–19. doi: 10.1007/s00109-015-1346-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115:680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang G, Xiang B, Ye S, Chrzanowska-Wodnicka M, Morris AJ, Gartner TK, Whiteheart SW, White GC, Smyth SS, Li Z. Distinct roles for Rap1b protein in platelet secretion and integrin αIIbβ3 outside-in signaling. J Biol Chem. 2011;286:39466–39477. doi: 10.1074/jbc.M111.239608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 45.Stolla M, Stefanini L, Roden RC, Chavez M, Hirsch J, Greene T, Ouellette TD, Maloney SF, Diamond SL, Poncz M, Woulfe DS, Bergmeier W. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: Implications for antiplatelet therapy. Blood. 2011;117:1005–1013. doi: 10.1182/blood-2010-07-297713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amirkhosravi A, Boulaftali Y, Robles-Carrillo L, Meyer T, McKenzie SE, Francis JL, Bergmeier W. Caldag-gefi deficiency protects mice from FcγRIIa-mediated thrombotic thrombocytopenia induced by CD40l and β2GPI immune complexes. J Thromb Haemost. 2014;12:2113–2119. doi: 10.1111/jth.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piatt R, Paul DS, Lee RH, McKenzie SE, Parise LV, Cowley DO, Cooley BC, Bergmeier W. Mice expressing low levels of CalDAG-GEFI exhibit markedly impaired platelet activation with minor impact on hemostasis. Arterioscler Thromb Vasc Biol. 2016;36:1838–1846. doi: 10.1161/ATVBAHA.116.307874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 50.Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003;23:2131–2137. doi: 10.1161/01.ATV.0000095974.95122.EC. [DOI] [PubMed] [Google Scholar]
- 51.Schulz C, Massberg S. Platelets in atherosclerosis and thrombosis. Handb Exp Pharmacol. 2012:111–133. doi: 10.1007/978-3-642-29423-5_5. [DOI] [PubMed] [Google Scholar]
- 52.Li D, Wang Y, Zhang L, Luo X, Li J, Chen X, Niu H, Wang K, Sun Y, Wang X, Yan Y, Chai W, Gartner TK, Liu J. Roles of purinergic receptor P2Y, G protein-coupled 12 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:e81–89. doi: 10.1161/ATVBAHA.111.239095. [DOI] [PubMed] [Google Scholar]
- 53.Boulaftali Y, Owens AP, Beale A, Piatt R, Casari C, Lee RH, Conley PB, Paul DS, Mackman N, Bergmeier W. CalDAG-GEFI deficiency reduces atherosclerotic lesion development in mice. Arterioscler Thromb Vasc Biol. 2016;36:792–799. doi: 10.1161/ATVBAHA.115.306347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spyridon M, Moraes LA, Jones CI, Sage T, Sasikumar P, Bucci G, Gibbins JM. LXR as a novel antithrombotic target. Blood. 2011;117:5751–5761. doi: 10.1182/blood-2010-09-306142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moraes LA, Unsworth AJ, Vaiyapuri S, Ali MS, Sasikumar P, Sage T, Flora GD, Bye AP, Kriek N, Dorchies E, Molendi-Coste O, Dombrowicz D, Staels B, Bishop-Bailey D, Gibbins JM. Farnesoid X receptor and its ligands inhibit the function of platelets. Arterioscler Thromb Vasc Biol. 2016;36:2324–2333. doi: 10.1161/ATVBAHA.116.308093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unsworth AJ, Bye AP, Tannetta DS, Desborough MJR, Kriek N, Sage T, Allan HE, Crescente M, Yaqoob P, Warner TD, Jones CI, Gibbins JM. Farnesoid X receptor and liver X receptor ligands initiate formation of coated platelets. Arterioscler Thromb Vasc Biol. 2017;37:1482–1493. doi: 10.1161/ATVBAHA.117.309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toker A, Newton AC. Cellular signaling: Pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 58.Chen X, Zhang Y, Wang Y, Li D, Zhang L, Wang K, Luo X, Yang Z, Wu Y, Liu J. PDK1 regulates platelet activation and arterial thrombosis. Blood. 2013;121:3718–3726. doi: 10.1182/blood-2012-10-461897. [DOI] [PubMed] [Google Scholar]
- 59.Dangelmaier C, Manne BK, Liverani E, Jin J, Bray P, Kunapuli SP. PDK1 selectively phosphorylates Thr(308) on Akt and contributes to human platelet functional responses. Thromb Haemost. 2014;111:508–517. doi: 10.1160/TH13-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Münzer P, Walker-Allgaier B, Geue S, et al. PDK1 determines collagen-dependent platelet Ca2+ signaling and is critical to development of ischemic stroke in vivo. Arterioscler Thromb Vasc Biol. 2016;36:1507–1516. doi: 10.1161/ATVBAHA.115.307105. [DOI] [PubMed] [Google Scholar]
- 61.Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG. Characterization of the protein kinase activity of TRPM7/CHAK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem. 2004;279:3708–3716. doi: 10.1074/jbc.M308820200. [DOI] [PubMed] [Google Scholar]
- 62.Stritt S, Nurden P, Favier R, et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg(2+) homeostasis and cytoskeletal architecture. Nat Commun. 2016;7:11097. doi: 10.1038/ncomms11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gotru SK, Chen W, Kraft P, et al. TRPM7 kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler Thromb Vasc Biol. 2018;38:344–352. doi: 10.1161/ATVBAHA.117.310391. [DOI] [PubMed] [Google Scholar]
- 64.Tourdot BE, Holinstat M. Targeting 12-lipoxygenase as a potential novel antiplatelet therapy. Trends Pharmacol Sci. 2017;38:1006–1015. doi: 10.1016/j.tips.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 65.Yeung J, Tourdot BE, Adili R, Green AR, Freedman CJ, Fernandez-Perez P, Yu J, Holman TR, Holinstat M. 12(s)-HETrE, a 12-lipoxygenase oxylipin of dihomo-γ-linolenic acid, inhibits thrombosis via Gαs signaling in platelets. Arterioscler Thromb Vasc Biol. 2016;36:2068–2077. doi: 10.1161/ATVBAHA.116.308050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tourdot BE, Adili R, Isingizwe ZR, Ebrahem M, Freedman JC, Holman TR, Holinstat M. 12-HETrE inhibits platelet reactivity and thrombosis in part through the prostacyclin receptor. Blood Adv. 2017;1:1124–1131. doi: 10.1182/bloodadvances.2017006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yeung J, Apopa PL, Vesci J, Stolla M, Rai G, Simeonov A, Jadhav A, Fernandez-Perez P, Maloney DJ, Boutaud O, Holman TR, Holinstat M. 12-lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb Haemost. 2013;110:569–581. doi: 10.1160/TH13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeung J, Tourdot BE, Fernandez-Perez P, Vesci J, Ren J, Smyrniotis CJ, Luci DK, Jadhav A, Simeonov A, Maloney DJ, Holman TR, McKenzie SE, Holinstat M. Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. 2014;124:2271–2279. doi: 10.1182/blood-2014-05-575878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adili R, Tourdot BE, Mast K, Yeung J, Freedman JC, Green A, Luci DK, Jadhav A, Simeonov A, Maloney DJ, Holman TR, Holinstat M. First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler Thromb Vasc Biol. 2017;37:1828–1839. doi: 10.1161/ATVBAHA.117.309868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, Violi F. Gp91phox-dependent expression of platelet CD40 ligand. Circulation. 2004;110:1326–1329. doi: 10.1161/01.CIR.0000134963.77201.55. [DOI] [PubMed] [Google Scholar]
- 71.Krötz F, Riexinger T, Buerkle MA, Nithipatikom K, Gloe T, Sohn HY, Campbell WB, Pohl U. Membrane-potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24:595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- 72.Jin RC, Mahoney CE, Coleman Anderson L, Ottaviano F, Croce K, Leopold JA, Zhang YY, Tang SS, Handy DE, Loscalzo J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 2011;123:1963–1973. doi: 10.1161/CIRCULATIONAHA.110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic-Terpo A, Shen B, Ushio-Fukai M, Cho J, Du X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–854. doi: 10.1161/ATVBAHA.116.307308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Y, Hu M, Luo D, Yue M, Wang S, Chen X, Zhou Y, Wang Y, Cai Y, Hu X, Ke Y, Yang Z, Hu H. Class III PI3K positively regulates platelet activation and thrombosis via PI(3)P-directed function of NADPH oxidase. Arterioscler Thromb Vasc Biol. 2017;37:2075–2086. doi: 10.1161/ATVBAHA.117.309751. [DOI] [PubMed] [Google Scholar]
- 75.Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Gaffney PR, Coadwell J, Chilvers ER, Hawkins PT, Stephens LR. Ptdins(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox) Nat Cell Biol. 2001;3:679–682. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 76.Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, Sindrilaru A, Scharffetter-Kochanek K, Rausch O, Stephens LR, Hawkins PT. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of escherichia coli or staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. doi: 10.1182/blood-2008-04-149450. [DOI] [PubMed] [Google Scholar]
- 77.Valet C, Levade M, Chicanne G, Bilanges B, Cabou C, Viaud J, Gratacap MP, Gaits-Iacovoni F, Vanhaesebroeck B, Payrastre B, Severin S. A dual role for the class III PI3K, VPS34, in platelet production and thrombus growth. Blood. 2017;130:2032–2042. doi: 10.1182/blood-2017-04-781641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Falet H. New insights into the versatile roles of platelet FLNA. Platelets. 2013;24:1–5. doi: 10.3109/09537104.2011.654004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feng S, Reséndiz JC, Lu X, Kroll MH. Filamin a binding to the cytoplasmic tail of glycoprotein Ibalpha regulates von willebrand factor-induced platelet activation. Blood. 2003;102:2122–2129. doi: 10.1182/blood-2002-12-3805. [DOI] [PubMed] [Google Scholar]
- 80.Liu J, Das M, Yang J, Ithychanda SS, Yakubenko VP, Plow EF, Qin J. Structural mechanism of integrin inactivation by filamin. Nat Struct Mol Biol. 2015;22:383–389. doi: 10.1038/nsmb.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Berrou E, Adam F, Lebret M, Planche V, Fergelot P, Issertial O, Coupry I, Bordet JC, Nurden P, Bonneau D, Colin E, Goizet C, Rosa JP, Bryckaert M. Gain-of-function mutation in filamin a potentiates platelet integrin αIIbβ3 activation. Arterioscler Thromb Vasc Biol. 2017;37:1087–1097. doi: 10.1161/ATVBAHA.117.309337. [DOI] [PubMed] [Google Scholar]
- 82.Lopez JJ, Albarrán L, Jardín I, Sanchez-Collado J, Redondo PC, Bermejo N, Bobe R, Smani T, Rosado JA. Filamin a modulates store-operated Ca2+ entry by regulating STIM1 (stromal interaction molecule 1)-Orai1 association in human platelets. Arterioscler Thromb Vasc Biol. 2018;38:386–397. doi: 10.1161/ATVBAHA.117.310139. [DOI] [PubMed] [Google Scholar]
- 83.Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, Di Paola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111:1257–1265. doi: 10.1182/blood-2007-05-092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32:2946–2955. doi: 10.1161/ATVBAHA.112.300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.White MJ, Schoenwaelder SM, Josefsson EC, Jarman KE, Henley KJ, James C, Debrincat MA, Jackson SP, Huang DC, Kile BT. Caspase-9 mediates the apoptotic death of megakaryocytes and platelets, but is dispensable for their generation and function. Blood. 2012;119:4283–4290. doi: 10.1182/blood-2011-11-394858. [DOI] [PubMed] [Google Scholar]
- 86.Choo HJ, Kholmukhamedov A, Zhou C, Jobe S. Inner mitochondrial membrane disruption links apoptotic and agonist-initiated phosphatidylserine externalization in platelets. Arterioscler Thromb Vasc Biol. 2017;37:1503–1512. doi: 10.1161/ATVBAHA.117.309473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 88.Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, Jan LY. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151:111–122. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A. 2015;112:12800–12805. doi: 10.1073/pnas.1516594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baig AA, Haining EJ, Geuss E, Beck S, Swieringa F, Wanitchakool P, Schuhmann MK, Stegner D, Kunzelmann K, Kleinschnitz C, Heemskerk JW, Braun A, Nieswandt B. TMEM16F-mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice-brief report. Arterioscler Thromb Vasc Biol. 2016;36:2152–2157. doi: 10.1161/ATVBAHA.116.307727. [DOI] [PubMed] [Google Scholar]
- 91.van Kruchten R, Mattheij NJ, Saunders C, Feijge MA, Swieringa F, Wolfs JL, Collins PW, Heemskerk JW, Bevers EM. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood. 2013;121:1850–1857. doi: 10.1182/blood-2012-09-454314. [DOI] [PubMed] [Google Scholar]