

Abstract
Autophagy is an essential catabolic process responsible for recycling of intracellular material and preserving cellular fidelity. Key to the autophagy pathway is the ubiquitin‐like conjugation system mediating lipidation of Atg8 proteins and their anchoring to autophagosomal membranes. While regulation of autophagy has been characterized at the level of transcription, protein interactions and post‐translational modifications, its translational regulation remains elusive. Here we describe a role for the conserved eukaryotic translation initiation factor 5A (eIF5A) in autophagy. Identified from a high‐throughput screen, we find that eIF5A is required for lipidation of LC3B and its paralogs and promotes autophagosome formation. This feature is evolutionarily conserved and results from the translation of the E2‐like ATG3 protein. Mechanistically, we identify an amino acid motif in ATG3 causing eIF5A dependency for its efficient translation. Our study identifies eIF5A as a key requirement for autophagosome formation and demonstrates the importance of translation in mediating efficient autophagy.
Keywords: ATG3, autophagy, eIF5A, translation
Subject Categories: Autophagy & Cell Death, Protein Biosynthesis & Quality Control
Introduction
Macroautophagy, hereafter referred to as autophagy, is an evolutionarily conserved catabolic process targeting intracellular components for lysosomal degradation 1. This process, which operates at basal levels in all eukaryotic cells, is essential for preserving cellular fidelity through nutrient recycling and by mediating effective clearance of damaged or superfluous cytoplasmic material. Intensive efforts have unveiled multiple components of the basic autophagy machinery that allow for highly controlled, fine‐tuned execution of this vesicular trafficking system 2, 3. The fundamental importance of autophagy is demonstrated by the fact that its dysregulation is implicated in multiple human diseases including neurodegeneration, infection and cancer 4, 5.
Key to autophagy is the formation of double‐membrane structures called phagophores, which sequester cellular components while growing to form sphere‐like, closed organelles called autophagosomes 1. Autophagosomes ultimately fuse with acidic, hydrolase‐containing lysosomes to form autolysosomes where cargo digestion occurs 3. The core autophagy machinery consists of a large group of autophagy‐related (ATG) proteins, most of which were originally identified in yeast, that work together through complex mechanisms to execute the various steps in the pathway 1, 6. Key to this machinery is the Atg8 family of ubiquitin‐like proteins, which associate with the growing phagophore through conjugation to the lipid phosphatidylethanolamine (PE) 7. While only one Atg8 exists in yeast, humans have six Atg8 homologs comprising two families: the microtubule‐associated protein 1 light chain 3 (MAP1LC3 or LC3) proteins including MAP1LC3A, MAP1LC3B, MAP1LC3C and the γ‐amino‐butyric acid (GABA) receptor‐associated (GABARAP) proteins including GABARAP, GABARAPL1 and GABARAPL2/GATE‐16 8. A unique and highly conserved ubiquitin‐like conjugation system involving the sequential action of the E1‐like ATG7, the E2‐like ATG3 and the E3‐like ATG12–ATG5–ATG16L1 complex mediates lipidation of LC3 and GABARAP proteins, thereby anchoring them to the membrane 9, 10. From a functional perspective, this ubiquitin‐like conjugation system is crucial in ensuring proper autophagosome formation 11 as well as for driving subsequent steps of autophagosome maturation, including cargo capture, membrane expansion and sealing and lysosome targeting/fusion 12, 13, 14, 15. The importance of the Atg3 enzyme, a main player in this pathway and catalyser of the lipid conjugation reaction, is emphasized by the finding that Atg3‐deficient mice display severe defects in autophagosome formation and die within 1 day after birth 11. While the molecular functions of ATG3 are well understood, the mechanisms governing its regulation remain unclear.
Emerging evidence of autophagy regulation by RNA‐binding proteins (RBPs) has recently been reviewed 16, including examples of post‐transcriptional regulation of key autophagy mRNAs via stability or translational control 17, 18, 19. In fact, RBPs comprise almost 10% of the human proteome and play key roles in most aspects of cellular function 20. Besides well‐described contributions of RBPs to major cellular processes including transcription, translation, RNA processing, localization and decay, more recent elucidation of RBP function revealed novel links to metabolic signalling pathways 21, 22, 23. In search for autophagy regulators among RBPs, we found the eukaryotic translation initiation factor 5A, eIF5A, initially characterized as a translation initiation factor 24, 25, but subsequently implicated in translation elongation 26, 27, 28. eIF5A is broadly conserved and contains the highly unusual amino acid hypusine, which is formed by a unique post‐translational modification of a specific lysine residue 29. Importantly, the hypusine residue has been ascribed a critical role in eIF5A‐mediated translation 26, 27, 30, 31. Yeast eIF5A and its bacterial ortholog EF‐P have been shown to specifically promote translation of troublesome poly‐proline motifs 27, 32, 33, 34, 35 leading to a model in which eIF5A/EF‐P binds to and prevents ribosomes from stalling during translation of geometrically challenging amino acid stretches 31, 36. Recent advances in yeast suggest that this does not only hold true for poly‐prolines, but also for a broader range of amino acid motifs 37, 38. eIF5A is highly conserved in higher eukaryotes including humans where it is also known to be hypusinated 39, yet little is known concerning the mechanisms for eIF5A function with the ribosome in protein synthesis of mammalian cells.
Here we present a high‐throughput comprehensive siRNA screen identifying RBPs, which affect the autophagy process. Among the top hits, we identify eIF5A as a positive effector of autophagy, through a mechanism ascribed to translation. We find that eIF5A depletion interferes with lipidation of LC3B and its family members and inhibits autophagosome formation. Importantly, these defects are effectively rescued by wild type, but not by a hypusination‐defective mutant of eIF5A. By employing a global proteomics‐based approach, we identify the E2‐like ATG3 protein as a direct translational target of eIF5A. We show that eIF5A, by ensuring efficient translation of ATG3, is required for LC3B lipidation. In addition, we identify a tripeptide motif in the ATG3 sequence, which contributes to its translational dependency on eIF5A. Collectively, our data shed light on a previously unknown mechanistic requirement for translation of a core autophagy protein, ATG3, necessary for a key biochemical step in the autophagy pathway.
Results
High‐throughput RNAi screen identifies eIF5A as a key autophagy effector
To identify RBPs impacting on autophagy, we performed an image‐based high‐throughput siRNA screen using an MCF‐7 human breast cancer cell line stably expressing the autophagosome marker GFP‐LC3B 40. We designed an siRNA library targeting 1,530 RBPs, selected by compiling the findings from three studies globally defining the RNA‐binding proteome 22, 41, 42 (Dataset EV1A). Cells were reverse transfected in 384‐well format with three individual siRNAs targeting each RBP for 72 h, fixed and analysed by high content microscopy and automated quantification of GFP‐LC3B puncta. The screen was conducted in both untreated and Torin‐1‐treated conditions, to address effects on both basal and induced autophagy, respectively (Fig 1A). The data were normalized to the median plate score, and siRNAs strongly decreasing cell viability were excluded, resulting in 1,420 RBP candidates subjected to statistical RSA analysis 43 (Dataset EV1B and C). In our initial filtering steps, candidates deregulating the number of GFP‐LC3B puncta were identified by applying both significance and fold change thresholds, resulting in the selection of 147 candidates from the basal autophagy screen and 176 candidates from the Torin‐1‐treated screen (Fig 1B, and Dataset EV1D and E; see Materials and Methods). Subsequent filtering of the candidates from both screens was done by manual curation, based on the degree of puncta deregulation, LogP values, number of scoring siRNAs, RNA‐binding potential and subcellular localization. This led to the selection of 23 candidates for our secondary validation screen, for which siRNAs either up‐ or downregulated GFP‐LC3B puncta (Fig 1B). Using the two best scoring siRNAs for each of these 23 RBP candidates, two validation screens were performed as in the initial screen, in both basal and Torin‐1‐induced conditions (Figs 1C and EV1). Reasoning that unspecific stress effects could cause an increase in GFP‐LC3B puncta, we chose to focus on proteins for which depletion caused a decrease in puncta, the majority of which were successfully validated in our secondary screens (Figs 1C and EV1). Among the strongest hits emerging from both untreated and treated secondary screens were CISD2, eIF5A, LARP1 and RC3H1. We chose to focus on the most prominent candidate from both validation screens, the evolutionarily conserved eukaryotic translation initiation factor 5A (eIF5A).
Figure 1. High‐throughput RNAi screen identifies eIF5A as an autophagy regulator.
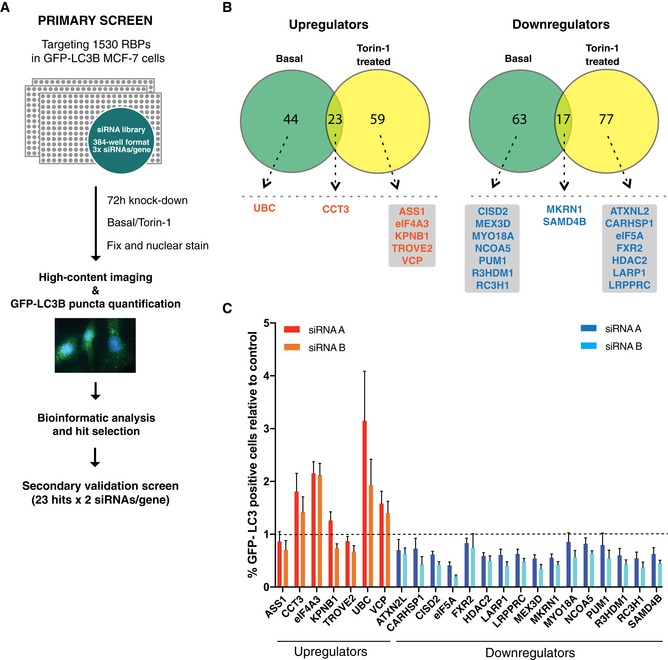
- Schematic overview of high‐throughput siRNA screening procedure performed in MCF‐7 cells stably expressing GFP‐LC3B.
- Venn diagrams showing distribution and overlap of significantly scoring RBP candidates identified from basal and Torin‐1‐treated screens based on statistical filtering by RSA analysis (see Materials and Methods for details and Dataset EV1 for full data sets). Dotted lines indicate selection of candidates based on LogP values, degree of puncta deregulation, number of scoring siRNAs and manual curation based on RNA‐binding potential and subcellular localization. Left: RBP candidates for which knockdown upregulated GFP‐LC3B puncta (orange). Right: RBP candidates for which knockdown downregulated GFP‐LC3B puncta (blue). Candidates are ordered alphabetically.
- Secondary validation screen (basal autophagy) for 23 hits from (B). Data shown are the percentage of GFP‐LC3B puncta‐positive cells relative to the scramble siRNA control (indicated by dashed line) and represent the mean + SEM from three biological replicates. The two best of three siRNAs from the primary screen were used and indicated as “siRNA A” and “siRNA B”. With the exception of ASS1, KPNB1, TROVE2 and FXR2, all candidates scored significantly (P < 0.05, Student's t‐test) in the expected direction relative to scramble. CCT3, ATXN2L, CARHSP1, MYO18A, NCOA5 and PUM1 scored with 1 siRNA, while EIF4A3, UBC, VCP, CISD2, eIF5A, HDAC2, LARP1, LRPPRC, MEX3D, MKRN1, R3HDM1, RC3H1, SAMD4B scored with both siRNAs.
Figure EV1. Secondary validation screen upon Torin‐1 induction of autophagy.
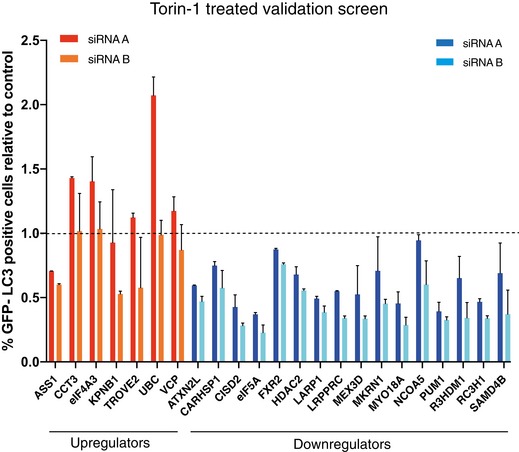
Secondary validation screen performed as in Fig 1C, but treated for 2 h with Torin‐1 prior to fixation. Data shown are the percentage of GFP‐LC3B puncta‐positive cells relative to the scramble siRNA control (indicated by dashed line) and represent the mean + SD from one representative experiment (n = 2).
eIF5A depletion reduces LC3/GABARAP lipidation and disrupts autophagosome formation
Prompted by the findings of our secondary validation screen, we focused our attention on eIF5A. Using two independent siRNAs, we confirmed that knockdown of eIF5A decreased the number of GFP‐LC3B puncta in basal conditions, during autophagy induction by Torin‐1 and after lysosomal inhibition by Bafilomycin A1 (Fig 2A and B). A clear effect of eIF5A knockdown in the face of mTORC1 inhibition by Torin‐1 indicates a likely function for eIF5A downstream or independent of mTORC1, further supported by the observation that mTOR activation levels remained unchanged after eIF5A depletion (Fig EV2A). In line with the initial findings from our screen and follow‐up validation experiments, we confirmed by Western blotting that eIF5A knockdown effectively decreased lipidated levels of LC3B (LC3B‐II) and its homologs, GABARAP (Fig 2C) and GATE‐16 (albeit to a lower degree; Fig EV2B), while LC3B, GABARAP and GATE‐16 mRNA levels remained largely unchanged (Fig EV2C). We confirmed that both siRNAs mediated a potent knockdown of eIF5A protein and mRNA (Figs 2C and EV2C), and as the siRNAs resulted in indistinguishable phenotypes, for subsequent experiments we used eIF5A si_B (from now on denoted as eIF5A si). The observed phenotype was not restricted to MCF‐7 cells, as lipidated LC3B was consistently decreased upon eIF5A depletion in a panel of human and mouse normal and cancer cell lines (Fig EV2D). Notably, we observed minor effects of eIF5A depletion on unlipidated levels of LC3B (LC3B‐I) in some experiments, which we believe to be a secondary consequence of altered lipidation status since this was not observed in the unlipidated pool of LC3B‐I from lipidation‐deficient ATG5 knockout (KO) cells (Fig EV2E). The effect of eIF5A in the presence of Bafilomycin A1, both on GFP‐LC3B puncta and LC3B‐II (Figs 2A and B, and EV2F), suggests that eIF5A plays a role in autophagosome formation rather than autophagosome turnover. By transmission electron microscopy, we confirmed a clear reduction in the number of mature autophagosomes after eIF5A depletion (Fig 2D and E) and, interestingly, the remaining population of autophagosomes in eIF5A‐depleted cells displayed a smaller size relative to control cells (Fig EV2G). Immunostaining for the pre‐autophagosomal marker ATG16L1 44 revealed a clear increase in the number of ATG16L1 puncta upon eIF5A knockdown, suggesting an accumulation of early or pre‐autophagosomal structures (Fig EV2H).
Figure 2. eIF5A regulates lipidation of ATG8 homologs and autophagosome formation.
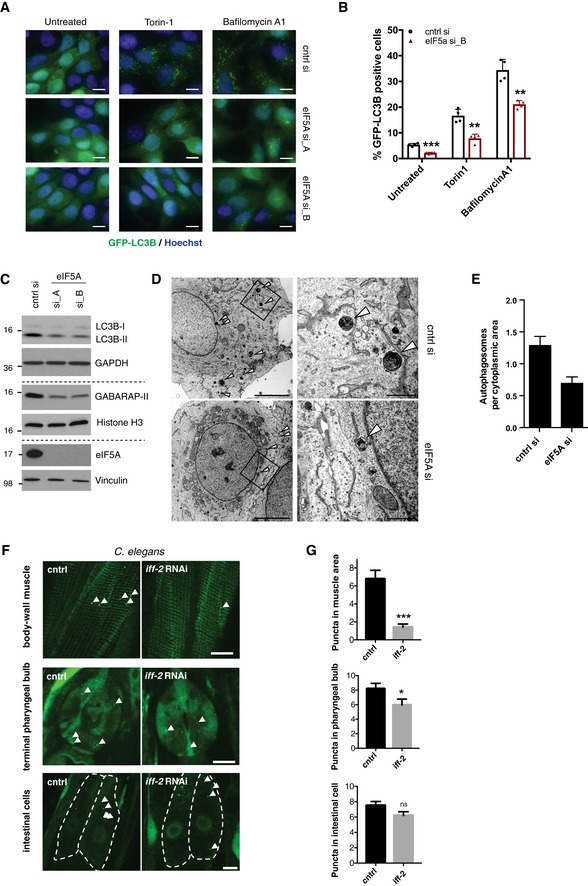
- Representative images of GFP‐LC3B puncta in MCF‐7 cells 72 h after transfection with cntrl and eIF5A siRNAs. For the last 2 h prior to fixation, cells were either left untreated or treated with Torin‐1 or Bafilomycin A1. Scale bars 10 μm.
- Quantification of GFP‐LC3B puncta in MCF‐7 cells 72 h after transfection. Cells were treated 2 h prior to fixation as in (A). Data are mean + standard deviation (SD) from one representative of three independent experiments. Each datapoint represents a technical replicate with values obtained from the quantification of > 1,500 cells. **P < 0.01, ***P < 0.001. Student's t‐test.
- Western blotting analysis of LC3B, GABARAP and eIF5A in MCF‐7 GFP‐LC3B cells transfected for 72 h with indicated siRNAs. A representative experiment is shown (n = 3). Vinculin, histone H3 and GAPDH were used as loading markers. Dashed lines indicate separate gels of the same samples.
- Transmission electron microscopy (TEM) images from MCF‐7 GFP‐LC3 cells after 72‐h transfection. Arrowheads indicate mature autophagosomes. Images (right) are enlargements of boxed area in overview images (left). Scale bars 5 μm (left), 1 μm (right).
- Quantification of autophagosomes per cytoplasmic area from TEM images. Data are mean + standard error of the mean (SEM) (n = 2) (30–35 cells/sample, counting a total of 90 autophagosomes in cntrl si and 54 autophagosomes in eIF5A si samples).
- Representative images from body‐wall muscle, terminal pharyngeal bulb and intestinal cells (individual cells are outlined) of Caenorhabditis elegans expressing GFP::LGG‐1 subjected to CTRL (empty vector) or iff‐2 RNAi. Arrowheads denote individual GFP::LGG‐1 puncta. Scale bars 10 μm.
- Quantification of GFP::LGG‐1 puncta in intestinal cells (n = 91), terminal pharyngeal bulbs (n = 43) and body‐wall muscle cell areas (n = 61–78) of animals subjected to CTRL (empty vector) or iff‐2 RNAi. Data are mean + SEM from four independent experiments. Student's t‐test: ns P > 0.05, *P < 0.05, ***P < 0.001.
Source data are available online for this figure.
Figure EV2. eIF5A depletion effects on specific autophagy markers.
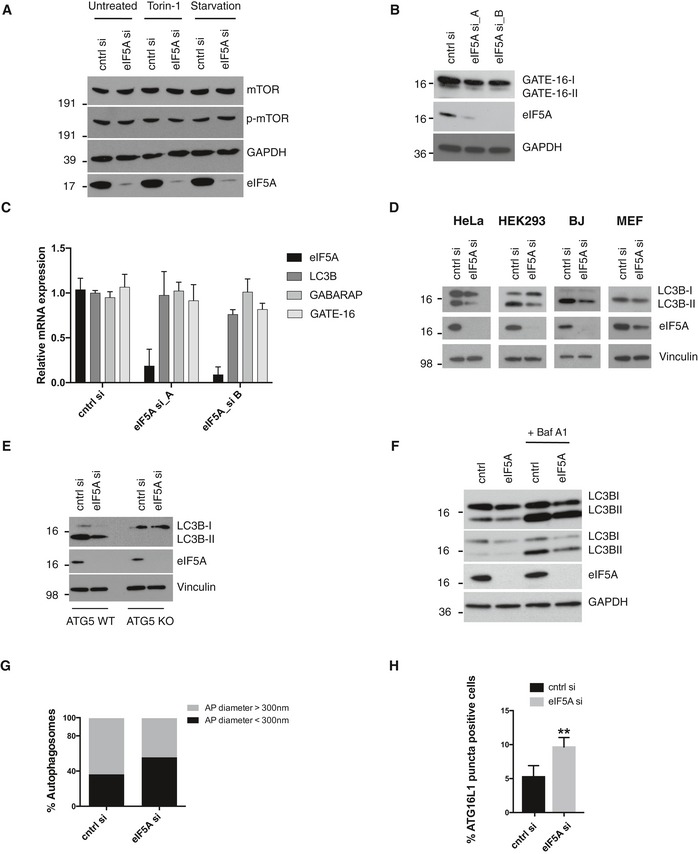
- Western blotting analysis of p‐mTOR (Ser 2448) in MCF‐7 GFP‐LC3B cells after indicated treatments (2 h) and siRNA transfections (72 h). A representative experiment is shown (n = 2).
- Western blot of MCF‐7 GFP‐LC3B cells after 72‐h transfection with indicated siRNAs. A representative experiment is shown (n = 3).
- qRT–PCR of LC3B, GABARAP and GATE‐16 mRNA levels in MCF‐7 GFP‐LC3B cells of indicated transcripts after 72‐h transfection with indicated siRNAs. Data represent the mean + SD (n = 3).
- Western blots of LC3B in indicated cells lines and siRNA transfections (72 h). A representative experiment is shown (n = 3).
- Western blot in ATG5 WT or ATG5 KO HeLa cells with indicated siRNAs transfections for 72 h. A representative experiment is shown (n = 3).
- Western blot of MCF‐7 cells with indicated siRNAs for 72 h. Bafilomycin treatment for the last 2 h prior to harvest. A representative experiment is shown (n = 3).
- Mature autophagosomes identified from TEM images were scored as having diameter of > 300 or < 300 nm, and their percentage‐wise distribution in cntrl or eIF5A siRNA‐treated cells is shown (n = 2).
- MCF‐7 cells were transfected for 72 h as indicated, fixed and immunostained for ATG16L1. Cells containing more that 5 puncta were defined as ATG16L1 puncta‐positive cells. Data represent the mean + SD (n = 3). Student's t‐test: **P < 0.01.
Source data are available online for this figure.
In order to address the potential evolutionary conservation of the observed phenotype, we employed a Caenorhabditis elegans strain stably expressing the GFP‐tagged Atg8 homolog lgg‐1 (GFP::LGG‐1) 45. eIF5A has two homologs in C. elegans, iff‐1 and iff‐2, where iff‐1 is specifically expressed in the germline and iff‐2 is more broadly expressed 46. We therefore focused on iff‐2, which we depleted by RNAi and quantified GFP::LGG‐1 puncta in the body‐wall muscle, terminal pharyngeal bulb (i.e. the foregut of the animal) and in the intestine. Interestingly, while there was a mild but insignificant trend in intestinal cells, iff‐2 depletion significantly reduced GFP::LGG‐1 puncta relative to control animals in the body‐wall muscle and terminal pharyngeal bulb (Fig 2F and G). Taken together, the effects of eIF5A depletion on GFP‐LC3B/LGG‐1 puncta, autophagosome number and lipidation of LC3/GABARAP family members across several cell lines and species strongly suggest a functionally conserved role for eIF5A in autophagosome formation.
Hypusination of eIF5A is required for its role in autophagy
eIF5A contains the highly unique amino acid, hypusine, formed by a post‐translational modification of a conserved lysine residue (K50 in humans), which is both important for eIF5A binding to the ribosome 47, 48, 49 and its role in translation 26, 27, 30, 31. In order to assess whether hypusination of eIF5A is required for autophagy, we created stable MCF‐7 cell lines expressing doxycycline‐inducible wild‐type (WT) eIF5A (“eIF5A WT”) or a hypusination‐defective K50A mutant (“eIF5A K50A”). We first verified the inducible expression and hypusination status in both cell lines (Figs 3A and EV3A). Interestingly, while 48‐h overexpression of eIF5A WT increased the level of GFP‐LC3B puncta, a significantly less pronounced effect was observed upon overexpression of eIF5A K50A (Fig 3B). To confirm this result in an independent manner, we treated MCF‐7 cells with N1‐guanyl‐1,7‐diaminoheptane (GC7), a potent and specific inhibitor of hypusination 50, 51, which decreased basal puncta levels and efficiently reduced the eIF5A‐mediated increase in GFP‐LC3B puncta (Fig 3B). By combining GC7 and eIF5A K50A expression, we observed an additive effect, likely due to GC7‐mediated inhibition of endogenous eIF5A present in the K50A cell line. Moreover, while re‐introduction of eIF5A WT effectively rescued the effects of siRNA‐mediated eIF5A depletion on GFP‐LC3B puncta and LC3B lipidation, the K50A mutant failed to do so (Fig 3C and D). Together, these results suggest that hypusination of eIF5A is important for its role in autophagy. Given the previously described importance of hypusine for eIF5A binding to the ribosome and for its role in translation, our results suggest that eIF5A is important for autophagy via its role in protein synthesis.
Figure 3. eIF5A hypusination is required for autophagy regulation.
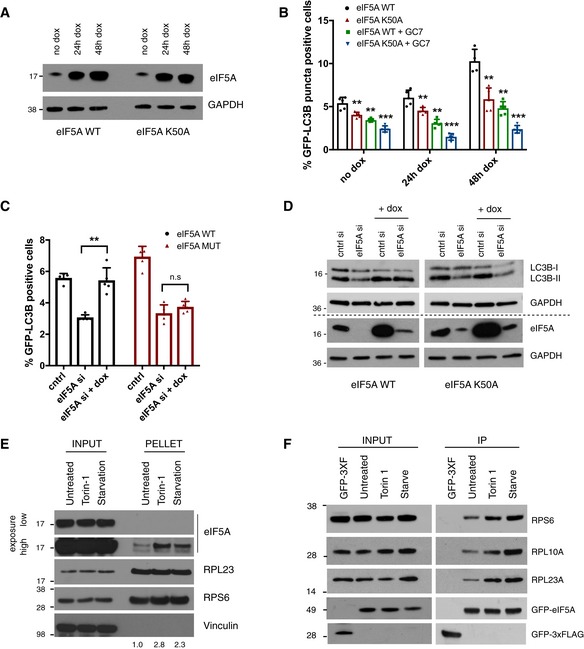
- Western blotting analysis of eIF5A levels in cell lines expressing WT and K50A eIF5A at indicated time points after doxycycline treatment. A representative experiment is shown (n = 3).
- Quantification of GFP‐LC3B puncta in MCF‐7 eIF5A WT or K50A cell lines at indicated time points of doxycycline treatment. For GC7 samples, treatment was given 24 h prior to fixation. Data are mean + standard deviation (SD) from one representative of three independent experiments. Each datapoint represents a technical replicate with values obtained from the quantification of > 1,500 cells. Statistical significance for each (no dox, 24 h dox and 48 h dox) was tested relative to corresponding eIF5A WT samples. **P < 0.01, ***P < 0.001. Student's t‐test.
- Quantification of GFP‐LC3B puncta in MCF‐7 eIF5A WT or K50A cell lines after indicated siRNA transfections (72 h) and doxycycline treatments (48 h). Data are mean + standard deviation (SD) from one representative of three independent experiments. Each datapoint represents a technical replicate with values obtained from the quantification of > 1,500 cells. Student's t‐test: ns P > 0.05, **P < 0.01.
- Western blot for LC3B in MCF‐7 eIF5A WT and K50A cell lines after 72‐h transfection with indicated siRNAs. Doxycycline was added for last 48 h of transfection. A representative experiment is shown (n = 3). Dashed lines indicate separate gels of the same samples.
- Western blotting analysis of eIF5A levels in pelleted ribosomes. Ribosomes were isolated through a 10% sucrose cushion from cells after indicated treatments (2 h prior to cell lysis) and analysed together with corresponding input samples. A representative experiment is shown (n = 3). Quantification of eIF5A levels relative to RPL23A in pellet is indicated below the figure (ImageJ software).
- Analysis of eIF5A interaction with the ribosome by Western blotting analysis. Inputs and eluates obtained after purification of GFP‐eIF5A were analysed with RPS6, RPL10A, RPL23A and GFP antibodies as indicated. A cell line expressing GFP‐3xFLAG was used as a negative control. A representative experiment is shown (n = 4).
Source data are available online for this figure.
Figure EV3. eIF5A hypusination, localization and ribosome binding.
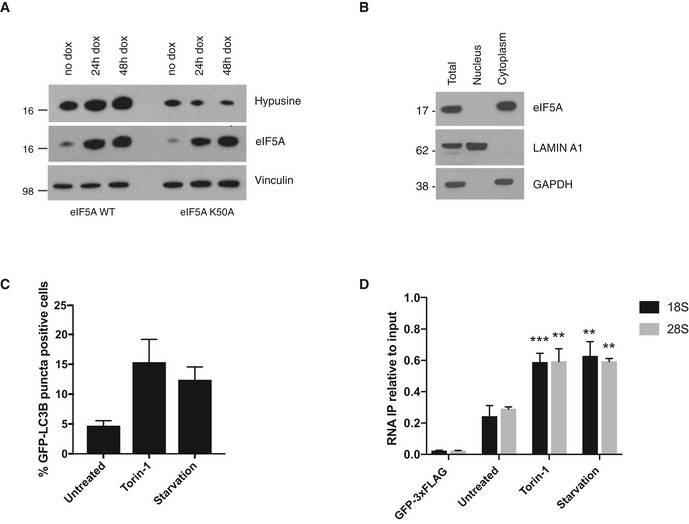
- Western blotting analysis of eIF5A and hypusine levels in cell lines expressing WT and K50A eIF5A at indicated time points after doxycycline treatment. A representative experiment is shown (n = 2).
- Nucleus/cytoplasmic fractionations were performed in MCF‐7 cells and subsequently analysed by Western blotting for eIF5A. Lamin A1 and GAPDH were used as nuclear and cytoplasmatic markers, respectively. A representative experiment is shown (n = 3).
- Quantification of GFP‐LC3B puncta after indicated treatments (2 h) in MCF‐7 GFP‐LC3B eIF5A WT inducible cells (used for ribosome purifications in Fig 3E). Data shown are mean + SD (n = 2).
- RNA inputs and eluates obtained from co‐purification of GFP‐eIF5A were analysed by qRT–PCR for 18S and 28S rRNA. A cell line expressing GFP‐3xFLAG was used as a negative control. Data are shown as eluates (RNA IP) relative to input and represent the mean + SEM (n = 4). Student's t‐test comparing treatments to untreated: **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
Association of eIF5A with ribosomes is enhanced by autophagy induction
Little is known concerning stress‐dependent dynamics of eIF5A in translational regulation, and thus, considering the highly stress‐responsive nature of autophagy, we questioned whether eIF5A itself responds to autophagy induction. We first confirmed that eIF5A is in fact located in the cytoplasm of MCF‐7 cells (Fig EV3B) and we then assayed its binding to ribosomes upon stimulation of autophagy by Torin‐1 treatment and starvation. While total protein levels of eIF5A remained unaffected by autophagy‐inducing treatments, biochemical purification of ribosomes and associated factors revealed an increased association of eIF5A with ribosomes after 2‐h starvation or Torin‐1 treatment, relative to untreated conditions (Fig 3E). Induction of autophagy by the above‐described treatments was confirmed in parallel by GFP‐LC3B puncta quantification (Fig EV3C). Further supporting these results, immunoprecipitation of GFP‐eIF5A showed specific retrieval of ribosomal proteins and ribosomal RNA (rRNA) from both small and large ribosomal subunits, which was enhanced upon autophagy induction (Figs 3F and EV3D). These data suggest that upon induction of autophagy, exemplified here by two independent modes of mTORC1 inhibition, eIF5A could be recruited to or stabilized at ribosomes. Alternatively, the enhanced association of eIF5A with ribosomes could reflect a change in the repertoire of translated mRNAs under these conditions.
eIF5A is required for translation of ATG3
Prompted by its previously described role as a translation factor, we assayed the effects of eIF5A depletion on global cellular translation in MCF‐7 cells. Fluorescently tagged O‐propargyl‐puromycin was incorporated into all newly translated proteins and analysed by image‐based quantification (see Materials and Methods). In parallel, the extent of eIF5A knockdown was monitored by immunofluorescence (Fig EV4). Interestingly, siRNA‐mediated knockdown of eIF5A for 72 h in MCF‐7 cells did not significantly affect global protein synthesis, in contrast to 5‐min treatment with the translational inhibitor cycloheximide (CHX; Fig 4A). These data support previous findings in yeast and human cells 52, 53, 54 and suggest a role for eIF5A in facilitating translation of a subset of proteins. In order to pin down more specific effects on protein synthesis, we performed liquid chromatography–mass spectrometry (LC‐MS) analysis of newly synthesized proteins in control and eIF5A‐depleted cells. In this approach, all newly synthesized proteins were pulse‐labelled for 2 h with the methionine analogue L‐azidohomoalanine (AHA), enriched using the Click‐iT enrichment resin and subsequently subjected to LC‐MS analysis (Fig 4B). Among the 2,308 detected newly synthesized proteins, 350 were significantly deregulated (> 1.5 fold change) upon eIF5A depletion (Fig 4C, Dataset EV2A and B, and Materials and Methods). Of these, 278 proteins were found to be downregulated in eIF5A‐depleted cells relative to the control, in line with the known role of eIF5A in stimulating translation 26, 27. To focus on translation‐related defects and to exclude potential effects of eIF5A on transcription or mRNA stability, we performed RNA sequencing of control and eIF5A siRNA‐transfected cells (Dataset EV3). Exclusion of genes significantly deregulated at the mRNA level further reduced the candidate list to a selection of 211 proteins, for which translation is likely hypersensitive to eIF5A levels. Focusing on autophagy‐relevant functions, we overlaid these candidates with the gene ontology defined autophagy‐related proteins (geneontology.org), leading us to the identification of a small subset of nine autophagy‐relevant proteins for which translation was specifically affected upon eIF5A depletion (Fig 4C and Dataset EV2C). Interestingly, this list included the ATG3 E2‐like enzyme, which is directly responsible for lipidation of Atg8 family proteins 11, 55, and the well‐known autophagy receptor protein SQSTM1/p62 13.
Figure EV4. eIF5A depletion from OPP assay.
Quantification of eIF5A knockdown in MCF‐7 cells from Fig 4A by immunostaining using Alexa 594‐coupled secondary antibody. Data are mean + SEM from four independent experiments.
Figure 4. eIF5A regulates translation of ATG3.
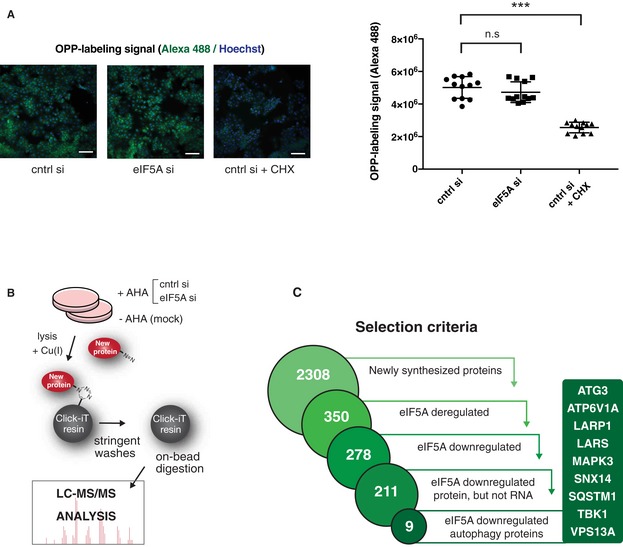
- Global translation measurement upon eIF5A depletion by pulse labelling of newly translated proteins with Alexa Fluor 488‐tagged O‐propargyl‐puromycin (OPP) followed by imaging and fluorescent quantification. MCF‐7 cells were transfected for 72 h, and a positive control was treated with cycloheximide (CHX) for 5 min. Representative images (left) show OPP (green) and Hoechst signals (blue). Quantification (right) shows mean values of fluorescent signal per well from four wells of three combined experiments. Error bars represent SD. Scale bars 100 μm. Student's t‐test: ns P > 0.05, ***P < 0.001.
- Schematic overview of labelling and mass spectrometry‐based analysis of newly translated proteins. Cells were transfected as indicated for 72 h and incubated 2 h with the methionine analogue L‐azidohomoalanine (+AHA) or left untreated (−AHA/mock). The Click‐iT chemistry‐based kit was used for covalent capture of newly synthesized proteins, which were subsequently subjected to LC‐MS analysis. The experiment was performed in biological triplicates.
- Flow diagram illustrating data analysis stages of newly synthesized protein LC‐MS described in (B). A set of newly synthesized proteins was defined using a fivefold enrichment threshold above mock (unlabelled −AHA) sample (2,308 proteins). Further filtering by applying P‐value and fold change thresholds (see Materials and Methods) identified 350 newly synthesized proteins deregulated by eIF5A of which 278 proteins were downregulated upon eIF5A depletion. Comparison to total RNA‐seq data and exclusion of proteins for which mRNA transcripts were deregulated by eIF5A narrowed down this selection to 211 candidates. Merging these with a set of 468 autophagy‐associated genes (geneontology.org) resulted in an overlap of nine autophagy‐related proteins.
eIF5A affects autophagy via ATG3 protein synthesis
Among the nine identified autophagy‐relevant MS hits (Fig 4C), we depleted ATG3 and SQSTM1/p62 by siRNA and found that while ATG3 knockdown clearly phenocopied eIF5A depletion with regard to LC3B lipidation (Fig EV5A), p62/SQSTM1 knockdown had no major effects on LC3B‐II levels and was thus excluded from further analysis (Fig EV5B). In accordance with results from our LC‐MS analysis, which revealed a 1.95‐fold reduction in nascent ATG3 protein levels (Dataset EV2C), we found that eIF5A depletion reduced the total pool of cellular ATG3 protein in MCF‐7 and HEK 293 cells (Figs 5A–C and EV5A), while validation of our RNA‐seq by qRT–PCR confirmed that ATG3 mRNA levels remained unchanged (Fig 5A). To further confirm that this effect was due to a defect in protein synthesis, we performed pulse‐labelling and coupling of newly synthesized proteins to biotin followed by streptavidin pull‐down and immunoblotting. These experiments confirmed that eIF5A‐depleted cells contained markedly less newly synthesized ATG3 relative to control cells (Fig 5B). Since the combined knockdown of eIF5A and ATG3 did not give rise to an additive or synergistic effect (Fig EV5A), this suggests a role for eIF5A function in the same pathway as ATG3. Importantly and further supporting this, we show that mild overexpression of ATG3 can effectively rescue the LC3B‐lipidation defect caused by eIF5A knockdown (Fig 5C). Collectively, these data suggest that eIF5A‐mediated effects on LC3B lipidation are caused, at least in part, by its role in ATG3 translation.
Figure EV5. eIF5A affects autophagy via ATG3.
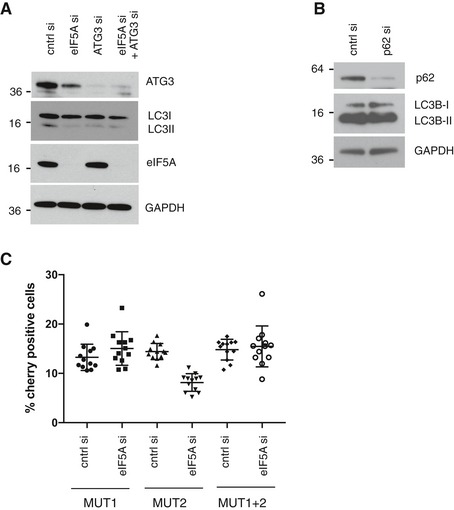
- Western blotting analysis of LC3B and ATG3 protein levels after 72‐h transfection with indicated siRNAs. Representative experiment is shown (n = 3).
- Western blotting analysis of effects of p62 depletion on LC3B lipidation in MCF‐7 cells after 72‐h transfection with indicated siRNAs. Representative experiment is shown (n = 2).
- Analysis of eIF5A effect on single or combined mutations of ATG3‐mCherry constructs assessed by imaging and automated quantification of mCherry fluorescent signal. HEK 293 cells were transfected with indicated siRNAs for 72 h. For the last 48 h, cells were transfected with indicated ATG3‐mCherry constructs. Data show quantification of mean mCherry fluorescence + SD from one representative experiment (n = 2).
Source data are available online for this figure.
Figure 5. Regulation of autophagy by eIF5A is mediated via ATG3 translation.
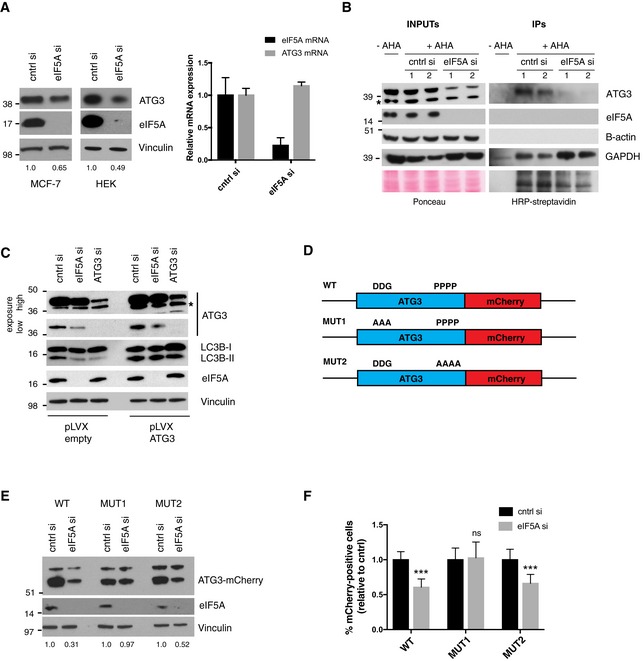
- Western blotting of ATG3 protein levels in HEK 293 and MCF‐7 cells (left) and qRT–PCR of ATG3 mRNA levels from MCF‐7 cells (right) after 72‐h transfection with cntrl and eIF5A siRNAs. For the Western blot, a representative experiment is shown (n = 3). Quantifications of ATG3 intensity relative to Vinculin are shown below the Western blot (ImageJ software). For the qRT–PCR, data shown are mean + SD (n = 3).
- Analysis of newly synthesized ATG3 levels upon depletion of eIF5A. Pulse‐labelling and coupling of newly synthesized proteins to biotin followed by streptavidin pull‐down and Western blotting; 1 and 2 indicate replicate samples from one experiment. A representative experiment is shown (n = 3). The asterisk indicates a nonspecific band.
- Western blot of HEK 293 cells transfected with pLVX‐empty or pLVX‐ATG3 constructs together with indicated siRNAs for 72 h. All samples were treated with doxycycline for 72 h to induce pLVX expression. A representative experiment is shown (n = 3). The asterisk indicates a nonspecific band.
- Schematic of ATG3‐mCherry WT and mutant constructs.
- Western blot of HEK 293 cells transfected with indicated siRNAs for 72 h. For the last 48 h, cells were transfected with indicated ATG3‐mCherry constructs from (D). ATG3‐mCherry is detected with anti‐mCherry Ab. A representative experiment is shown (n = 3), and the full‐length ATG3‐mCherry band intensity (lower band) is quantified relative to Vinculin and shown below the figure (ImageJ software).
- HEK 293 cells transfected as described in (E), fixed and imaged for automated quantification of cytoplasmic mCherry fluorescence. Percentage of mCherry positive cells was defined based on arbitrary fluorescent intensity cut‐offs. Data show the mean fluorescent signal + SEM relative to control from three independent experiments. Student's t‐test: ns P > 0.05, ***P < 0.001.
Source data are available online for this figure.
A tripeptide motif in ATG3 contributes to its eIF5A dependency
eIF5A/EF‐P has previously been shown to alleviate translational stalling of the ribosome at hard‐to‐translate motifs in yeast and bacteria 27, 32, 33. While these studies initially focused on poly‐prolines, eIF5A was more recently shown in yeast to be generally required for translation, with a broader selection of motifs displaying hyperdependency on the factor. In particular, Schuller et al 37 identified 29 tripeptide motifs, which cause strong ribosome pausing (at least 10‐fold) in the absence of eIF5A. Based on this, we searched the amino acid (aa) sequence of ATG3 and identified two potential stalling motifs: DDG at position 104–106 and PPPP at position 254–257. To assess their functional importance, we utilized a reporter construct containing the full‐length ATG3 C‐terminally coupled to a mCherry fluorophore (Fig 5D, “WT”). We then mutated each of the respective motifs DDG→AAA (“MUT1”) and PPPP→AAAA (“MUT2”) and evaluated the sensitivity of these constructs to eIF5A depletion by monitoring ATG3‐mCherry translation (Fig 5D). Interestingly, while the MUT2 construct remained sensitive to eIF5A depletion to a similar extent as the WT construct, the MUT1 construct displayed clearly decreased sensitivity to eIF5A knockdown, as determined by immunoblotting for ATG3‐mCherry protein expression (Fig 5E) and by microscopy‐based quantification of ATG3‐mCherry fluorescence (Fig 5F). In addition, we generated a reporter construct containing both mutations (MUT1 and MUT2), which behaved similarly to the DDG→AAA single mutant (Fig EV5C). These data indicate that the DDG motif in ATG3 may represent a hard‐to‐translate motif, which causes eIF5A dependency for its efficient translation. Thus, our findings not only identify eIF5A as a novel translational requirement of autophagy, but also uncover previously unknown mechanistic aspects of eIF5A‐mediated translation in human cells.
Collectively, our data support a model in which eIF5A displays enhanced ribosome association upon induction of autophagy, where it assists in translation of ATG3 through the DDG motif, which in turn facilitates lipidation of LC3B and its family members to promote autophagosome formation (Fig 6).
Figure 6. Model for eIF5A‐mediated regulation of autophagy.
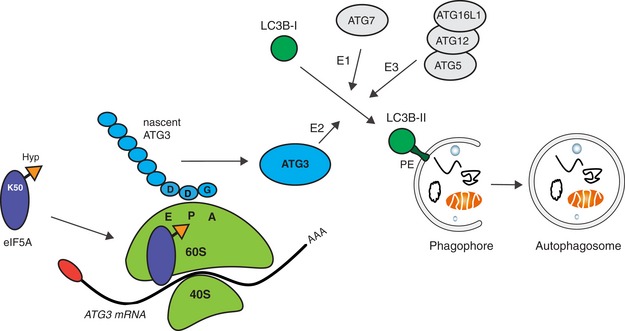
During autophagosome formation, cytosolic LC3B‐I is conjugated to the lipid phosphatidylethanolamine (PE), forming LC3B‐II which is anchored to the phagophore. This is mediated via the sequential action of the E1‐like ATG7, E2‐like ATG3 and E3‐like ATG5–ATG12–ATG16L1 complex. eIF5A, via its hypusine residue, assists the ribosome in translating the ATG3 protein at its DDG motif. This increases the efficiency of ATG3 production and facilitates LC3B lipidation and autophagosome formation. eIF5A association with ribosomes is enhanced upon autophagy induction.
Discussion
The autophagy process is tightly controlled at multiple levels. This ensures that a fine‐tuned balance is maintained to manage the cellular needs for quality control and nutrient availability, while at the same time preventing excessive degradation of crucial cellular components 3. Although the regulatory mechanisms governing this control are well studied in terms of protein interactions, post‐translational modifications and transcriptional regulation of ATG proteins 56, 57, 58, the regulation of ATG protein production at the translational level is not well investigated. The data presented here reveal a novel requirement for ATG3 translation, helping us to understand the complexity of this fundamental process.
Prompted by recent literature assigning novel functions to RBPs with links to metabolic signalling pathways including autophagy 16, 21, 22, 23, we here present a high‐throughput screen identifying several RBP candidates with functional effects in autophagy regulation. Our screen not only sheds light on novel regulatory mechanisms of autophagy regulation, but also provides a strong resource for the research community. A top candidate from this screen was the highly conserved translation factor, eIF5A, found to positively impact autophagy. Specifically, we show that eIF5A is a limiting factor for lipidation of LC3B and its mammalian paralogs GABARAP and GATE‐16, and hence required for proper autophagosome formation. Our proteomics approach elucidated the key finding that the translation of the E2‐like enzyme ATG3, a crucial factor in autophagosome formation 11, 59, 60, is hyperdependent on eIF5A. Indeed, it is known that depletion of ATG3 blocks accumulation of LC3B puncta and renders cells incapable of forming lipidated LC3B (LC3B‐II). Moreover, it has previously been shown that Atg3 KO mice display hampered membrane elongation and closure, resulting both in malformation and reduced number of autophagosomes 11, 60. Likewise, mutation of Atg3 in yeast results in inefficient expansion of the isolation membrane and leads to the formation of smaller autophagosomes 59. Our own observations strongly support these findings by establishing that eIF5A depletion and thereby reduced ATG3 translation not only inhibits lipidation of LC3B and its paralogs and decreases autophagosome number, but also causes a reduction in autophagosome size, as revealed by electron microscopy experiments. In addition, the increase in ATG16L1 puncta seen upon eIF5A depletion suggests the accumulation of pre‐autophagosomal intermediate structures, similar to what has been previously observed upon ATG3 depletion 11, 60. Indeed, the Atg12–Atg5–Atg16L complex is localized on the isolation membrane during elongation, but disassociates again before completion of mature autophagosomes 44. While we observe lipidation phenotypes in a panel of mammalian cells, including human and mouse normal and cancer cell lines, we additionally find that depletion of the eIF5A homolog iff‐2 in C. elegans decreases the number of GFP::LGG‐1 puncta in the body‐wall muscle and terminal pharyngeal bulb. Thus, although it is important to acknowledge the likely role of eIF5A in several cellular processes, our data strongly advocate for the evolutionary conservation, and thus broad functional importance, of this newly described role for eIF5A.
eIF5A has mainly been studied as a somewhat inert regulator of translation, and to our knowledge, little is known concerning the dynamics of eIF5A function or for instance whether it displays differential expression, activity and/or subcellular localization during conditions of cellular stress. One study showed that eIF5A contributes to stress granule assembly and that it differentially modulates translation in the absence or presence of arsenite‐induced stress 53. Interestingly, while mTORC1 inhibition via starvation or Torin‐1 treatment did not affect eIF5A expression levels, we observed an increase of eIF5A in ribosome‐enriched fractions and increased eIF5A‐mediated retrieval of ribosomal proteins and rRNA under these conditions. The reason for this enhanced association is unknown and will require further investigation. This result underscores the physiological relevance of our findings and leads us to propose a model in which eIF5A displays increased binding to ribosomes under conditions of autophagy induction where it is needed to assist the translation likely of several proteins including ATG3. Whether this enhanced ribosomal binding could result from an active recruitment or increased stabilization of eIF5A remains unknown. The increased production of ATG3 protein resulting from this in turn mediates a positive regulation of the autophagy response via increased lipidation of Atg8 family proteins, enabling efficient autophagosome formation (Fig 6).
eIF5A has been proposed to play an essential role in facilitating slow or difficult reactions encountered at the ribosome peptidyl‐transferase centre during translation 27, 36, 61. Notably, its hypusination stabilizes the association of eIF5A with the ribosome 47, 48, 49 and has been ascribed a key role in eIF5A‐mediated translation 26, 27, 30, 31. Moreover, eIF5A is proposed to be particularly critical for the translation of poor ribosome substrates or geometrically challenging amino acid motifs 30, 31. Interestingly, we found that inhibition of eIF5A hypusination by K50 mutation or GC7 treatment effectively impaired the induction of GFP‐LC3B puncta mediated by eIF5A overexpression. Moreover, the LC3B lipidation defect was efficiently rescued by WT eIF5A but not by its hypusination‐defective mutant. Hence, in contrast to previous indications 62, these findings collectively suggest the importance of this highly unique post‐translational modification. Remarkably, spermidine, which is the donor for hypusination, is also well characterized as an autophagy inducer 63. Yet, it remains unknown whether spermidine‐induced autophagy can, to some extent, be attributed to activation of eIF5A via hypusination. Working in Drosophila, Patel et al 64 hypothesized a role for eIF5A and the deoxyhypusine hydrolase homolog nero in autophagy. In contrast to our data in mammalian cell lines and C. elegans, loss of eIF5A or lack of its hypusination in larvae resulted in the appearance of Atg8‐positive structures perhaps indicating stress induction. However, mechanistic aspects of these findings remain unclear.
In agreement with previous studies from both yeast and human cells 52, 53, 54, we did not detect noticeable effects of eIF5A depletion on global translation in our cellular system, supporting the idea that eIF5A is less important for general protein synthesis, and instead required for more specific translation of a subset of mRNAs. One defining characteristic of such mRNAs is the amino acid stretches they encode. Indeed, initial functional studies of yeast eIF5A and its bacterial ortholog EF‐P focused on hard‐to‐translate poly‐proline stretches, which were shown to cause ribosome stalling in the absence of eIF5A/EF‐P 27, 32, 33. Two recent studies, utilizing transcriptome‐wide ribosome profiling and 5′‐phosphorylated RNA sequencing, suggest that there are, at least in yeast, several additional eIF5A‐dependent motifs 37, 38. In fact, among 29 identified tripeptide pause motifs, 18 do not contain more than one proline, and four (DVG, DDG, GGT, RDK) are completely devoid of proline 37. Since the details of eIF5A function in translation have not been fully addressed in mammalian cells, the importance of poly‐prolines for eIF5A function in mammals remains unknown. Surprisingly, our study shows that while four consecutive prolines in the sequence of ATG3 had little or no effect on ATG3 regulation by eIF5A, mutation of a motif devoid of prolines, DDG, clearly reduced the sensitivity of ATG3 to eIF5A depletion. Interestingly, poly‐proline enrichment was neither found among total proteome changes occurring in HeLa cells after knockdown of eIF5A 52 nor in our own nascent proteomics data (not shown). Undoubtedly, future efforts including in‐depth ribosome profiling analysis in mammalian cells will help us to unravel these mechanistic details further. While the DDG motif is not fully conserved in C. elegans Atg3, there are two additional potential eIF5A‐sensitive tripeptide motifs present the C. elegans Atg3 homolog. Thus, it is possible that iff‐2 acts via an another motif or an alternative mechanism to affect autophagy in C. elegans.
While the vast majority of studies exploring core autophagy regulation via ATG proteins has focused mainly on their molecular actions, less attention has been given to mechanisms ensuring their adequate levels of production. More elaborate insight has been obtained with regard to transcriptional control of ATG transcripts 57, 65, 66, 67, though only a small handful of studies have addressed aspects of translational regulation of ATG proteins 17, 68. Importantly, the highly dynamic nature of autophagy, its importance in stress response and the potentially detrimental consequences of its dysregulation all underscore the necessity for its tightly coordinated control at multiple levels. Our findings highlight novel insight towards the translational requirements for LC3B lipidation and autophagosome formation.
Materials and Methods
Oligonucleotides, plasmids and cloning
Oligonucleotides and plasmids used in the study are listed in Table EV1. Plasmids: pLVX_eIF5A_WT and pLVX_GFP_eIF5A were constructed with In‐Fusion HD Cloning Kit (Clontech), as described by the manufacturer, using primers listed in Table EV1 and a pCEFL‐GFP‐eIF5A 69 plasmid as a template. pLVX_eIF5A_K50A was generated using the QuickChange II XL Site‐Directed Mutagenesis Kit (Promega) according to the manufacturers' protocol and mutagenesis primers listed in Table EV1. pLVX_ATG3 was generated with In‐Fusion HD Cloning Kit (Clontech), as described by the manufacturer, using primers listed in Table EV1 and mCherry‐ATG3‐N‐18 plasmid as a template. ATG3_mCherry_MUT1 and ATG3_mCherry_MUT2 were generated by site‐directed mutagenesis with oligonucleotides listed in Table EV1, using mCherry‐ATG3‐N‐18 as a template. Mutagenesis was conducted using QuickChange II XL Site‐Directed Mutagenesis Kit (Promega) according to the manufacturers' protocol, or by an analogous in‐house approach using CloneAmp PCR amplification (Clontech) and Dpn1 digestion (NEB). pLVX‐GFP‐3XFLAG was constructed by fusing a triple FLAG tag to the C‐terminus of eGFP using In‐Fusion HD Cloning Kit (Clontech).
Sequences of all generated constructs were confirmed by Sanger sequencing.
Cell culture, transfections, lentiviral transduction and treatments
MCF‐7 GFP‐LC3B 40 were propagated in RPMI 1640 (Invitrogen) supplemented with 6% foetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin (P/S). HEK 293, HeLa, BJ and MEF were propagated in DMEM supplemented with 10% FBS and P/S. The screen and all follow‐up experiments were performed in MCF‐7 GFP‐LC3B cells unless otherwise indicated. HEK 293 cells were used for plasmid transfections due to higher transfection efficiency than MCF‐7 GFP‐LC3B. Stable MCF‐7 GFP‐LC3B cells expressing doxycycline‐inducible eIF5A WT or eIF5A K50A or ATG3, as well as MCF‐7 GFP‐eIF5A and MCF‐7 GFP‐3xFLAG cells, were generated by lentiviral transduction as follows: HEK cells were transfected with relevant pLVX constructs together with pAX8 (packaging) and pCMV‐VSVG (envelope) plasmids, and virus‐containing supernatants were harvested after 24 h. Supernatants were filtered through a 0.45 μm filter, supplemented with 8 μg/ml polybrene (Sigma) and added to pre‐seeded target cells. Puromycin selection (2 μg/ml) was initiated 24 h after virus infection. For Fig EV5B, SQSTM1/p62 and control siRNAs were purchased from Sigma and were transfected at 50 nM and re‐transfected 48 h later. All other siRNAs were Silencer Select chemistry (Thermo Fisher Scientific), and transfections were performed with 10 nM (except for primary screens which used 5 nM siRNA) using Lipofectamine 2000 or RNAi max (Invitrogen) as transfection reagents according to manufacturer's protocol. Treatment concentrations are as follows: Doxycycline 100 ng/ml, GC7 100 μM, Torin‐1 250 nM, Bafilomycin A1 100 nM. For starvation cells were treated with Hank's balanced salt solution (HBBS). ATG5 WT and CRISPR KO HeLa cells were generated by lentiviral transduction using previously described NTC and ATG5 CRISPR constructs 70.
Antibodies and Western blotting
Unless otherwise indicated, cells were lysed in radioimmune precipitation buffer (150 mM NaCl, 1% Nonidet P‐40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris–HCl, pH 8, 2 mM EDTA) containing 1 mM dithiothreitol, 1 mM Pefabloc (Roche Applied Science), Complete Mini Protease Inhibitor Cocktail (Roche Applied Science) and Phosphatase Inhibitor Cocktail (Roche Applied Science). Protein was separated on a 4–12% NuPAGE Bis–Tris gel or a 16% Tris‐Glycine gel and transferred to a nitrocellulose membrane or polyvinylidene difluoride membrane. Primary antibodies used are as follows: LC3B (nanotools, 1:200) (LC3B CST, 1:1,000), GABARAP (Abgent 1:1,000), GATE‐16 (MBL, 1:1,000), ATG3 (Sigma, 1:500), Vinculin (Sigma, 1:100,000), GAPDH (Santacruz, 1:20,000), eIF5A (Santacruz, 1:500), Lamin A1 (Santacruz, 1:1,000), histone H3 (Abcam 1:10,000), p62 (MBL, 1:5,000), RFP/Cherry (Rockland, 1:6,000), p‐mTOR (Ser 2448) (CST, 1:1,000), mTOR (Cell signaling, 1:1,000), RPL23A (Abcam, 1:50,000), RPS6 (CST, 1:1,000), RPL10A (Santacruz, 1:2,000), Hypusine (Merck Millipore 1:5,000). For cellular fractionations, the Nuclei EZ lysis buffer (Sigma) was used according to manufacturer's instructions.
qRT–PCR analysis
Total RNA from transfected cells was isolated using Trizol reagent (Invitrogen). qRT–PCR was performed using the Fast SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences for genes of interest and housekeeping genes are shown in Table EV1.
High‐throughput siRNA screen
RNAi screen was performed using a custom‐designed Silencer® Select siRNAs (Thermo Fisher Scientific) library designed to target 1,530 predicted RBPs chosen by compiling data from three recent studies 22, 41, 42. MCF‐7 GFP‐LC3B cells were reverse transfected with 5 nM siRNA for 72 h in 384‐well format, and each RBP was targeted by three independent siRNAs. For the last 2 h prior to fixation, cells were either left untreated or treated with 250 nM Torin‐1. Cells were then fixed in 3.7% formaldehyde, and nuclei were stained with Hoechst (33342, Thermo Fisher Scientific). Liquid handling was performed using the a Microlab STARlet liquid handling workstation (Hamilton Robotics). Image acquisition was done using an InCell2200 automated microscope equipped with a 20× Nikon objective (GE Healthcare). Ten pictures were taken per well (counting > 1,500 cells per well) and analysed using the InCell Analyzer Workstation 3.7.3 software (GE Healthcare). Nuclei were segmented based on the Hoechst signal, and cells were segmented from the GFP channel. Cells with more than either 5 (untreated screen) or 10 (treated screen) GFP‐LC3B puncta were considered GFP‐LC3B puncta‐positive cells. Statistical analysis was performed with a previously described statistical pipeline 71 utilizing Redundant siRNA Activity (RSA) analysis for hit scoring 43. RSA assigned P‐values (LogP) per gene were based on the score of individual siRNAs, which was derived from the percentage of puncta‐positive cells (signal), normalized to median plate intensity (Dataset EV1). siRNAs targeting the known autophagy regulators, Raptor and Beclin‐1, were used to define thresholds in the analysis. Hits altering cell number by more than 2SD were excluded from further analysis, and the following cut‐off was used to extract the most significantly changed candidates: Score ±1.5 relative to control siRNAs and LogP < −1.0 if all three siRNA scoring, or LogP < −1.5 if 2 siRNA scoring. Following this initial selection, a multi‐factor manual curation was performed based on LogP values, the degree of puncta deregulation, number of scoring siRNAs, RNA‐binding potential and subcellular localization. Validation screens were performed essentially as described above, but scaled up to 96‐well format and using 10 nM siRNA. For each of the 23 hits, two best of three siRNAs were used and siRNA sequences are listed in Table EV1.
Newly synthesized protein analysis
Newly synthesized protein labelling—liquid chromatography–mass spectrometry (LC‐MS) analysis
Capture of newly synthesized proteins was carried out using the Click‐iT® Protein Enrichment Kit (Thermo Fisher Scientific, C10416). MCF‐7 GFP‐LC3B cells were transfected with control and eIF5A siRNAs for 72 h, and methionine‐free media supplemented with Click‐iT® AHA (L‐azidohomoalanine) (or no AHA for mock sample) was added for the last 2 h at a final concentration of 40 μM to label freshly made proteins. The experiment was performed in three biological replicates. The cells were lysed and labelled proteins were enriched according to the manufacturer's protocol. Tryptic peptides were identified by LC‐MS using an EASY‐nLC 1000 (Thermo Scientific) coupled to a Q Exactive HF (Thermo Scientific) equipped with a nanoelectrospray ion source. Peptides were separated on an in‐house packed column of ReproSil‐Pur C18‐AQ, 3 μm resin (Dr Maisch, GmbH) using a 120‐min gradient of solvent A (0.5% acetic acid) and solvent B (80% acetonitrile in 0.5% acetic acid) and a flow of 250 nl/min. The mass spectrometer was operated in positive ion mode with a top 12 data‐dependent acquisition, a resolution of 60,000 (at 400 m/z), a scan range of 300–1,700 m/z and an AGC target of 3e6 for the MS survey. MS/MS was performed at a scan range of 200–2,000 m/z using a resolution of 30,000 (at 400 m/z), an AGC target of 1e5, an intensity threshold of 1e5 and an isolation window of 1.2 m/z. Further parameters included an exclusion time of 45 s and a maximum injection time for survey and MS/MS of 15 and 45 ms, respectively.
The raw files obtained from LC‐MS were processed using the MaxQuant software 72 version 1.5.3.30 which facilitates both protein identification and quantification. Peak lists were searched against the human UniProt database version 2016.08 using the Andromeda search engine incorporated in MaxQuant with a tolerance level of 7 ppm for MS and 20 ppm for MS/MS. In group‐specific parameters, trypsin was chosen as digestion enzyme with max 2 missed cleavages allowed. Variable modifications included methionine oxidation, protein N‐terminal acetylation, deamidation of asparagine and glutamine and the box “Re‐quantify” was checked. In global parameters, carbamidomethylation of cysteine was set as fixed modification, minimum peptide length was set to 7, and the box match between runs was checked with a match time window of 0.7 min and an alignment time window of 20 min. For protein and peptide identification, the FDR was set to 1% based on the target‐decoy approach and protein quantifications were based on a minimum ratio count of 2 using both unmodified and proteins modified with the variable modifications mentioned above for quantification.
Individual peptide intensity values were normalized to sum of peptide intensities per sample to correct for minor differences in amounts of loaded material. Statistical analysis was conducted in R using samr package (“Two class unpaired”, nperms = 1,000). Only proteins detected with at least 2 unique peptides in each replicate of cntrl si or eIF5A si, and with median total peptide intensity fivefold above the mock sample were used in the analysis (Fig 4C). Fold change values of median peptide intensities were calculated for three replicates, and pseudo intensity counts of 1 were added for median values equal to zero. Significantly deregulated hits upon eIF5A depletion were selected based on P‐value < 0.1 and fold change ±1.5 relative to the control sample. A subset hits downregulated by eIF5A was further merged with RNA‐seq data to discard genes significantly deregulated upon eIF5A depletion (FDR < 0.05). A set of 211 translationally downregulated genes was then merged with 468 proteins associated with the “autophagy” gene ontology term extracted for AmiGo2 (geneontology.org) 73. This filtering step resulted in identification of nine proteins, of which translation is potentially eIF5A‐dependent.
Nascent protein labelling—biotin pull‐down/Western blotting analysis
Validations of the LC‐MS results of newly synthesized proteins were carried out by Western blotting analysis. MCF‐7 GFP‐LC3B stably overexpressing ATG3 protein upon doxycycline induction were used in order to increase the pull‐down efficiency of freshly translated ATG3. Cells were cultured on 15‐cm2 dishes and subjected to 72‐h transfection using control and eIF5A siRNAs. Expression of ATG3 was induced for 24 h by doxycycline (400 ng/ml), and proteins were labelled with Click‐iT® AHA in methionine‐free medium for 4 h prior to harvesting. Click‐iT® Protein Reaction Buffer Kit (Thermo Fisher Scientific, C10276) and biotin alkyne (Thermo Fisher Scientific, B10185) were used to label protein according to the manufacturer. Cell pellets were resuspended in 300 μl of lysis buffer (50 mM Tris–HCl pH 8.0, 1% SDS) in the presence of protease inhibitors (Roche) and incubated 10 min on ice. Cell lysates were sonicated using Branson sonicator (3 × 10 s, 10% amplitude) and centrifuged at 15,000 g, at 4°C for 10 min. At least 200 μg of total protein was labelled and precipitated according to the manufacturer. Two labelling reactions were carried out for each sample to increase the final outcome. Proteins were resuspended in 50 μl lysis buffer by shaking at 1,200 rpm for 10 min at 30°C in the presence of protease inhibitors, and two labelling reactions were pooled. Sample volumes were adjusted to 1 ml with Tris–HCl pH 8.0 (final conc. 50 mM Tris–HCl, 0.1% SDS), incubated for 10 min at 30°C and centrifuged at 15,000 g at 25°C for 5 min to remove undissolved material. Supernatants were incubated with 100 μl of Dynabeads™ MyOne™ Streptavidin C1 beads on a wheel rotator for 60 min at room temperature. After five washes with 1 ml of 50 mM Tris–HCl/0.1% SDS buffer proteins were eluted in 1.5× NuPAGE SDS sample buffer containing 20 mM DTT and analysed by Western blotting.
Library construction, sequencing and data treatment
Total RNA from three biological replicates was purified, and libraries were built using the Illumina TruSeq kit (RPHMR12126) according to the manufacturer's protocol. The quality and quantity of the library were assessed by the 2100 expert Bioanalyzer (Agilent), by Qubit 2.0 fluorometer (Invitrogen) and by ABI StepOnePlus Real‐Time PCR. The libraries were sequenced on the Illumina NextSeq500 platform to generate 50‐nt‐long reads using single‐end. Raw RNA‐seq data were subjected to quality control filters applying threshold of Phred (quality) ≥ 25. Prior to mapping, Illumina adapters were removed and the reads were trimmed using cutadapt. Read mapping was performed with bowtie2 74 (version: 2.2.4, using flag: –very‐sensitive‐local) using the human genome version 19 (hg19). Counting of reads was performed using Bam files in HTSeq (using gencode hg19 gene annotations). Read counts were analysed by DESeq2 75 using default parameters in R environment.
OPP protein synthesis assay
Effects of eIF5A knockdown on global protein synthesis were assessed using the Click‐iT™ Plus OPP Alexa Fluor™ 488 Protein Synthesis Assay Kit (C10428 Thermo Fisher Scientific) according to manufacturer's instructions. In brief, MCF‐7 eGFP‐LC3B cells were reverse transfected for 72 h and treated with 20 μM Click‐it OPP for 30 min prior to fixation in 3.7% formaldehyde and permeabilization in 0.5% Triton X‐100. Click‐it reaction was performed using Alexa fluor 488 according to manufacturer's instructions and nuclei were stained with Hoechst. Image acquisition was done using an InCell2200 automated microscope equipped with a 20× Nikon objective (GE Healthcare). Six pictures were taken per well (counting > 1,500 cells per well) and fluorescent quantification and analysis was done using the InCell Analyzer Workstation 3.7.3 software (GE Healthcare). Cycloheximide treatment was for 5 min (100 μg/ml).
Analysis of ribosome‐associated eIF5A
Ribosome purification through 10% sucrose cushion by ultracentrifugation
MCF‐7 cells were left untreated or treated with Torin‐1 (250 nM) or starved in Hank's balanced salt solution (HBBS) for 2 h. Cycloheximide was added to media for 3 min at final concentration 100 μg/μl. Cells were washed with ice‐cold PBS containing cycloheximide and collected by centrifugation for 5 min at 300 g. Cell pellets were resuspended in 300 μl of lysis buffer containing 20 mM HEPES pH 7.4, 100 mM KOAc, 5 mM Mg(OAc)2, 2 mM DTT, 0.5% Triton X‐100, 0.25 mM spermine, 100 μg/ml cycloheximide, protease inhibitors (Roche, without EDTA) and RNase inhibitors (NEB). After 5‐min incubation on ice followed by gentle pipetting, the samples were centrifuged for 10 min at 16,000 g at 4°C. Concentrations of lysates were adjusted based on A260/A280 measurements. Input samples were collected, and 250 μl of the remaining lysate was layered on a 250 μl 10% sucrose cushion prepared in same buffer as above but excluding Triton X‐100. Samples were spun at 287,582 g (90,000 rpm, TLA 120.2 rotor) for 30 min at 4°C. Pellets were washed by addition and subsequent removal of lysis buffer without Triton X‐100. Pellets were resuspended in 40 μl of 1.5× NuPAGE sample loading buffer containing 10 mM TCEP and analysed by Western blotting.
Co‐immunoprecipitation (co‐IP) of GFP‐eIF5A
MCF‐7 cells stably expressing GFP‐eIF5A or a control GFP‐3XFLAG construct were used in the co‐immunoprecipitate experiment. The cells were either left untreated (GFP‐3xFLAG or “Untreated” control) or treated as it is explained in the above section. Cell lysates were prepared and clarified as for the cushion experiment. Input samples were collected, and 850 μl of the remaining lysates was used for co‐IP of eIF5A. The lysates were incubated with 10 μl slurry of magnetic agarose GFP‐Trap beads (Chromotek) for 90 min at 4°C. After washing steps in the lysis buffer, elution of protein material was performed with 25 μl of 1.5× NuPAGE loading buffer supplemented with TCEP. Inputs (10 μl) and eluates (20 μl) were analysed by Western blotting. For isolation of co‐purified RNA, standard Trizol‐based purification was performed and samples subjected to qRT–PCR analysis.
GFP‐LC3B and ATG16L1 puncta quantification
For GFP‐LC3B puncta analysis, MCF‐7 GFP‐LC3B cells were reverse transfected in 96‐well plates with indicated siRNAs for 72 h. For the last 2 h prior to fixation, cells were either left untreated or treated with 250 nM Torin‐1 or 100 nM Bafilomycin A1. Cells were fixed in 4% formaldehyde and nuclei were stained with Hoechst (33342, Thermo Fisher Scientific). Image acquisition and analysis was automated and done essentially as described above for the high‐throughput screen. Cells with more than 10 GFP‐LC3B puncta were considered GFP‐LC3 puncta‐positive cells. For ATG16L1 puncta, cells were additionally stained with the ATG16L1 Ab (PM040 MBL, 1:200) and puncta analysis was performed similarly to GFP‐LC3B puncta, defining ATG16L1 puncta‐positive cells as those containing more than 5 puncta per cell.
C. elegans maintenance and RNAi
Caenorhabditis elegans strain DA2123 (adIs2122[gfp::lgg‐1 + rol‐6]) 45 was maintained and cultured under standard conditions at 20°C on the standard Escherichia coli strain OP50 76.
Gene inactivation was achieved by feeding C. elegans with E. coli strain HT115 expressing dsRNA targeting iff‐2 or the empty vector (L4440) for control. The iff‐2 RNAi clone was obtained from the Ahringer RNAi library 77, and the empty vector (L4440) is from Dr. Andrew Fire. The iff‐2 RNAi clone was verified by sequencing. For RNAi experiments, HT115 bacteria were grown in LB liquid culture medium containing 0.1 mg/ml carbenicillin (Carb; BioPioneer), and 80‐μl aliquots of bacterial suspension were spotted onto 6‐cm Nematode Growth Medium (NGM)/Carb plates. Bacteria were allowed to grow for 1–2 days. For induction of dsRNA expression, 80 μl of a solution containing 0.1 M IPTG (Promega) and 50 μg/ml Carb was placed directly onto the lawn. C. elegans eggs were manually transferred onto NGM/Carb plates seeded with dsRNA‐expressing or control bacteria. To increase the efficacy of the gene inactivation of iff‐2, animals were kept on RNAi plates for 2–4 generations, depending on strength of phenotype. Specifically, newly laid eggs were again manually transferred onto NGM/Carb plates seeded with dsRNA‐expressing or control bacteria. To confirm the efficacy of the iff‐2 RNAi animals were analysed for reduced brood size. The iff‐2(RNAi) animals that had a reduction in progeny production of about 50–75% were used for subsequent experiments. RNAi efficacy was variable between independent experiments and was achieved after 2–4 generations of growth on dsRNA‐expressing bacteria.
Quantification of GFP::LGG‐1 puncta in C. elegans
Caenorhabditis elegans were mounted live on a 2% agarose pad in M9 medium containing 0.1% NaN3 and imaged using Zeiss Imager Z1 including apotome.2 at 100× magnification with a Hamamatsu orca flash 4LT camera and Zen 2.3 software. For image acquisition, the Z‐position was selected as follows: visible body‐wall muscle striation, visible intestinal nuclei and visible lumen of the terminal pharyngeal bulb. Puncta were quantified from images as follows: for body‐wall muscle, punctae in one 1,000 μm2 area per 0.6‐μm slice per animal; for the intestine, the number of punctae per cell per 0.6‐μm slice (2–3 intestinal cells visible per animal); and for the pharynx, the number of punctae in the terminal pharyngeal bulb. Data were collected in four biological replicates, and the total number of animals analysed is denoted in the figure legend.
Transmission electron microscopy
MCF‐7 GFP‐LC3B cells were transfected for 72 h and fixed in 2% v/v glutaraldehyde in 0.05 M sodium phosphate buffer (pH 7.2) for 24 h. Samples were rinsed three times in 0.15 M sodium cacodylate buffer (pH 7.2) and subsequently post‐fixed in 1% w/v OsO4 in 0.12 M sodium cacodylate buffer (pH 7.2) for 2 h. The specimens were dehydrated in a graded series of ethanol, transferred to propylene oxide and embedded in Epon according to standard procedures. Sections, B80‐nm thick, were cut with a Reichert‐Jung Ultracut E microtome and collected on copper grids with Formvar supporting membranes. Sections were stained with uranyl acetate and lead citrate. Imaging was done on a Phillips CM 100 BioTWIN transmission electron microscope. The number of mature autophagosomes per cytoplasmic area was quantified counting 30–35 cells per sample and assessed by size by determining diameter length and classifying autophagosomes as small (diameter < 300 nm) or large (diameter > 300 nm).
Statistical analysis
All statistical analysis was performed on at least three independent replicates. Unless otherwise mentioned, results are shown as mean + SD or mean + SEM and statistical significance was determined by a two‐tailed Student's t‐test for two group comparison. Significance in all figures is indicated as follows: ns P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Data availability
Raw data for the RNA‐seq are deposited to GEO (GSE104604). Raw data for proteomics experiments are deposited to PRIDE ProteomeXchange (PXD008874).
Author contributions
ML, AHL and LBF designed and interpreted experiments and wrote the manuscript. ML, IT and LBF performed experiments, except C. elegans experiments, which were designed and performed by CK in MH's laboratory. ML, LMH and LBF analysed the data. JSA supervised LMH.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We thank Roland Baumgartner for siRNA library design, Cornelia Steinhauer and Elin Pietras for assistance with screening and automated quantifications, Sudeep Sahadevan for analysis of RNA‐seq data, Bettina Mentz for technical assistance, Sharon Tooze for commenting the manuscript, Kevin Ryan for providing pLenti‐CRISPR NTC and ATG5 constructs 70 and Myung Hee Park for providing the pCEFL‐GFP‐eIF5A vector. Michal Lubas was supported by a grant from The Danish Cancer Society (R72‐A4642). Work in the Lund laboratory was supported by funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007‐2013/under REA Grant agreement 607720, the Danish Council for Independent Research (Sapere Aude program), the Novo Nordisk Foundation, the Lundbeck Foundation, the Danish Cancer Society and the EU COST Action Transautophagy (CA15138). Work in MH's laboratory is funded by the National Institute on Aging (R01AG038664) and National Institute of General Medical Sciences (R01GM117466). MH is a Julie Martin Mid‐Career Awardee in Aging Research supported by The Ellison Medical Foundation and American Federation for Aging Research.
EMBO Reports (2018) 19: e46072
Contributor Information
Anders H Lund, Email: anders.lund@bric.ku.dk.
Lisa B Frankel, Email: lisa.frankel@bric.ku.dk.
References
- 1. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635 [DOI] [PubMed] [Google Scholar]
- 3. Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774 [DOI] [PubMed] [Google Scholar]
- 4. Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24: 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368: 651–662 [DOI] [PubMed] [Google Scholar]
- 6. Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- 7. Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy‐related ubiquitin‐like protein family. Genome Biol 12: 226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wild P, McEwan DG, Dikic I (2014) The LC3 interactome at a glance. J Cell Sci 127: 3–9 [DOI] [PubMed] [Google Scholar]
- 9. Nakatogawa H (2013) Two ubiquitin‐like conjugation systems that mediate membrane formation during autophagy. Essays Biochem 55: 39–50 [DOI] [PubMed] [Google Scholar]
- 10. Geng J, Klionsky DJ (2008) The Atg8 and Atg12 ubiquitin‐like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 9: 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y et al (2008) The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 19: 4762–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xie Z, Nair U, Klionsky DJ (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19: 3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- 14. Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome‐lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215: 857–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354: 1036–1041 [DOI] [PubMed] [Google Scholar]
- 16. Frankel LB, Lubas M, Lund AH (2017) Emerging connections between RNA and autophagy. Autophagy 13: 3–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D'Agostino VG et al (2013) Autophagy activation clears ELAVL1/HuR‐mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One 8: e69563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim C, Kim W, Lee H, Ji E, Choe YJ, Martindale JL, Akamatsu W, Okano H, Kim HS, Nam SW et al (2014) The RNA‐binding protein HuD regulates autophagosome formation in pancreatic beta cells by promoting autophagy‐related gene 5 expression. J Biol Chem 289: 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rojas‐Rios P, Chartier A, Pierson S, Severac D, Dantec C, Busseau I, Simonelig M (2015) Translational control of autophagy by Orb in the Drosophila germline. Dev Cell 35: 622–631 [DOI] [PubMed] [Google Scholar]
- 20. Gerstberger S, Hafner M, Tuschl T (2014) A census of human RNA‐binding proteins. Nat Rev Genet 15: 829–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castello A, Hentze MW, Preiss T (2015) Metabolic enzymes enjoying new partnerships as RNA‐binding proteins. Trends Endocrinol Metab 26: 746–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM et al (2012) Insights into RNA biology from an atlas of mammalian mRNA‐binding proteins. Cell 149: 1393–1406 [DOI] [PubMed] [Google Scholar]
- 23. Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume AM, Schwarzl T, Curk T, Foehr S, Huber W et al (2015) The RNA‐binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun 6: 10127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schreier MH, Erni B, Staehelin T (1977) Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors. J Mol Biol 116: 727–753 [DOI] [PubMed] [Google Scholar]
- 25. Kemper WM, Berry KW, Merrick WC (1976) Purification and properties of rabbit reticulocyte protein synthesis initiation factors M2Balpha and M2Bbeta. J Biol Chem 251: 5551–5557 [PubMed] [Google Scholar]
- 26. Saini P, Eyler DE, Green R, Dever TE (2009) Hypusine‐containing protein eIF5A promotes translation elongation. Nature 459: 118–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE (2013) eIF5A promotes translation of polyproline motifs. Mol Cell 51: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gregio AP, Cano VP, Avaca JS, Valentini SR, Zanelli CF (2009) eIF5A has a function in the elongation step of translation in yeast. Biochem Biophys Res Commun 380: 785–790 [DOI] [PubMed] [Google Scholar]
- 29. Wolff EC, Kang KR, Kim YS, Park MH (2007) Posttranslational synthesis of hypusine: evolutionary progression and specificity of the hypusine modification. Amino Acids 33: 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shin BS, Katoh T, Gutierrez E, Kim JR, Suga H, Dever TE (2017) Amino acid substrates impose polyamine, eIF5A, or hypusine requirement for peptide synthesis. Nucleic Acids Res 45: 8392–8402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidt C, Becker T, Heuer A, Braunger K, Shanmuganathan V, Pech M, Berninghausen O, Wilson DN, Beckmann R (2016) Structure of the hypusinylated eukaryotic translation factor eIF‐5A bound to the ribosome. Nucleic Acids Res 44: 1944–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K (2013) Translation elongation factor EF‐P alleviates ribosome stalling at polyproline stretches. Science 339: 82–85 [DOI] [PubMed] [Google Scholar]
- 33. Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV (2013) EF‐P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339: 85–88 [DOI] [PubMed] [Google Scholar]
- 34. Wohlgemuth I, Brenner S, Beringer M, Rodnina MV (2008) Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem 283: 32229–32235 [DOI] [PubMed] [Google Scholar]
- 35. Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC (2009) Slow peptide bond formation by proline and other N‐alkylamino acids in translation. Proc Natl Acad Sci USA 106: 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buskirk AR, Green R (2017) Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos Trans R Soc Lond B Biol Sci 372: 20160183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schuller AP, Wu CC, Dever TE, Buskirk AR, Green R (2017) eIF5A functions globally in translation elongation and termination. Mol Cell 66: 194–205 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pelechano V, Alepuz P (2017) eIF5A facilitates translation termination globally and promotes the elongation of many non polyproline‐specific tripeptide sequences. Nucleic Acids Res 45: 7326–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cano VS, Jeon GA, Johansson HE, Henderson CA, Park JH, Valentini SR, Hershey JW, Park MH (2008) Mutational analyses of human eIF5A‐1–identification of amino acid residues critical for eIF5A activity and hypusine modification. FEBS J 275: 44–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoyer‐Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R et al (2007) Control of macroautophagy by calcium, calmodulin‐dependent kinase kinase‐beta, and Bcl‐2. Mol Cell 25: 193–205 [DOI] [PubMed] [Google Scholar]
- 41. Kwon SC, Yi H, Eichelbaum K, Fohr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN (2013) The RNA‐binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20: 1122–1130 [DOI] [PubMed] [Google Scholar]
- 42. Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold‐Brown D, Drew K, Milek M et al (2012) The mRNA‐bound proteome and its global occupancy profile on protein‐coding transcripts. Mol Cell 46: 674–690 [DOI] [PubMed] [Google Scholar]
- 43. Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y et al (2007) A probability‐based approach for the analysis of large‐scale RNAi screens. Nat Methods 4: 847–849 [DOI] [PubMed] [Google Scholar]
- 44. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T (2003) Mouse Apg16L, a novel WD‐repeat protein, targets to the autophagic isolation membrane with the Apg12‐Apg5 conjugate. J Cell Sci 116: 1679–1688 [DOI] [PubMed] [Google Scholar]
- 45. Kang C, You YJ, Avery L (2007) Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21: 2161–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanazawa M, Kawasaki I, Kunitomo H, Gengyo‐Ando K, Bennett KL, Mitani S, Iino Y (2004) The Caenorhabditis elegans eukaryotic initiation factor 5A homologue, IFF‐1, is required for germ cell proliferation, gametogenesis and localization of the P‐granule component PGL‐1. Mech Dev 121: 213–224 [DOI] [PubMed] [Google Scholar]
- 47. Zanelli CF, Maragno AL, Gregio AP, Komili S, Pandolfi JR, Mestriner CA, Lustri WR, Valentini SR (2006) eIF5A binds to translational machinery components and affects translation in yeast. Biochem Biophys Res Commun 348: 1358–1366 [DOI] [PubMed] [Google Scholar]
- 48. Melnikov S, Mailliot J, Shin BS, Rigger L, Yusupova G, Micura R, Dever TE, Yusupov M (2016) Crystal structure of hypusine‐containing translation factor eIF5A bound to a rotated eukaryotic ribosome. J Mol Biol 428: 3570–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jao DL, Chen KY (2006) Tandem affinity purification revealed the hypusine‐dependent binding of eukaryotic initiation factor 5A to the translating 80S ribosomal complex. J Cell Biochem 97: 583–598 [DOI] [PubMed] [Google Scholar]
- 50. Lee Y, Kim HK, Park HE, Park MH, Joe YA (2002) Effect of N1‐guanyl‐1,7‐diaminoheptane, an inhibitor of deoxyhypusine synthase, on endothelial cell growth, differentiation and apoptosis. Mol Cell Biochem 237: 69–76 [DOI] [PubMed] [Google Scholar]
- 51. Jakus J, Wolff EC, Park MH, Folk JE (1993) Features of the spermidine‐binding site of deoxyhypusine synthase as derived from inhibition studies. Effective inhibition by bis‐ and mono‐guanylated diamines and polyamines. J Biol Chem 268: 13151–13159 [PubMed] [Google Scholar]
- 52. Mandal A, Mandal S, Park MH (2016) Global quantitative proteomics reveal up‐regulation of endoplasmic reticulum stress response proteins upon depletion of eIF5A in HeLa cells. Sci Rep 6: 25795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li CH, Ohn T, Ivanov P, Tisdale S, Anderson P (2010) eIF5A promotes translation elongation, polysome disassembly and stress granule assembly. PLoS One 5: e9942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang HA, Hershey JW (1994) Effect of initiation factor eIF‐5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae . J Biol Chem 269: 3934–3940 [PubMed] [Google Scholar]
- 55. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M et al (2000) A ubiquitin‐like system mediates protein lipidation. Nature 408: 488–492 [DOI] [PubMed] [Google Scholar]
- 56. Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, Tang D (2015) Posttranslational modification of autophagy‐related proteins in macroautophagy. Autophagy 11: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pietrocola F, Izzo V, Niso‐Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G (2013) Regulation of autophagy by stress‐responsive transcription factors. Semin Cancer Biol 23: 310–322 [DOI] [PubMed] [Google Scholar]
- 58. Behrends C, Sowa ME, Gygi SP, Harper JW (2010) Network organization of the human autophagy system. Nature 466: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sakoh‐Nakatogawa M, Kirisako H, Nakatogawa H, Ohsumi Y (2015) Localization of Atg3 to autophagy‐related membranes and its enhancement by the Atg8‐family interacting motif to promote expansion of the membranes. FEBS Lett 589: 744–749 [DOI] [PubMed] [Google Scholar]
- 60. Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, Melia TJ (2014) Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane‐curvature‐sensing domain in Atg3. Nat Cell Biol 16: 415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mateyak MK, Kinzy TG (2017) Breaking the silos of protein synthesis. Trends Biochem Sci 42: 587–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oliverio S, Corazzari M, Sestito C, Piredda L, Ippolito G, Piacentini M (2014) The spermidine analogue GC7 (N1‐guanyl‐1,7‐diamineoheptane) induces autophagy through a mechanism not involving the hypusination of eIF5A. Amino Acids 46: 2767–2776 [DOI] [PubMed] [Google Scholar]
- 63. Bhukel A, Madeo F, Sigrist SJ (2017) Spermidine boosts autophagy to protect from synapse aging. Autophagy 13: 444–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Patel PH, Costa‐Mattioli M, Schulze KL, Bellen HJ (2009) The Drosophila deoxyhypusine hydroxylase homologue nero and its target eIF5A are required for cell growth and the regulation of autophagy. J Cell Biol 185: 1181–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH (2016) AMPK‐SKP2‐CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, Boyd DD (2013) ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell 50: 16–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim HD, Kong E, Kim Y, Chang JS, Kim J (2017) RACK1 depletion in the ribosome induces selective translation for non‐canonical autophagy. Cell Death Dis 8: e2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee SB, Park JH, Kaevel J, Sramkova M, Weigert R, Park MH (2009) The effect of hypusine modification on the intracellular localization of eIF5A. Biochem Biophys Res Commun 383: 497–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. O'Prey J, Sakamaki J, Baudot AD, New M, Van Acker T, Tooze SA, Long JS, Ryan KM (2017) Application of CRISPR/Cas9 to autophagy research. Methods Enzymol 588: 79–108 [DOI] [PubMed] [Google Scholar]
- 71. Poulsen EG, Nielsen SV, Pietras EJ, Johansen JV, Steinhauer C, Hartmann‐Petersen R (2016) High‐throughput siRNA screening applied to the ubiquitin‐proteasome system. Methods Mol Biol 1449: 421–439 [DOI] [PubMed] [Google Scholar]
- 72. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 73. Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, The AmiGO Hub, The Web Presence Working Group (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25: 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kamath RS, Ahringer J (2003) Genome‐wide RNAi screening in Caenorhabditis elegans . Methods 30: 313–321 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Data Availability Statement
Raw data for the RNA‐seq are deposited to GEO (GSE104604). Raw data for proteomics experiments are deposited to PRIDE ProteomeXchange (PXD008874).