

Abstract
The production of type I interferon (IFN) is essential for cellular barrier functions and innate and adaptive antiviral immunity. In response to virus infections, RNA receptors RIG‐I and MDA5 stimulate a mitochondria‐localized signaling apparatus that uses TRAF family ubiquitin ligase proteins to activate master transcription regulators IRF3 and NFκB, driving IFN and antiviral target gene expression. Data indicate that a third RNA receptor, LGP2, acts as a negative regulator of antiviral signaling by interfering with TRAF family proteins. Disruption of LGP2 expression in cells results in earlier and overactive transcriptional responses to virus or dsRNA. LGP2 associates with the C‐terminus of TRAF2, TRAF3, TRAF5, and TRAF6 and interferes with TRAF ubiquitin ligase activity. TRAF interference is independent of LGP2 ATP hydrolysis, RNA binding, or its C‐terminal domain, and LGP2 can regulate TRAF‐mediated signaling pathways in trans, including IL‐1β, TNFα, and cGAMP. These findings provide a unique mechanism for LGP2 negative regulation through TRAF suppression and extend the potential impact of LGP2 negative regulation beyond the IFN antiviral response.
Keywords: innate immunity, interferon, LGP2, RIG‐I‐like receptors, TRAF
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The production of and response to type I interferon (IFN) are essential for protection against virus infection, and control both innate and adaptive immune responses. IFN restricts virus replication by inducing expression of antiviral effectors that suppress host and viral RNA transcription or protein translation, initiate RNA degradation, induce growth arrest, activate apoptosis, and trigger autophagy that together facilitate infection clearance. IFN also acts as an immune modulator, regulating dendritic cells, monocytes, and other professional immune cells to counteract infections and stimulate lasting immunity 1, 2, 3, 4, 5. While essential for controlling virus replication, chronic production and exposure to IFN can result in autoimmune diseases and chronic inflammatory responses, and influence therapeutic efficacy of chemotherapy, radiotherapy, and immunotherapy 6, 7, 8, 9. To ensure rapid and transient responses and to prevent these adverse outcomes, homeostatic regulation of antiviral responses includes endogenous negative regulators that attenuate IFN production and response.
IFN production is triggered by virus infections or intrinsic defects that cause accumulation of RNA species in the cytoplasm. The RNAs are detected by a family of three pattern‐recognition receptor sensor proteins, RIG‐I, MDA5, and LGP2. All three RIG‐I‐like receptor (RLR) proteins share conserved DECH‐box helicase and C‐terminal domain (CTD) regions that have intrinsic dsRNA binding and ATP hydrolysis activities 10, 11, 12, 13. RIG‐I and MDA5 contain tandem caspase activation and recruitment domain (CARD) regions, interaction domains that mediate contact with downstream signaling proteins. RNA‐activated RLRs initiate the oligomerization of an essential mitochondrial antiviral signaling protein, MAVS, that serves as a scaffold for the activation of TNF receptor‐associated factor (TRAF) family proteins, TRAF2, TRAF3, TRAF5, and TRAF6 14, 15. TRAF proteins catalyze the assembly of K63‐linked ubiquitin chains that are required for the activation of serine kinases, IKKα, IKKβ, IKKγ, IKKε, and TBK1 16, 17. Kinase activation triggers the phosphorylation and nuclear import of IRF3 and NFκB, driving the production of primary antiviral effectors including IFNs 18. For IRF3 activation, MAVS itself becomes phosphorylated by activated IKKs and/or TBK1 on serine 442 and directly recruits IRF3 for subsequent kinase activation. The precise means of MAVS‐mediated NFκB activation has not been elucidated, but many pathways to NFκB activation rely on TRAF‐mediated ubiquitination to initiate IKK kinase activity that results in alleviation of IκB‐mediated repression and NFκB activity 17, 19.
LGP2 is unique among the RLR proteins, as it is associated with both activation and inhibition of antiviral signaling 20. LGP2 lacks CARDs but can participate in antiviral signaling as a positive regulator of MDA5–dsRNA interactions 21, 22, 23, 24, 25, 26. This positive regulation by LGP2 occurs upstream of MAVS and absolutely requires both ATP hydrolysis and RNA binding activities 24, 26.
As a negative regulator, LGP2 can suppress both MDA5‐dependent and RIG‐I‐dependent signal transduction 13, 23, 27, 28, 29, 30, 31, but the precise targets and mechanisms underlying LGP2 interference are poorly understood. Distinct mechanistic interpretations for the observed feedback inhibition have been suggested 13, 29, 31, but the observation that LGP2 negative regulation remains intact in the absence of either RNA binding or ATP hydrolysis activity 26, 32 invited further inquiry into the molecular targets and mechanisms of LGP2 inhibition. Experiments demonstrate that LGP2 can interfere with TRAF‐mediated signaling downstream of MAVS. Data indicate that LGP2 can co‐precipitate with TRAF protein C‐termini, disrupting ubiquitin ligase activity and restricting IRF3 and NFκB activation. Although LGP2 is a component of the RLR‐MAVS‐IFN system, results demonstrate its TRAF‐suppressing activity can disengage NFκB activation induced by MAVS‐independent signaling via TNFα, IL‐1β, and cGAMP. These findings identify novel targets for LGP2 negative regulation and a previously unrecognized means for antagonizing TRAF‐mediated signaling.
Results
LGP2 suppresses antiviral signaling
The ability of LGP2 to antagonize antiviral signaling is apparent in transcription suppression assays. Sendai virus infection is a potent inducer of IFNβ promoter transcription, but LGP2 expression prevents IFNβ reporter gene activity (Fig 1A and EV1). The primary activated transcription factors contributing to IFNβ gene induction are IRF3 and NFκB, which bind to positive regulatory domain (PRD) elements of the IFNβ enhancer, PRDIII/I, and PRDII, respectively 33, 34, 35. Virus infection activated gene expression from both PRDIII/I and PRDII, but LGP2 expression dramatically reduced IRF3 and NFκB transcriptional responses to baseline levels (Fig 1B and C). Similarly, the induction of endogenous IFNβ mRNA, the IRF3 target gene, ISG56, and the NFκB target gene, TNFα, was suppressed by LGP2 expression (Fig 1D–F). To complement these expression experiments, fibroblasts derived from LGP2‐deficient mice 36 were mock‐infected or infected with Sendai virus for 4 h, and endogenous IFNβ, ISG56, TNFα, and IL6 mRNAs were measured by RT–qPCR (Fig 1G–J). Infection robustly activated these mRNAs, and loss of LGP2 significantly increased mRNA levels by twofold to threefold, consistent with derepression due to the absence of LGP2 negative regulation. Together, these data support negative regulatory functions of LGP2 that suppress both IRF3 and NFκB signaling.
Figure 1. LGP2 suppresses virus‐induced antiviral gene expression.
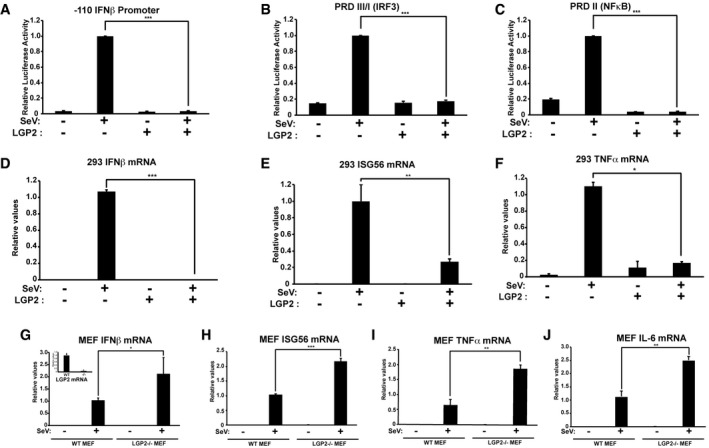
- HEK293 cells were transfected with IFNβ promoter reporter gene and 500 ng LGP2 expression plasmid (+) or salmon sperm DNA (−), then infected with 200 HA units per ml of SeV for 6 h prior to luciferase assays.
- Similar to (A), but using PRDIII/I (IRF3) reporter gene.
- Similar to (A), but using PRDII (NFκB) reporter gene.
- Cells were transfected with empty vector (−) or 500 ng LGP2 expression plasmid (+), then stimulated with 200 HA units per ml of SeV for 8 h prior to RNA isolation and RT–qPCR with IFNβ‐specific primers or control GAPDH.
- Similar to (D), but using primers for ISG56.
- Similar to (D), but using primers for TNFα.
- Using WT and LGP2−/− MEFs, mRNA was isolated after 4 h SeV infection and subjected to RT–qPCR with murine IFNβ‐specific primers or control GAPDH. Inset graph illustrates LGP2 mRNA level to verify knockout.
- Similar to (G), but using primers for ISG56.
- Similar to (G), but using primers for TNFα.
- Similar to (G), but using primers for IL‐6.
Figure EV1. Immunoblots for luciferase assays.
Immunoblot loading controls for reporter gene assays in Figs 1A–C, 2A–H, 4A–D, 5A, C and D, and 7B.Source data are available online for this figure.
LGP2 inhibits signaling downstream of MAVS
To more clearly define the target(s) for LGP2 negative regulation, experiments were conducted using the synthetic dsRNA, poly(I:C) to stimulate RLR/MAVS signaling. Poly(I:C) transfection activated expression of the IFNβ enhancer (Fig 2A), or the substituent PRDII and PRDIII/I reporters (Fig 2B and C). Expression of LGP2 in these assays results in a concentration‐dependent decrease in dsRNA signaling. To determine whether the LGP2 suppression occurs at the level of RNA detection by RLRs upstream of MAVS activation or antagonizes a step downstream of RLR function, expression of MAVS was used to activate antiviral signaling in the absence of either virus infection or RLR engagement 30. Expression of MAVS can potently induce downstream responses and results in activation of the complete IFNβ enhancer (Fig 2D), or the PRDII and PRDIII/I reporters (Fig 2E and F). Expression of LGP2 in these assays results in a concentration‐dependent decrease in MAVS‐dependent signaling, demonstrating LGP2 interference with both IRF3 and NFκB occurs downstream of MAVS. To verify the specificity of LGP2 suppression and rule out off‐target interference with the reporter gene assay, similar experiments were conducted using IFNα‐activated ISRE luciferase or IFNγ‐activated GAS luciferase assays. No LGP2 interference was observed with IFNα or IFNγ signaling (Fig 2G and H). These experiments localize LGP2 antagonism to a step downstream of the MAVS signaling.
Figure 2. LGP2 inhibits signaling downstream of MAVS.
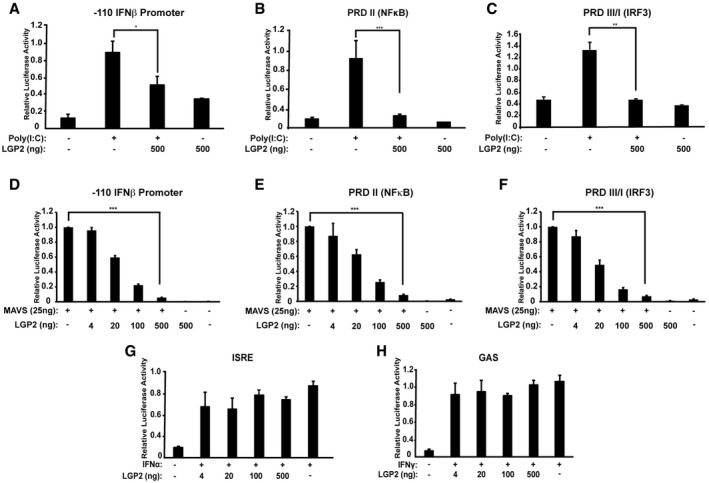
- IFNβ‐reporter gene assay with 6 h poly(I:C) (5 μg/ml) stimulation with or without LGP2 expression. 2fTGH cells were harvested for luciferase assays.
- Similar to (A), but using PRDII (NFκB) reporter gene.
- Similar to (A), but using PRDIII/I (IRF3) reporter gene.
- IFNβ‐reporter gene assay with MAVS expressed with or without LGP2 titration. Cells were harvested 24 h post‐transfection for luciferase assays.
- Similar to (D), but using PRDII (NFκB) reporter gene.
- Similar to (D), but using PRDIII/I (IRF3) reporter gene.
- ISRE‐reporter gene assay with 6‐h IFNα stimulation with or without LGP2 titration.
- GAS‐reporter gene assay with 6‐h IFNγ stimulation with or without LGP2 titration.
LGP2 regulates signaling to IRF3 and NFκB
IRF3 and NFκB pathway activation was directly examined in intact or LGP2‐deficient mouse and human cells. In mouse embryo fibroblasts, Sendai virus infection induced IRF3 Ser396 phosphorylation within 4 h, and this was reduced by 6 h. In MEFs lacking LGP2, IRF3 Ser396 phosphorylation was dramatically increased and remained detectable at 6 h postinfection (Fig 3A). Virus‐activated phosphorylation of IκBα at Ser32 was also tested and found to be detectable in the LGP2‐/‐ MEFs even at steady state. Nonetheless, IκBα phosphorylation dramatically increased in the absence of LGP2 and was observed to rise until 8 h postinfection (Fig 3B).
Figure 3. LGP2 inhibits signal transduction to IRF3 and NFκB.
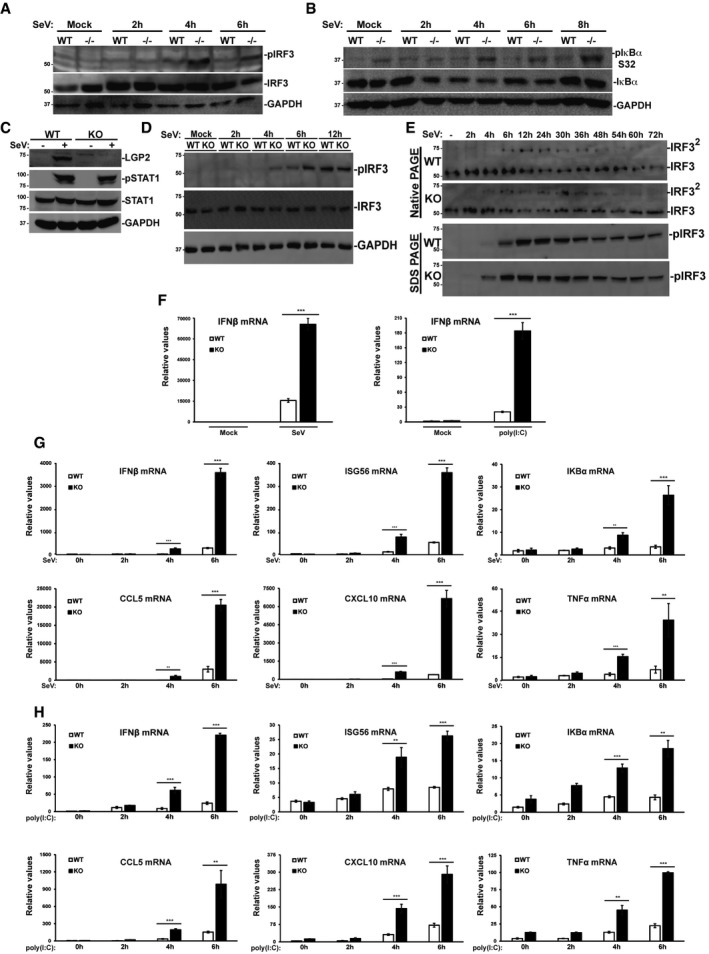
- WT and LGP2−/− MEFs were stimulated with 200 HA units per ml of SeV at indicated time points, and cell lysates were subjected to immunoblotting with antibodies that recognize phospho‐IRF3 (S396), total IRF3, and GAPDH.
- Similar to (A), but probing with antibodies for phospho‐IκBα (S32), total IκBα, and GAPDH.
- Immunoblot of WT and LGP2‐deficient (KO) HEK293 cells. Cells were mock‐infected (−) or infected with SeV (+) for 24 h prior to analysis of LGP2, STAT1 phosphotyrosine 701 (pSTAT1), total STAT1, and control GAPDH.
- WT and LGP2 KO cells were subjected to a time course of SeV infection prior to analysis of IRF3 phosphorylation at S396 (pIRF3).
- Similar to (D), but lysates were subjected to native PAGE to detect IRF3 dimerization (IRF32), and to SDS–PAGE to detect IRF3 S396 phosphorylation (pIRF3).
- WT and LGP2 KO cells were infected with SeV for 24 h or treated with poly(I:C) for 6 h. RNA was isolated and subjected to RT–qPCR with IFNβ‐specific primers or control GAPDH.
- WT and LGP2 KO cells were subjected to a time course of SeV infection prior to RNA isolation and RT–qPCR.
- Similar to (G), except cells were treated with poly(I:C).
To test the veracity of these findings in human cells, CRISPR/Cas9 was used to create HEK293 cells lacking LGP2 expression. Disruption of LGP2 does not alter overall cellular antiviral response, as indicated by intact phosphorylation of STAT1 tyrosine 701 (Fig 3C). Absence of LGP2 was found to derepress the response to Sendai virus infection, leading to altered kinetics of IRF3 Ser396 phosphorylation (Fig 3D) and IRF3 dimerization (Fig 3E). Phosphorylation and dimerization of IRF3 were detected 2 h earlier in the LGP2‐knockout cells, confirming in human cells the negative role of LGP2 in antiviral signaling observed in mouse cells.
The ability of LGP2 deficiency to alter antiviral transcription was tested in the CRISPR cell system for both Sendai virus infection and poly(I:C) transfection (Fig 3F–H). IFNβ mRNA level following 24 h of Sendai virus or 6 h of poly(I:C) was greatly increased in the absence of LGP2. Time‐course analysis for Sendai virus infection (Fig 3G) and poly(I:C) transfection (Fig 3H) correlate well with the observed differential transcription factor activation. All tested inducible mRNAs accumulated to higher levels in the absence of LGP2 within the 4–6 h time course, confirming the biological outcome of hyperactive transcription factors IRF3 and NFκB.
LGP2 targets TRAF signaling
Signal transduction between MAVS and IRF3/NFκB is mediated by TRAF family proteins. The ability of LGP2 to influence TRAF activation was tested, and strong activation of the PRDII reporter, indicating stimulation of NFκB‐mediated transcription, was induced by expression of TRAF2, TRAF3, TRAF5, or TRAF6 (Fig 4A–D). Titration of LGP2 resulted in a dramatic concentration‐dependent decrease in PRDII‐dependent signaling, demonstrating potent interference with TRAF‐mediated NFκB activation. None of the TRAFs were found to activate the PRDIII/I reporter, reflecting an unfulfilled requirement for MAVS to activate IRF3 37 (Fig 4E–H). Consequently, TRAF protein activation of the IFNβ promoter was weak due to the absence of IRF3, but in all cases, any induced IFNβ promoter signal was eliminated by LGP2 expression (Fig 4I–L). In the absence of MAVS, LGP2 expression disrupts TRAF‐dependent NFκB activation.
Figure 4. LGP2 inhibits TRAF‐mediated NFκB activation.
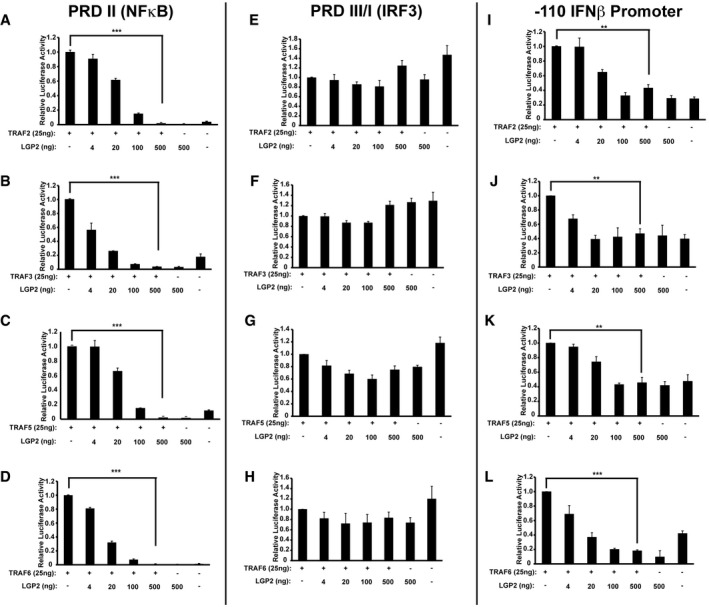
-
A–DPRD II (NFκB)‐reporter gene assays with TRAF2, TRAF3, TRAF5, or TRAF6 expressed with or without LGP2 titration. Cells were harvested 24 h post‐transfection for luciferase assays. Bars represent average values (n = 3) with ± standard deviation. Corresponding immunoblots in Fig EV1.
-
E–HSimilar to (A), but using PRDIII/I (IRF3) reporter gene.
-
I–LSimilar to (A), but using ‐110 IFNβ‐ promoter reporter gene.
LGP2 inhibits MAVS‐independent, TRAF‐mediated signaling
As TRAF proteins mediate NFκB activation triggered by many immune and inflammatory signals, we examined diverse contexts to more generally test the ability of LGP2 to inhibit TRAF‐dependent signal transduction. Stimulation with TNFα activates NFκB signaling through TRADD adaptor‐mediated TRAF2/TRAF5 recruitment 38, 39, IL‐1β activates NFκB through MyD88 adaptor‐mediated TRAF6 recruitment 40, and the cGAMP receptor, STING, induces NFκB via TRAF6 41, 42. Treatment of cells with TNFα robustly induced PRDII/NFκB activity, and it was suppressed by LGP2 expression (Fig 5A). To confirm the reporter gene analysis, the Ser32 phosphorylation status of endogenous IκBα was examined (Fig 5B). TNFα induced robust IκBα phosphorylation, but LGP2 expression interfered with TNFα‐mediated IκBα phosphorylation. Similarly, NFκB signaling induced by IL‐1β or cGAMP/STING pathways was suppressed by LGP2 (Fig 5C and D). These results indicate that LGP2 interference with TRAF activity disrupts kinase regulation and demonstrate LGP2's ability to disrupt TRAF‐mediated signaling irrespective of the stimulatory ligand or specific TRAF adaptor, and in the absence of MAVS (Fig EV2C). LGP2 can function as a trans‐inhibitor of TRAF signaling.
Figure 5. LGP2 inhibits MAVS‐independent TRAF‐mediated signaling.
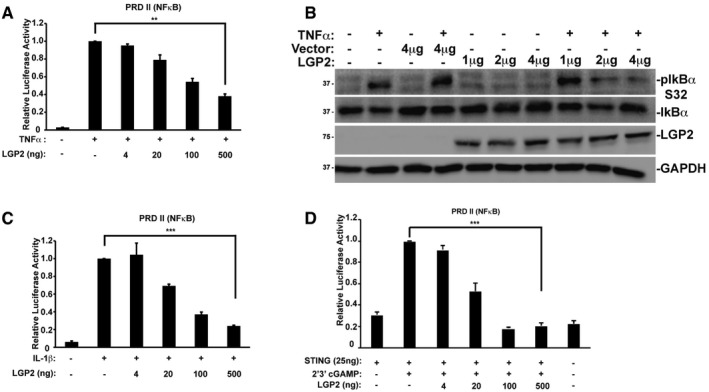
- NFκB‐reporter gene assay with HEK293 cells treated (+) for 6 h with TNFα (10 ng/ml) with or without LGP2 plasmid titration as indicated. Bars represent average values (n = 3) ± standard deviation. Corresponding immunoblots in Fig EV1.
- HeLa cells transfected with indicated amounts of LGP2 or empty vector were treated with TNFα (10 ng/ml) for 5 min. Cell lysates were subjected to immunoblotting with antibodies that recognize phospho‐IκBα (ser32), total IκBα, FLAG‐tagged LGP2, and GAPDH.
- Similar to (A), but treating HEK293 cells with IL‐1β (10 ng/ml)
- Similar to (A), but STING was co‐transfected with indicated amounts of the LGP2 plasmid prior to stimulation with 2′3′ cGAMP (20 μg/ml) for 24 h in HEK293 cells.
Figure EV2. Supplementary and control experiments.
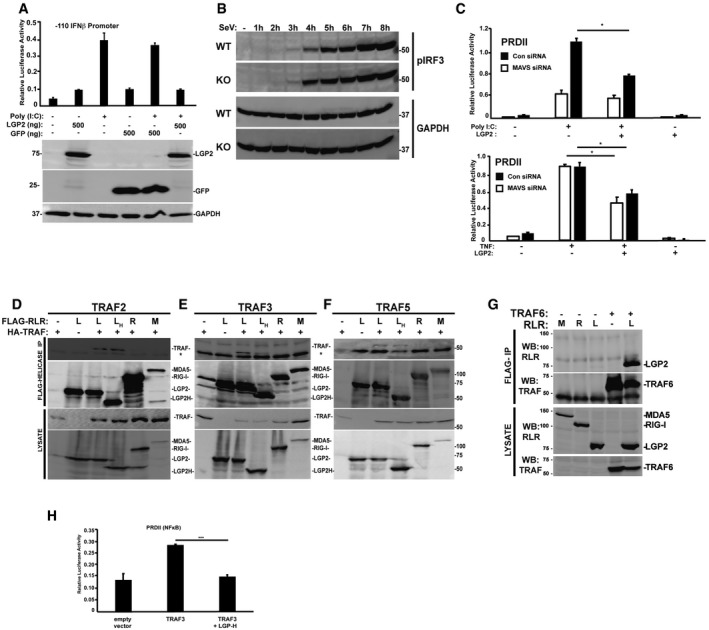
- IFNβ‐reporter gene assay in HEK293 cells treated (+) for 6 h with poly(I:C) (5 μg/ml) with or without LGP2 and GFP plasmid titration as indicated. Bars represent average values (n = 3) ± standard deviation.
- WT and LGP2 KO cells were subjected to an hourly time course of SeV infection prior to analysis of IRF3 phosphorylation at S396 (pIRF3).
- NFκB‐reporter gene assay in HEK293 cells that were transfected with MAVS siRNA or non‐targeting control (Con) siRNA and plasmids expressing FLAG‐tagged LGP2 for 48 h. Cells were then stimulated for 6 h with either poly(I:C) (5 μg/ml) or TNFα (10 ng/ml). Bars represent average values (n = 2) with ± standard deviation. *P ≤ 0.05 by two‐tailed Student's t‐test.
- Cells were transfected with plasmids expressing FLAG‐tagged LGP2 (L), LGP2‐H (H), RIG‐I (R), and MDA5 (M) and HA‐tagged TRAF2 as indicated. Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (RLR Helicases) and HA (TRAF2) (n = 3). Asterisk (*) denotes non‐specific cross‐reactivity.
- Similar to (D), but using HA‐tagged TRAF3 (n = 3).
- Similar to (D), but using HA‐tagged TRAF5 (n = 3).
- Cells were transfected with plasmids expressing HA‐tagged MDA5 (M), RIG‐I (R), LGP2 (L), and FLAG‐Vector (−) as indicated. Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (TRAF) and HA (LGP2) (n = 3).
- Plot of TRAF3 inhibition by LGP2‐H from Fig 7B without mathematical normalization.
Source data are available online for this figure.
LGP2 targets the TRAF C‐terminal MATH domain
To investigate the means of LGP2‐mediated TRAF inhibition, co‐immunoprecipitation assays were performed with LGP2 and TRAF6 (Fig 6). In addition to full‐length TRAF6, co‐precipitation of a series of truncations that dissected the protein along known structural domain boundaries was tested (Fig 6A). LGP2 was found to co‐precipitate with TRAF6, but RIG‐I did not (Fig 6B). Co‐precipitation of LGP2 was observed only with protein fragments containing the TRAF‐C domain (Fig 6B; TRAF6 residues 352–522, also known as the MATH domain 43). An analogous set of full‐length, N‐terminal, or C‐terminal fragments of TRAF2, TRAF3, and TRAF5 were also tested for LGP2 co‐precipitation, revealing in all cases that the TRAF protein C‐terminal domain is necessary and sufficient for LGP2 interaction (Fig 6C). This highly conserved TRAF domain apparently forms a conserved target for LGP2 interference.
Figure 6. LGP2 co‐precipitation with TRAF C‐terminal MATH domain.
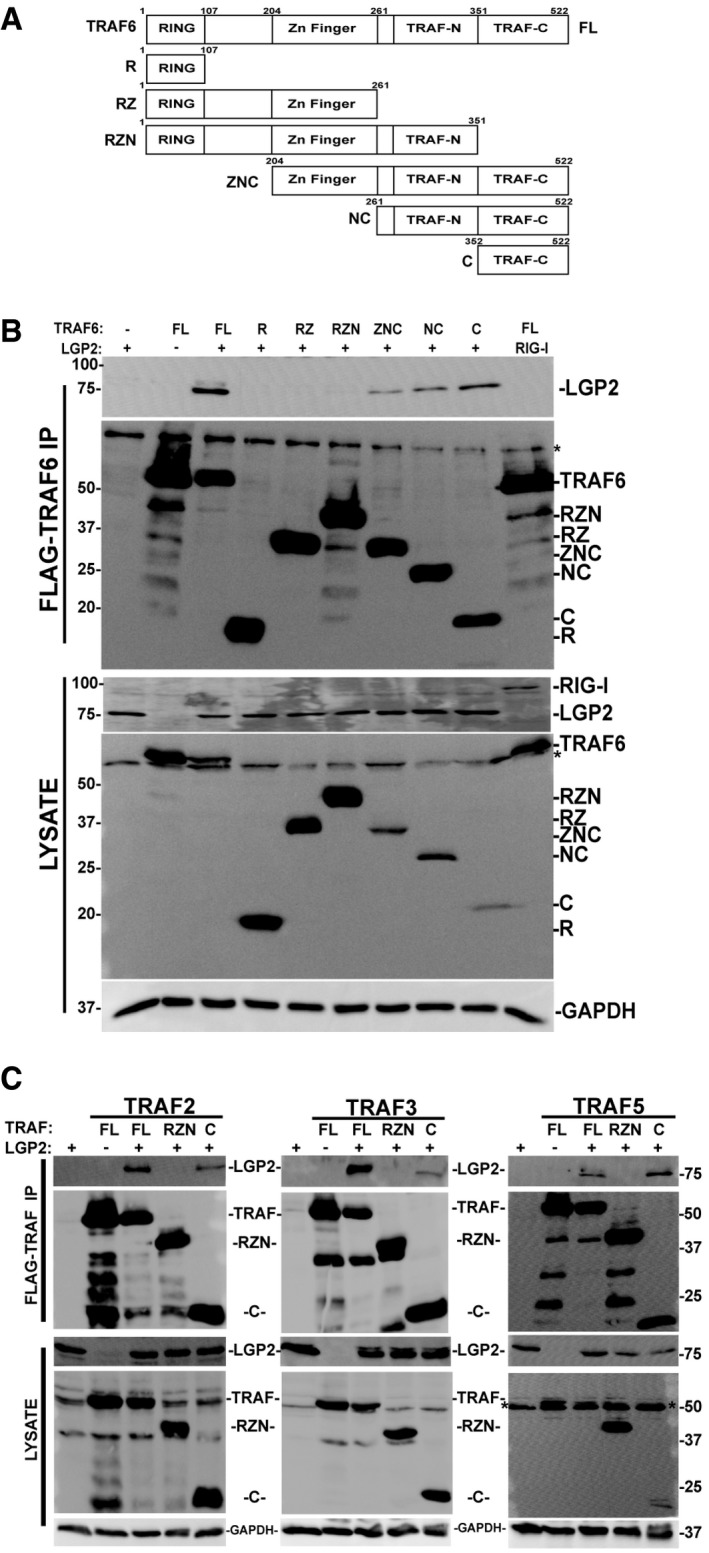
- Diagram illustrating the domain structure of full‐length TRAF6 and the fragments generated for this study.
- Cells were transfected with FLAG‐tagged full‐length TRAF6 or TRAF6 fragments along with HA‐tagged LGP2, and lysates were subjected to FLAG immunoaffinity purification and detection of co‐precipitation was carried out by anti‐HA immunoblot for LGP2 (or RIG‐I as a control), anti‐FLAG immunoblot for TRAF6, and anti‐GAPDH control (n = 5). * indicates non‐specific cross‐reactivity.
- Similar to (B), testing the interaction with full‐length, N‐ and C‐terminal truncations of TRAF2, TRAF3, and TRAF5 with LGP2. All full‐length TRAFs and their C‐terminal domains co‐precipitate LGP2. * indicates non‐specific cross‐reactivity (n = 3 for all experiments).
Source data are available online for this figure.
Novel means of LGP2 TRAF suppression
TRAF proteins are previously unrecognized regulatory targets for LGP2. Earlier work has suggested that LGP2 RNA binding or CTD is required for repression 13, 29, 31. To test the importance of LGP2 catalytic activities in TRAF repression, the TRAF binding and interference of a truncated LGP2 protein were characterized. LGP2‐H (residues 1–546) consists only of the helicase domain without the CTD, and it lacks both RNA binding and ATPase activities (Fig 7A). Despite these defects, LGP2‐H retained the ability to suppress NFκB activation induced by either MAVS or TRAFs (Fig 7B). In addition, co‐precipitation assays revealed both LGP2 and LGP2‐H co‐purified with all of the TRAF proteins (Fig 7C). The TRAF co‐precipitation is specific for LGP2, as neither MDA5 nor RIG‐I was found to co‐precipitate with any TRAF tested (Fig 7C), though it remains possible the co‐precipitation represents an indirect interaction. Immunoprecipitation of TRAF6 revealed slower migrating species typically indicative of auto‐modification by endogenous ubiquitin chains, and the observed ladder was greatly diminished by the presence of LGP2 or LGP2‐H but not RIG‐I nor MDA5. The specificity of LGP2 (and LGP2‐H) co‐precipitation was verified in reverse immunoprecipitations, both by alternating antisera for immunoprecipitation and blotting (Fig 7D) and by swapping tags (Figs 7E and EV2D–F). These data indicate that LGP2 can associate with diverse TRAF proteins and disrupt TRAF6 ubiquitin ligase activity.
Figure 7. TRAF suppression by catalytically inactive LGP2.
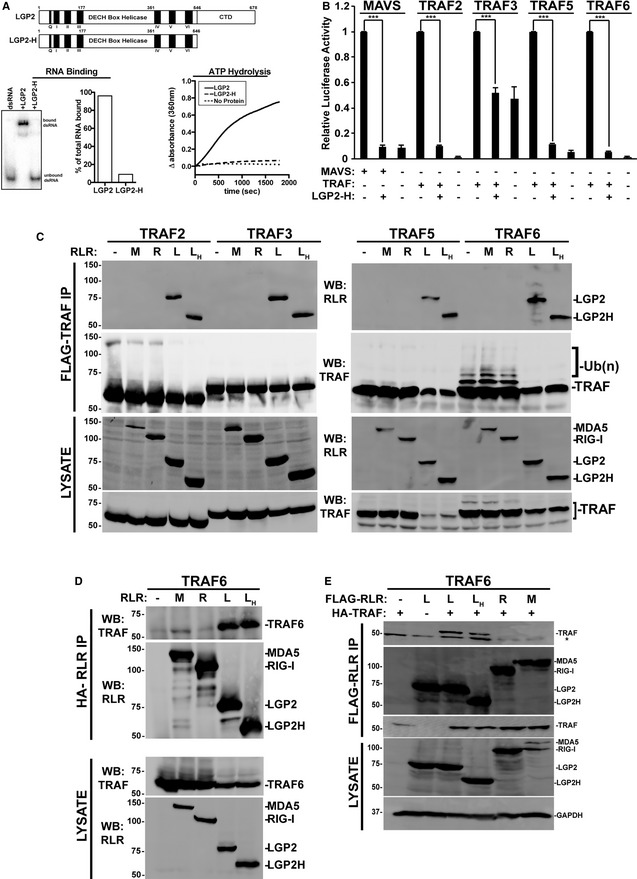
- Diagram illustrating the domain structure of full‐length LGP2 and LGP2‐H (top). Analysis of LGP2‐H catalytic properties. RNA binding was tested by mobility shift assay and quantified by phosphor imaging (bottom left). ATP hydrolysis was directly measured for both proteins at 100 nM (bottom right).
- NFκB‐reporter gene assay with MAVS or indicated TRAFs expressed with or without LGP2‐H. Cells were harvested 24 h post‐transfection prior to luciferase assays. Bars represent normalized mean values (n = 3) ± standard deviation. ***P ≤ 0.0005 by two‐tailed Student's t‐test. Corresponding immunoblots in Fig EV1.
- Cells were transfected with plasmids expressing HA‐tagged MDA5 (M), RIG‐I (R), LGP2 (L), LGP2‐H (LH) and FLAG‐tagged TRAF2, 3, 5, and 6 as indicated (n = 3). Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (TRAF) and HA (LGP2).
- Reverse co‐immunoprecipitation. Similar to (C), except cell lysates were subjected to HA immunoprecipitation and immunoblotting with antisera for FLAG (TRAF6) and HA (LGP2).
- Reverse co‐immunoprecipitation. Similar to (C), except cells were transfected with plasmids expressing HA‐tagged TRAF6 and FLAG‐tagged RLRs as indicated. Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (RLR) and HA (TRAF6) (n = 3). Asterisk (*) indicates non‐specific cross‐reactivity.
Source data are available online for this figure.
LGP2 interference with TRAF ubiquitin ligase activity
TRAF proteins activate NFκB and IRF3 signaling by creating K63‐linked ubiquitin chains, modifications to themselves, and to relevant cellular target proteins including IKKγ/NEMO 44, 45, 46. Interference with TRAF E3 activity prevents IKK activation and abrogates downstream NFκB signaling 47. TRAF ubiquitin ligase activity was tested in the presence or absence of LGP2. For TRAF2 and especially TRAF6, slower migrating species were observed in the absence of transfected ubiquitin, indicating self‐modification by endogenous ubiquitin. Additional samples included an expression vector for ubiquitin which enhanced the detection of ubiquitin‐modified TRAF proteins. Importantly, all of the observed ubiquitin modifications were substantially diminished by the presence of LGP2 (Fig 8A). Parallel experiments were done with a ubiquitin variant in which all lysines were mutated to arginine, except for K63 (Fig 8B). Again, modification by K63‐only ubiquitin was observed as slower migrating species, and in all cases, these forms were suppressed in the presence of LGP2. TRAF6‐dependent ligation is inhibited by LGP2 and LGP‐H, but not MDA5 or RIG‐I, even in the presence of exogenous ubiquitin (Fig 8C). These results indicate that LGP2 interferes with diverse TRAF proteins to disrupt ubiquitin ligase activity and interfere with NFκB signaling.
Figure 8. LGP2 interference with TRAF ubiquitin ligase activity.
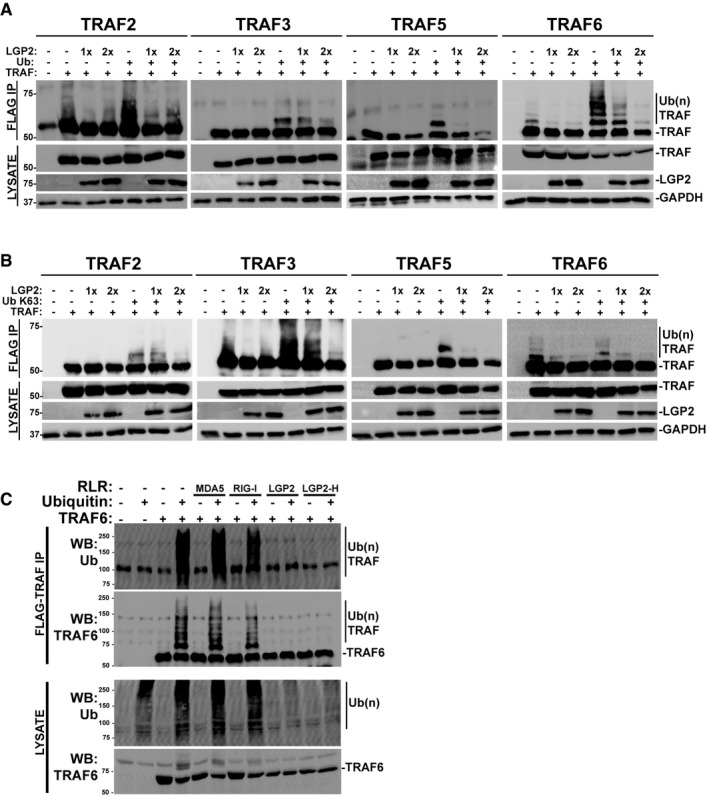
- Cells were transfected with plasmids expressing HA‐tagged LGP2, FLAG‐tagged TRAF2, 3, 5, 6, and ubiquitin (Ub) as indicated (n = 3). Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (TRAF), LGP2, and GAPDH.
- Similar to (A), but using K63‐only variant of ubiquitin (Ub K63) (n = 3).
- Cells were transfected with plasmids expressing HA‐tagged MDA5, RIG‐I, LGP2, LGP2‐H, FLAG‐tagged TRAF6, and ubiquitin (Ub) as indicated. Cell lysates were subjected to FLAG immunoaffinity purification and immunoblotting with antisera for FLAG (TRAF) and ubiquitin (n = 3).
Source data are available online for this figure.
Discussion
The data presented here describe a new means of negative regulation by the innate immune regulator, LGP2, that targets TRAF proteins and their ubiquitin ligase activities. The results provide a mechanistic explanation for LGP2's role as an inhibitor of signaling induced by viruses, cytosolic dsRNA, or the activity of RLRs and MAVS pathways. Data here indicate that LGP2 negative regulation is retained in the absence of ATP hydrolysis or RNA binding and is able to be mediated by a truncated LGP2 variant missing the C‐terminal domain.
Loss of LGP2 in both mouse and human cells results in hypersensitive and overactive antiviral signaling and IRF3/NFκB transcription, and results indicate that LGP2 can co‐precipitate with and inhibit all of the TRAF proteins that have been associated with MAVS signaling, preventing ubiquitin ligase activity. Interaction domain mapping revealed that LGP2 targets the TRAF protein C‐terminal domain. This domain, referred to as the TRAF‐C domain or the “meprin and TRAF homology (MATH)” domain 43, is a fold of seven anti‐parallel helices that is broadly represented among eukaryotes and participates in protein–protein interactions. The TRAF/MATH domain is known to be important for TRAF oligomerization, association with transmembrane receptor intracellular domains, or engaging downstream signaling machinery including partners such as MAVS 48, TRADD 38, and CD40 49, 50. As many protein families encompassing a TRAF/MATH domain are involved in protein processing and ubiquitination, it has been suggested that TRAF/MATH domains co‐evolved with proteolysis pathways in eukaryotes 51.
As a member of the RLR family of signaling proteins, LGP2 is a component of the MAVS‐mediated antiviral signaling circuit, but results show that TRAF disruption by LGP2 does not formally require MAVS and remains suppressive in cells with MAVS knockdown (Fig EV2C). Diverse MAVS‐independent NFκB activation pathways, including TNFα, IL‐1β, and cGAMP/STING, are suppressed by LGP2 expression (Fig 5). All of these signaling pathways share a common requirement for TRAF ubiquitin ligase activity leading to NFκB activation, but they access their cognate TRAFs through distinct signaling adaptors 38, 39, 40, 41, 52, 53. This result suggests the possibility of crosstalk regulation by LGP2 expression downstream of antiviral signals regulating TRAF sensitivity to limit NFκB responses in allied immune response pathways.
Consistent with these findings, all of the reported biological responses attributed to LGP2 negative regulation in vivo can be readily connected to TRAF/NFκB signal transduction. In one report of LGP2‐deficient mice, heightened NFκB activity was observed in the absence of LGP2 following poly(I:C) stimulation 25. An independent knockout mouse strain uncovered a role for LGP2 in the control of CD8+ T‐cell survival and the regulation of death‐receptor signaling in the context of virus infections 36. The role for TRAF interference in LGP2‐mediated T‐cell survival function was not examined, but LGP2 regulation of NFκB pathways may provide a mechanistic basis for this phenomenon, as T‐cell receptor and death receptors activate TRAF‐mediated NFκB signaling pathways 19 and TCR ligation induces LGP2 expression.
In humans, LGP2 has been characterized as an important regulator of cellular responses to ionizing radiation 54. LGP2 expression confers radioresistance to susceptible cells, and patient tumors with elevated LGP2 expression were associated with more adverse clinical outcomes following radiation therapy. NFκB activation is a known consequence of ionizing radiation 55, and we speculate this is likely to be subject to regulation by LGP2.
The broad use of TRAF proteins to activate NFκB in diverse immune and inflammatory processes extends LGP2 regulation of TRAF‐mediated NFκB activation well beyond the antiviral system. Results demonstrate LGP2 is able to interfere in trans with NFκB activation by TNFα, IL‐1β, and cGAMP/STING, and it is predicted to regulate many other TRAF pathways. This highlights potential cross‐regulation between antiviral responses and inflammatory signals. It is tempting to speculate that the TRAF inhibitory actions of LGP2 could be harnessed for the relief of interferonopathic systemic autoimmunity, arthritis, inflammatory bowel diseases, asthma, atherosclerosis, cancer, and other conditions characterized by hyperactive IKK or NFκB signaling.
Materials and Methods
Cells, viruses, and cytokines
HEK293 cells (ATCC) and mouse embryonic fibroblasts (MEFs; gift of Dr. M. Gale Jr., University of Washington, Seattle, WA) were grown in DMEM (Invitrogen, cat# 11965118) supplemented with 10% cosmic calf serum (CCS, Hyclone, cat# 25200114) and 1% penicillin–streptomycin (Invitrogen, cat# 15140122). Cells are routinely tested for mycoplasma contamination and regularly restored from early‐passage frozen stocks. MEFs were used before the sixth passage. Infection of cells with Sendai virus (Sendai Virus, Cantell strain, 3×108 pfu/ml, 40 HA units per μl) was performed at 200 HA units per 1 ml in serum‐free media. After 1 h, cells were washed, placed in growth medium supplemented with 2% CCS, and harvested at the indicated time points. Treatment of cells with recombinant human tumor necrosis factor alpha (TNFα, R&D systems, cat# 210‐TA‐005) and human interleukin‐1β (IL‐1β, Cell Signaling) was performed using 10 ng per 1 ml in 10% CCS media. 2′3′ cGAMP (Invivogen, cat# tlrl‐cga23) was used at 20 μg/ml.
For generation and analysis of CRISPR KO cells, gRNA target sequences were identified in exon 3 of LGP2 genomic DNA using Target Finder (http://crispr.mit.edu/). The gRNAs with the top three scores and least off‐target binding were selected from exon3:
gRNA#1 (GAGCTTCGGTCCTACCAAT), gRNA#2 (CTTCGGTCCTACCAATGGG), gRNA#3 (GGGTCTTCCCGGCACCCGT). Each gRNA was then incorporated into a 455‐bp DNA fragment synthesized as a gBlock (IDT) and cloned into pCR‐Blunt II‐TOPO vector (Invitrogen, cat# 450245). All three gRNAs and hCas9 (Addgene #41815) were transfected into cells and selected with 500 μg/ml G418. Genetic edit was validated by sequencing and by mismatch‐cleavage assay (IndelCheck, GeneCopoeia, cat# ICPE‐050). Positive clones were ultimately screened by immunoblot with a LGP2 antibody after virus induction (Proteintech, cat# 11355‐1‐AP).
Plasmids, immunoprecipitations, and immunoblotting
LGP2 and MDA5 cDNAs were cloned into a mammalian expression plasmid (p3XFLAG‐CMV‐10) with an amino‐terminal 3× FLAG epitope tag. MAVS and LGP2 cDNAs were cloned into a mammalian expression plasmid (pEF‐HA) with an amino‐terminal HA epitope tag. GFP and MAVS were cloned into a mammalian expression plasmid (pEF‐FLAG) with an amino‐terminal FLAG epitope tag. Point mutations were made using Agilent's Quikchange mutagenesis Lightning kit (cat# 210519) and confirmed by DNA sequencing. Expression vectors for TRAF2, TRAF5, and TRAF6 were provided by Dr. Z.J. Chen (UT Southwestern Medical Center, Dallas, TX). Plasmid pcDNA3.1‐FLAG‐TRAF3 was provided by Dr. J. Hiscott (VGTI Florida). Plasmid pcDNA3‐FLAG‐STING was provided by Dr. R. Weichselbaum, University of Chicago.
To construct FLAG‐tagged TRAF6 N‐terminal truncation fragments, PCR‐amplified segments of TRAF6 cDNAs (ZNC, aa 204–522; NC, aa 261–522; C, aa 352–522) were subcloned into the BamHI and NotI sites into plasmid pEF‐FLAG to generate an N‐terminal in‐frame epitope tag. TRAF6 C‐terminal truncation fragments were made using stop‐codon insertion mutagenesis with QuikChange II XL mutagenesis kit (Agilent, cat# 200522). Stop codons were inserted at aa 107 (R), aa 261 (RZ), and aa 351 (RZN). Similar methods were used to create FLAG‐tagged TRAF2, TRAF3, and TRAF5 RZN and C fragments.
For co‐immunoprecipitation experiments, FLAG‐tagged and HA‐tagged plasmids were transfected into HEK293 cells by the calcium phosphate method. Twenty‐four hours later, cells were harvested by first washing with cold phosphate‐buffered saline and then lysed with whole cell extract buffer (WCEB) consisting of 50 mM Tris, 280 mM NaCl, 0.5% NP‐40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM DTT, 2.5 mM sodium vanadate, and protease inhibitors. Cell lysates were then precleared with Sepharose beads. A percentage of the cleared lysates was reserved for analysis and the remainder incubated with FLAG M2 affinity beads (Sigma, cat# A2220) overnight and washed 3× with WCEB, eluted with SDS sample buffer, then separated by SDS–PAGE and processed for immunoblotting. For immunoblotting, the separated proteins were transferred to nitrocellulose and probed with commercial primary antibodies recognizing FLAG (Sigma, cat# F3165), HA (Sigma, cat# H3663), phospho‐IRF3 (Ser396, Cell Signaling, cat# 4947), IRF3 (Santa Cruz Biotechnology, cat# sc‐9082), phospho‐IκBα (IκBα, Ser32, Cell Signaling, cat# 14D4), IκBα (Cell Signaling), ubiquitin (Ub, Santa Cruz Biotechnology, cat# sc‐8017), DHX58 (LGP2, Abcam, cat# ab67270), or GAPDH (Santa Cruz Biotechnology, cat# sc‐47724). Proteins were visualized by enhanced chemiluminescence (Perkin Elmer, cat# NEL105001EA) and imaged using a UVP BioSpectrum MultiSpectral Imaging System. Digital files are printed on thermal paper for laboratory notebook records and exported to Adobe Photoshop and Illustrator for cropping and figure assembly. Intact digital files are only subjected to minor contrast and quality modifications and applied to entire image and shown intact without lane splicing. For HA immunoprecipitations, cells were lysed with WCEB and incubated overnight with 20 μg of anti‐HA antibody (Sigma, cat# H3663), followed by addition of 50 μl protein A/G PLUS agarose beads (Santa Cruz, cat# sc‐2003) for 6 h. Beads were washed 4× with WCEB, 1× with PBS, eluted with SDS sample buffer, and analyzed by SDS–PAGE. Figures indicate 3, 4, 5 biological replicates, and the figure presented is a representative of those replicates.
Native PAGE for IRF3 dimerization was performed as described by 56. The gel was prerun with 25 mM Tris and 192 mM glycine, pH 8.4 with and without 1% deoxycholate for 30 min at 40 mA on ice. 10 μg protein in native sample buffer was loaded immediately at the end of the prerun and the gel run at 25 mA for 60 min. Immunoblotting was performed with antisera for IRF3.
Reporter gene assays
HEK293 cells were transfected with the −110 IFNβ luciferase reporter gene, or reporters for NFκB (4× PRDII) or IRF3 (3× PRDIII/I) along with Renilla luciferase vector and other expression vectors as indicated. In samples where < 600 ng of DNA was transfected, DNA amounts were equalized using salmon sperm DNA (Invitrogen, cat# 15632‐011) in order to bring the total amount of DNA to 600 ng. At 24 h post‐transfection, cells were harvested or stimulated for 6 h with SeV, poly(I:C) (5 μg/ml), IFNα (1,000 units/ml), or IFNγ (50 ng/ml) and 24 h with 2′3′ cGAMP (10 μg/ml, Invivogen) and assayed for firefly and Renilla luciferase activities. Luciferase activity was measured using the Dual‐Luciferase Reporter Assay System (Promega, cat# E1960). Relative luciferase activity was calculated by dividing the firefly luciferase values by those of the Renilla luciferase. Data are plotted as mean values, with error bars representing standard deviation. Figures indicate two to three biological replicates, the figure presented is a representative of those replicates, and statistical analysis was done using a two‐tailed Student's t‐test.
RT–qPCR
Total RNA was extracted using the TRIzol Reagent (Invitrogen, cat# 15596018). Samples were treated with DNAse I (Invitrogen, cat# AM2224), and 1–5 μg of total RNA was primed with random primers and reverse transcribed using SuperScript III (Invitrogen, cat# 18080085). Gene expression was measured by quantitative real‐time PCR (qPCR) using the Mx3005P SYBR Green real‐time PCR system (Agilent) and normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Data are representative of multiple experiments and plotted as mean values of technical replicates with error bars representing standard deviation in technical replicates. Primers used for human mRNAs:
CCL5 (F: 5′‐CTGCTTTGCCTACATTGCCC‐3′, R: 5′‐TCGGGTGACAAAGACGACTG‐3′), CXCL10 (F: 5′‐AGCAGAGGAACCTCCAGTCT‐3′, R: 5′‐ATGCAGGTACAGCGTACAG‐3′), GAPDH (F: 5′‐ACAGTCAGCCGCATCTTCTT‐3′, R: 5′‐ACGACCAAATCCGTTGACTC‐3′), ISG56 (F: 5′‐CAGAACGGCTGCCTAATTT‐3′, R: 5′‐GGCCTTTCAGGTGTTTCAC‐3′), IFNβ (F: 5′‐ACGCCGCATTGACCATCTAT‐3′, R: 5′‐AGCCAGGAGGTTCTCAACAA‐3′), IκBα (F: 5′‐GATCCGCCAGGTGAAGGG‐3′, R: 5′‐GCAATTTCTGGCTGGTTGG‐3′), TNFα (F: 5′‐GCCCATGTTGTAGCAAACCC‐3′, R: 5′‐TATCTCTCAGCTCCACGCCA‐3′). Primers used for mouse mRNAs:
IFNβ (F: 5′‐CTGTTTTCCTTTGACCTTTCAAATG‐3′, R: 5′‐GAAGACCTGTCAGTTGATGCC‐3′); GAPDH (F: 5′‐CTTCAGAGTGGAATACTGTTGC‐3′, R: 5′‐GCATACATTTCTAATGTACTGTGTC‐3′); LGP2 (F: 5′‐GCTGGGTGGTGGTGGC‐3′, R: 5′‐GAACTCACTCTGTAGACCAGG‐3′); IL‐6 (F: 5′‐CCGGAGAGGAGACTTCACAG‐3′, R: 5′‐TCCACG ATTTCCCAGAGAAC‐3′). TNFα (F: 5′‐TAGCCAGGAGGGAGAACAGA‐3′, R: 5′‐TTTTCTGGAGGGAGATGTGG‐3′).
Protein purification, ATP hydrolysis, and RNA binding assays
Flag‐tagged LGP2 and LGP2‐H were expressed and purified from recombinant baculoviruses, and ATP hydrolysis assessed using the Enz‐Chek Phosphate Assay Kit (Molecular Probes, cat# E6646) according to the manufacturer's protocol with 100 nM protein concentrations as previously described in Bruns et al 26. RNA binding was measured using electrophoretic mobility shift assay with 10 pmol protein and 2 pmol of an end‐labeled 25‐bp dsRNA probe, and signals were quantified by phosphor image analysis.
RNA interference
Individual wells of a 24‐well culture dish containing 2fTGH cells were transfected with 60 pmol (20 μM) of siRNA with a NFκB (PRDII) luciferase reporter and Renilla luciferase plasmid using Lipofectamine 2000 (Invitrogen, cat# 11668‐500) following the manufacturer's recommendations. SMARTpool:onTARGETplus siRNAs specific for MAVS (catalog # L‐024237‐00) or non‐targeted control (ON‐TARGETplus Non‐targeting pool, catalog # D001810‐10) was obtained from Dharmacon. Experiments were performed in triplicate. Treatment with poly(I:C) (5 μg/ml) for 6 h followed by reporter gene analysis was performed 48 h after transfection.
Statistical analysis
For luciferase reporter gene assays, figures are representative of ≥ 3 biological replicates and each of these experiments contains three technical replicates. Data are plotted as mean values of these replicates, with error bars representing standard deviation calculated using a two‐tailed Student's t‐test. The two‐tailed Student's t‐test is a standard statistical test for measuring the significance of the results from these assays. P values calculated from the two‐tailed Student's t‐test conform to the standards set by the field.
For RT–qPCR, data presented are representative of ≥ 3 independent experiments and plotted as mean values of technical replicates, averaged before any statistical inference test is performed, with error bars representing standard deviation in technical replicates. Statistical analysis was done using a two‐tailed Student's t‐test.
Author contributions
J‐PP, JJL, RM, KRR, KQ, and AMB designed and conducted experiments; CMH designed experiments and analyzed results; J‐PP, JJL, RM, KRR, and CMH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We are grateful to the Horvath Lab for their guidance and helpful comments on this work and the manuscript, Michael Gale, John Errett, and Nanette Crochet for providing LGP2−/− MEFs, James Chen, Feng Du, John Hiscott, and Samar Bel Hadj for TRAF cDNAs, and Ralph Weichselbaum and Diana Ranoa for the STING expression vector. Research on RLRs in the Horvath Lab was supported by NIH grants AI073919, AI50707, and GM111652 to CMH. JJL is supported by Cellular and Molecular Basis of Disease Training Grant (NIH T32 GM008061), KQ was assisted by a research grant from the NU office of Undergraduate Research, and KRR was supported by the Northwestern University Rappaport Award for Research Excellence.
EMBO Reports (2018) 19: e45176
References
- 1. McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A (2015) Type I interferons in infectious disease. Nat Rev Immunol 15: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tough DF (2012) Modulation of T‐cell function by type I interferon. Immunol Cell Biol 90: 492–497 [DOI] [PubMed] [Google Scholar]
- 3. Kiefer K, Oropallo MA, Cancro MP, Marshak‐Rothstein A (2012) Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 90: 498–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trinchieri G (2010) Type I interferon: friend or foe? J Exp Med 207: 2053–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tough DF, Borrow P, Sprent J (1996) Induction of bystander T cell proliferation by viruses and type I interferon in vivo [see comments]. Science 272: 1947–1950 [DOI] [PubMed] [Google Scholar]
- 6. Crow YJ (2015) Type I interferonopathies: mendelian type I interferon up‐regulation. Curr Opin Immunol 32: 7–12 [DOI] [PubMed] [Google Scholar]
- 7. Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- 8. Parker BS, Rautela J, Hertzog PJ (2016) Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 16: 131–144 [DOI] [PubMed] [Google Scholar]
- 9. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G (2015) Type I interferons in anticancer immunity. Nat Rev Immunol 15: 405–414 [DOI] [PubMed] [Google Scholar]
- 10. Bruns AM, Horvath CM (2012) Activation of RIG‐I‐like receptor signal transduction. Crit Rev Biochem Mol Biol 47: 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruns AM, Horvath CM (2014) Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev 25: 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bamming D, Horvath CM (2009) Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG‐I, and LGP2. J Biol Chem 284: 9700–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S et al (2005) Shared and unique functions of the DExD/H‐box helicases RIG‐I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175: 2851–2858 [DOI] [PubMed] [Google Scholar]
- 14. Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ (2013) MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2: e00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paz S, Vilasco M, Werden SJ, Arguello M, Joseph‐Pillai D, Zhao T, Nguyen TL, Sun Q, Meurs EF, Lin R et al (2011) A functional C‐terminal TRAF3‐binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res 21: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang XD, Sun SC (2015) Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev 266: 56–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pineda G, Ea CK, Chen ZJ (2007) Ubiquitination and TRAF signaling. Adv Exp Med Biol 597: 80–92 [DOI] [PubMed] [Google Scholar]
- 18. Freaney JE, Kim R, Mandhana R, Horvath CM (2013) Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep 4: 959–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie P (2013) TRAF molecules in cell signaling and in human diseases. J Mol Signal 8: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodriguez KR, Bruns AM, Horvath CM (2014) MDA5 and LGP2: accomplices and antagonists of antiviral signal transduction. J Virol 88: 8194–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruns AM, Horvath CM (2015) LGP2 synergy with MDA5 in RLR‐mediated RNA recognition and antiviral signaling. Cytokine 74: 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deddouche S, Goubau D, Rehwinkel J, Chakravarty P, Begum S, Maillard PV, Borg A, Matthews N, Feng Q, van Kuppeveld FJM et al (2014) Identification of an LGP2‐associated MDA5 agonist in picornavirus‐infected cells. Elife 3: e01535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bruns AM, Leser GP, Lamb RA, Horvath CM (2014) The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5‐RNA interaction and filament assembly. Mol Cell 55: 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O (2010) LGP2 is a positive regulator of RIG‐I‐ and MDA5‐mediated antiviral responses. Proc Natl Acad Sci USA 107: 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN (2007) Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol 178: 6444–6455 [DOI] [PubMed] [Google Scholar]
- 26. Bruns AM, Pollpeter D, Hadizadeh N, Myong S, Marko JF, Horvath CM (2013) ATP hydrolysis enhances RNA recognition and antiviral signal transduction by the innate immune sensor, laboratory of genetics and physiology 2 (LGP2). J Biol Chem 288: 938–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Si‐Tahar M, Blanc F, Furio L, Chopy D, Balloy V, Lafon M, Chignard M, Fiette L, Langa F, Charneau P et al (2014) Protective role of LGP2 in influenza virus pathogenesis. J Infect Dis 210: 214–223 [DOI] [PubMed] [Google Scholar]
- 28. Malur M, Gale M, Krug RM (2012) LGP2 downregulates interferon production during infection with seasonal human influenza A viruses that activate interferon regulatory factor 3. J Virol 86: 10733–10738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M Jr (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG‐I and LGP2. Proc Natl Acad Sci USA 104: 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komuro A, Horvath CM (2006) RNA‐ and virus‐independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol 80: 12332–12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA (2005) The RNA helicase Lgp2 inhibits TLR‐independent sensing of viral replication by retinoic acid‐inducible gene‐I. J Immunol 175: 5260–5268 [DOI] [PubMed] [Google Scholar]
- 32. Rodriguez KR, Horvath CM (2014) Paramyxovirus V protein interaction with the antiviral sensor LGP2 disrupts MDA5 signaling enhancement but is not relevant to LGP2‐mediated RLR signaling inhibition. J Virol 88: 8180–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thanos D, Maniatis T (1995) Virus induction of human IFNb gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100 [DOI] [PubMed] [Google Scholar]
- 34. Du W, Thanos D, Maniatis T (1993) Mechanisms of transcriptional synergism between distinct virus‐inducible enhancer elements. Cell 74: 887–898 [DOI] [PubMed] [Google Scholar]
- 35. Maniatis T, Whitemore L‐A, Du W, Fan C‐M, Keller AD, Palombella VJ, Thanos D (1992) Positive and negative control of human interferon beta gene expression In Transcriptional regulation, Part 2, McKnight SL, Yamamoto KR. (eds), pp 1193–1220. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- 36. Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, McMillan A, Diamond MS, Clark EA, Bevan MJ et al (2012) The RIG‐I‐like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 37: 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV et al (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630 [DOI] [PubMed] [Google Scholar]
- 38. Hsu H, Shu HB, Pan MG, Goeddel DV (1996) TRADD‐TRAF2 and TRADD‐FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84: 299–308 [DOI] [PubMed] [Google Scholar]
- 39. Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV (1996) TRAF6 is a signal transducer for interleukin‐1. Nature 383: 443–446 [DOI] [PubMed] [Google Scholar]
- 40. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z (1997) MyD88: an adapter that recruits IRAK to the IL‐1 receptor complex. Immunity 7: 837–847 [DOI] [PubMed] [Google Scholar]
- 41. Abe T, Barber GN (2014) Cytosolic‐DNA‐mediated, STING‐dependent proinflammatory gene induction necessitates canonical NF‐kappaB activation through TBK1. J Virol 88: 5328–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cai X, Chiu YH, Chen ZJ (2014) The cGAS‐cGAMP‐STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54: 289–296 [DOI] [PubMed] [Google Scholar]
- 43. Uren AG, Vaux DL (1996) TRAF proteins and meprins share a conserved domain. Trends Biochem Sci 21: 244–245 [PubMed] [Google Scholar]
- 44. Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A (2009) NEMO specifically recognizes K63‐linked poly‐ubiquitin chains through a new bipartite ubiquitin‐binding domain. EMBO J 28: 2885–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D et al (2009) Specific recognition of linear ubiquitin chains by NEMO is important for NF‐kappaB activation. Cell 136: 1098–1109 [DOI] [PubMed] [Google Scholar]
- 46. Yoshikawa A, Sato Y, Yamashita M, Mimura H, Yamagata A, Fukai S (2009) Crystal structure of the NEMO ubiquitin‐binding domain in complex with Lys 63‐linked di‐ubiquitin. FEBS Lett 583: 3317–3322 [DOI] [PubMed] [Google Scholar]
- 47. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin‐conjugating enzyme complex and a unique polyubiquitin chain. Cell 103: 351–361 [DOI] [PubMed] [Google Scholar]
- 48. Shi Z, Zhang Z, Zhang Z, Wang Y, Li C, Wang X, He F, Sun L, Jiao S, Shi W et al (2015) Structural insights into mitochondrial antiviral signaling protein (MAVS)‐tumor necrosis factor receptor‐associated factor 6 (TRAF6) signaling. J Biol Chem 290: 26811–26820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ni CZ, Welsh K, Leo E, Chiou CK, Wu H, Reed JC, Ely KR (2000) Molecular basis for CD40 signaling mediated by TRAF3. Proc Natl Acad Sci USA 97: 10395–10399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McWhirter SM, Pullen SS, Holton JM, Crute JJ, Kehry MR, Alber T (1999) Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc Natl Acad Sci USA 96: 8408–8413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zapata JM, Martinez‐Garcia V, Lefebvre S (2007) Phylogeny of the TRAF/MATH domain. Adv Exp Med Biol 597: 1–24 [DOI] [PubMed] [Google Scholar]
- 52. Muzio M, Ni J, Feng P, Dixit VM (1997) IRAK (Pelle) family member IRAK‐2 and MyD88 as proximal mediators of IL‐1 signaling. Science 278: 1612–1615 [DOI] [PubMed] [Google Scholar]
- 53. Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K et al (2001) Critical roles of TRAF2 and TRAF5 in tumor necrosis factor‐induced NF‐kappa B activation and protection from cell death. J Biol Chem 276: 36530–36534 [DOI] [PubMed] [Google Scholar]
- 54. Widau RC, Parekh AD, Ranck MC, Golden DW, Kumar KA, Sood RF, Pitroda SP, Liao Z, Huang X, Darga TE et al (2014) RIG‐I‐like receptor LGP2 protects tumor cells from ionizing radiation. Proc Natl Acad Sci USA 111: E484–E491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D (1991) Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest 88: 691–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robitaille AC, Mariani MK, Fortin A, Grandvaux N (2016) A high resolution method to monitor phosphorylation‐dependent activation of IRF3. J Vis Exp 107: e53723 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8