

Abstract
Cellular senescence is a unique cell fate characterized by stable proliferative arrest and the extensive production and secretion of various inflammatory proteins, a phenomenon known as the senescence‐associated secretory phenotype (SASP). The molecular mechanisms responsible for generating a SASP in response to senescent stimuli remain largely obscure. Here, using unbiased gene expression profiling, we discover that the scavenger receptor CD36 is rapidly upregulated in multiple cell types in response to replicative, oncogenic, and chemical senescent stimuli. Moreover, ectopic CD36 expression in dividing mammalian cells is sufficient to initiate the production of a large subset of the known SASP components via activation of canonical Src–p38–NF‐κB signaling, resulting in the onset of a full senescent state. The secretome is further shown to be ligand‐dependent, as amyloid‐beta (Aβ) is sufficient to drive CD36‐dependent NF‐κB and SASP activation. Finally, loss‐of‐function experiments revealed a strict requirement for CD36 in secretory molecule production during conventional senescence reprogramming. Taken together, these results uncover the Aβ–CD36–NF‐κB signaling axis as an important regulator of the senescent cell fate via induction of the SASP.
Keywords: aging, amyloid‐beta, cellular senescence, inflammation, SASP
Subject Categories: Ageing, Signal Transduction
Introduction
Cellular senescence, defined as a state of irreversible cell cycle arrest, was discovered in 1962 when Dr. Leonard Hayflick observed that upon prolonged cell culture, human diploid fibroblasts indefinitely lose their ability to proliferate 1, 2. In the ensuing decades, senescence has been increasingly appreciated for its physiological functions in vivo, with important roles during embryonic development and normal aging and in multiple pathological conditions including fibrosis and cancer 3, 4, 5, 6, 7. In addition to persistent replicative stress, various other types of stimuli, including DNA damage, oncogene activation, oxidative stress, and telomere dysfunction, are known to induce senescence in various cellular contexts 8. Moreover, the administration of specific chemical agents, such as doxorubicin and erlotinib, is sufficient to induce cellular senescence in certain cancer and normal epithelial cell types, respectively 9, 10.
Across these various induction stimuli, senescent cells are currently thought to share two major molecular features. First, senescent cells have increased expression of at least one cyclin‐dependent kinase (CDK) inhibitor, typically p16 or p21, which functions to activate the Rb tumor suppressor, resulting in cell cycle arrest 8. Second, senescent cells exhibit a unique secretory profile, termed the senescence‐associated secretory phenotype (SASP). Upon senescence initiation, two transcription factors normally present in immune cells, NF‐κB and CEBPβ, are activated to promote the transcription of a set of relatively conserved pro‐inflammatory cytokines, chemokines, growth factors, and proteases 9, 10, 11. Some canonical signal transduction cascades, such as the mTOR and p38 MAPK pathways, have been shown to stimulate NF‐κB and SASP formation during senescence 12, 13. However, the upstream inputs that trigger the activation of those pathways in order to produce the SASP remain largely unknown.
CD36 is a multi‐ligand scavenger receptor expressed in various mammalian cell types that functions in a context‐dependent manner. Previous studies have identified diverse CD36 ligands including collagen, thrombospondin, and various lipoproteins and lipids that bind CD36 in order to regulate vascular and adipose homeostasis 14, 15, 16, 17. In contrast, when expressed in macrophages and microglia cells, CD36 can generate a strong pro‐inflammatory response through its interaction with secreted amyloid‐beta 1–42 (Aβ) or oxidized low‐density lipoprotein (oxLDL). Upon Aβ or oxLDL binding, CD36 stimulates MAPK signaling through Src family kinase activation, leading to the activation of NF‐κB and subsequent cytokine and chemokine transcriptional initiation 18. However, the presence of this CD36‐dependent pro‐inflammatory signaling axis outside the immune system has not been previously described.
In this study, we show that CD36 expression is rapidly and robustly induced in a variety of senescent states and cell types. Molecular analysis further revealed that the interaction of upregulated CD36 with its ligand Aβ is sufficient to promote Src–MAPK–NF‐κB pathway activation and establishment of a SASP. Importantly, sustained exposure of CD36 to ligand drives pro‐inflammatory cytokine production and cell cycle arrest in order to establish the overall senescent state in both epithelial cells and fibroblasts. Finally, loss‐of‐function experiments demonstrate that CD36 is strictly required for NF‐κB phosphorylation and SASP initiation and maintenance during both oncogene‐ and chemical‐induced senescence. Taken together, we identify CD36 as a novel SASP modulator involved in both senescence‐associated secretome activation and the formation of a comprehensive senescent state.
Results and Discussion
CD36 is induced in multiple senescence contexts
In previous work, we showed that targeted chemical inhibition of the epidermal growth factor receptor (EGFR) in primary human bronchial epithelial (HBE) cells is sufficient to trigger a comprehensive senescent phenotype within 3 days 9. Taking advantage of the efficiency of this method, we developed an unbiased gene expression profiling approach to compare senescent HBE cells with their proliferating counterparts in order to identify novel signaling molecules that function to regulate senescence initiation and the SASP. HBE cells, treated with either erlotinib or DMSO, were incubated with the fluorescent substrate C12FDG for the senescence‐associated beta‐galactosidase (SA‐βGal), and senescent cells were purified using flow cytometry as previously described 19. The biological triplicated samples of senescent and proliferating cells were then screened by transcriptional profiling to identify genes differentially expressed during the early phase of senescence establishment (Fig 1A). This method revealed 331 genes with at least 1.5‐fold transcriptional induction or suppression at 18 h after erlotinib‐mediated senescence initiation (in press). Of these, 10 candidates were chosen for further investigation based on their plasma membrane localization (GeneCards confidence > 4), and time‐dependent mRNA upregulation was validated by qPCR (Fig EV1A). As an indication that our method can detect bona fide senescence regulators, this subset of membrane proteins included the interleukin‐1 receptor (IL‐1R) and Notch3, both of which have been previously shown to modulate senescence but not SASP initiation per se 20, 21. However, among the candidates, the scavenger receptor CD36 stood out as the one with the strongest and most rapid induction (Fig EV1A). In fact, CD36 mRNA expression was upregulated approximately ten‐ and thirty‐fold at the 6‐ and 48‐h time points, respectively (Fig 1B). Normally at 6 h, most features of the senescent phenotype are not yet evident and signal transduction is still in its initiation phase. Thus, we hypothesized that CD36 might be functional in the senescence programming process.
Figure 1. CD36 is induced in multiple senescence contexts.
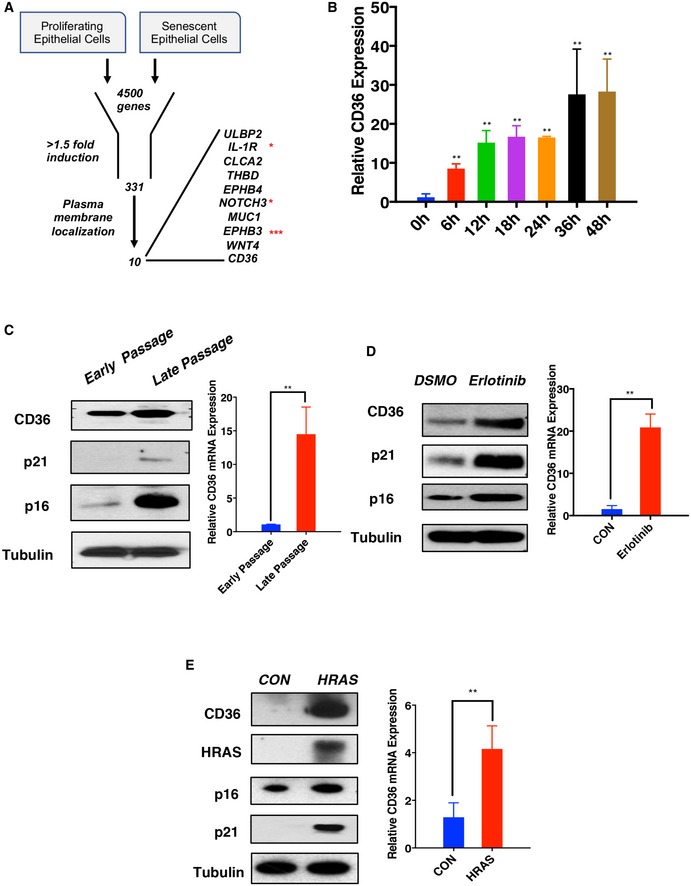
- Schematic of gene expression profiling approach to identify novel regulators of the SASP. Proliferating HBE cells were treated with DMSO or 1 μM erlotinib for 18 h, after which cell lysates were collected and analyzed using gene expression arrays. A total of 331 genes were significantly upregulated in senescent cells (1.5‐fold cutoff threshold). Further protein localization analysis revealed a group of 10 surface proteins among this subset. * indicates proteins previously reported to regulate cellular senescence. *** indicates the candidate with the fastest and most robust upregulation.
- CD36 mRNA expression time‐course analysis. Proliferating HBE cells were treated with DMSO or 1 μM erlotinib. Lysates were collected at 0, 4, 12, 18, 24, 36, and 48 h post‐treatment. qPCR was performed to measure CD36 mRNA expression normalized to β‐actin. Experiments were replicated three times independently (n = 3). Data are reported as the mean ± SEM. **P < 0.01 compared with control group, one‐way ANOVA.
- CD36 mRNA and protein analysis during replicative senescence. IMR90 cells were collected at passages 27 (early) and 70 (late) for CD36 expression analysis by qPCR and immunoblotting. The immunoblot figures are a representative image of at least three independent experiments (n = 3). qPCR results are normalized to β‐actin. Data are reported as the mean ± SEM. P‐values were calculated based on at least three independent experiments (n = 3). **P < 0.01, Student's t‐test.
- CD36 mRNA and protein analysis in erlotinib‐induced senescence. Proliferating HBE cells were treated with either 1 μM erlotinib or vehicle (DMSO) for 48 h prior to sample collection for CD36 expression analysis by qPCR and immunoblotting. Immunoblot images are representative of five independent experiments (n = 5). qPCR results are normalized to β‐actin (n = 5). Data are reported as the mean ± SEM. P‐values were calculated based on five independent experiments. **P < 0.01, Student's t‐test.
- CD36 mRNA and protein analysis during HRAS‐induced senescence. Proliferating IMR90 cells were transfected with Tet‐on HRAS or a control plasmid. After puromycin selection, cells were further treated with 1 μg/ml doxycycline for 7 days to fully establish senescence. Samples were then analyzed by qPCR and immunoblotting. Immunoblot figures are representative of three independent experiments (n = 3). qPCR results are normalized to β‐actin (n = 3). Data are reported as the mean ± SEM. P‐values were calculated based on three independent experiments. **P < 0.01, Student's t‐test.
Figure EV1. CD36 is rapidly upregulated upon senescence induction.

-
ACandidate gene expression in senescent HBE cells. Proliferating HBE cells were treated with DMSO or 1 μM erlotinib, and lysates were collected at 0, 4, 12, 18, 24, 36 and 48 h post‐treatment. Samples were analyzed by qPCR using primers specifically recognizing the indicated transcripts. Each measurement was normalized to β‐actin (mean ± SEM, n = 3).
-
BCD36 expression analysis using GEO datasets. CD36 expression in control (proliferating) and senescent IMR90 fibroblasts was obtained from publicly available replicative (GSE53356) and oncogene‐induced (GSE75207) senescence datasets, as indicated. Data are reported as means ± SEM. **P < 0.01, Student's t‐test.
-
C, DIMR90 replicative senescence proliferation and SA‐βGal staining analysis. IMR90 cells were cultured to 27 (early) and 70 (late) passages and collected. (C) Representative flow cytometry (left) and quantification (right) of cell proliferation. Cells were treated with EdU for 2 h prior to analysis by flow cytometry. (D) Representative SA‐βGal staining images (left) and quantification (right) of passage 27 (early) and 70 (late) IMR90 cells. Scale bars, 50 μm. Data are reported as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test.
-
E, FErlotinib‐induced HBE cell senescence. (E) Representative flow cytometry (left) and quantification (right) of EdU incorporation. Proliferating HBE cells were treated with DMSO or 1 μM erlotinib for 3 days. Cells were then treated with EdU for 2 h and analyzed by flow cytometry. (F) Representative SA‐βGal staining images (left) and quantification (right) of DMSO or erlotinib‐treated HBE cells. Data are reported as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test.
-
G, HHRAS‐induced IMR90 cell senescence. Proliferating IMR90 cells were infected with HRAS or control viruses for 9 days, and samples were collected for proliferation and SA‐βGal staining assays. (G) Representative flow cytometry (left) and quantification (right) of cell proliferation. Cells were treated with EdU for 2 h prior to analysis. (H) Representative SA‐βGal staining images (left) and quantification (right) of HRAS and control IMR90 cells. Scale bars, 50 μm. Data are reported as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test.
-
ICD36 expression in organs from aged (30 months old) and young (1 month old) mice. Lung, liver, and muscle tissues as indicated were collected and analyzed for CD36 expression by qPCR. Results were normalized to 18S rRNA. Data are reported as the mean ± S.D. (n = 3 technical replicates). **P < 0.01, Student's t‐test
To determine whether CD36 is also induced in other senescence models, we analyzed CD36 expression in replicative and oncogene‐induced senescent fibroblasts using the publicly available Gene Expression Omnibus (GEO) database. In agreement with our chemical‐induced senescence results, CD36 was found to also be significantly upregulated in both of these microarray datasets (Fig EV1B).
To further validate our microarray results, we replicated and confirmed the models of erlotinib‐induced, oncogene‐induced, and replicative senescence using canonical senescence markers (Fig EV1C–H). We then measured CD36 expression in those senescent contexts. Consistently, a strong induction of CD36 mRNA and protein was observed in senescent cells induced by all three stimuli (Fig 1C–E). It is known that senescent cells accumulate within aged tissues 22, and this phenomenon is conserved across different species and proposed to be functionally responsible for the development of major aging‐related phenotypes 23. To assess whether CD36 expression is correlated with senescent cell accumulation in aging organs, we measured CD36 expression in lung, liver, and muscle tissue of both young (1 month) and old (30 months) mice. Importantly, all tissue types tested contained elevated CD36 expression. Moreover, in lung tissue, which is the origin of HBE cells, CD36 had a ~100‐fold induction in aged mice, indicating a strong correlation between CD36 induction and bronchial cell senescence and aging (Fig EV1I). Taken together, these results demonstrate that CD36 expression is consistently upregulated in a wide range of cell and tissue types by various senescent stimuli including replicative stress, oncogene activation, EGFR inhibition, and the natural aging process.
Short‐term CD36 expression initiates production of secretory molecules
Based on its strong induction during the early stages of senescence initiation, we hypothesized that CD36 might have a functional impact on senescence programming. To investigate its possible sufficiency in inducing cellular senescence, we ectopically expressed CD36 in HBE cells using a Tet‐on inducible expression system. Since there are no published reports describing a role for CD36 in cellular senescence, we first examined established features of the senescent phenotype including cell proliferation, CDK inhibitor expression (p16 and p21), SA‐βGal activity, and the SASP. First, we performed SA‐βGal staining (Fig 2A), 5‐ethynyl‐2A‐deoxyuridine (EdU) incorporation (Fig 2B), and p16 and p21 Western blot assays (Fig 2C) using HBE cells engineered to overexpress CD36 for 7 days or control cells. However, these proliferation‐related assays failed to reveal significant differences between CD36‐expressing and control HBE cells at the 7‐day time point, indicating that forced CD36 expression is insufficient to drive cell cycle arrest in the short term.
Figure 2. Short‐term CD36 upregulation initiates NF‐κB signaling and secretome establishment in epithelial cells.

- Representative images of SA‐βGal staining (left) and quantification (right) of short‐term CD36‐expressing or control HBE cells. HBE cells were infected with Tet‐on CD36 or a Tet‐on control virus. Cells were then treated with 1 μg/ml doxycycline for 7 days. Subsequently, cells were fixed and stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM; n = 3. N.S., not significant, Student's t‐test.
- Flow cytometry (left) and quantification (right) of short‐term CD36‐overexpressing HBE cell proliferation. HBE cells overexpressing Tet‐on CD36 or Tet‐on control were treated with 1 μg/ml doxycycline for 7 days. Cells were then treated with EdU for 2 h and analyzed by flow cytometry. Data are reported as the mean ± SEM; n = 3. P‐values were calculated based on at least three independent experiments. N.S., not significant, Student's t‐test.
- Cyclin‐dependent kinase inhibitors (p16 and p21) of short‐term CD36‐expressing HBE cells. Whole‐cell lysates of control and CD36‐overexpressing HBE cells (7 days) were collected and subsequently immunoblotted with the indicated antibodies. Blots are representative of three independent biological replicates (n = 3).
- Signal transduction analysis of short‐term CD36‐expressing HBE cells. Whole‐cell lysates of control and CD36‐overexpressing HBE cells (7 days) were collected and subsequently immunoblotted with the indicated antibodies. Blots are representative of four independent biological replicates (n = 4).
- NF‐κB luciferase reporter assay of short‐term CD36‐expressing HBE cells. Luciferase reporters were transfected into control and CD36‐overexpressing HBE cells (4 days). Luciferase reporter assays were then executed at day 7. Data are reported as the mean ± SEM; n = 3. P‐values were calculated based on three independent experiments. **P < 0.01, Student's t‐test.
- SASP transcriptional analysis of short‐term CD36 expression in HBE cells. HBE cells from three independent donors were infected with Tet‐on CD36 or Tet‐on control and treated with 1 μg/ml doxycycline for 7 days. Samples were then collected and analyzed by qPCR. Results were normalized to control HBE cells. Red and blue colors represent up‐ and downregulated transcripts, respectively. P‐values were calculated based on three cell donors analyzed in independent experiments. **P < 0.01; *P < 0.05; Student's t‐test.
Since CD36 is known to activate an inflammatory phenotype in certain immune system cell types 15, 16, 18, 24, we next considered the possibility that it might have a similar function during senescence reprogramming. SASP has been shown to be mediated through the activity of the NF‐κB transcription factor complex, and a conventional indicator of NF‐κB activation is the phosphorylation status of its functional subunit p65; we therefore measured p65 phosphorylation in CD36‐overexpressing and control cells by Western blotting 25. Interestingly, these assays revealed increased phosphorylation of p65 along with activation of its upstream tyrosine kinase c‐Src and MAP kinase p38 (Fig 2D). Luciferase reporter assays further verified the activation of NF‐κB signaling in CD36‐expressing HBE cells (Fig 2E). Since the conventional role of NF‐κB is regulating cytokine production and secretion, the finding of CD36 driving NF‐κB activation suggests that CD36 might promote the SASP through stimulating the Src–p38–NF‐κB axis. To comprehensively explore a relationship between CD36 and the SASP, we performed quantitative PCR (qPCR)‐based profiling analysis of 78 molecules previously reported as components of the SASP 26. Strikingly, across HBE cells from three independent human donors, 22 of these molecules showed a statistically significant (> 1.5‐fold) upregulation upon ectopic CD36 expression (Fig 2F). Among these, many well‐recognized conventional SASP components were found to be consistently produced upon CD36 expression, including interleukin 6 (IL‐6) and interleukin 8 (IL‐8) 11, 25, 27, 28. These results demonstrate that short‐term expression of CD36 in HBE cells, while unable to trigger a cell cycle exit, can promote NF‐κB signaling and production of a large set of SASP components.
Long‐term CD36 expression promotes a comprehensive senescent phenotype
Some senescent stimuli, such as replicative exhaustion, require a period of weeks to yield cell cycle arrest 29. To further assess a possible effect of CD36 on the cell cycle, HBE cells expressing CD36 were maintained for a period of 14 days. Interestingly, prolonged CD36 expression led to a striking phenotype of cell cycle exit that was associated with increased levels of cyclin‐dependent kinase inhibitors and SA‐βGal activity (Fig 3A–C). Further signaling pathway analysis revealed a sustained activity of the Src–p38–NF‐κB axis, indicating that long‐term activation of CD36‐dependent pro‐inflammatory signaling might be responsible for proliferative arrest (Fig 3D–E). Overall, these results suggest that long‐term CD36‐dependent SASP signaling activation can trigger a comprehensive senescence phenotype in primary human epithelial cells.
Figure 3. Long‐term CD36 ectopic expression triggers a comprehensive senescent phenotype in epithelial cells.

- Representative images of SA‐βGal staining (left) and quantification (right) for long‐term CD36‐expressing and control HBE cells. HBE cells were infected with Tet‐on CD36 or Tet‐on control viruses and subsequently treated with 1 μg/ml doxycycline for 14 days. At day 14, cells were fixed and stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM; n = 4. **P < 0.01, Student's t‐test.
- Flow cytometry analysis (left) and quantification (right) of HBE cell proliferation after long‐term CD36 overexpression. HBE cells were infected with Tet‐on CD36 or Tet‐on control and treated with 1 μg/ml doxycycline for 14 days. At day 14, cells were treated with EdU for 2 h and analyzed by flow cytometry. Data are reported as the mean ± SEM; n = 3. P‐values were calculated based on three independent experiments. **P < 0.01, Student's t‐test.
- Cyclin‐dependent kinase inhibitor (p16 and p21) analysis of long‐term CD36‐expressing HBE cells. Whole‐cell lysates from control or CD36‐overexpressing HBE cells (14 days) were collected and subsequently immunoblotted with the indicated antibodies. Blots are representative of three independent biological replicates.
- Signal transduction analysis of long‐term CD36‐expressing HBE cells. Whole‐cell lysates of control and CD36‐overexpressing HBE cells (14 days) were collected and immunoblotted with the indicated antibodies. Blots are representative of three independent biological replicates.
- NF‐κB luciferase reporter assay of long‐term CD36‐expressing HBE cells. Luciferase reporters were transfected into control or CD36‐overexpressing HBE cells (11 days), and luciferase activity was measured at day 14. Data are reported as the mean ± SEM; n = 3. P‐values were calculated based on three independent experiments. **P < 0.01, Student's t‐test.
Based on the fact that CD36 is broadly induced during epithelial cell and fibroblast senescence and is sufficient to promote cellular senescence in HBE cells, we next asked whether the effects of CD36 on NF‐κB activation and cell cycle arrest are conserved across cell types. To investigate this, we ectopically expressed CD36 for 7 days in IMR90 human diploid fibroblasts. This short‐term CD36 overexpression in IMR90 cells did not produce significant NF‐κB activation (Fig 4A), cell cycle arrest (Fig 4C), or SA‐βGal activity (Fig 4E). However, as in HBE cells, upon extended (17 days) CD36 expression, we observed a significantly decreased proliferative capability (Fig 4D) associated with increased SA‐βGal staining (Fig 4F) and increased levels of p16, p21, and activated forms of NF‐κB signaling pathway components (Fig 4B). Together, these findings suggest that the CD36–NF‐κB–SASP signaling cascade exists in both human epithelial cells and diploid fibroblasts and prolonged exposure to this signaling is capable of inducing stable cell cycle arrest and appearance of a senescent phenotype.
Figure 4. Long‐term CD36 expression triggers a comprehensive senescence phenotype in human diploid fibroblasts.
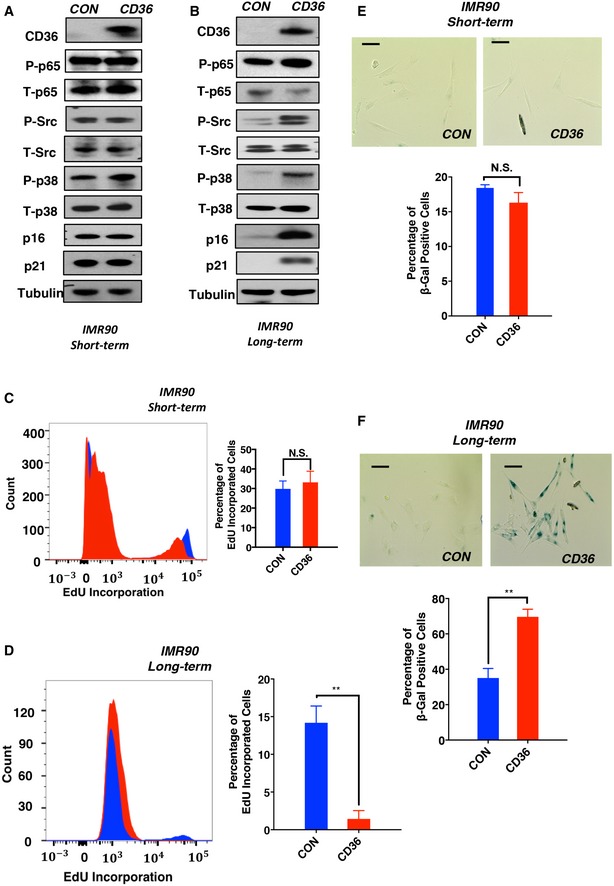
-
A, BCyclin‐dependent kinase inhibitor (p16 and p21) and signal transduction analysis of short‐term (A) and long‐term (B) CD36‐expressing IMR90 cells. Whole‐cell lysates of control and CD36‐overexpressing IMR90 cells (7 or 17 days) were collected and immunoblotted with the indicated antibodies. Blots are representative of three independent biological replicates.
-
C, DProliferation of IMR90 cells after short‐ (C) or long‐term (D) CD36 expression. Representative flow cytometry (left) and quantification (right) of IMR90 cell proliferation. IMR90 cells were infected with Tet‐on CD36 or Tet‐on control viruses and treated with 1 μg/ml doxycycline for 7 or 17 days. Cells were then treated with EdU for 2 h and analyzed by flow cytometry. Data are reported as the mean ± SEM; n = 3. P‐values were calculated based on three independent experiments. N.S., not significant; **P < 0.01; Student's t‐test.
-
E, FSA‐βGal staining of IMR90 cells after short‐ (E) or long‐term (F) expression of CD36. Representative SA‐βGal staining images (above) and quantification (below) are shown. IMR90 cells were infected with Tet‐on CD36 or Tet‐on control viruses and then treated with 1 μg/ml doxycycline for 7 or 17 days, after which cells were fixed and stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM; n =3. N.S., not significant; **P < 0.01; Student's t‐test.
The SASP comprises a combination of secreted molecules that collectively function to reinforce the overall senescent phenotype 27, 28. To investigate the biological function of the CD36‐dependent SASP, we applied conditioned media from long‐term CD36‐expressing or control IMR90 cells to naïve fibroblasts. Following 10 days of culture, a strong senescent phenotype was observed only in the group treated with supernatant from CD36‐expressing cells (Fig EV2A–C), suggesting that upon CD36‐mediated NF‐κB activation, fibroblasts secrete a cohort of cytokines which act to accelerate the onset of senescence. To test whether NF‐κB and its downstream SASP components are the major factors driving cell cycle arrest, we administered the NF‐κB inhibitor Bay 11‐7082 to block CD36‐dependent NF‐κB‐mediated cytokine production and secretion and then measured fibroblast senescence. Importantly, NF‐κB inhibition resulted in diminished SA‐βGal staining, reduced p16 and p21 expression, and partially restored proliferative capacity in long‐term CD36‐expressing IMR90 cells (Fig EV2D–F). Based on these results, we conclude that long‐term activation of the CD36–Src–p38–NF‐κB signaling axis is sufficient to drive a comprehensive senescent phenotype.
Figure EV2. NF‐κB‐dependent SASP production is required for comprehensive senescence phenotype establishment.
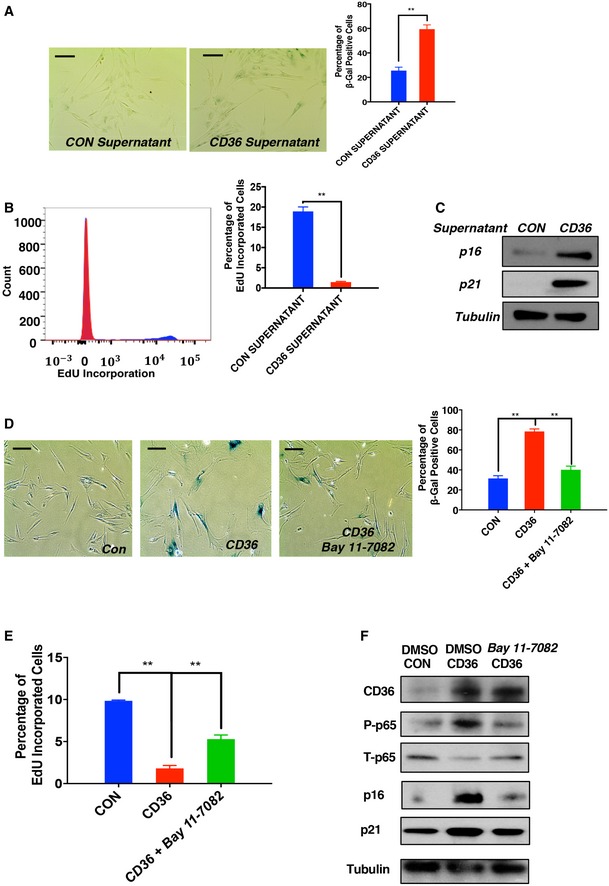
- Representative images (left) and quantification (right) of SA‐βGal‐stained IMR90 cells cultured with control or CD36 supernatant. Supernatants from IMR90 cells ectopically expressing CD36 or control plasmid for 9 days were collected and applied to naïve IMR90 cells. Supernatants added to naïve IMR90 cells were changed every 3 days up to day 9, at which point cells were fixed and stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test.
- Representative flow cytometry (left) and quantification (right) of control or CD36 supernatant‐treated IMR90 cells. Cells described in (A) were further treated with EdU for 2 h and analyzed by flow cytometry. Data are reported as the mean ± SEM (n = 3). **P < 0.01, Student's t‐test.
- Expression of the cyclin‐dependent kinase inhibitors p21 and p16 was analyzed in control or CD36 supernatant‐treated IMR90 cells as described in (A). Blots shown are representative of three independent experiments.
- Representative images (left) and quantification (right) of SA‐βGal staining for long‐term CD36‐expressing IMR90 cells treated with DMSO or a selective NF‐κB pharmacological inhibitor. Proliferating IMR90 cells were infected with Tet‐on CD36 or control viruses and treated with 1 μg/ml doxycycline. At day 7, cells were further treated with DMSO or 100 nM Bay 11‐7082. Samples were fixed on day 15 and subsequently stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM (n = 4). **P < 0.01, one‐way ANOVA.
- Proliferation analysis of long‐term CD36‐expressing IMR90 cells treated with DMSO or NF‐κB inhibitor. IMR90 cell cultures described in (D) were treated with EdU for 2 h and analyzed by flow cytometry. Data are reported as the mean ± SEM (n = 4). **P < 0.01, one‐way ANOVA.
- Cyclin‐dependent kinase expression analysis of long‐term CD36‐expressing IMR90 cells treated with DMSO or NF‐κB inhibitor. Lysates from samples described in (D) were collected and immunoblotted with the indicated antibodies. Blots shown are representative of three independent biological replicates.
Next, we explored the involvement of individual SASP components in CD36‐driven cell cycle arrest. Both paracrine signaling and autocrine signaling are known to contribute to the senescent process, and canonical SASP cytokines such as IL‐6 and IL‐8 have been shown to promote fibroblast proliferative arrest 21, 27, 28. IL‐6 and IL‐8 are among the secreted factors upregulated in HBE cells in response to ectopic CD36 expression (Fig 2F). To test whether these cytokines are capable of driving epithelial cell senescence, we treated HBE cells with recombinant IL‐6 or IL‐8 for 9 days, a procedure that resulted in increased SA‐βGal activity (Fig EV3A), reduced proliferative potential (Fig EV3B), and mild but consistent upregulation of p16 and p21 (Fig EV3C). Consistent with previous reports, IL‐6 administration produced a strong senescent phenotype in IMR90 fibroblasts (Fig EV3D), indicating cell type‐specific differences in the ability of individual SASP components to induce senescence. These results suggest that at early time points, CD36 functions to drive NF‐κB‐mediated secretion of canonical SASP components, which in turn act in a feed‐forward manner to promote stable cell cycle arrest and establish the senescent state.
Figure EV3. Long‐term exposure to CD36‐dependent SASP components accelerates HBE cellular senescence.
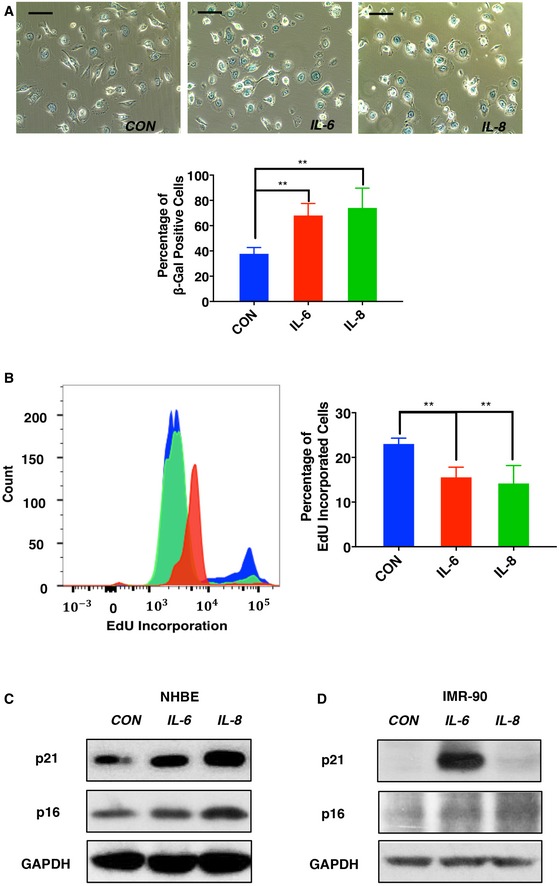
- Representative images (above) and quantification (below) of SA‐βGal staining of IL‐6‐ and IL‐8‐treated HBE cells. HBE cells were treated with CD36‐dependent cytokines IL‐6, IL‐8, or PBS at a concentration of 50 ng/ml for 9 days. Cells were then fixed and stained for SA‐βGal activity. Scale bars, 50 μm. Data are reported as the mean ± SEM (n = 3). **P < 0.01, one‐way ANOVA.
- Proliferation analysis of IL‐6‐, IL‐8‐, or PBS‐treated HBE cells. HBE cells were cultured with recombinant cytokines as described in (A). Then, cells were further treated with 10 μM EdU for 2 h prior to collection, staining, and analysis by flow cytometry. Data are presented as the mean ± SEM of three biological replicates. P‐values were calculated based on three independent experiments. **P < 0.01, one‐way ANOVA.
- Expression of cyclin‐dependent kinase inhibitors p21 and p16 was analyzed using the lysates from samples as described in (A). Blots shown are representative of three independent biological replicates.
- IMR90 fibroblasts were treated with 50 ng/ml IL‐6, IL‐8, or PBS for 9 days. Cell lysates were then immunoblotted using antibodies recognizing p16, p21, or GAPDH, as indicated.
Ligands are required for CD36‐dependent SASP production and senescence establishment
Like many other cell surface receptors, CD36's signaling activity requires an interaction with its cognate ligand. Extracellular oxidized low‐density lipoprotein (oxLDL) and amyloid‐beta 1–42 (Aβ) are known to bind CD36 to drive Src–p38‐dependent cytokine and chemokine expression in immune cell types 16, 18, 24. To identify ligands involved in stimulating CD36‐dependent NF‐κB signaling in HBE cells and fibroblasts, we ectopically expressed CD36 in HBE cells and supplemented them with purified oxLDL or Aβ for 24 h, using phospho‐p65 as a NF‐κB activation readout. As shown in Fig EV4A, the addition of oxLDL had no measurable effect on NF‐κB activation in either the presence or absence of CD36 overexpression, indicating that this ligand is unable to activate inflammatory signaling in this particular cell context, perhaps due to the absence of certain co‐receptors or intracellular adaptor proteins. In contrast, supplementation with recombinant Aβ produced strong NF‐κB activity when combined with CD36 expression. Importantly, Aβ addition alone stimulated lesser but detectable levels of p65 phosphorylation, likely due to low levels of CD36 present under basal conditions (Fig EV4A). Aβ supplementation was also sufficient to induce NF‐κB activation in CD36‐expressing fibroblasts (Fig EV4B), suggesting that Aβ is the major ligand driving the CD36–NF‐κB‐dependent SASP in cultured human cells.
Figure EV4. Aβ 1–42 is a potent CD36‐dependent SASP activator in both IMR90 and HBE cells.
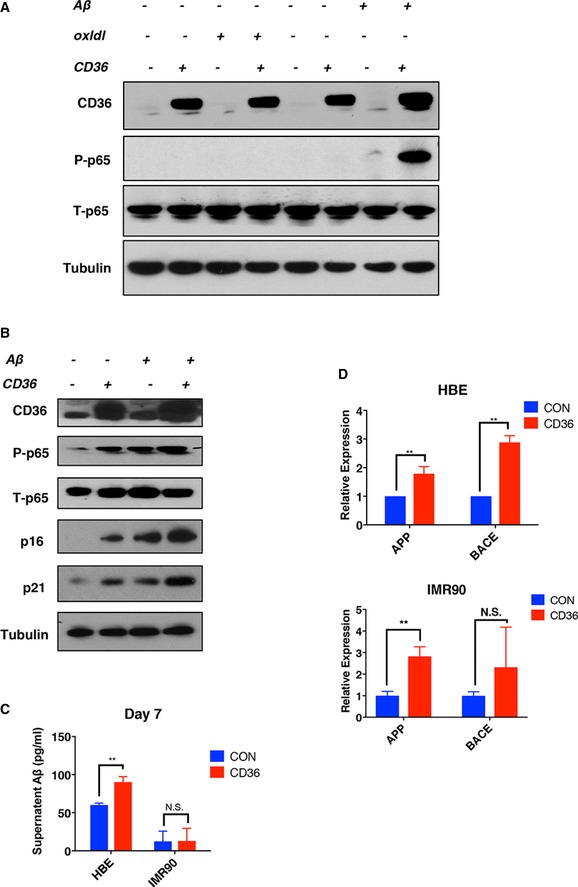
-
A, BEffects of CD36 expression and ligand administration on NF‐κB phosphorylation. HBE (A) or IMR90 (B) cells were infected with Tet‐on CD36 or a control plasmid. After selection, CD36 was induced with 1 μM doxycycline for 24 h in the presence of ligand, as indicated. Samples were then collected and blotted with the indicated antibodies.
-
CAβ 1–42 analysis of IMR90 and HBE cell supernatants by ELISA. IMR90 and HBE cells were plated at 40% confluency and cultured in standard medium for 2 days prior to supernatant collection. Samples were then subjected to ELISA to measure Aβ concentration (mean ± SEM, n = 4). **P < 0.01; N.S., not significant; Student's t‐test.
-
DAPP and BACE expression in HBE and IMR90 cells ectopically expressing CD36. HBE and IMR90 cells were infected with Tet‐on CD36 or a control plasmid. After selection, CD36 was induced by adding 1 μM doxycycline for 7 days. Cells were then collected and analyzed for APP and BACE mRNA expression by qPCR. Results are normalized to β‐actin. Data are reported as the mean ± SEM. P‐values were calculated based on at least three independent experiments. **P < 0.01; N.S., not significant; Student's t‐test.
If CD36 is involved in the generation of a SASP and Aβ is necessary for CD36‐dependent signaling, then this ligand should be detectable under standard cell culture conditions. To examine this, we measured Aβ levels in HBE cell culture medium by ELISA and found that Aβ is indeed present at a concentration of approximately 75 pg/ml (Fig EV4C). In parallel assays, IMR90 fibroblast cultures, which do not activate NF‐κB signaling in response to short‐term CD36 expression (Fig 4A), do not contain measurable levels of extracellular Aβ at the 7‐day time point (Fig EV4C). Aβ production is regulated at several levels including by transcription of amyloid precursor protein (APP) mRNA as well as post‐translational cleavage by multiple proteolytic secretase proteins including beta‐secretase 1 (BACE1) 30, 31. To investigate Aβ production, we directly measured the expression levels of APP and BACE1 in CD36‐expressing HBE cells and IMR90 cells. Interestingly, we noted an induction of APP and BACE1 at the 7‐day time point in both cell types, suggesting the presence of a positive feedback loop from CD36 signaling to generate its ligand Aβ (Fig EV4D). Based on these results, we conclude that ligand‐dependent receptor activation is necessary for CD36's SASP‐ and senescence‐promoting activity.
CD36‐dependent secretory molecule transcription and senescence establishment require ligand–receptor interactions
The results presented above indicate that expression of CD36 and its ligands is sufficient to trigger a NF‐κB‐mediated SASP, and prolonged signaling through this pathway can promote a comprehensive senescence phenotype. This prompted us to ask whether CD36 is required for SASP establishment and cell cycle arrest during conventional senescence reprogramming. To address this, we first blocked the induction of CD36 using small‐hairpin RNAs and then induced HBE cell senescence by administering the chemical inhibitor erlotinib (Fig 5A). Although CD36 knockdown did not rescue the cell cycle arrest or SA‐βGal activity phenotypes caused by EGFR inhibition, we observed a major impairment in Src–p38–NF‐κB activation after senescence induction when CD36 upregulation was suppressed (Fig 5B and C). Consistent with this, a large cohort of SASP components was not induced during senescence after CD36 knockdown (Fig 5D), and this subset largely coincides with the group of pro‐inflammatory molecules induced by ectopic CD36 expression (Fig 2F). To determine whether CD36 is also necessary for SASP generation in other senescent contexts, we performed similar experiments using fibroblasts triggered to senescence by HRAS activation and also observed decreased levels of active NF‐κB (Fig 5E) upon CD36 knockdown. These results demonstrate a functional requirement of CD36 for NF‐κB‐mediated SASP establishment in response to multiple senescent stimuli.
Figure 5. CD36 is required for NF‐κB activation and SASP establishment during erlotinib‐induced senescence programming.
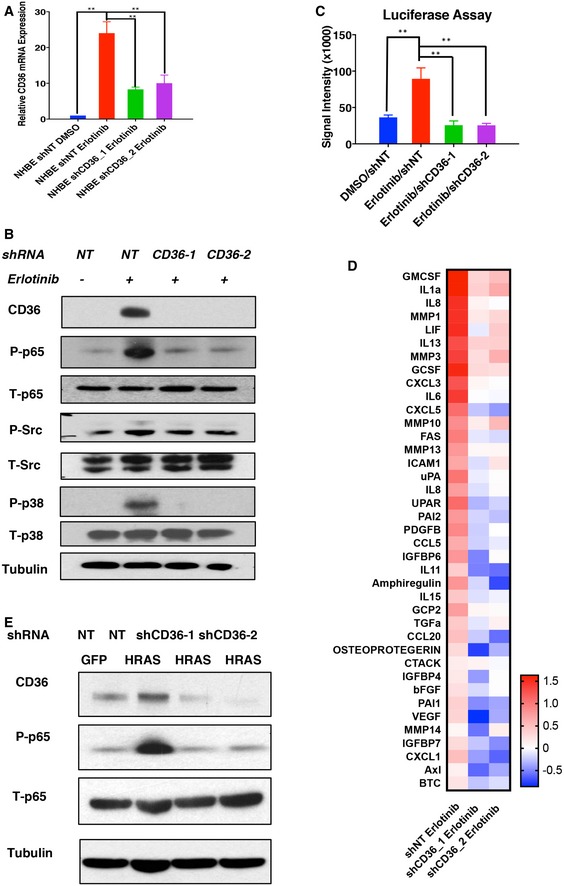
- Validation of CD36 shRNA knockdown efficiency during erlotinib‐induced senescence. Proliferating shNT and shCD36 (#1 and #2) HBE cells were treated with DMSO or 1 μM erlotinib for 48 h. Cell lysates were then collected, and CD36 expression was measured by qPCR. CD36 expression was normalized to β‐actin (mean ± SEM; n = 3). **P < 0.01, one‐way ANOVA.
- Signal transduction analysis after CD36 knockdown in erlotinib‐treated HBE cells. Proliferating shNT and shCD36 (#1 and #2) HBE cells were treated with DMSO or 1 μM erlotinib for 48 h. Cell lysates were then collected and blotted using the indicated antibodies. Blots are representative of three independent biological replicates.
- NF‐κB luciferase reporter assay of erlotinib‐induced senescent HBE cells after CD36 knockdown. Luciferase reporters were transfected into proliferating shNT and shCD36 (#1 and #2) HBE cells (day 0). On day 1, cells were treated with DMSO or 1 μM erlotinib, and luciferase reporter assays were performed on day 3. Signal intensities are reported as means ± SEM (n = 3). **P < 0.01, one‐way ANOVA.
- SASP transcriptional analysis upon CD36 knockdown in erlotinib‐treated HBE cells. Proliferating shNT and shCD36 (#1 and #2) HBE cells were treated with DMSO or 1 μM erlotinib for 48 h. Cell lysates were then collected and subjected to qPCR‐based SASP profiling. qPCR results are normalized to β‐actin (n = 3). Red and blue colors represent up‐ and downregulated transcripts, respectively.
- NF‐κB phosphorylation analysis after CD36 knockdown in oncogene‐induced senescence. HRAS‐induced senescent IMR90 cells (4 days) were transfected with shNT or shCD36 (#1 and #2). Protein lysates were collected on day 9 and subsequently immunoblotted with the indicated antibodies. Blots shown are representative of three independent biological replicates.
In this study, we discovered the CD36 receptor and its ligand Aβ as novel regulators of cellular senescence via their cooperative ability to initiate a SASP. Although the NF‐κB and CEBPβ transcription factors are previously known to be major mediators of the SASP and their immediate upstream signaling cascades are well characterized, the identity of the specific molecular trigger responsible for activating these pathways during the onset of senescence has remained elusive 26. It is important to note that CD36 is previously known for its ability to stimulate cytokine and chemokine production in immune cell types such as monocytes 16, 18. Here, we propose that the ability of senescent cells to adopt an immune‐like secretory phenotype largely stems from their capacity to upregulate the expression of CD36 in response to various senescent stimuli. Further research will be required to determine the precise molecular mechanisms responsible for CD36 transcriptional upregulation during senescence induction.
Based on results obtained using epithelial and fibroblast model systems, we conclude that ligand‐mediated CD36 activation is necessary for expression of the full‐senescence secretome. In particular, interaction with Aβ appears to be a critical event in this process. In both HBE and IMR90 cells, APP and BACE are upregulated during senescence resulting in sufficient levels of Aβ to activate CD36 and generate a SASP (Fig EV4C). Recent in vivo analysis has identified Aβ as mediating phenotypic changes in intestinal epithelial cells, and it will be interesting to determine whether a similar relationship holds for fibroblast cells and the molecular mechanisms responsible for such lineage‐specific distinctions 32. Because Aβ is most well characterized as the primary component of the amyloid plagues found in the brains of Alzheimer's patients, an intriguing area for future investigation concerns the possible relationship between Aβ accumulation in neurodegenerative diseases and processes related to cellular senescence and inflammation 33, 34.
Several recent reports have implicated the cyclic GMP‐AMP synthase (cGAS)–STING cytosolic DNA sensing pathway in SASP induction 35, 36, 37. In brief, these studies show that multiple types of senescent cells engage the cGAS–STING pathway through recognizing cytosolic DNA fragments, resulting in IRF3‐mediated IFN‐β upregulation and the subsequent production of many known SASP components. Like the CD36 pathway reported here, cytosolic DNA sensing allows senescent cells to co‐opt signaling cascades normally utilized by the immune system in order to generate an inflammatory phenotype. However, two major differences between these distinct SASP regulatory pathways should be noted. First, CD36‐mediated cytokine and chemokine production proceeds directly through NF‐κB and does not involve an IFN‐β intermediate. Thus, CD36 is likely to be a more direct and rapid pathway for SASP activation in response to senescent stimuli. Indeed, nearly ten‐fold CD36 upregulation was observed here only 6 h after senescence induction (Fig 1B). Second, the CD36‐mediated secretome does not require the presence of cytosolic DNA but instead depends on an extracellular CD36‐activating ligand. Therefore, the CD36–NF‐κB–SASP pathway might be predicted to be utilized by a broader range of senescent cell types, perhaps including those known to be involved in embryonic development 4, 37, 38.
In summary, here we report a critical role for the scavenger receptor CD36 in the early initiation of the SASP in response to multiple senescent stimuli. Through this study, we have also demonstrated the dynamic nature of the senescent program, from the initiation of the process to the establishment of the senescent state dependent on its enforcement by multiple secreted components. Although the two distinct features of cellular senescence, cell cycle exit and the SASP, are mediated by different signaling mechanisms, they are closely linked by the pathway identified in our study. Given the expanding appreciation of the relationship between cellular senescence and the aging process, future studies should aim to address the role of ligand‐dependent CD36 activation as it relates to the SASP in various age‐related diseases including neurodegeneration and cancer.
Materials and Methods
Cell culture
Primary human bronchial epithelial (HBE) cells were obtained from deceased donors under an approved protocol of the University of North Carolina Biomedical Institutional Review Board (03‐1396). Primary cells were cultured on plastic dishes coated with bovine collagen in basal epithelial growth medium (Lonza). Primary human diploid fibroblasts (IMR90) were cultured in Eagle's minimum essential medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were cultured from Passage 0 (HBE) or Passage 32 (IMR90) and expanded through subsequent splitting and freezing. 293T cells were maintained in Dulbecco's modified Eagle's medium with 10% FBS. To chemically induce senescence, HBE cells were treated with erlotinib as we previously reported 9.
Plasmids and lentiviral production
CD36 and HRAS G12V sequences were cloned into a Tet‐on inducible PCDH_Teton_one plasmid. The CD36 small‐hairpin RNA was constructed in PLKO.1‐puro lentiviral vector (Sigma‐Aldrich). The small‐hairpin RNA sequences were as follows:
shCD36_1: CCGGCCGACGTTAATCTGAAAGGAACTCGAGTTCCTTTCAGATTAACGTCGGTTTTTG;
shCD36_2: CCGGCCTGCTTATCCAGAAGACAATCTCGAGATTGTCTTCTGGATAAGCAGGTTTTTG.
For each transfection assay, 12 μg of each HRAS, CD36 overexpression, or shRNA plasmid together with 9 μg of ps‐PAX2 and 3 μg of vesicular stomatitis virus G glycoprotein (VSV‐G) expressing plasmids was co‐transfected into 293T cells using Lipofectamine 2000 (Thermo Fisher Scientific) according the manufacturer's instructions. Viruses were collected 48 h post‐transfection. For both HBE and IMR90 cell infections, once the cells reached 40% confluence, virus particles combined with 4 μg/ml polybrene were introduced into the medium for 18 h. Twenty‐four hours later, 1 μg/ml of puromycin was applied for selection. After 3 days of puromycin selection, cell lines were considered to be established.
EdU staining
Once cells reached 50% confluence, they were first treated with 10 μM EdU for 2 h at 37°C. Subsequent fixation and staining were performed using the Click‐iT EdU flow cytometry assay kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
Quantitative PCR
Total mRNA was extracted using RNeasy Mini Kit (QIAGEN) and reverse‐transcribed into cDNA with iScript™ Reverse Transcription Supermix (BIO‐RAD). Quantitative PCR (qPCR) was then performed using SYBR Green Real‐Time PCR Master Mixes (Thermo Fisher Scientific) based on the manufacturer's protocol.
Western blotting
Cells were trypsinized, washed, centrifuged, and collected as a cell pellet. Total protein was then extracted using RIPA Lysis and Extraction Buffer supplemented with protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific). Protein lysates were incubated on ice for 30 min and then centrifuged at 270 g for 10 min. Supernatants were then collected and boiled in sodium dodecyl sulfate (SDS) loading buffer for 10 min. Thirty micrograms of each protein sample was separated on a denaturing SDS gel, and proteins were subsequently transferred to a nitrocellulose membrane (Millipore). Membranes were blocked in 5% bovine serum albumin in Tris‐buffered saline with 0.1% Tween‐20 (TBST) and probed with the following antibodies: primary antibodies anti‐CD36 (H300; Santa Cruz, RRID: AB_2072518), p21 (12D1; Cell Signaling Technology, RRID: AB_11217627), NF‐κB p65 (D14E12; Cell Signaling Technology, RRID: AB_10859369), Phosphorylated‐NF‐κB p65 (93H1; Cell Signaling Technology, RRID: AB_10827881), Phosphorylated‐p38 (D3F9; Cell Signaling Technology, RRID: AB_2139685), Phosphorylated‐Src (Tyr416; Cell Signaling Technology, RRID: AB_10860245), and β‐Tubulin (Cell Signaling Technology, RRID: AB_2210545), and corresponding horseradish peroxidase‐labeled mouse and rabbit secondary antibodies (Invitrogen). Secondary antibodies were visualized using a Pierce enhanced chemiluminescent Western blotting substrate kit (Thermo Fisher Scientific).
SA‐βGal assays
SA‐βGal assays were performed as previously described 39. In brief, control or experimental HBE cells or IMR90 cells were washed twice in PBS and then fixed using 4% formaldehyde and 0.2% glutaraldehyde in PBS for 5 min at room temperature in the dark. Cells were subsequently stained with 40 mM citric acid/sodium phosphate buffer (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, 2 mM MgCl2, and 1 mg/ml X‐Gal, or with senescence beta‐galactosidase staining kit (Cell Signaling Technology), typically for 4–12 h at 37°C. Microscopic analyses were performed using an Olympus CK40 microscope (Center Valley) with a DP20 camera. For each experimental and control group, five views were randomly selected and captured.
Luciferase reporter assays
HBE cells were transfected with 12 μg of a NF‐κB‐driving luciferase plasmid [40] at day 0. Cells were lysed 72 h post‐transfection and analyzed for firefly luciferase activity using the Dual Luciferase Assay system (Promega) on a VICTOR3 multilabel plate reader (PerkinElmer).
Statistical analysis
Unless otherwise indicated, Student's t‐tests were used for all statistical analyses. Data are reported as mean ± standard error (SEM).
Author contributions
MC, TY, RC, HX, LY, YD, CCP, and ZT performed experiments. MC, TY, RC, LY, PBA, Q‐JL, and X‐FW analyzed the data. PBA, Q‐JL, and X‐FW provided guidance and/or senior supervision. MC, PBA, and X‐FW wrote the manuscript. MC and TY prepared the figures under supervision from PBA and X‐FW. All authors provided input and corrections to the preparation of the manuscript and figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Drs. Pengyuan Yang, Yun Zhang, Jing Hu, and Geoff Markowitz for valuable discussions and suggestions. We thank Dr. Siqi Liu for SASP profiling data analysis. This work was supported by CA154586, CA164791 to XFW and CA190991 to QJL from the NIH.
EMBO Reports (2018) 19: e45274
Contributor Information
Qi‐Jing Li, Email: qi-jing.li@duke.edu.
Xiao‐Fan Wang, Email: xiao.fan.wang@duke.edu.
References
- 1. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24: 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25: 36 [DOI] [PubMed] [Google Scholar]
- 3. Munoz‐Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496 [DOI] [PubMed] [Google Scholar]
- 4. Munoz‐Espin D, Canamero M, Maraver A, Gomez‐Lopez G, Contreras J, Murillo‐Cuesta S, Rodriguez‐Baeza A, Varela‐Nieto I, Ruberte J, Collado M et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118 [DOI] [PubMed] [Google Scholar]
- 5. Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, Vainchenker W, Constantinescu SN, Giraudier S (2010) A senescence‐like cell‐cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol 8: e1000476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon‐Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco MA, Manns MP et al (2017) Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 16: 7 [DOI] [PubMed] [Google Scholar]
- 8. Campisi J, d'Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740 [DOI] [PubMed] [Google Scholar]
- 9. Alexander PB, Yuan L, Yang P, Sun T, Chen R, Xiang H, Chen J, Wu H, Radiloff DR, Wang XF (2015) EGF promotes mammalian cell growth by suppressing cellular senescence. Cell Res 25: 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang MY, Lin PM, Liu YC, Hsiao HH, Yang WC, Hsu JF, Hsu CM, Lin SF (2012) Induction of cellular senescence by doxorubicin is associated with upregulated miR‐375 and induction of autophagy in K562 cells. PLoS One 7: e37205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A et al (2015) mTOR regulates MAPKAPK2 translation to control the senescence‐associated secretory phenotype. Nat Cell Biol 17: 1205–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J 30: 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pepino MY, Kuda O, Samovski D, Abumrad NA (2014) Structure‐function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr 34: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Canton J, Neculai D, Grinstein S (2013) Scavenger receptors in homeostasis and immunity. Nat Rev Immunol 13: 621–634 [DOI] [PubMed] [Google Scholar]
- 16. Silverstein RL, Febbraio M (2009) CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2: re3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ (2005) Response to Staphylococcus aureus requires CD36‐mediated phagocytosis triggered by the COOH‐terminal cytoplasmic domain. J Cell Biol 170: 477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart CR, Stuart LM, Wilkinson K, Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA et al (2010) CD36 ligands promote sterile inflammation through assembly of a Toll‐like receptor 4 and 6 heterodimer. Nat Immunol 11: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Debacq‐Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O (2009) Protocols to detect senescence‐associated beta‐galactosidase (SA‐betagal) activity, a biomarker of senescent cells in culture and in vivo . Nat Protoc 4: 1798–1806 [DOI] [PubMed] [Google Scholar]
- 20. Cui H, Kong Y, Xu M, Zhang H (2013) Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res 73: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orjaloa AV, Bhaumika D, Genglera BK, Scotta GK, Campisi J (2009) Cell surface‐bound IL‐1α is an upstream regulator of the senescence‐associated IL‐6:IL‐8 cytokine network. Proc Natl Acad Sci 106: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006) Cellular senescence in aging primates. Science 311: 1257 [DOI] [PubMed] [Google Scholar]
- 23. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Febbraio M, Hajjar DP, Silverstein RL (2001) CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 108: 785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoesel B, Schmid JA (2013) The complexity of NF‐kappaB signaling in inflammation and cancer. Mol Cancer 12: 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N et al (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018 [DOI] [PubMed] [Google Scholar]
- 28. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 133: 1019–1031 [DOI] [PubMed] [Google Scholar]
- 29. Campisi J (1997) The biology of replicative senescence. Eur J Cancer 33: 6 [DOI] [PubMed] [Google Scholar]
- 30. Page RM, Baumann K, Tomioka M, Perez‐Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H et al (2008) Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease‐associated presenilin mutations and gamma‐secretase modulation. J Biol Chem 283: 677–683 [DOI] [PubMed] [Google Scholar]
- 31. Chow VW, Mattson MP, Wong PC, Gleichmann M (2010) An overview of APP processing enzymes and products. Neuromolecular Med 12: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puig KL, Manocha GD, Combs CK (2015) Amyloid precursor protein mediated changes in intestinal epithelial phenotype in vitro . PLoS One 10: e0119534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murphy MP, LeVine H (2010) Alzheimer's disease and the Amyloid‐β peptide. J Alzheimers Dis 19: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diaz‐Moreno M, Hortiguela R, Goncalves A, Garcia‐Carpio I, Manich G, Garcia‐Bermudez E, Moreno‐Estelles M, Eguiluz C, Vilaplana J, Pelegri C et al (2013) Abeta increases neural stem cell activity in senescence‐accelerated SAMP8 mice. Neurobiol Aging 34: 2623–2638 [DOI] [PubMed] [Google Scholar]
- 35. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang H, Wang H, Ren J, Chen Q, Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 114: E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun L, Wu J, Du F, Chen X, Chen JZ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130 [DOI] [PubMed] [Google Scholar]
- 39. Xiang H, Yuan L, Gao X, Alexander PB, Lopez O, Lau C, Ding Y, Chong M, Sun T, Chen R et al (2017) UHRF1 is required for basal stem cell proliferation in response to airway injury. Cell Discov 3: 17019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File