

Abstract
Background
A global consensus on the loss of skeletal muscle mass and function in humans refers as sarcopenia and cachexia including diabetes, obesity, renal failure, and osteoporosis. Despite a current improvement of sarcopenia or cachexia with exercise training and supportive therapies, alternative and specific managements are needed to discover for whom are unable or unwilling to embark on these treatments. Alendronate is a widely used drug for osteoporosis in the elderly and postmenopausal women. Osteopenic menopausal women with 6 months of alendronate therapy have been observed to improve not only lumbar bone mineral density but also handgrip strength. However, the effect and mechanism of alendronate on muscle strength still remain unclear. Here, we investigated the therapeutic potential and the molecular mechanism of alendronate on the loss of muscle mass and strength in vitro and in vivo.
Methods
Mouse myoblasts and primary human skeletal muscle‐derived progenitor cells were used to assess the effects of low‐dose alendronate (0.1–1 μM) combined with or without dexamethasone on myotube hypertrophy and myogenic differentiation. Moreover, we also evaluated the effects of low‐dose alendronate (0.5 and 1 mg/kg) by oral administration on the limb muscle function and morphology of mice with denervation‐induced muscle atrophy and glycerol‐induced muscle injury.
Results
Alendronate inhibited dexamethasone‐induced myotube atrophy and myogenic differentiation inhibition in mouse myoblasts and primary human skeletal muscle‐derived progenitor cells. Alendronate significantly abrogated dexamethasone‐up‐regulated sirtuin‐3 (SIRT3), but not SIRT1, protein expression in myotubes. Both SIRT3 inhibitor AKG7 and SIRT3‐siRNA transfection could also reverse dexamethasone‐up‐regulated atrogin‐1 and SIRT3 protein expressions. Animal studies showed that low‐dose alendronate by oral administration ameliorated the muscular malfunction in mouse models of denervation‐induced muscle atrophy and glycerol‐induced muscle injury with a negative regulation of SIRT3 expression.
Conclusions
The putative mechanism by which muscle atrophy was improved with alendronate might be through the SIRT3 down‐regulation. These findings suggest that alendronate can be a promising therapeutic strategy for management of muscle wasting‐related diseases and sarcopenia.
Keywords: Alendronate, Skeletal muscle, Atrophy, Dysfunction, Sirtuin3
Introduction
There is a growing global health awareness of the risk factors of skeletal muscle in many physiological and disease processes, including skeletal muscle mass declines with ageing (sarcopenia), obesity, other chronic diseases,1, 2 and muscle growth retardation.3 Loss of skeletal muscle is related to the frailty of skeletal muscle function by weakness, fatigue, disability, and glucose intolerance. Moreover, muscle fibre atrophy has been reported to be an obvious histological phenomenon of people with sarcopenia and muscle wasting diseases.4 Muscle growth retardation related to low birth weight of foetus has also been shown to be associated with myogenic inhibition by several risk factors, including polycyclic aromatic hydrocarbons and arsenic.5, 6 An appropriate remedy for skeletal muscle atrophy and dysfunction needs to be discovered. To date, pharmacological treatments of sarcopenia and disease‐related muscle atrophy and dysfunction are focused on elevation of the exercise and physical activity, nutritional supplement, hormone therapy, and vitamin D.7, 8, 9, 10 However, some people with sarcopenia or disease‐induced muscle atrophy and dysfunction are unable or unwilling to embark on exercise training or other supportive therapies after diagnosis. Therefore, alternative managements are needed to find out for the clinical efficacy of sarcopenia and disease‐related muscle atrophy and dysfunction.
Alendronate (ALN), the most popular first‐line bisphosphonate drug, has been widely used for the treatment of osteoporosis by increasing bone density and micro‐architecture and prohibiting bone resorption in the elderly and postmenopausal women.11, 12 Due to the hazardous duet of the decline in bone mineral density (osteoporosis) and muscle mass (sarcopenia) leading to frailty, recent reports on the treatment of the osteoporosis with ALN or other bisphosphonates have also indicated that these anti‐osteoporosis drugs provided a positive effect on muscle mass and strength in osteoporotic patients.13, 14, 15 However, Widrick et al. have found that ALN can improve bone loss but has no independent effect on muscle function in ovariectomized rats, an animal model of post‐menopausal osteoporosis.16 Therefore, the effect and mechanism of ALN on skeletal muscle function still remain to be clarified and in need of further investigation. In addition, potential therapeutic interventions for cachexia, ageing or other muscle wasting‐related diseases have been extensively discovered. Trimetazidine, a metabolic modulator, elicited a significant increase in muscle strength in sarcopenic mice.17 A proteasome inhibitor, bortezomib, has been suggested to prevent skeletal muscle from wasting in cancer cachexia‐induced muscle loss rats.18 Enoki et al. have suggested that indoxyl sulfate is an aetiology of chronic kidney disease‐associated sarcopenia and indoxyl sulfate‐targeted intervention, AST‐120, could prevent from chronic kidney disease‐induced muscle atrophy and lower exercise endurance.19 Wendowski et al. have also indicated that dihydrotestosterone could rescue an age‐dependent reduction in protein synthesis of skeletal muscle fibres in sarcopenic mice.20 A natural product, salidroside, sourced from Rhodiola rosea L., has been found to exhibit potential therapy against cancer‐associated cachexia‐induced limb muscle loss in tumour‐borne BALB/c mice, including gastrocnemius (Gascn) muscles.21 Moreover, anamorelin, a ghrelin‐analog, could show an increase in muscle mass in cancer anorexia‐cachexia syndrome.22 It is critical to discover the potential interventions against muscle wasting from sarcopenia, cachexia, and other diseases.
In the present study, the aim was to characterize the action and molecular mechanism of ALN‐inhibited skeletal muscle atrophy and regeneration impairment in cultured cell models [mouse myoblasts and human skeletal muscle‐derived progenitor cells (HSMPCs)] and mouse models of denervation‐induced skeletal muscle atrophy and glycerol‐induced muscle injury (a model for muscle regeneration in response to injury).
Materials and methods
Animals
Five to 6‐week‐old male ICR mice were purchased from the animal centre of the College of Medicine, National Taiwan University, Taipei, Taiwan, and housed in the controlled vivarium (22 ± 2°C and 40–60% relative humidity with a cycle of 12 h light and 12 h dark) with free access to food and water. All animal protocols of this study were conformed to the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources)23 and approved by the Institution Animal Care and Use Committee of College of Medicine, National Taiwan University, Taipei, Taiwan. All animals were handled with humane treatment and alleviated the suffering while experimental challenges. The 48 mice were divided into eight groups (n = 6/group) including (a) sham control mice for both denervation and glycerol injection, (b) mice with or without ALN (0.5 and 1 mg/kg) (Merck Millipore, Temecula, CA, USA) administration for 1 month until 7 days after denervation prior to sacrifice, and (c) mice with or without ALN (0.5 and 1 mg/kg) administration for 1 month until 5 days after glycerol‐induced muscle injury prior to sacrifice.
Denervation model
The mice were anaesthetized with inhalational application of a mixture gas of isoflurane (3%) (Baxter Healthcare of Puerto Rico, Guayama, PR, USA) and oxygen (97%), and denervation was achieved by surgical transection of the sciatic nerve from right hind limb as previously described by MacDonald et al.24 Control mice were sham operated without removal of the sciatic nerve.
Induction of muscle regeneration
The mice were intramuscularly injected 100 μL glycerol (50% vol/vol) (Avantor Performance Materials, Center Valley, PA, USA) into monolateral soleus muscles under inhalational anaesthesia with a mixture gas of isoflurane (3%) (Baxter Healthcare of Puerto Rico, Guayama, PR, USA) and oxygen (97%) as described by Chiu et al.25
Muscle fatigue task
Muscular endurance was measured by the muscle fatigue task as previously described by Chiu et al.25 Briefly, the mice were accommodated to training prior to the fatigue task by using an accelerating rota‐rod (Ugo Basile, Varese, Italy). The mice were given the training trial with the rota‐rod at a constant speed of 13 r.p.m. for 15 min. After 15 min of rest, the mice were replaced on the rota‐rod adjusted to accelerate from 13 to 25 r.p.m. in 180 s for 15 min. The next day, each mice received the muscle fatigue task on the rota‐rod operated at speeds ramping from 13 to 25 r.p.m. in 180 s and maintained at 25 r.p.m. for 30 min intertrial interval. Latency to fall off the rota‐rod of each individual mouse was measured. The fatigued mouse was identified while the mouse fell off 4 times within 1 min, and the task terminated.
Sampling
The mice were anaesthetised and euthanized after the fatigue task. Soleus and Gascn muscles were dissected, harvested, weighed, and fixed in 4% paraformaldehyde (Sigma, St. Louis, MO, USA) at room temperature overnight for following histological analysis.
Assessment of muscle tension
Isometric contraction of isolated soleus muscles was assessed as described by Liu et al.26 with modification. Briefly, harvested soleus muscles were suspended in the modified Krebs solution (130.6 mM NaCl, 4.8 mM potassium chloride (KCl), 2.5 mM CaCl2, 1.2 mM MgSO4, 12.5 mM NaHCO3, and 11.1 mM glucose, pH = 7.4) constantly gassed with 5% CO2 and 95% O2 at 37 ± 0.5°C in a 10 mL organ bath. The soleus muscle was stimulated with a Grass S88 (Astro‐Med Inc., West Warwick, RI, USA) constant voltage stimulator (rectangular pulses of 0.5 ms width at 0.2 Hz) or 30 mM KCl to evoke single twitch responses and the contracture. The muscle was loaded with a resting tension of 1 g, and the changes of tension were recorded with an isometric transducer (Mantracourt Electronics Limited, Devon, UK) and a high‐resolution laboratory data recorder (e‐corder) (eDAQ Pty Ltd, Denistone East, NSW, Australia). In some experiments, the KCl‐induced tonic contracture was detected in isolated soleus muscles. The entry of external calcium has been suggested to play an essential role in the potassium‐induced tonic contracture.27
Histological assessment
Fixed skeletal muscles imbedded in paraffin were sectioned into 4 μm sections. Histological examination was assessed by using haematoxylin and eosin staining. For assessment of the collagen deposition during muscle regeneration, the paraffin‐embedded soleus muscle sections were stained with Masson's trichrome as described previously.25 Immunohistochemistry was performed on fixed soleus muscle sections for Atrogin‐1 and SIRT‐3 staining with anti‐mouse Atrogin‐1 rabbit polyclonal antibody and anti‐mouse SIRT‐3 rabbit polyclonal antibody (Abcam, Cambridge, UK). A SuperPicture horseradish peroxidase polymer conjugate kit (Invitrogen, Carlsbad, CA, USA) was used to signal visualization according to the manufacturer's instructions, and sections were counterstained with haematoxylin. Histology images were collected with an optical Nikon Eclipse TS100 microscope equipped with a Nikon D5100 digital camera. Cross‐sectional area (CSA) and myofibres with central nuclei of individual myofibres were counted and calculated by using the ImageJ 1.48 software (National Institutes of Health, Bethesda, MD, USA) in five random fields of each section. The regenerative capacity of injured skeletal muscles was determined by the ratio of the numbers of myofibres with a centrally located nucleus over the total numbers of myofibres.
C2C12 mouse myoblasts
C2C12 mouse myoblasts were acquired from American Type Culture Collection (Manassas, VA, USA) and grown in proliferation medium composed of Dulbecco's modified Eagle's medium (Gibco, Bethesda, MD, USA) supplemented with 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin/amphotericin B (Biological Industries, Cromwell, CT, USA) in 5% CO2 at 37°C.
Isolation and culture of primary human skeletal muscle‐derived progenitor cells
Human tissue sampling of the present study was reviewed and approved by the National Taiwan University Hospital Institutional Review Board for clinical investigation and collected after written informed consent from all participating subjects. The process of isolation and culture of primary HSMPCs was described previously.25 Briefly, human skeletal muscle biopsies (~0.2 g) were collected from 10 rectus muscles of patients under orthopaedic surgery (mean age, 64 years; range, 34 to 81 years, both male and female). The minced segregate muscle was processed for HSMPC isolation within a few hours by serial enzymatic dissociation (0.5% type XI collagenase for 1 h, Sigma; 2.4 IU/mL dispase for 45 min, Invitrogen; 0.2% trypsin–EDTA for 15 min, Biological Industries). The cell suspension was passed through a 70 μm mesh filter and seeded in a collagen‐coated dish in proliferative growth medium [20% foetal bovine serum (Gibco), 1% PSA (Biological Industrial), and 5 ng/mL basic fibroblast growth factor (ProSpec, East Brunswick, NJ, USA) in Ham's F‐10 (Gibco)]. After serial transfers and pre‐plates based on different adhesion abilities between fibroblasts and muscle‐derived progenitor cells as described previously,25 the isolated HSMPCs with desmin‐positive (as a myogenic progenitor marker)28 staining were used for myogenic differentiation.
Myogenic differentiation and differentiated myotube treatment
C2C12 myoblasts and HSMPCs were seeded on six‐well plates in growth media and induced to differentiate by replacement of nutrient mixture F‐12K Ham solution (Gibco)/MCDB201 (Sigma) (1:1) with 2% horse serum (Gibco). The differentiated medium was freshly exchanged every 24 h with or without dexamethasone (Dexa) (Sigma) (1 μM) or ALN (0.1–1 μM) treatment during the 4 days of differentiation. To investigate muscle anti‐atrophic effects of ALN in vitro, C2C12 and HSMPC differentiated myotubes on the fourth day of differentiation were treated with Dexa (1 μM) or ALN (0.1–1 μM) for 2 days. Effects of ALN on the cellular signalling pathway of Dexa‐induced myogenic inhibition and atrophic myotube formation were determined by a pharmacological inhibitor of SIRT3 (AGK7) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and small interfering RNA (siRNA) of SIRT3 (Invitrogen) transfection. AKG7 was added for 1 h followed by treatment with Dexa and ALN in C2C12 and HSMPC myogenic differentiation and myotubes. Transfection was performed a day before treatment with Dexa and ALN in C2C12 and HSMPC myogenic differentiation and myotubes by using the lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions. A non‐targeting siRNA was used as a negative control (Invitrogen).
Morphological myotube analysis
To analyse myotube formation and atrophic myotubes, five visual fields of haematoxylin and eosin staining were chosen for calculation of multinucleated myotube formation and diameters under an optical Nikon Eclipse TS100 microscope equipped with a Nikon D5100 digital camera. The percentage of myotubes that contained the indicated number of nuclei and myotube diameters were calculated by using the ImageJ 1.48 software (National Institutes of Health, Bethesda, MD, USA).
Immunoblotting
Immunoblotting was performed as described previously.25 Briefly, cell protein concentration was quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Following electrophoresis, proteins were transferred on to polyvinylidene difluoride membranes blocked with 5% non‐fat powdered milk and subsequently incubated overnight at 4°C with anti‐Atrogin‐1 (Abcam), anti‐SIRT1, anti‐SIRT3 (Cell Signalling Technology, Danvers, MA, USA), anti‐myosin heavy chain (MHC), and anti‐β actin (Santa Cruz Biotechnology). The signals were detected by using horseradish peroxidase‐conjugated secondary antibodies (Cell Signalling Technology) and revealed with an enhanced chemiluminescence reagent (BioRad Laboratories, Redmond, WA, USA). The gel band quantitative densitometric analysis was quantified by the ImageJ 1.48 software (National Institutes of Health, Bethesda, MD, USA).
Statistics
All data are presented as mean ± standard errors of the means of at least three independent experiments. The statistical significance of differences in the means of experimental groups was analysed by one‐way analysis of variance and unpaired two‐tailed Student's t‐test with a significance threshold of 0.05 via GraphPad Prism software.
Results
Alendronate promotes hypertrophy and ameliorate dexamethasone dexa‐induced atrophy in C2C12 and human skeletal muscle‐derived progenitor cell‐derived myotubes
As a generally applied model of skeletal muscle atrophy in vitro, the C2C12 murine myoblasts and primary HSMPCs were induced to differentiate into myotubes. The myotube atrophy and protein degradation were induced by Dexa.29, 30, 31 As shown in Figure 1A and B, ALN (0.1–1 μM) dose‐dependently promoted the enlargement in diameters of myotubes, which were over 20 and 40 μm of myotubes differentiated from C2C12 myoblast and HSMPCs, respectively. Moreover, treatment with 1 μM Dexa for 24 h caused morphological alteration and reduced diameter in myotubes differentiated from both C2C12 myoblasts (Figure 2A) and HSMPCs (Figure 2B), which could be significantly inhibited by ALN (1 μM) treatment. The total numbers of myotubes were not changed by Dexa or ALN treatment (total number of myotubes, control: 56.0 ± 2.2; Dexa: 51.4 ± 1.8; Dexa + ALN: 59.3 ± 2.2, n = 4, P > 0.05).
Figure 1.

Alendronate (ALN) promoted myotube hypertrophy in C2C12 myoblasts and primary human skeletal muscle‐derived progenitor cells (HSMPCs). C2C12 myoblasts (A) and primary HSMPCs (B) were differentiated for 4 days under differentiation medium, followed by an additional 24 h with ALN (0.1–1 μM). The representative haematoxylin and eosin stained myotubes and the frequency distribution of myotube diameter were shown. All data are presented as means ± standard errors of the means for at least three independent experiments. The solid arrows indicated the multi‐nuclei myotubes. Scale bar = 100 μm. *P < 0.05 as compared with control group; #P < 0.05 as compared with dexamethasone group.
Figure 2.

Alendronate (ALN) protected myotube from atrophy induced by dexamethasone in C2C12 myoblasts and primary human skeletal muscle‐derived progenitor cells (HSMPCs). C2C12 myoblasts (A) and primary HSMPCs (B) were differentiated for 4 days under differentiation medium, followed by an additional 24 h with ALN (1 μM) in the presence or absence of dexamethasone (1 μM). The representative haematoxylin and eosin stained myotubes and the frequency distribution of myotube diameter were shown. All data are presented as means ± standard errors of the means for at least three independent experiments. The solid arrows indicated the multi‐nuclei myotubes. Scale bar = 100 μm. *P < 0.05 as compared with control group; #P < 0.05 as compared with dexamethasone group.
Alendronate prevents Dexa‐induced atrophy in C2C12‐derived and human skeletal muscle‐derived progenitor cell‐derived myotubes via SIRT3 mediation
Muscle atrophy in vitro is ordinarily determined by both the reduction of myotube diameters and the induction of ubiquitin ligase muscle atrophy F‐box or Atrogin‐1 expressions, which drive skeletal muscle specific protein degradation.32 As shown in Figure 3A, myotubes were treated with 1 μM Dexa in the presence or absence of 0.1–1 μM ALN for 24 h. Dexamethasone significantly elevated the Atrogin‐1 protein expressions in both C2C12‐derived [Figure 3A(a)] and HSMPC‐derived [Figure 3A(b)] myotubes, which could be significantly reversed by the treatment of ALN in a dose‐dependent manner.
Figure 3.

Alendronate (ALN) inhibited dexamethasone (Dexa)‐induced Atrogin‐1 and SIRT3 protein expressions in myotubes. C2C12 myoblasts and primary human skeletal muscle‐derived progenitor cells (HSMPCs) were differentiated for 4 days under differentiation medium, followed by an additional 24 h with ALN (0.1–1 μM) in the presence or absence of Dexa (1 μM). Western blot analysis of protein expressions of Atrogin‐1 (A), SIRT3 (B), and SIRT1 (C) in myotubes from C2C12 myoblasts (a) and primary HSMPCs (b) was shown. All data are presented as means ± standard errors of the means for at least three independent experiments. *P < 0.05 as compared with control group; #P < 0.05 as compared with Dexa group.
Sirtuins (SIRT), a family of nicotinamide adenine dinucleotide (NAD+)‐dependent deacetylases with seven members (SIRT1–SIRT7), have been performed to play an important role in skeletal muscle metabolism in which both SIRT1 and SIRT3 were the most extensively studied.33 As shown in Figure 3B(a) and 3B(b), Dexa significantly up‐regulated the SIRT3 protein expression in both C2C12‐derived and HSMPC‐derived myotubes, which could be abrogated by the treatment of ALN in a dose‐dependent manner. Dexamethasone could also up‐regulate the SIRT1 protein expression in C2C12‐derived myotubes, but ALN did not affect this Dexa‐up‐regulated SIRT1 expression [Figure 3C(a)]. Dexamethasone did not induce the SIRT1 protein expression in HSMPC‐derived myotubes [Figure 3C(b)]. In addition, treatment with 1 μM Dexa for 24 h caused morphological alteration and reduced diameter in myotubes differentiated from C2C12 myoblasts, which could be significantly inhibited by ALN treatment at higher concentrations (5 and 10 μM) (Figure 4A). Alendronate treatment at higher concentrations (5 and 10 μM) could also ameliorate Dexa‐induced Atrogin‐1 [Figure 4B(a)] and SIRT3 [Figure 4B(b)] protein expressions in C2C12‐derived myotubes, while higher concentrations of ALN did not affect Dexa‐up‐regulated SIRT1 protein expression [Figure 4B(c)]. Moreover, we used the pharmacological SIRT3 inhibitor (AKG7) and small interfering RNA of SIRT3 (SIRT3 siRNA) to determine whether the down‐regulation of SIRT3 could reverse the Dexa‐induced myotube atrophy. As shown in Figure 5, both AKG7 [Figure 5A(a) and B(a)] and SIRT3 siRNA [Figure 5A(b) and B(b)] significantly reversed the atrophic effect of Dexa on the C2C12‐derived myotubes by down‐regulating protein expressions of Atrogin‐1 (Figure 5A) and SIRT3 (Figure 5B), whereas there were no observed effects of both AGK7 and SIRT3 siRNA on the protein expressions of SIRT1 in the Dexa‐induced atrophic myotubes (Figure 5C). In addition, resveratrol, an SIRT3 activator, could blockade the inhibitory effect of ALN on the Dexa‐induced atrophic myotubes by up‐regulating protein expressions of Atrogin‐1 [Figure 5D(a)] and SIRT3 [Figure 5D(b)] but not SIRT1 [Figure 5D(c)]. These results suggest that the anti‐atrophic activity of ALN on Dexa‐treated C2C12‐derived and HSMPC‐derived myotubes may be through the regulation of SIRT3 signalling.
Figure 4.
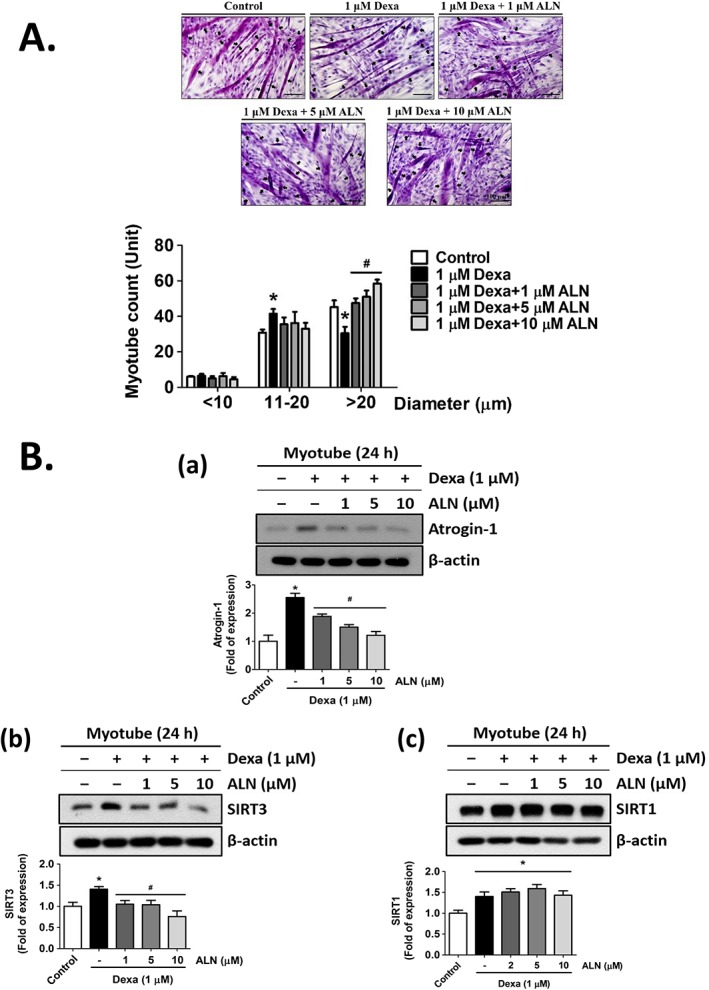
Alendronate (ALN) at a higher concentrations protected myotube from atrophy induced by dexamethasone (Dexa) in C2C12 myoblasts. C2C12 myoblasts were differentiated for 4 days under differentiation medium, followed by an additional 24 h with Dexa (1 μM) in the presence or absence of ALN (1, 5, and 10 μM). (A) The representative haematoxylin and eosin stained myotubes and the frequency distribution of myotube diameter were shown. (B) Western blot analysis of protein expressions of (a) Atrogin‐1, (b) SIRT3, and (c) SIRT1 in myotubes from C2C12 myoblasts was shown. All data are presented as means ± standard errors of the means for at least three independent experiments. The solid arrows indicated the multi‐nuclei myotubes. Scale bar = 100 μm. *P < 0.05 as compared with control group; #P < 0.05 as compared with Dexa group.
Figure 5.

Both SIRT3 inhibitor AKG7 treatment and SIRT3‐siRNA transfection suppressed dexamethasone (Dexa)‐induced Atrogin‐1 and SIRT3 protein expressions in myotubes. Fully differentiated C2C12 myotubes (day 4) were pre‐treated with AKG7 (1 μM) or transfected with scramble or SIRT3 siRNA (50 nM) or resveratrol (RSV, 100 μM) and subsequently treated with Dexa (1 μM) for 24 h in the presence or absence of alendronate (1 μM). Western blot analysis of protein expressions of Atrogin‐1 (A), SIRT3 (B), and SIRT1 (C) in myotubes with AKG pre‐treatment (a) or SIRT3 siRNA transfection (b) was shown. (D) Western blot analysis of protein expressions of Atrogin‐1 (a), SIRT3 (b), and SIRT1 (c) in myotubes with RSV pre‐treatment was shown. All data are presented as means ± standard errors of the means for at least three independent experiments. *P < 0.05 as compared with control group; #P < 0.05 as compared with Dexa group; &P < 0.05 as compared with Dexa + ALN group.
Alendronate ameliorate Dexa‐diminished myogenesis of C2C12 myoblasts and primary human skeletal muscle‐derived progenitor cells
In response to skeletal muscle damage, quiescent mononucleated myogenic cells, called satellite or progenitor cells, are activated and initiated to proceed myogenic differentiation as revealing the skeletal muscle regeneration and functional recovery.34 Besides, the signalling regulation has been shown the similar correspondence between skeletal muscle atrophy and retarded regeneratio.35 Therefore, we next investigated whether ALN reversed Dexa‐induced myogenic differentiation inhibition in C2C12 myoblasts and primary HSMPCs. Dexamethasone (1 μM) treatment in C2C12 myoblasts (Figure 6A) and HSMPCs (Figure 6B) subjected to 4 days of differentiation morphologically exhibited a significant reduction in multinucleated myotube formation (the most frequent distribution of nuclei of and >5 unit in both C2C12 myoblasts and primary HSMPCs). Alendronate (1 μM) could greatly reverse the Dexa‐induced impaired effect on myotube formation during myogenesis of C2C12 myoblasts and HSMPCs. The protein expressions of MHC, a myogenesis marker, were also significantly down‐regulated by Dexa treatment in C2C12 myoblasts [Figure 5C(a)] and primary HSMPCs [Figure 6C(b)], accompanying SIRT3 up‐regulation. Alendronate significantly reversed the down‐regulation of MHC protein expression and the up‐regulation of SIRT3 protein expression in Dexa‐treated C2C12 myoblasts [Figure 6C(a)] and HSMPCs [Figure 6C(b)] during myogenic differentiation. Furthermore, SIRT3 siRNA could also significantly reverse the effect on the suppression/promotion of protein expressions of MHC/SIRT3 in Dexa‐treated C2C12 myoblasts during the myogenic differentiation (Figure 6D).
Figure 6.

Alendronate (ALN) suppressed dexamethasone (Dexa)‐induced myogenic differentiation inhibition and SIRT3 expression in C2C12 myoblasts and primary human skeletal muscle‐derived progenitor cells (HSMPCs). C2C12 myoblasts and primary HSMPCs were cultured in growth medium or differentiation medium for 4 days with ALN (0.1–1 μM) in the presence or absence of Dexa (1 μM). (A and B) Representative haematoxylin and eosin E stained myotube formation during myogenic differentiation of C2C12 myoblasts (A) and primary HSMPCs (B). (C) Western blot analysis of the protein expressions of myosin heavy chain (MHC) and SIRT3 in C2C12 myoblasts (a) and primary HSMPCs (b) during myogenic differentiation was shown. (D) Western blot analysis of the protein expressions of MHC and SIRT3 in C2C12 myoblasts with or without scramble or SIRT3 siRNA (50 nM) transfection during myogenic differentiation was shown. All data are presented as means ± standard errors of the means for at least three independent experiments. The solid arrows indicated the multi‐nuclei myotubes. Scale bar = 100 μm. *P < 0.05 as compared with control group; #P < 0.05 as compared with Dexa group.
Alendronate ameliorate muscle wasting/weakness and lower regenerative capacity in the mouse models of denervation and glycerol‐induced skeletal muscle injury
The sciatic nerve‐denervated mice shown a significantly lower skeletal muscle endurance analysed by the muscle fatigue task with a rota‐rod apparatus (Figure 7A) and a significantly lower soleus [Figure 7B(a)] and Gascn [Figure 7B(b)] muscle mass, which could be effectively reversed by ALN (0.5 or 1 mg/kg) treatment. Besides, the significantly lower isometric contraction and KCl‐induced tonic contracture were shown in soleus muscles from denervated mice as compared with control group, which could be significantly reversed by 1 mg/kg ALN treatment (Figure 7C). Denervated mice featured an obvious decrease in average soleus [Figure 7D(a)] and Gascn [Figure 7D(b)] myofibre CSA with a leftward shift in the representative frequency distribution histogram of soleus and Gascn myofibre CSA (the most frequent distribution of myofibre CSA of <600 μm2 in soleus muscles and 600–1199 μm2 in Gascn muscles) as compared with the sham control mice, the most frequent distribution of myofibre CSA of 600–1199 μm2 in soleus muscles [Figure 7D(a)] and >2400 μm2 in Gascn muscles [Figure 7D(b)]. Treatment with ALN significantly ameliorated the distribution of myofibre CSA in sciatic nerve‐denervated mice (Figure 7D).
Figure 7.

Alendronate (ALN) treatment protected skeletal muscle from denervation‐induced atrophy in mice. Alendronate (0.5 and 1 mg/kg) or vehicle was provided 4 weeks in mice, and mice were kept administration of ALN until 7 days after sham operation or sciatic nerve resection (DN) from right hind limb. (A) Muscle endurance was determined as latency to fall in the muscle fatigue after denervation. (B) The average muscle weights of soleus muscles (a) and gastrocnemius (Gascn) muscles (b) were measured. (C) The isometric contractions of soleus muscles evoked by a voltage stimulator and potassium chloride‐induced tonic contracture were shown. (D) The representative haematoxylin and eosin stained muscle sections (i), the average cross‐sectional area (CSA) of myofibres (ii), and the frequency distribution of myofibres (iii) in soleus muscles (a) and Gascn (b) were shown. The average CSA of myofibre was evaluated from random five visual fields of each group. The frequency distribution of myofibre CSA from each group was presented as percent fibres/size of fibres. Cross‐sectional area was calculated by the ImageJ analysis software. Data are presented as means ± standard errors of the means, n = 6 of each group. Scale bar = 100 μm. *P < 0.05 as compared with sham control group; #P < 0.05 as compared with DN group.
Next, we investigated whether ALN reversed the muscle atrophy‐related signals observed in denervated mice. As shown in Figure 8, the soleus muscle isolated from denervated mice showed exacerbation of atrophy accompanying with an increased protein expressions of Atrogin‐1 and SIRT3 by the immunohistochemistry staining. Reversion of atrophic phenotype by ALN treatment was performed due to blockade of Atrogin‐1 and SIRT3 signalling molecules in the soleus muscles.
Figure 8.

Alendronate (ALN) treatment down‐regulated the expressions of Atrogin‐1 and SIRT3 in the soleus muscles of denervated mice. Alendronate (0.5 and 1 mg/kg) or vehicle was provided 4 weeks in mice, and mice were kept administration of ALN until 7 days after sham operation or sciatic nerve resection from right hind limb. The representative immunohistochemical images for the expressions of Atrogin‐1 and SIRT3 in the soleus muscles isolated from each group were shown (n = 4 of each group).
To assess the restored potential of ALN for regenerative capacity, hind leg soleus muscles of the mice were injected with glycerol to induce skeletal muscle injury. At 5 days post injury, glycerol‐injured mice were unable to sustain 30 min of rota‐rod running (Figure 9A) and showed a lower soleus muscle mass (Figure 9B) as compared with non‐glycerol‐injured mice. Besides, ALN treatment could significantly improve the lower skeletal muscle endurance and exhibited an elevated trend in soleus muscle mass. A serious decline in myofibres with centrally located nuclei characterized as early regenerating myofibres was manifested in glycerol‐injured mice, which could be significantly reversed by administration with ALN (0.5 and 1 mg/kg) coupling with the decrease in the protein expression of SIRT3 [Figure 9C(a)]. Unexpectedly, the CSA of myofibres exhibited a significant reduction in glycerol‐injured mice, which could not be significantly reversed by administration with ALN at neither 0.5 mg/kg nor 1 mg/kg [Figure 9C(b) and 9C(c)]. An increased protein expression of SIRT3 by the immunohistochemistry staining shown in soleus muscles isolated from glycerol‐injured mice could be inhibited by ALN treatment (Figure 9D). Moreover, it has been reported that myofibres are substituted for fatty infiltrate and collagen until skeletal muscle eventually losing the regenerative capacity.36 As shown in Figure 8E, Masson's trichrome staining showed the collagen accumulation in the glycerol‐injured soleus muscle, which could be effectively ameliorated by ALN treatment.
Figure 9.

Alendronate treatment protects skeletal muscle from glycerol‐impaired regenerative capacity in mice. Alendronate (ALN, 0.5 and 1 mg/kg) or vehicle was provided 4 weeks in mice, and mice were kept administration of ALN until 5 days after sham operation or glycerol (Gly; 50% vol/vol)‐induced muscle injury from right hind limb. (A) Muscle endurance was determined as latency to fall in the muscle fatigue after 5 days of Gly injection. (B) The average muscle weight of soleus muscles was measured. (C) Representative haematoxylin and eosin stained sections of soleus muscles with Gly‐induced muscle injury (a), quantification of the percent of soleus myofibres containing central nuclei (b), the average cross‐sectional area (CSA) of myofibres (c), and the frequency distribution of myofibres of each group (d) were shown. Data are presented as means ± standard errors of the means, n = 6 of each group. Scale bar = 100 μm. *P < 0.05 as compared with sham control group; #P < 0.05 as compared with Gly group. (D) The representative immunohistochemical images for the expressions of SIRT3 in the soleus muscles isolated from each group were shown (n = 4 of each group). (E) The representative Masson's trichrome‐stained sections of soleus muscles isolated from each group were shown (n = 4 of each group).
Discussion
The present study demonstrated for the first time that low‐dose ALN inhibited muscle atrophy and promoted muscle regenerative capacity via SIRT3 down‐regulation in vitro and in vivo. We also postulated that the reversibility of ALN in muscle atrophy might be linked to the stimulation of hypertrophic myotube formation. We used a well‐established murine model of skeletal muscle atrophy‐denervation‐induced muscle atrophy24 instead of using an osteoporotic murine model to investigate the effect of ALN on muscle wasting. We hypothesized that this anti‐osteoporosis drug could effectively not only enhance skeletal muscle mass and strength in patients with osteoporosis15 but also improve skeletal muscle loss and dysfunction from cachexia, sarcopenia, and other muscle wasting‐related diseases. These findings suggest that ALN can be a promising therapeutic strategy for management of muscle wasting‐related diseases and sarcopenia.
The effects of bisphosphonates osteoporosis drugs on muscle growth and function are controversial. Yoon et al. have indicated that pamidronate, a member of bisphosphonates, provided positive effects on both bone (an increase in bone growing and strength) and skeletal muscle function (an increase in grip strength) in Mdx mice, an animal model used as a surrogate for Duchenne muscular dystrophy.37 Børsheim et al. have suggested that pamidronate attenuates muscle loss as indicated by a positive net balance of muscle protein synthesis and breakdown and preserve muscle strength in post‐burned paediatric patients.38 However, the considerable controversy also exists concerning the positive effects of bisphosphonates on skeletal muscle preservation. Uchiyama et al. have indicated that the skeletal muscle CSA around femur in 32 osteoporotic patients with 3 years of bisphosphonate (ALN, risedronate, or minodronate) was smaller than that of the bone mineral density‐matched control.39 Widrick et al. have also shown that ALN, at a dosage shown to improve bone loss, did not affect the skeletal muscle function in ovariectomized rats.6 Moreover, the results from Shiomi et al. found that ALN (at a higher concentration of 100 μM) disrupted proliferation and myogenic differentiation in primary cultured human myogenic cells.40 These negative results still remain some concerns for dosage and sample size. In the present study, our results exhibited a beneficial effect on myotube atrophy and myogenic differentiation in C2C12 myoblasts and HSMPCs at relative lower concentrations (1 μM) of ALN. We also found that ALN monotherapy could attenuate skeletal muscle atrophy in the denervated mice and improve the skeletal muscle regenerative capacity in mice with glycerol‐induced skeletal muscle injury. These results are consistent with the positive observations of ALN on muscle function in the previous studies. Furthermore, we also suggested that the possible mechanism of the fact may be through SIRT3 inhibition.
Glucocorticoids such as Dexa are steroid hormones widely used against inflammatory and immunosuppressive activities. However, high doses and prolonged administration may induce adverse lateral effects including hyperglycaemia, insulin resistance, osteoporosis, and skeletal muscle weakness/wasting, as well called ‘steroid myopathy’.41 Its characteristics represent lower skeletal muscle endurance, poor skeletal muscle regeneration, and skeletal muscle atrophy mainly in the proximal skeletal muscles of upper or lower limbs and neck flexors.42, 43 The mechanisms of glucocorticoids on myopathy have been suggested to affect the IGF/PI3K/Akt‐dependent mTOR pathway, resulting in increased atrogin‐1 and MURF1 protein expressions for muscle degradation/myotube atrophy, and to modulate myogenic regulatory factors, interfering with myogenic differentiation process for suppression of muscle myogenesis/myotube formation.44, 45 In the clinical presentation of steroid myopathy, muscle weakness has been found in 2.4% to 21% of patients with exogenous glucocorticoid administration.46 Miller has suggested that rapid onset of clinically significant muscle weakness can be observed within 2 weeks after the initiation of over 10 mg/day prednisone administration.47 To date, reducing steroid dose and endurance exercise training have been applied as the treatment of glucocorticoid excess state‐induced muscle weakness.46, 48 Moreover, many factors have contributed to ageing—or chronic kidney disease‐related loss of muscle mass and strength, including physical inactivity, a disruption of several positive regulators of muscle hypertrophy (e.g. the Akt pathway), and hormonal changes (e.g. testosterone, vitamin D, and parathyroid hormone).49, 50 However, in the present study, we found that an anti‐osteoporotic drug, ALN at low doses, could effectively reverse the impaired effects of Dexa on the muscle hypertrophy and the regenerative capacity via SIRT3 down‐regulation in vitro and muscle degeneration therapy in vivo, suggesting that ALN may be provided as a novel potential remedy for steroid myopathy in the clinic in the future.
Sirtuin proteins are mammalian homologues of yeast silent information regulator 2 encoding NAD+‐dependent class III histone deacetylases.51, 52 The mammalian silent information regulator 2 family comprised seven orthologues (SIRT1–SIRT7), which all have a conserved catalytic core NAD+‐binding domain but have diverse substrates, subcellular locations, and functions, which have gained growing attention for their connections with many biological processes including cellular metabolism regulation, neuroprotection, apoptosis, inflammation, and cancer progression.53, 54, 55, 56 In skeletal muscle metabolism, both SIRT1 and SIRT3 are the most studied members of SIRT family serving as an important role in skeletal muscle energy homeostasis. Fulco et al. have reported that SIRT1 is shown as a negative regulator to skeletal muscle differentiation by associating with the complex PCAF/GCN5.57 Although SIRT1 may inhibit myogenic differentiation, Lee and Goldberg have suggested that SIRT1 activation can block muscle atrophy by suppressing FoxO expressions and activating PGC‐1α to combat muscle wasting and restore muscle mass from muscle‐wasting related diseases.58 On the other hand, Jing et al. reported that deletion of SIRT3 contributes to impair glucose oxidation in skeletal muscle, which is correlated with a decrease in pyruvate dehydrogenase activity and accumulation of pyruvate metabolites to inhibit fatty acid oxidation.59 Despite a positive modulator of SIRT3 in skeletal muscle oxidative capacity, Lin et al. have been indicated that SIRT3 activation may improve muscle metabolic function under caloric restriction condition but it may unfortunately contribute to reduce muscle mass through up‐regulation of FOXO1/MuRF‐1 atrophic signalling pathway.60 In the present study, our results showed that ALN could ameliorate muscle atrophy and promote skeletal muscle regenerative capacity due to SIRT3 regulation but not SIRT1 regulation. We found that SIRT3 protein expressions were activated after muscle injury (denervation or glycerol‐induced muscle injury) in vitro and in vivo and ALN could effectively ameliorate muscle atrophy and poor regenerative capacity through suppressing SIRT3 protein expressions.
In the current study, SIRT3 regulation was performed in both skeletal muscle atrophy and myogenic differentiation. This observation postulated that muscle fibre atrophy may be associated with retardation of skeletal muscle regeneration, which results from lower satellite cell number in myofibres. Van der Velden et al. have suggested that muscle regrowth from muscle atrophy facilitates myogenic differentiation dependently through the inactivation of muscle‐specific glycogen synthase kinase‐3β.61 Our previous study has been shown that advanced glycation end products performed similar regulative mechanism between muscle atrophy and impairment of muscle regeneration through receptor for advanced glycation end product‐mediated AMPK‐down‐regulated Akt signalling pathway in diabetic animals and cultured skeletal muscle cells.25 These findings support our postulation that muscle fibre atrophy correlates with retardation of skeletal muscle regeneration through the similar regulative mechanism. In the present study, inhibition of SIRT3 protein expressions by ALN could be applied to amelioration of both skeletal muscle atrophy and impairment of skeletal muscle regeneration. However, the detailed mechanism participated in the positive regulation of muscle satellite cell/stem cell homeostasis by ALN remains to be further elucidated.
Sirtuin‐1 and SIRT3 have also been reported to exert several organ protection from fibrosis, including kidney, lung, liver, and heart. Yang et al. have reported that resveratrol, a well‐known SIRT1 activator, could ameliorate inflammation and fibrosis in rats with unilateral ureteral obstruction‐induced renal failure.62 Huang et al. have provided the evidence that overexpression of SIRT1 could attenuate extracellular matrix expression through the down‐regulation of TGF‐β1/Smad3 pathway in cultured mesangial cells.63 With regard to regulation of SIRT3 involved in fibrosis, Sundaresan et al. have reported that SIRT3‐knockout mice were shown tissue fibrosis of several organs including lungs, kidney, liver, and heart, whereas there was not shown tissue fibrosis of these organs in SIRT3 overexpressing mice.64 Controversially, in the present study, we found the fibrotic feature shown in glycerol‐injured soleus muscles accompanying with SIRT3 activation, which ALN could reverse the collagen infiltration with down‐regulation of SIRT3 protein expression. This different regulatory effect of SIRT3 on fibrosis from the study of Sundaresan et al. may be due to different target organs and different injured models. However, further investigation is required to evaluate the detail molecular mechanism between ALN and collagen production under SIRT3 regulation in injured skeletal muscles.
In conclusion, the present study demonstrates for the first time that osteoporosis drug ALN can significantly prevent muscle wasting in vitro and in denervation‐induced and glycerol‐induced myopathy animal models through a SIRT‐3 down‐regulation. However, there are some limitations for our animal models. As a result, the denervation‐induced and glycerol‐induced myopathy animal models used in this study seem not to mimic the muscle wasting‐related diseases in human. Further investigation for the action and mechanism of ALN on osteoporosis‐induced or ageing‐induced skeletal muscle wasting animal models needs to be considered.
Author contributions
All authors approved the final version to be published. Study conception and design: S.‐H.L., R.‐S.Y., and C.‐K.C. Acquisition of data: C.‐Y.C. and H.‐C.C. Analysis and interpretation of data: C.‐Y.C. and H.‐C.C. Provide reagents and technical support: D.‐C.C. and R.‐S.Y. Wrote the paper: C.‐Y.C., C.‐K.C., and S.‐H.L.
Conflict of interest
Hsien‐Chun Chiu, Chen‐Yuan Chiu, Rong‐Sen Yang, Ding‐Cheng Chan, Shing‐Hwa Liu, and Chih‐Kang Chiang stated that they have no conflict of interest.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.65
This study was supported by grants from the Ministry of Science and Technology of Taiwan (MOST105‐2314‐B‐002‐025‐MY3) and the National Taiwan University Hospital (NTUH105‐S2953).
Chiu, H.‐C. , Chiu, C.‐Y. , Yang, R.‐S. , Chan, D.‐C. , Liu, S.‐H. , and Chiang, C.‐K. (2018) Preventing muscle wasting by osteoporosis drug alendronate in vitro and in myopathy models via sirtuin‐3 down‐regulation. Journal of Cachexia, Sarcopenia and Muscle, 9: 585–602. doi: 10.1002/jcsm.12289.
Contributor Information
Shing‐Hwa Liu, Email: shinghwaliu@ntu.edu.tw.
Chih‐Kang Chiang, Email: ckchiang@ntu.edu.tw.
References
- 1. Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 2006;61:1059–1064. [DOI] [PubMed] [Google Scholar]
- 2. Bosy‐Westphal A, Müller MJ. Identification of skeletal muscle mass depletion across age and BMI groups in health and disease—there is need for a unified definition. Int J Obes (Lond) 2015;39:379–386. [DOI] [PubMed] [Google Scholar]
- 3. Bernstein IM, Plociennik K, Stahle S, Badger GJ, Secker‐Walker R. Impact of maternal cigarette smoking on fetal growth and body composition. Am J Obstet Gynecol 2000;183:883–886. [DOI] [PubMed] [Google Scholar]
- 4. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;4:58–74. [DOI] [PubMed] [Google Scholar]
- 5. Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. Arsenic inhibits myogenic differentiation and muscle regeneration. Environ Health Perspect 2010;18:949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiu CY, Yen YP, Tsai KS, Yang RS, Liu SH. Low‐dose benzo(a)pyrene and its epoxide metabolite inhibit myogenic differentiation in human skeletal muscle‐derived progenitor cells. Toxicol Sci 2014;38:344–353. [DOI] [PubMed] [Google Scholar]
- 7. Volpato S, Bianchi L, Cherubini A, Landi F, Maggio M, Savino E, et al. Prevalence and clinical correlates of sarcopenia in community‐dwelling older people: application of the EWGSOP definition and diagnostic algorithm. J Gerontol A Biol Sci Med Sci 2014;9:438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tipton KD, Rasmussen BB, Miller SL, Wolf SE, Owens‐Stovall SK, Petrini BE, et al. Timing of amino acid‐carbohydrate ingestion alters anabolic response of muscle to resistance exercise. Am J Physiol Endocrinol Metab 2001;281:E197–E206. [DOI] [PubMed] [Google Scholar]
- 9. Dionne IJ, Kinaman KA, Poehlman ET. Sarcopenia and muscle function during menopause and hormone‐replacement therapy. J Nutr Health Aging 2000;4:156–161. [PubMed] [Google Scholar]
- 10. Bischoff HA, Borchers M, Gudat F, Duermueller U, Theiler R, Stähelin HB, et al. In situ detection of 1,25‐dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem J 2001;33:19–24. [DOI] [PubMed] [Google Scholar]
- 11. Delmas PD. Treatment of postmenopausal osteoporosis. Lancet 2002;359:2018–2026. [DOI] [PubMed] [Google Scholar]
- 12. NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy . Osteoporosis prevention, diagnosis, and therapy. JAMA 2001;285:785–795.11176917 [Google Scholar]
- 13. Montpetit K, Plotkin H, Rauch F, Bilodeau N, Cloutier S, Rabzel M, et al. Rapid increase in grip force after start of pamidronate therapy in children and adolescents with severe osteogenesis imperfecta. Pediatrics 2003;111:e601–e603. [DOI] [PubMed] [Google Scholar]
- 14. Park JH, Park KH, Cho S, Choi YS, Seo SK, Lee BS, et al. Concomitant increase in muscle strength and bone mineral density with decreasing IL‐6 levels after combination therapy with alendronate and calcitriol in postmenopausal women. Menopause 2013;20:747–753. [DOI] [PubMed] [Google Scholar]
- 15. Harada A, Ito S, Matsui Y, Sakai Y, Takemura M, Tokuda H, et al. Effect of alendronate on muscle mass: investigation in patients with osteoporosis. Osteoporos Sarcopenia 2015;1:53–58. [Google Scholar]
- 16. Widrick JJ, Fuchs R, Maddalozzo GF, Marley K, Snow C. Relative effects of exercise training and alendronate treatment on skeletal muscle function of ovariectomized rats. Menopause 2007;14:528–534. [DOI] [PubMed] [Google Scholar]
- 17. Ferraro E, Pin F, Gorini S, Pontecorvo L, Ferri A, Mollace V, et al. Improvement of skeletal muscle performance in ageing by the metabolic modulator trimetazidine. J Cachexia Sarcopenia Muscle 2016;7:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Penna F, Bonetto A, Aversa Z, Minero VG, Rossi Fanelli F, Costelli P, Muscaritoli M. Effect of the specific proteasome inhibitor bortezomib on cancer‐related muscle wasting. J Cachexia Sarcopenia Muscle 2016;7:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, et al. Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 2017; [Epub ahead of print]. doi: https://doi.org/10.1002/jcsm.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wendowski O, Redshaw Z, Mutungi G. Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres. J Cachexia Sarcopenia Muscle 2017;8:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X, Wu Y, Yang T, Wei M, Wang Y, Deng X, et al. Salidroside alleviates cachexia symptoms in mouse models of cancer cachexia via activating mTOR signalling. J Cachexia Sarcopenia Muscle 2016;7:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anker SD, Coats AJ, Morley JE. Evidence for partial pharmaceutical reversal of the cancer anorexia‐cachexia syndrome: the case of anamorelin. J Cachexia Sarcopenia Muscle 2015;6:275–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Institute of Laboratory Animal Resources . Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 2011. [Google Scholar]
- 24. MacDonald EM, Andres‐Mateos E, Mejias R, Simmers JL, Mi R, Park JS, et al. Denervation atrophy is independent from Akt and mTOR activation and is not rescued by myostatin inhibition. Dis Model Mech 2014;7:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chiu CY, Yang RS, Sheu ML, Chan DC, Yang TH, Tsai KS, et al. Advanced glycation end‐products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE‐mediated, AMPK‐down‐regulated, Akt pathway. J Pathol 2016;238:470–482. [DOI] [PubMed] [Google Scholar]
- 26. Liu SH, Fu WM, Lin‐Shiau SY. Studies on the inhibition by chlorpromazine of myotonia induced by ion channel modulators in mouse skeletal muscle. Eur J Pharmacol 1993;231:23–30. [DOI] [PubMed] [Google Scholar]
- 27. Sato T, Aoyama T, Ono H. Mechanisms of slow contracture induced by potassium and caffeine in skeletal muscle of the dog. Jpn J Pharmacol 1984;34:147–152. [DOI] [PubMed] [Google Scholar]
- 28. Asakura A, Seale P, Girgis‐Gabardo A, Rudnicki MA. Myogenic specification of side population cells in skeletal muscle. J Cell Biol 2002;159:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lokireddy S, Mouly V, Butler‐Browne G, Gluckman PD, Sharma M, Kambadur R, et al. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin‐proteasome pathway‐mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol 2011;301:C1316–C1324. [DOI] [PubMed] [Google Scholar]
- 30. Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 2013;123:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Son YH, Lee SJ, Lee KB, Lee JH, Jeong EM, Chung SG, et al. Dexamethasone downregulates caveolin‐1 causing muscle atrophy via inhibited insulin signaling. J Endocrinol 2015;225:27–37. [DOI] [PubMed] [Google Scholar]
- 32. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 33. Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev 2012;92:1479–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004;84:209–238. [DOI] [PubMed] [Google Scholar]
- 35. Verdijk LB, Dirks ML, Snijders T, Prompers JJ, Beelen M, Jonkers RA, et al. Reduced satellite cell numbers with spinal cord injury and aging in humans. Med Sci Sports Exerc 2012;44:2322–2330. [DOI] [PubMed] [Google Scholar]
- 36. Radley HG, De Luca A, Lynch GS, Grounds MD. Duchenne muscular dystrophy: focus on pharmaceutical and nutritional interventions. Int J Biochem Cell Biol 2007;39:469–477. [DOI] [PubMed] [Google Scholar]
- 37. Yoon SH, Sugamori KS, Grynpas MD, Mitchell J. Positive effects of bisphosphonates on bone and muscle in a mouse model of Duchenne muscular dystrophy. Neuromuscul Disord 2016;26:73–84. [DOI] [PubMed] [Google Scholar]
- 38. Børsheim E, Herndon DN, Hawkins HK, Suman OE, Cotter M, Klein GL, et al. Pamidronate attenuates muscle loss after pediatric burn injury. J Bone Miner Res 2014;9:1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uchiyama S, Ikegami S, Kamimura M, Mukaiyama K, Nakamura Y, Nonaka K, et al. The skeletal muscle cross sectional area in long‐term bisphosphonate users is smaller than that of bone mineral density‐matched controls with increased serum pentosidine concentrations. Bone 2015;5:84–87. [DOI] [PubMed] [Google Scholar]
- 40. Shiomi K, Nagata Y, Kiyono T, Harada A, Hashimoto N. Differential impact of the bisphosphonate alendronate on undifferentiated and terminally differentiated human myogenic cells. J Pharm Pharmacol 2014;66:418–427. [DOI] [PubMed] [Google Scholar]
- 41. Ramamoorthy S, Cidlowski JA. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev 2013;24:41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Annane D. What is the evidence for harm of neuromuscular blockade and corticosteroid use in the intensive care unit? Semin Respir Crit Care Med 2016;37:51–56. [DOI] [PubMed] [Google Scholar]
- 43. Hermans G, Van den Berghe G. Clinical review: intensive care unit acquired weakness. Crit Care 2015;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome‐wide analysis of glucocorticoid receptor‐binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A 2012;109:11160–11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim J, Park MY, Kim HK, Park Y, Whang KY. Cortisone and dexamethasone inhibit myogenesis by modulating the AKT/mTOR signaling pathway in C2C12. Biosci Biotechnol Biochem 2016. [Epub ahead of print];80:2093–2099. [DOI] [PubMed] [Google Scholar]
- 46. Alshekhlee A, Kaminski HJ, Ruff RL. Neuromuscular manifestations of endocrine disorders. Neurol Clin 2002;20:35–58. [DOI] [PubMed] [Google Scholar]
- 47. Miller ML. Glucocorticoid‐induced myopathy. version 16.3., 2009.
- 48. LaPier TK. Glucocorticoid‐induced muscle atrophy. The role of exercise in treatment and prevention. J Cardiopulm Rehabil 1997;17:76–84. [DOI] [PubMed] [Google Scholar]
- 49. Sakuma K, Yamaguchi A. Sarcopenic obesity and endocrinal adaptation with age. Int J Endocrinol 2013;2013:204164;1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Molina P, Carrero JJ, Bover J, Chauveau P, Mazzaferro S, Torres PU. European Renal Nutrition (ERN) and Chronic Kidney Disease‐Mineral and Bone Disorder (CKD‐MBD) Working Groups of the European Renal Association‐European Dialysis Transplant Association (ERA‐EDTA). Vitamin D, a modulator of musculoskeletal health in chronic kidney disease. J Cachexia Sarcopenia Muscle 2017; [Epub ahead of print]. doi: https://doi.org/10.1002/jcsm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature 2000;403:795–800. [DOI] [PubMed] [Google Scholar]
- 52. Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2‐like proteins. Biochem Biophys Res Commun 2000;273:793–798. [DOI] [PubMed] [Google Scholar]
- 53. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2‐like proteins (sirtuins) metabolize NAD and may have protein ADP‐ribosyltransferase activity. Biochem Biophys Res Commun 1999;260:273–279. [DOI] [PubMed] [Google Scholar]
- 54. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 2005;16:4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol 2007;21:1745–1755. [DOI] [PubMed] [Google Scholar]
- 56. Mellini P, Valente S, Mai A. Sirtuin modulators: an updated patent review (2012‐2014). Expert Opin Ther Pat 2015;25:5–15. [DOI] [PubMed] [Google Scholar]
- 57. Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell 2003;12:51–62. [DOI] [PubMed] [Google Scholar]
- 58. Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem 2013;288:30515–30526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013;62:3404–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin L, Chen K, Abdel Khalek W, Ward JL 3rd, Yang H, Béatrice Chabi, et al. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS One 2014;9(1):e85636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van der Velden JL, Langen RC, Kelders MC, Willems J, Wouters EF, Janssen‐Heininger YM, et al. Myogenic differentiation during regrowth of atrophied skeletal muscle is associated with inactivation of GSK‐3beta. Am J Physiol Cell Physiol 2007;292:C1636–C1644. [DOI] [PubMed] [Google Scholar]
- 62. Yang SY, Lin SL, Chen YM, Wu VC, Yang WS, Wu KD. Downregulation of angiotensin type 1 receptor and nuclear factor‐κB by sirtuin 1 contributes to renoprotection in unilateral ureteral obstruction. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang XZ, Wen D, Zhang M, Xie Q, Ma L, Guan Y, et al. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF‐β/Smad3 pathway. J Cell Biochem 2014;115:996–1005. [DOI] [PubMed] [Google Scholar]
- 64. Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, et al. SIRT3 blocks aging‐associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol Cell Biol 2015;36:678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]