

Abstract
Truncating mutations in PRNP have been associated with heterogeneous phenotypes ranging from chronic diarrhea and neuropathy to dementia, either rapidly or slowly progressive. We identified novel PRNP stop‐codon mutations (p.Y163X, p.Y169X) in two Italian kindreds. Disease typically presented in the third or fourth decade with progressive autonomic failure and diarrhea. Moreover, one proband (p.Y163X) developed late cognitive decline, whereas some of his relatives presented with isolated cognitive and psychiatric symptoms. Our results strengthen the link between PRNP truncating mutations and systemic abnormal PrP deposition and support a wider application of PRNP screening to include unsolved cases of familial autonomic neuropathy.
Introduction
Human prion diseases are neurodegenerative disorders characterized by tissue accumulation of a misfolded form (PrPSc) of the cellular prion protein (PrPC). They comprise three major phenotypes, namely Creutzfeldt‐Jakob disease (CJD), by far the most common, fatal insomnia (FI) and Gerstmann‐Straüssler‐Scheinker syndrome (GSS).1, 2 At variance with CJD and FI, GSS manifests a much slower clinical course and is mainly characterized by amyloid deposition rather than spongiform change in the affected brain tissue.2, 3 This major phenotypic difference directly reflects the composition and topology of PrPSc aggregates. While in CJD and FI PrPSc accumulates as a full‐length or N‐terminally truncated fragments retaining the glycophosphatidylinositol (GPI) membrane anchor attaching the protein to the cell membrane,4 in GSS PrPSc mainly deposits in the extracellular space as unglycosylated anchorless fragments.2, 3 GSS was originally described as a parenchymal brain amyloidosis linked to missense or insertional mutations in the prion protein gene (PRNP).2 More recently, however, the discovery of premature truncating mutations in PRNP has expanded the phenotypic spectrum of anchorless PrP amyloidosis. The description of a PRNP stop‐codon mutation (p.Y145X) in a Japanese with dementia and both parenchymal and vascular PrP‐amyloid,5, 6 was followed by the findings of similar mutations at codons 160 (p.Q160X),7, 8 226 (p.Y226X),9 and 227 (p.Y227X)9 in patients with progressive dementia and pure PrP‐cerebral amyloid angiopathy (PrP‐CAA), GSS or mixed GSS/PrP‐CAA pathology.7, 9, 10 Most significantly, PRNP truncating mutations at codons 163 (p.Y163X) and 203 (p.D203X), have been recently found in patients with chronic diarrhea, progressive autonomic failure, and peripheral polyneuropathy in association with widespread abnormal PrP deposition in peripheral organs and the CNS.11, 12 Based on the multiple organ system PrP deposition and the amyloid properties of the protein aggregates in the brain, the term PrP systemic amyloidosis was introduced, irrespectively of the demonstration of the classical pathological hallmarks of systemic amyloidosis.11, 13, 14
Here, we describe two Italian families with several affected members carrying novel truncating mutations in PRNP associated with progressive autonomic failure and variable cognitive impairment.
Subjects and Methods
The study was performed according to the Helsinki Declaration and approved by local ethics committee. Written informed consent was obtained from all subjects.
Genetic Analysis
PRNP (Ref. Seq. NM_000311) open reading frame was analyzed according to previously described procedures.9
Clinical history
Subjects IV‐2A (proband 1) and V‐1B in family 1, and subject III‐6 (proband 2) and III‐5 in family 2 underwent a thorough clinical investigation (Fig. 1A–C). For deceased patients, clinical data were obtained from medical notes or sought from relatives.
Figure 1.
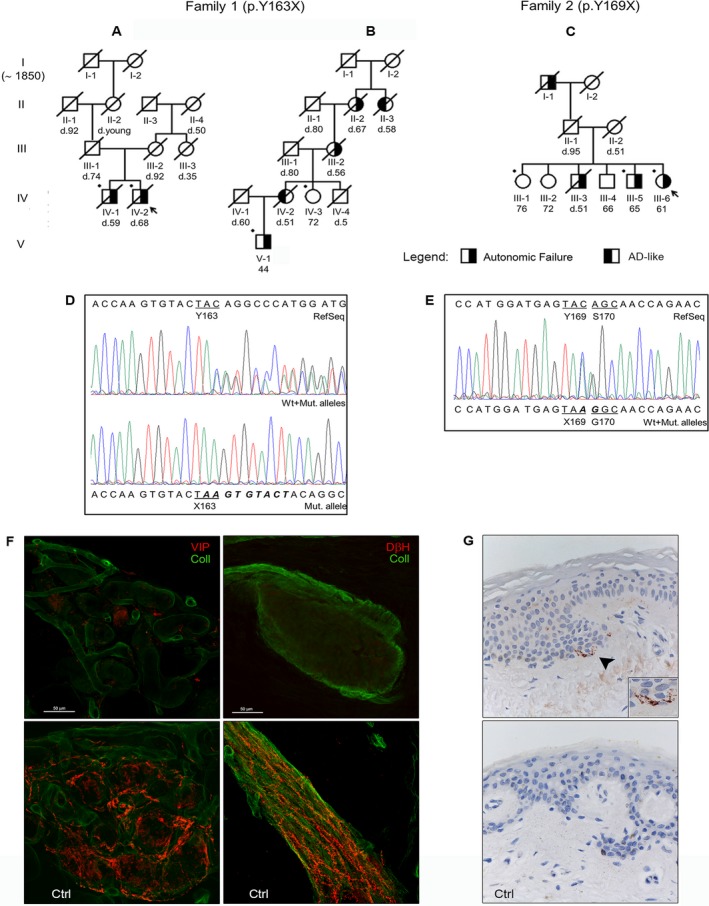
Pedigree of family 1 (A,B) and family 2 (C). Arrows point out the probands, while bullets indicate the subjects who underwent genetic analysis. Although the patient's ancestors of family 1 were traced back to 1850, there was no common founder for family branches A and B; however, the two were likely connected because of the restricted geographical residence area (two cities in adjacent provinces with a cumulative population of ~40,000 people) and the identity of the peculiar genetic mutation. (D and E) Sanger sequencing electropherograms showing the nucleotide sequence surrounding the two mutations (c.478_479insAAGTGTACT resulting in the p.Y163X mutation on the left; c.507C>A determining the p.Y169X mutation on the right). (F) A confocal study (magnification ×400) of autonomic patterns of innervation in the proband 1 (upper boxes) and a control subject. The leg autonomic innervation of sweat gland (left) and arrector pilorum muscle (right) were analyzed. Nerve fibers are marked in red by specific autonomic markers for sweat gland (VIP, vasoactive intestinal peptide) and arrector pilorum muscle (DβH, dopamine‐beta‐hydroxylase), whereas the collagen staining is shown in green. The innervation analysis revealed an almost total loss of cholinergic and noradrenergic fibres in proband 1 compared to the control subject. (G) Immunohistochemical detection of abnormal PrP in proband 1 (upper box) and in a control subject (magnification ×400). PrP staining with primary antibody 3F4 reveals fine, punctate deposits (arrowhead) in the upper dermis at the transition to epidermis only in the patient (the box in the lower‐right corner shows the PrP deposits at a higher magnification).
Clinical investigations
Cardiovascular autonomic function was investigated by means of head‐up tilt test (HUTT), Valsalva maneuver, deep breathing and cold face. Nerve conduction studies were carried out in at least one motor and one sensory nerve and the sympathetic skin response (SSR) evaluated from the palm of the hand and the sole of the foot bilaterally.
Cognitive function was explored by the Mini Mental State Examination and specific neuropsychological batteries for memory, visual‐spatial, attention, and executive performances. Conventional brain MRIs, including diffusion imaging, and voxel proton MR spectroscopy were performed according to standard protocols.
Skin studies
Studies of skin innervation were performed in 3‐mm punch biopsies from thigh and calf, according to previously published procedures.15 For PrP immunohistochemistry, paraffin sections from formalin‐fixed tissue blocks were processed using the monoclonal antibody 3F4, according to published protocols.16
CSF biomarkers
The 14‐3‐3 protein was measured semi‐quantitatively by immunoblotting, whereas total‐tau and neurofilament light (Nfl‐L) proteins were quantitatively analyzed using commercially available kits based on a sandwich ELISA method, as described.17 Prion RT‐QuIC was performed as described.18
Results
PRNP analysis showed a 9‐base pair duplicated fragment, c.478_479insAAGTGTACT, resulting in a p.Y163X stop‐codon mutation in family 1 and a c.507C>A variant determining a p.Y169X stop‐codon mutation associated with the in‐cis missense c.508A>G (p.S170G) in family 2 (Fig. 1D–E). The mutation was in‐cis with the valine allele at codon 129 in family 1 and with the methionine allele in family 2.
Proband 1 (Fig. 1A) had medical history unremarkable until the age of 45, when he started complaining of abdominal pain, chronic diarrhea and progressive weight loss eventually requiring parenteral nutrition. In the subsequent 10 years, the patient suffered urinary retention and presented syncopal episodes while standing and during urination. Since 63, he also complained of memory difficulties and confusion. At age of 66, neurologic examination disclosed short‐memory lapses, sluggish reacting pupils, diffuse brisk deep tendon reflexes, apallesthesia at the lower limbs, positive Romberg sign, ataxic gait, and segmental or generalized, both spontaneous and evoked, myoclonus.
Proband 2 (Fig. 1C) started to complain of abdominal pain, chronic diarrhea, hyporexia and early satiety at age of 40. In the next 20 years, she progressively lost about 50 kg and required parenteral nutrition, developed urinary incontinence, bladder and rectal prolapses, and suffered episodes of dizziness and gait unsteadiness, often immediately after standing. At 61, neurological examination revealed claw‐feet, reduced deep tendon reflexes at lower limbs, bilateral ptosis, and a mild strabismus.
Clinical findings in other affected members and the results of instrumental and laboratory investigations of all subjects are summarized in Tables 1 and 2. Autonomic tests revealed the impairment of both sympathetic and parasympathetic autonomic nervous system, fulfilling the diagnostic criteria for orthostatic hypotension at HUTT in all tested patients.
Table 1.
Main clinical findings in subjects belonging to families 1 and 2
| Case | Age at onset | Symptom/s at onset | Clinical phenotype | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| diarrhea | OH | urinary dysfunction | sweating disorders | sexual dysfunction | cognitive impairment | psychiatric symptoms | axonal neuropathy | |||
| FAMILY 1 (p.Y163X) | ||||||||||
| Proband 1 | 45 | diarrhea | +++ | +++ | ++ | ++ | + | ++ | ||
| IV‐1A | 56 | neurogenic bladder | + | +++ | ++ | ++ | ||||
| III‐1A | n.a. | urinary retention | yes | |||||||
| V‐1B | 33 | diarrhea | +++ | ++ | ++ | ++ | ++ | +/− | ||
| IV‐2B | 35 | insomnia | + | + | +++ | +++ | ||||
| III‐2B | 30 | diarrhea | +++ | |||||||
| II‐2B | n.a. | n.a. | yes | |||||||
| II‐3B | n.a. | psychiatric, cognitive | yes | yes | ||||||
| FAMILY 2 (p.Y169X) | ||||||||||
| Proband 2 | 40 | diarrhea | +++ | +++ | ++ | +/− | ||||
| III‐5 | 40 | diarrhea, neurogenic bladder, syncope | +++ | +++ | +++ | ++ | ++ | ++ | ||
| III‐3 | 40 | diarrhea | yes | yes | ||||||
| I‐1 | n.a. | diarrhea | yes | |||||||
[OH: orthostatic hypotension; source of clinical history: clinical evaluation for proband 1 and 2, V‐1B, III‐5; medical record for IV‐1A, IV‐2B, III‐2B; amnestic for III‐1A, II‐2B, II‐3B, III‐3, I‐1].
+mild; ++moderate; +++severe; n.a.:not available.
Table 2.
Results of laboratory investigations
| Family 1 (p.Y163X) | Family 2 (p.Y169X) | |||
|---|---|---|---|---|
| Proband 1 | Subject V‐1B | Proband 2 | Subject III‐5 | |
| Cardiovascular reflexes | – | |||
| HUTT (3rd min): | pathological | pathological | pathological | |
| ΔSBP, ΔDBP (mmHg); ΔHR (bpm) | −50, −30; 8 | −3, −17; – | −54, −38; −4 | |
| Valsalva ratio | 1.01 | – | 1.06 | |
| Overshoot (mmHg) | absent | 1.27 | absent | |
| Deep breathing: | absent arrhythmia | – | absent arrhythmia | |
| Cold Face (3rd min): | ||||
| ΔSBP, ΔDBP (mmHg); ΔHR (bpm) | 5, −4; −2 | – | 23, 15; −2 | |
| Electromyography | sensorimotor axonal PNP | normal | normal | sensorimotor axonal PNP |
| Skin sympathetic response | absent | absent | absent | absent |
| Cognitive dysfunction | mild global impairment (MMSE 23/30) with memory and visual attention dysfunction | globally normal (MMSE 30/30), but visuo‐spatial task impairment | globally normal (MMSE 26/30), but visual memory dysfunction | – |
| Electroencephalography | mild slowing | – | normal | – |
| Brain MRIa | nonspecific WM hyperintensities | normal | nonspecific WM hyperintensities | – |
| H1‐MRSa | normal | – | normal | – |
| Skin innervation analysis | autonomic SFN | autonomic and somatic SFN | autonomic and somatic SFN | – |
| Skin PrP immunostaining | positive | – | negative | – |
| Cerebrospinal fluid analysis | – | – | – | |
| Total proteins (mg/dL) | 215 | |||
| 14.3.3 protein assay | positive | |||
| Total tau protein (pg/mL) | 17,900 | |||
| Nf‐L (pg/mL) | 10,200 | |||
| Prion RT‐QuIC | negative | |||
Performed at the age of 66 in proband 1 and at the age of 60 in proband 2. – , Not done. [HUTT, head‐up tilt test; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate (bpm, beats per minute); PNP, polyneuropathy; MMSE, Mini Mental State Examinantion (normal value >24); WM, white matter; SFN, small fiber neuropathy; H1‐MRS, brain single voxel proton magnetic resonance spectroscopy (thalamus and cerebellum were analyzed in proband 1, parietal and occipital white matter in proband 2); Nf‐L, neurofilament light chain protein; prion RT‐QuIC, detection of prion seeding activity using real‐time quaking induced conversion assay].
Neurophysiologic studies revealed a sensorimotor axonal polyneuropathy in two out of four tested patients and the absence of SSR in all. Neuropsychological tests showed, in proband 1, a selective deficit of visual attention, verbal memory and logical reasoning at age of 65 progressing to a multi‐domain mild cognitive impairment 1 year later. Moreover, they revealed a selective deficit of visual memory or visuo‐spatial performances in other tested patients. Brain MRIs, including proton spectroscopy of the thalamus and cerebellum (proband 1) or the parieto‐occipital white matter (proband 2), were uninformative in all examined patients. Skin innervation analysis revealed a severe small fiber neuropathy with somatic and/or autonomic involvement (Fig. 1F). PrP immunohistochemistry disclosed a fine punctate PrP staining in the upper dermis next to the transition to epidermis (Fig. 1G). At variance, skin punch biopsies from two patients with probable sCJD carrying MM and MV at codon 129 showed no abnormal PrP deposits (data not shown).
The CSF obtained from proband 1 was slightly xantochromic, and showed an increase in total proteins. CSF biomarker assays revealed a positive 14‐3‐3 test, markedly elevated total‐tau and Nfl‐L levels, and a negative prion RT‐QuIC (Table 2).
Discussion
We have characterized the phenotypic traits of an inherited prion disease linked to two novel PRNP truncating mutations at codons 163 and 169 affecting several members across multiple generations of two Italian families. The disease showed a typical onset in the third or fourth decade and a mean disease duration of 15–20 years. The hallmarks of the clinical phenotype included a generalized autonomic failure with chronic diarrhea, weight loss, and neurogenic OH and neuropathy. Beside this typical presentation, however, cognitive impairment was an early feature in a minority of cases, in one family.
In genetic prion disease linked to stop‐codon PRNP mutations, the anchorless prion protein may spread through the interstitial fluids and vascular compartments.13 This was elegantly shown in experimental studies using transgenic mice expressing a prion protein lacking the GPI anchor (GPI−/−), which demonstrated that the anchorless PrP is secreted from the cells and accumulates in both brain and peripheral organs after scrapie infection, assuming the characteristics of a systemic amyloidosis.19
The fact that PRNP truncations in humans result in heterogeneous phenotypes ranging from dementia to chronic diarrhea and peripheral neuropathy appears to be in line with the above observations in animal models, although the significant phenotypic heterogeneity between patients carrying different PRNP truncations remains unexplained.5, 6, 7, 8, 9, 10, 11, 12, 20
Dementia in patients carrying PRNP stop‐codons has been linked to a GSS‐like neuropathologic phenotype with abundant plaques and neurofibrillary tangles (NFT) and/or to PrP‐CAA.6, 7, 9, 20 Interestingly, widespread PrP plaques and a substantial amount of tau‐related disease in the form of NFT and neuropil threads also characterized the patients with the phenotype of chronic diarrhea with autonomic failure.11 In this context, our observation of a minority of patients with early cognitive impairment suggests that the timing of CNS involvement may vary even within subjects carrying the same PRNP truncating mutation.
The polymorphic codon 129 in the PRNP represents the strongest susceptibility locus in sporadic and acquired prion disease and significantly modulates the phenotype of all forms of prion disease. As in other families with PRNP stop‐codons,6, 7, 20 both age at onset and clinical phenotype in our patients were not affected by the genotype at codon 129 either in the mutated or wild‐type allele. Moreover, the lack of disease in obligate carriers in family 2 strongly suggests that the penetrance of the truncating PRNP mutations may be incomplete, as previously reported in a family carrying the PRNP p.Y160X.20
The possibility of a prion‐related systemic amyloidosis in patients presenting with slowly progressive symptoms and signs of autonomic and sensory neuropathy is still poorly recognized. Indeed, the diagnostic delay in our patients was remarkable. Most patients are initially referred to a gastroenterologist, and unhelpful tests and misdiagnoses are common, especially in the early clinical stages. Thus, PRNP genetic testing should be recommended in all patients presenting with insidious generalized autonomic failure, and a positive family history of autonomic failure and/or cognitive decline. Moreover, the significance and potential diagnostic role of the unexpected increase in CSF neurodegenerative biomarkers we documented in a single patient should be addressed.
In conclusion, based on clinical findings (i.e., peripheral autonomic neuropathy) and the demonstration of PrP deposition in the skin, the present data strengthen the link between PRNP truncating mutations and systemic PrP amyloidosis. They also consolidate current knowledge on the clinical expression of these mutations, confirming that dementia and autonomic failure may coexist in the same family and support a wider application of PRNP screening to include unsolved cases of familial autonomic polyneuropathy.
Authors Contributions
S.C., R.R., P.C., and P.P. contributed to conception and design of the study. S.C., S.B., R.R., A.B.S., C.G., S.P., G.C.B., R.D.A., C.T., R.L., V.D., L.P., P.C., and P.P. contributed to acquisition and/or data analysis. S.C., S.B., and P.P. contributed to drafting the text and preparing the figure. S.C. and S.B. contributed equally to this work.
Conflict of Interest
Nothing to report.
Acknowledgments
We are indebted to the patients, their families, and the physicians who helped us collect clinical information. We particularly thank Giovanna Vaula (Città della Salute e della Scienza University Hospital, Turin, Italy) and Anna Maria Cauda for their stimulating support to continue research on this rare disease. Anne Collins edited the English text. The study was supported by the Italian Ministry of Health, grant RF2011‐02351092, and by the Gino Galletti Foundation.
Funding Information
The study was supported by the Italian Ministry of Health, grant RF2011‐02351092, and by the Gino Galletti Foundation.
Funding Statement
This work was funded by Italian Ministry of Health grant RF2011‐02351092; Gino Galletti Foundation grant .
Contributor Information
Sabina Capellari, Email: sabina.capellari@unibo.it.
Piero Parchi, Email: piero.parchi@unibo.it.
References
- 1. Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 2012;50:20–45. [PubMed] [Google Scholar]
- 2. Ghetti B, Tagliavini F, Takao M, et al. Hereditary prion protein amyloidoses. Clin Lab Med 2003;23:65–85. [DOI] [PubMed] [Google Scholar]
- 3. Parchi P, Chen SG, Brown P, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann‐Sträussler‐Scheinker disease. Proc Natl Acad Sci USA 1998;95:8322–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stahl N, Baldwin MA, Burlingame AL, et al. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990;29:8879–8884. [DOI] [PubMed] [Google Scholar]
- 5. Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in gerstmann‐straussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun 1993;192:525–531. [DOI] [PubMed] [Google Scholar]
- 6. Ghetti B, Piccardo P, Frangione B, et al. Prion protein amyloidosis. Brain Pathol 1996;6:127–145. [DOI] [PubMed] [Google Scholar]
- 7. Jayadev S, Nochlin D, Poorkaj P, et al. Familial prion disease with Alzheimer disease‐like tau pathology and clinical phenotype. Ann Neurol 2011;69:712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guerreiro R, Brás J, Wojtas A, et al. Nonsense mutation in PRNP associated with clinical Alzheimer's disease. Neurobiol Aging 2014;35:2656.e13–2656.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP . Acta Neuropathol 2010;119:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009;118:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mead S, Gandhi S, Beck J, et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med 2013;369:1904–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuzono K, Ikeda Y, Liu W, et al. A novel familial prion disease causing pan‐autonomic‐sensory neuropathy and cognitive impairment. Eur J Neurol 2013;20:e67–e69. [DOI] [PubMed] [Google Scholar]
- 13. Mead S, Reilly MM. A new prion disease: relationship with central and peripheral amyloidoses. Nat Rev Neurol 2015;11:90–97. [DOI] [PubMed] [Google Scholar]
- 14. Blancas‐Mejía LM, Ramirez‐Alvarado M. Systemic amyloidoses. Annu Rev Biochem 2013;82:745–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donadio V, Incensi A, Giannoccaro MP, et al. Peripheral autonomic neuropathy: diagnostic contribution of skin biopsy. J Neuropathol Exp Neurol 2012;71:1000–1008. [DOI] [PubMed] [Google Scholar]
- 16. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–233. [PubMed] [Google Scholar]
- 17. Lattanzio F, Abu‐Rumeileh S, Franceschini A, et al. Prion‐specific and surrogate CSF biomarkers in Creutzfeldt‐Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p‐tau and Aβ42 levels. Acta Neuropathol 2017;133:559–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franceschini A, Baiardi S, Hughson AG, et al. High diagnostic value of second generation CSF RT‐QuIC across the wide spectrum of CJD prions. Sci Rep 2017;. https://doi.org/10.1038/s41598-017-10922-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trifilo MJ, Yajima T, Gu Y, et al. Prion‐induced amyloid heart disease with high blood infectivity in transgenic mice. Science 2006;313:94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fong JC, Rojas JC, Bang J, et al. Genetic prion disease caused by PRNP Q160X mutation presenting with an orbitofrontal syndrome, cyclic diarrhea, and peripheral neuropathy. J Alzheimers Dis 2017;55:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]