

Abstract
Rho‐associated kinase (ROCK) is an emerging target in acute ischemic stroke. Early pre‐hospital treatment with ROCK inhibitors may improve their efficacy, but their antithrombotic effects raise safety concerns in hemorrhagic stroke, precluding use prior to neuroimaging. Therefore, we tested whether ROCK inhibition affects the bleeding times, and worsens hematoma volume in a model of intracerebral hemorrhage (ICH) induced by intrastriatal collagenase injection in mice. Tail bleeding time was measured 1 h after treatment with isoform‐nonselective inhibitor fasudil, or ROCK2‐selective inhibitor KD025, or their vehicles. In the ICH model, treatments were administered 1 h after collagenase injection. Although KD025 but not fasudil prolonged the tail bleeding times, neither drug expanded the volume of ICH or worsened neurological deficits at 48 h compared with vehicle. Although more testing is needed in aged animals and comorbid models such as diabetes, these results suggest ROCK inhibitors may be safe for pre‐hospital administration in acute stroke.
Introduction
Rho‐associated kinases (ROCKs) control numerous cell functions relevant for cerebrovascular diseases, including vascular smooth muscle contraction, endothelial nitric oxide synthesis, and inflammation.1 Upregulation of ROCK activity is associated with vascular dysfunction. For almost two decades ROCK has been explored as a therapeutic target in cerebrovascular diseases. In experimental focal cerebral ischemia, ROCK inhibition has been uniformly efficacious.1, 2, 3, 4
In general, earlier treatment onset affords greater efficacy in focal cerebral ischemia. Although pre‐hospital administration greatly shortens the time to treatment,5 clinical exam alone is often not sufficient to accurately identify the etiology of acute neurological deficits in the field.6 For example, the need to distinguish ischemic from hemorrhagic stroke by neuroimaging is arguably the biggest contributor to delay of thrombolysis. Treatments deemed to be safe in both ischemic or hemorrhagic stroke could be administered in the field without delay for further diagnostic testing.
ROCK inhibition has antithrombotic and vasodilator effects that can be beneficial in acute ischemic stroke.1, 4, 7, 8 However, the same effects can potentially be harmful in primary intracerebral hemorrhage (ICH). We have recently shown that ROCK2‐selective inhibitor KD025 does not exacerbate hemorrhagic transformation in a mouse model of transient focal cerebral ischemia.2 Our primary aim in this study was to directly test whether ROCK inhibitors can be safely administered in the field in patients with acute neurological deficits suspected of stroke, prior to imaging to rule out a primary ICH. To this end, we tested the effect of two different ROCK inhibitors on hematoma volume in a widely used mouse model of collagenase‐induced primary ICH.
Material and Methods
Animals
All experiments were conducted according to protocols approved by the Animal Research Committee of Massachusetts General Hospital and NIH Guide for the Care and Use of Laboratory Animals. We followed the ARRIVE Guidelines for reporting animal research.9 Male CD1 mice (3–4 months, ~24–28 g; Charles River Laboratory, Wilmington, MA) were fed ad libitum. The animals’ health status was monitored throughout the experiments by a health surveillance program.
Procedure and treatments
ICH was induced by striatal collagenase injection.10 Mice were anesthetized with isoflurane and placed in a stereotaxic frame. A 30‐gauge needle was inserted into the striatum (from bregma: 2 mm lateral, 1 mm anterior, 3 mm ventral) through a burr hole, and 0.05 U of collagenase (Type IV; Sigma) was injected in 1 μL of saline over two minutes using an infusion pump. This dose was chosen to yield an intermediate hematoma volume based on prior literature.10 Sham groups received saline injections of the same volume. The needle was left in place for two minutes, and slowly removed. Naïve mice did not undergo any injections. Sixty minutes after collagenase injection, mice were treated with isoform non‐selective ROCK inhibitor fasudil (IC50 2–10 μmol/L11; 10 mg/kg in saline, intraperitoneal; Tocris, Bristol, UK) or ROCK2‐selective inhibitor KD025 (IC50 of 60 nmol/L12; 200 mg/kg in 0.4% cyclodextrin suspension, oral gavage; kindly provided by Kadmon, New York, NY), and compared to their respective vehicles. These doses were selected as the most efficacious in previous studies.2, 3, 4 Although the half‐life of fasudil is under 30 min after intraperitoneal administration, its active metabolite hydroxyfasudil has a longer half‐life (~1 h).13 KD025 has a plasma half‐life of 4 h.2
Experimental design and outcome measures
The investigator providing the treatment was blinded as to which treatment was being administered. Neurological tests were performed daily, and mice were euthanized forty‐eight hours after collagenase injection to measure hematoma volume. Investigators blinded to the treatment groups performed all assessments. Primary endpoint was hematoma volume. Secondary endpoints were edema, wire grip and pole test performance, and mortality.
Experimental groups
Group sizes were pre‐estimated to detect a 25% change in ICH volume with 80% power (α = 0.05), based on pilot experiments with fasudil and KD025. Experimental groups included naïve + vehicle (n = 8), naïve + fasudil (n = 4), naïve + KD025 (n = 4), sham + vehicle (n = 8), sham + fasudil (n = 4), sham + KD025 (n = 4), collagenase + vehicle (n = 20 and 16 as controls for collagenase + fasudil and collagenase + KD025, respectively), collagenase + fasudil (n = 20), collagenase + KD025 (n = 16). Mice were randomly allocated to each treatment group. Fasudil and KD025 experiments were separated by several months in time. Naïve mice did not differ from sham‐injected animals in wire‐grip test performance; therefore, these were pooled into a single control group for statistical comparisons. We excluded three collagenase + saline and one collagenase + fasudil animals that developed no ICH, as a sign of failure of collagenase injection one hour prior to any treatment. In addition, hematoma volume and edema could not be assessed due to a technical failure in tissue preparation in four collagenase + saline and three collagenase + fasudil animals; however, wire‐grip and pole tests were performed in these animals and included in the analyses.
Functional outcome assessment
Neurological deficits were quantified at 24 and 48 h in a blinded manner using the pole test and wire grip tests as previously described with minor modifications.14, 15 Preoperative training was done for 2 days. For wire grip test, both wire grip score and the latency to reach the bottom were recorded. Mice were picked up by the tail and placed on a taut metal wire suspended between two upright bars 50 cm above a padded table. The test was scored as: 0, if the mouse was unable to remain on the wire for more than 30 sec; 1, if the mouse failed to hold on to the wire with both forepaws and hindpaws; 2, if the mouse held on to the wire with both forepaws and hindpaws but not the tail; 3, if the mouse used its tail along with both forepaws and both hindpaws; 4, if the mouse moved along the wire on all four paws plus tail; 5, if mouse that scored 4 points also ambulated down one of the posts supporting the wire. For pole test, the mouse was placed head‐upward on the top of a vertical rough‐surfaced pole (diameter 8 mm; height 55 cm). The time the mouse took to turn completely head downwards (“time to turn”) and the total time it took to descend down and reach the floor after starting its turn (“time to bottom”) were recorded. Both tests were repeated twice and averaged.
Tissue outcome assessment
After neurological testing, brains were harvested and cut into 20 μm‐thick coronal cryosections every 500 μm between 2 mm anterior and 4.5 mm posterior to bregma, and stained with 3,3′‐diaminobenzidine tetrahydrochloride (DAB) as previously described.16 Briefly, Mouse brains were fresh frozen in isopentane precooled on dry ice. Tissue sections were prepared on a Microm 505E Cryostat and mounted onto ionized glass slides. Tissue sections were fixed in 100 percent ethanol and rinsed briefly in PBS. Sections were incubated in DAB solution, prepared using a commercial kit (Vector Laboratories, Burlingame CA) according to manufacturer instructions. Reaction was terminated by washing 3 times in an abundant volume of distilled water. Sections were then serially dehydrated in 50%, 75%, 95%, and 100% ethanol for 2 min each. Sections were cleared in xylene for 5 min and coverslipped using Permount (Fisher Scientific, Waltham MA). Sections were then counterstained with hematoxylin‐eosin. Hematoma area was measured on each section (ImageJ, National Institutes of Health, USA) and integrated across the brain.
Bleeding time
Mice were treated with KD025 or fasudil as above one hour before testing, anesthetized with isoflurane. Approximately 5–6 mm of the tail tip was cut off and tails were immersed in 0.9% isotonic saline at 37°C. The length of time required for bleeding to stop (defined as no blood flow for 1 min) was measured, as previously reported.16, 17
Statistics
Data were statistically tested using two‐way ANOVA for repeated measures (time) followed by post‐hoc Bonferroni's multiple comparisons test, chi‐square test, and unpaired t‐test, and indicated in figure legends. All statistical analyses were performed using Prism 6 (GraphPad Software, La Jolla, CA). Data in figures are shown as mean ± standard error, or whisker (full range) and box (interquartile range) plots, along with median (hortizontal line) and means (+) and individual data points. Data in text are expressed as mean ± standard error.
Results
Treatment with KD025 1 h prior to testing prolonged the tail bleeding time by more than threefold compared with vehicle (963 ± 169 vs. 295 ± 71 sec, respectively, n = 6 each; P = 0.0045; t‐test; Fig. 1). In contrast, fasudil had no effect compared with vehicle (332 ± 81 vs. 434 ± 57 sec, respectively, n = 6 each; P = 0.3276; t‐test). Sham (saline) intrastriatal injections did not produce detectable ICH, and did not significantly affect the pole test or wire‐grip test performance compared to naïve mice with no injections (data not shown). Collagenase injection induced ICH that was for the most part limited to the striatum (Fig. 2A). Mortality was two (12%) in collagenase + saline, three (19%) in collagenase + KD025, and four (21%) in collagenase + fasudil group (P = 0.32; χ 2), all within 24 h of collagenase injection; examination of mice with premature mortality did not reveal larger hematoma sizes compared to the rest of the cohorts (not shown) arguing against mortality bias in tissue and neurological outcomes. Compared to vehicle controls, neither fasudil nor KD025 changed the cross sectional area or the volume of ICH as the primary outcome endpoints (Fig. 2B). Of note, the overall hematoma volumes were larger in the experiment testing fasudil (upper graphs) compared with the experiment testing KD025 (P = 0.04; two‐way ANOVA), most likely owing to different vehicles, collagenase potency, or to the substantial time gap between cohorts. ICH worsened neurological performance compared with baseline (Fig. 3). Compared to vehicle controls, neither fasudil nor KD025 worsened the neurological deficits on wire grip or pole test.
Figure 1.
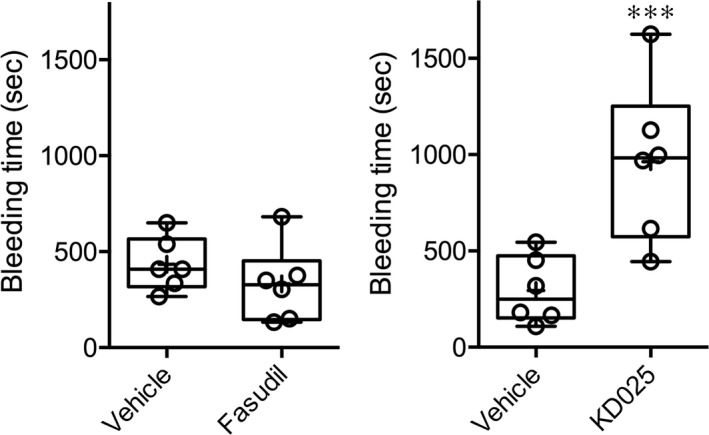
Tail bleeding times in mice treated with isoform‐nonselective ROCK inhibitor fasudil or ROCK2‐selective inhibitor KD025. KD025 but not fasudil significantly prolonged the bleeding time (***P = 0.0045; t‐test). Individual data points are also shown.
Figure 2.
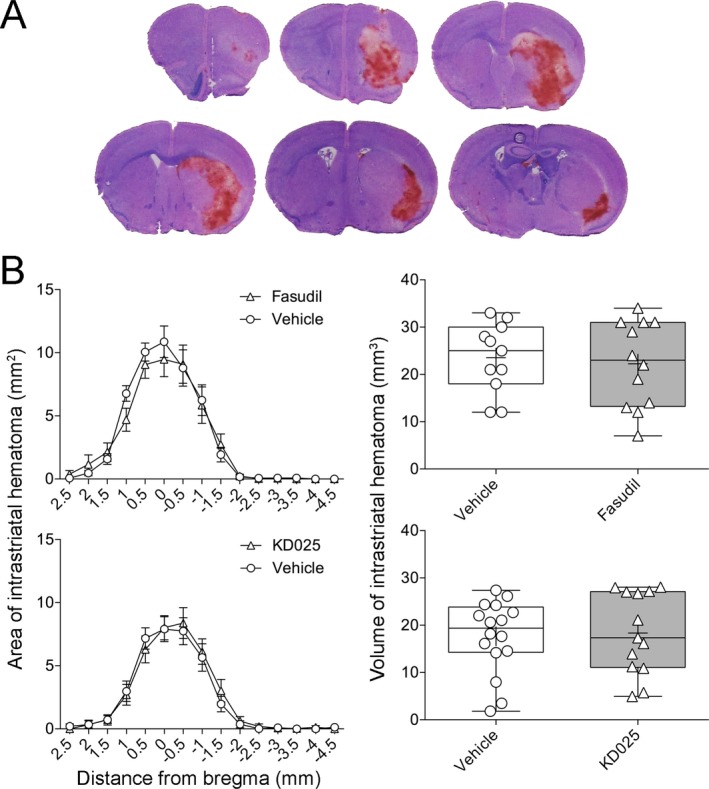
Tissue outcomes after collagenase‐induced intracerebral hemorrhage treated with isoform‐nonselective ROCK inhibitor fasudil or ROCK2‐selective inhibitor KD025. Fasudil and KD025 experiments were separated by several months in time. (A) Representative sections through the hematoma 48 h after intrastriatal collagenase injection. (B) Anteroposterior hematoma areas on each coronal section (left panel) and integrated hematoma volumes (right panel) did not differ between ROCK inhibitor and saline groups (two‐way ANOVA followed by Sidak's post‐hoc multiple comparisons, and unpaired t‐test, respectively).
Figure 3.
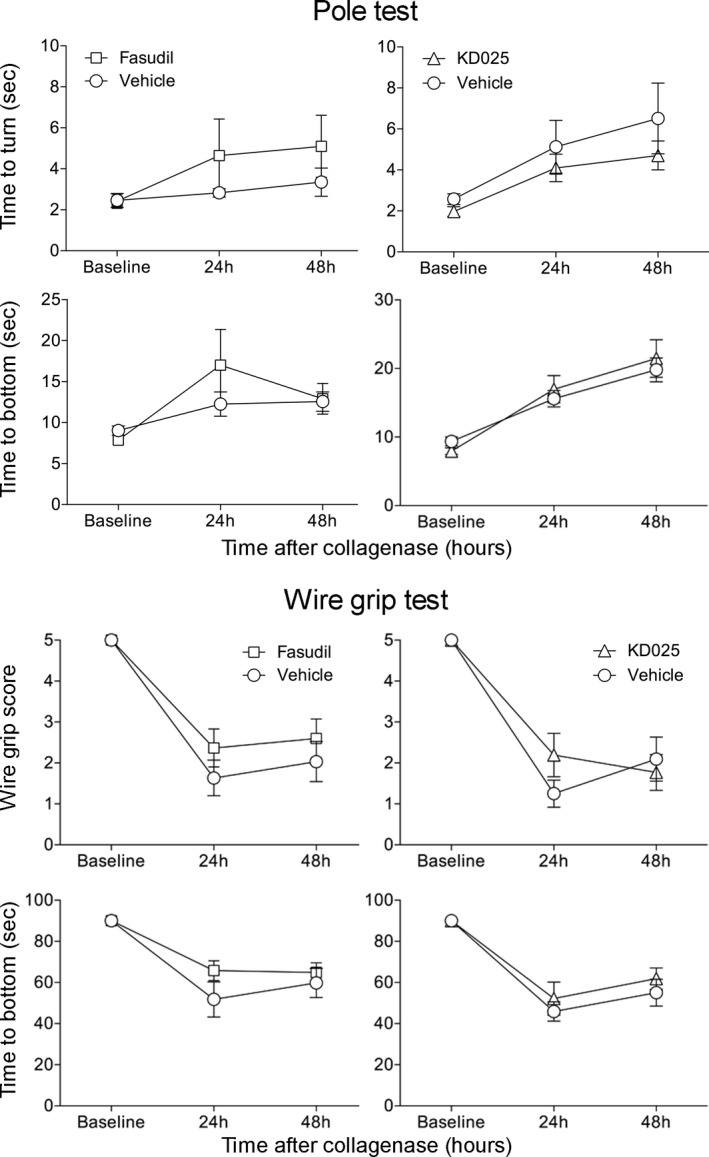
Neurological outcomes after collagenase‐induced intracerebral hemorrhage treated with isoform‐nonselective ROCK inhibitor fasudil or ROCK2‐selective inhibitor KD025. There was a significant worsening of neurological function after ICH. Fasudil or KD025 did not significantly alter pole test and wire‐grip test scores and times compared with vehicle‐treated mice (two‐way ANOVA for repeated measures followed by Bonferroni post‐hoc comparisons).
Discussion
We undertook this study to specifically examine whether ROCK inhibitors administered in a pre‐hospital, pre‐imaging setting for acute stroke might worsen the outcome if the final diagnosis is ICH rather than ischemic stroke. This is because ROCK inhibition relaxes vascular smooth muscle, dilates cerebral vessels, and may inhibit platelet aggregation,1, 8 potentially worsening ICH.1 Our results show that neither fasudil (isoform‐nonselective ROCK inhibitor) nor KD025 (selective ROCK2 inhibitor), at doses efficacious in ischemic stroke, had a significant effect on hematoma volume or neurological outcome compared with vehicle, when administered acutely during an actively evolving ICH, alleviating these concerns. Although these ROCK inhibitors appeared to be inert in acute ICH, it is possible that the potential harmful effects of ROCK inhibition in ICH (e.g., vasodilator, antithrombotic) might have offset the potential beneficial effects (e.g., anti‐inflammatory, anti‐edema, anti‐hypertensive).1, 4
We found that ROCK2 inhibitor KD025, but not isoform‐nonselective inhibitor fasudil, prolonged the bleeding time, consistent with previous reports.8, 17 It is unclear why fasudil does not prolong the bleeding time, but its hypotensive effect may be one explanation. Indirect evidence from other models suggests that ROCK inhibitors do not exacerbate hemorrhage risk. For example, fasudil has been shown to ameliorate blood‐brain barrier (BBB) disruption, vascular endothelial growth factor and matrix metalloproteinase activation, and hemorrhagic transformation caused by tissue plasminogen activator (tPA) treatment after transient middle cerebral artery occlusion in rats and mice.17, 18, 19 Despite the prolonged bleeding times in our study, in an in vitro human BBB model, KD025 was more potent than fasudil in preventing tPA‐ and plasminogen‐induced BBB disruption under both hypoxic and normoxic conditions,20 and did not worsen hemorrhagic transformation after focal cerebral ischemia in mice.2 Although these data do not directly pertain to primary intracerebral hemorrhage, taken together with our findings, they further support the safety of ROCK inhibition in this setting.
We selected a collagenase model of ICH over autologous blood injection because the latter was not appropriate to test the effect of ROCK inhibition on hematoma volume. The progressive ICH growth in the collagenase model mimics the expanding nature of hematoma in humans.21, 22 Moreover, neurological deficits typically recover quickly after autologous blood injection in contrast with the prolonged deficits in patients.10, 22, 23 We did not examine late time points because our primary endpoint hematoma volume reaches a plateau within 48 h, and peak neurological deficits in experimental ICH occur within the first few days of injury and dissipate significantly within a week.24, 25, 26 Importantly, lack of effect on hematoma growth by ROCK inhibitors was not due to a ceiling effect because hematoma volumes in our dataset showed a fair amount of scatter. Additionally, larger hematoma volumes have been reported in anticoagulated mice, and in pilot experiments, doubling the dose of collagenase yielded significantly larger hematomas (50 ± 3 mm3) and mortality (60%) in a pilot cohort (n = 5; data not shown). Therefore, while there are limitations to our experiments, we think they approximate the clinical ICH reasonably well.
Our primary aim was to simulate field administration of ROCK inhibitors by emergency medical personnel shortly after the onset of acute neurological deficits in stroke of unknown mechanism (i.e., pre‐hospital, pre‐CT). In the collagenase model of ICH, hemorrhage starts within an hour and tends to plateau within 6 h.10, 27 Therefore, we chose to administer the treatments 1 h after collagenase injection. Peak plasma KD025 concentrations are reached around 1 h after oral dosing, and significant concentrations persist at 9 h.2 Therefore, our timing of KD025 treatment well covered the hyperacute stage of hematoma growth. Fasudil has a shorter plasma half‐life, but since our primary aim was to test whether ROCK inhibitors are safe to use in the pre‐hospital setting, where a patient would only receive one dose in the field prior to arriving to the emergency room for a CT, we chose not to repeat the fasudil dose in our study. Nevertheless, the relatively short plasma half‐life of fasudil (~30 min) does not necessarily reflect its biological half‐life (e.g., tissue concentrations, target engagement). Numerous studies used a single dose or once a day dosing of fasudil, and found robust efficacy, for example, in focal cerebral ischemia.1, 3, 4, 28 Of course, more work is needed to test whether ROCK inhibition is efficacious in ICH, including earlier treatment, repeat dosing, and dose escalation. Overall, our treatment regimen ensured sufficient exposure to ROCK inhibitors throughout the period of maximum hematoma growth, making it unlikely that any detrimental effects were missed.
Overall, our study suggests that acute stroke patients can potentially be safely treated with ROCK inhibitors in the pre‐hospital setting as well as in cases where hemorrhagic transformation of the infarct is a risk. However, further translational studies are needed to determine the safety of ROCK inhibition in females and aged animals, in comorbid models such as diabetes and hypertension, and in other stroke mimics. Testing potential acute stroke therapies for their safety in ICH will be a critical strategy to facilitate clinical trials targeting ultra‐early interventions in a pre‐hospital setting.
Author Contribution
MA and CA conceived and designed the study. MA, TQ, HS, PF, and HHK performed the experiments and acquired the data. All authors analyzed and interpreted the data. MA and CA drafted the article. All authors revised it critically for important intellectual content, and all authors approved the version to be published.
Conflict of Interest
There is no financial or other relationship that might lead to a perceived conflict of interest.
Acknowledgements
This work was in part supported by NIH (NS055104), the Fondation Leducq, the Heitman Foundation, and the Ellison Foundation.
Funding Information
This work was in part supported by NIH (NS055104), the Fondation Leducq, the Heitman Foundation, and the Ellison Foundation.
Funding Statement
This work was funded by NIH grant NS055104; Fondation Leducq grant ; Heitman Foundation grant ; Ellison Foundation grant .
Contributor Information
Murtaza Akhter, Email: murtazaakhter@gmail.com.
Cenk Ayata, Email: cayata@mgh.harvard.edu.
References
- 1. Shin HK, Salomone S, Ayata C. Targeting cerebrovascular Rho‐kinase in stroke. Expert Opin Ther Targets 2008;12:1547–1564. [DOI] [PubMed] [Google Scholar]
- 2. Lee JH, Zheng Y, von Bornstadt D, et al. Selective ROCK2 inhibition in focal cerebral ischemia. Ann Clin Transl Neurol 2014;1:2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin HK, Huang PL, Ayata C. Rho‐kinase inhibition improves ischemic perfusion deficit in hyperlipidemic mice. J Cereb Blood Flow Metab 2014;34:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shin HK, Salomone S, Potts EM, et al. Rho‐kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab 2007;27:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saver JL, Starkman S, Eckstein M, et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N Engl J Med 2015;372:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Besson G, Robert C, Hommel M, Perret J. Is it clinically possible to distinguish nonhemorrhagic infarct from hemorrhagic stroke? Stroke 1995;26:1205–1209. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki Y, Yamamoto M, Wada H, et al. Agonist‐induced regulation of myosin phosphatase activity in human platelets through activation of Rho‐kinase. Blood 1999;93:3408–3417. [PubMed] [Google Scholar]
- 8. Sladojevic N, Oh GT, Kim HH, et al. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2‐deficient platelets. Cardiovasc Res 2017;113:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Foerch C, Arai K, Jin G, et al. Experimental model of warfarin‐associated intracerebral hemorrhage. Stroke 2008;39:3397–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 2000;351(Pt 1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boerma M, Fu Q, Wang J, et al. Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin. Blood Coagul Fibrinolysis 2008;19:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Satoh S, Utsunomiya T, Tsurui K, et al. Pharmacological profile of hydroxy fasudil as a selective rho kinase inhibitor on ischemic brain damage. Life Sci 2001;69:1441–1453. [DOI] [PubMed] [Google Scholar]
- 14. Matsuura K, Kabuto H, Makino H, Ogawa N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods 1997;73:45–48. [DOI] [PubMed] [Google Scholar]
- 15. Bermpohl D, You Z, Korsmeyer SJ, et al. Traumatic brain injury in mice deficient in Bid: effects on histopathology and functional outcome. J Cereb Blood Flow Metab 2006;26:625–633. [DOI] [PubMed] [Google Scholar]
- 16. Wasserman JK, Yang H, Schlichter LC. Glial responses, neuron death and lesion resolution after intracerebral hemorrhage in young vs. aged rats. Eur J Neurosci 2008;28:1316–1328. [DOI] [PubMed] [Google Scholar]
- 17. Ishiguro M, Kawasaki K, Suzuki Y, et al. A Rho kinase (ROCK) inhibitor, fasudil, prevents matrix metalloproteinase‐9‐related hemorrhagic transformation in mice treated with tissue plasminogen activator. Neuroscience 2012;18:302–312. [DOI] [PubMed] [Google Scholar]
- 18. Wang L, Fan W, Cai P, et al. Recombinant ADAMTS13 reduces tissue plasminogen activator‐induced hemorrhage after stroke in mice. Ann Neurol 2013;73:189–198. [DOI] [PubMed] [Google Scholar]
- 19. Fukuta T, Asai T, Yanagida Y, et al. Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke. FASEB J 2017;31:1879–1890. [DOI] [PubMed] [Google Scholar]
- 20. Niego B, Lee N, Larsson P, et al. Selective inhibition of brain endothelial Rho‐kinase‐2 provides optimal protection of an in vitro blood‐brain barrier from tissue‐type plasminogen activator and plasmin. PLoS ONE 2017;12:e0177332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu W, Gao Y, Chang CF, et al. Mouse models of intracerebral hemorrhage in ventricle, cortex, and hippocampus by injections of autologous blood or collagenase. PLoS ONE 2014;9:e97423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. MacLellan CL, Silasi G, Poon CC, et al. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab 2008;28:516–525. [DOI] [PubMed] [Google Scholar]
- 23. Brott T, Broderick J, Kothari R, et al. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke 1997;28:1–5. [DOI] [PubMed] [Google Scholar]
- 24. Matsushita H, Hijioka M, Hisatsune A, et al. MRI‐based analysis of intracerebral hemorrhage in mice reveals relationship between hematoma expansion and the severity of symptoms. PLoS ONE 2013;8:e67691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu L, Barfejani AH, Qin T, et al. Fingolimod exerts neuroprotective effects in a mouse model of intracerebral hemorrhage. Brain Res 2014;25:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Munakata M, Shirakawa H, Nagayasu K, et al. Transient receptor potential canonical 3 inhibitor Pyr3 improves outcomes and attenuates astrogliosis after intracerebral hemorrhage in mice. Stroke 2013;44:1981–1987. [DOI] [PubMed] [Google Scholar]
- 27. Tanaka Y, Marumo T, Shibuta H, et al. Serum S100B, brain edema, and hematoma formation in a rat model of collagenase‐induced hemorrhagic stroke. Brain Res Bull 2009;78:158–163. [DOI] [PubMed] [Google Scholar]
- 28. Vesterinen HM, Currie GL, Carter S, et al. Systematic review and stratified meta‐analysis of the efficacy of RhoA and Rho kinase inhibitors in animal models of ischaemic stroke. Syst Rev 2013;2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]