

ABSTRACT
The human gut is home to trillions of bacteria and provides the scaffold for one of the most complex microbial ecosystems in nature. Inflammatory bowel diseases, such as Crohn's disease, involve a compositional shift in the microbial constituents of this ecosystem with a marked expansion of Enterobacteriaceae, particularly Escherichia coli. Adherent-invasive E. coli (AIEC) strains are frequently isolated from the biopsies of Crohn's patients, where their ability to elicit inflammation suggests a possible role in Crohn's pathology. Here, we consider the origins of the AIEC pathovar and discuss how risk factors associated with Crohn's disease might influence AIEC colonization dynamics within the host to alter the overall disease potential of the microbial community.
KEYWORDS: Gut inflammation, nososymbiocity, dysbiosis
Introduction
Crohn's disease (CD) is an inflammatory bowel condition with increasing incidence worldwide, most recently expanding into newly industrialized nations in South America, Asia, and the Middle East.1 In Canada, CD incidence is among the highest in the world where about 20 per 100,000 individuals are diagnosed annually with the disease.2 The protracted and often refractory course of CD constitutes a significant societal burden, both in direct medical costs to the health care system, and also in indirect costs that often exceed health spending.1
Transmural inflammation and ulceration are common signatures of CD and about half of patients develop extraintestinal symptoms as well.2 An abnormal immune response to commensal gut microbes is believed to be the driving force for CD-associated inflammation, yet the triggers of this aberrant immune response remain unclear.3 Hampering the understanding of CD etiology is a complex web of host and environmental factors that interact in indistinct ways to drive disease progression. Genome-wide association studies have established genetic links to CD; however, only ∼12% of CD patients have a family history of the disorder4,5 and no single gene variant comes close to full penetrance in affected individuals. These findings stress the importance of non-genetic factors in disease onset and progression. It is tempting to speculate that the non-hereditary factors have propelled the high levels of CD in various countries, yet more studies are required to investigate this hypothesis.
CD is now considered a global disease. The sharp rise in CD in newly industrialized countries again emphasizes the involvement of a constellation of non-hereditary factors.1 This may include the consumption of diets high in fat and low in fiber,6 smoking,7 and the use of certain medications including oral contraceptives, aspirin, and non-steroidal anti-inflammatory drugs.2 The use of antibiotics, particularly during childhood, is linked to an increased risk of new onset CD.8 Despite the obvious beneficial role of antibiotics in combating bacterial infections, their indiscriminate activity also disturbs the balanced partnership between the human host and the gut microbiome, now recognized to preside over diverse states of health and disease in our bodies.9 Multiple studies have used metagenomics and 16s RNA profiling to examine the composition of gut bacteria during CD. This work as led to the broad conclusion that the microbiome of CD patients is compositionally different compared to healthy subjects.10-13 Changes in microbial abundance, also known as dysbiosis, can markedly alter human immune responses, thus disturbing gut homeostasis and possibly leading to disease. Dysbiosis in CD is characterized by a loss of keystone species in the phyla Firmicutes and Bacteriodetes and the enrichment of Actinobacteria and Gammaproteobacteria.14,15
Nearly 20 years ago, a newly described pathovar of the species Escherichia coli was isolated from CD patients in the laboratory of the late Dr. Darfeuille-Michaud, who made seminal contributions to AIEC research during her lifetime.16 These E. coli were referred to as adherent-invasive E. coli (AIEC), reflecting their ability to adhere to gut epithelial cells and their unusual ability to invade into mucosal epithelial cells. This moniker differentiates AIEC from other better-described E. coli pathovars like enteropathogenic E. coli, enterohemorrhagic E. coli, and enteroinvasive E. coli (EIEC).17 Despite the common ability of AIEC and EIEC to invade and replicate within intestinal epithelial cells, a closer scrutiny reveals a clear distinction between the two pathovars at the genomic level. In addition to the distinctive biochemical differences between the two pathotypes, AIEC lacks the typical invasins and pathogenicity islands found in EIEC. AIEC are now known to have an intracellular lifestyle where they can induce inflammatory pathways in host cells.18,19 Numerous studies have confirmed that AIEC are enriched in humans with CD, where they are about six-times more likely to be isolated from ileal and colonic samples compared to healthy controls and represent the dominant bacterial species present.16,20-25 Attention around the potential role of AIEC in the pathophysiology of CD is growing; however much remains to be learned about the host-pathogen interactions that govern AIEC infection biology.
Where does adherent-invasive Escherichia coli come from?
E. coli is a diverse bacterial species whose members range from seemingly innocuous commensal strains to quite dangerous human pathogens. Pathovar designations are used to classify E. coli into groups with unique molecular mechanisms that govern their pathogenic behavior.26 The genetic determinants that help define E. coli pathovars (including serotype, toxins, and virulence factors) represent the basic tenets for their identification and facilitate the tracking of their evolutionary history.27,28 While much is known about the evolution of many E. coli pathotypes, the origin of the AIEC group is less clear. One key challenge in defining the AIEC pathovar is that the genetic factors conferring the adherent-invasive phenotype are not fully defined. Consequently, the identification of AIEC is done based on a series of in vitro phenotype assays that are laborious, time-consuming, and somewhat non-standardized. Also, virulence determinants that define other E. coli pathovars at the genetic level (i.e. Shiga toxin, type III secretion systems) are not found in AIEC.17 While this fact can be used as exclusion criteria when attempting to classify isolates of E. coli from patients, a molecular genetic signature that distinguishes the AIEC pathovar remains elusive. In a recent study, comparative whole-genome analysis of 14 AIEC strains identified a potential subgroup within the B2 phylotype that appeared more similar due to three genetic insertions that differentiated them from commensal E. coli.29 A separate study by a different group did not identify a readily distinguishable genomic signature among 11 different B2 phylotype AIEC strains although these strains were from a different geographic locale.22 As shown in different genomic studies, AIEC appear to be most closely related to extraintestinal E. coli strains such as UPEC, APEC, and ExPEC, which are also among the B2 clade.17,29,30 Furthermore, many virulence factors were found to be shared between AIEC and UPEC, including genes that are required for iron acquisition and transport.17 Together, these findings suggest that AIEC do not arise by parallel evolution and clonal expansion as described for the notorious O157:H7 enterohemorrhagic E. coli.31
At this point, available evidence suggests that AIEC can evolve from diverse founder populations and use genetically distinct mechanisms to attain the ‘AIEC phenotype’. This is reminiscent of the genetic variability that exists among uropathogenic E. coli (UPEC), which also lacks a common genetic signature.32 UPEC are members of the so-called ‘extraintestinal pathogenic E. coli’ to which AIEC are also closely related.17 The selective drivers of AIEC's evolutionary trajectory remain obscure, yet they likely originate within the host. Given that individual host environments can drive adaptive bacterial evolution,33 an important question is whether unique host environments (i.e. different CD patients; biogeography within a single individual; or combinatorial risk factors, for example) might select for AIEC emergence and outgrowth. Considering that research is now mobilizing towards AIEC as a new therapeutic frontier in CD,34-39 efforts to understand the selective pressures driving AIEC evolution should be redoubled.
Factors mediating AIEC virulence
Despite the genetic diversity of AIEC, several strains do share common virulence factors, albeit not unique to AIEC. One of the defining features of AIEC is their ability to adhere to intestinal epithelial cells, which is likely facilitated by several bacterial surface structures. For example, surface expression of chitin-binding domains by AIEC was found to mediate their adherence to the chitinase-like receptors on the intestinal epithelium.40 Additionally, long polar fimbriae mediate AIEC attachment to Peyer's patches, allowing AIEC to localize to the terminal ileum from where they are often isolated.41 And finally, AIEC type I pili can bind carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) via FimH, the terminal subunit of these surface appendages.42,43 Unlike commensal E. coli strains, FimH in AIEC has accumulated a small number of non-synonymous mutations that may facilitate binding to CEACAM6.44 Notably, the abundance of CEACAM6 in the ileum of CD patients43 makes FimH a plausible target for pathoadaptation during AIEC evolution, and is the focus of most of the anti-adhesive strategies in therapeutic development in this space. Other host surface receptors that were found to be upregulated in the ileum of CD patients include the endoplasmic reticulum stress response glycoprotein Gp96. This host protein may be clinically relevant because it has been shown to interact with AIEC OmpA, a member of a family of outer membrane porins in Gram-negative bacteria.45 Given that both CEACAM6 and Gp96 are more abundant during inflammation, the expression of inflammation itself may promote the redistribution of AIEC to sites proximal to the epithelial surface, a process seen by the host as a danger signal that initiates even greater inflammatory responses.46
Other virulence factors in AIEC may help dictate its distribution within a host. These include GipA, a putative transcription factor involved in AIEC transcytosis of Peyer's patches similar to its function in the enteric pathogen Salmonella enterica serovar Typhimurium.47,48 Access to the gut epithelium may be facilitated by Vat-AIEC, a vacuolating toxin related to one found in avian pathogenic E.coli that has proteolytic activity towards mucin.49 It is noteworthy that both Vat-AIEC and long polar fimbriae were upregulated in the presence of bile salts. Although little is known about the chemical cues that direct AIEC gene regulation in the host, this finding infers the presence of potential regulatory circuits that have evolved to detect and respond to chemical signals that are unique to the gut environment. AIEC are able to invade, persist, and in some cases replicate within intestinal epithelial cells.50 Multiple AIEC strains encode an invasin, called IbeA that is important for invasion of Caco-2 and M-cells, as well as survival within macrophages51 but it should be noted that a full accounting of how AIEC invades into cells is ongoing. Although IbeA is the only invasin identified in AIEC, the IbeA mutant strain was still able to invade epithelial cells, thus suggesting the possible presence of other uncharacterized invasins. Furthermore, the genomes of multiple AIEC strains encode for type VI secretion system (T6SS) components.29 The T6SS is a surface structure related to contractile phage tails and used by Gram-negative bacteria to translocate effector proteins into both bacterial and eukaryotic cells. Although originally defined for its role in inter-bacterial competition,52 the T6SS is now recognized to have wider target specificity. For example, in some bacteria the T6SS is involved in dampening host immunity,53 promoting intracellular growth in macrophages,54 and interacting with the host microtubule network to promote bacterial internalization into host cells.55 It remains to be determined whether type VI secretion contributes to AIEC pathogenesis, but given the role mediated by this machinery in other species, investigation of it in AIEC is warranted.
Antimicrobial peptides are host defense peptides that are capable of killing microbes by disrupting their membrane integrity and are often enriched in the gut during inflammation.56 Many enteric pathogens can evade killing by these bactericidal molecules using enzymatic modification systems that alter surface chemistry of lipopolysaccharide, and by outer membrane proteases that cleave dibasic sites within the core of these cationic peptides. AIEC strain NRG857c, one of two prototype strains that have been fully sequenced, has high levels of resistance towards several antimicrobial peptides that are common in the gut.57 Resistance to this arm of innate host defense in this particular strain is mediated by a plasmid-based genomic island (PI-6) that encodes an outer membrane protease (ArlC) and a Mig-14 family protein (ArlA). Mig-14 homologs are involved in antimicrobial peptide resistance in multiple bacteria, yet their mode of action is not fully understood.
The diversity of virulence factors displayed by multiple AIEC strains, some of them shared with ExPEC, suggests that members of this pathovar have evolved different strategies to colonize their hosts. This is also in keeping with the genetic variability that exists within the AIEC. It remains to be determined whether the host environment might favor AIEC strains that harbor a specific set of virulence determinants. We believe that an important next step in our understanding of AIEC is to broaden the scope of genomic studies to both bacteria and host. This could reveal potential associations between bacterial genotypes and host genetic backgrounds from which the strains arose.
A polymicrobial view of AIEC pathogenesis: The enigmatic role of acute infectious gastroenteritis in the long-term risk of CD onset
Crohn's disease is more common in individuals following acute infectious gastroenteritis caused by Salmonella and other enteric pathogens, sometimes with onset times on the order of years after the infectious episode.58-60 The mechanistic basis for this long-term risk association following an acute infectious stimulus is enigmatic. The interaction between AIEC and their host seem to bring about multiple states, ranging from commensalism to pathogenic; however, the triggers for transitioning between these states are not known. The pathogenic potential of a microbe is often influenced by synergistic and antagonistic interactions with other microbes that collectively influence the community's pathogenic potential, known as nososymbiocity.61 Thus, disruption of gut homeostasis may be a key trigger that increases the overall disease potential of the microbial community.61 The well known ability of enteric pathogens to induce gut inflammation and disrupt the microbial community therein led us to question whether acute infectious gastroenteritis could be a trigger for the emergence of keystone pathogens like AIEC.
To address this question, we leveraged a previous model for chronic AIEC colonization that we developed in a range of inbred mouse lines.62 This resulted in two new polymicrobial infection models to measure host comorbidities related to acute gastroenteritis in mice colonized with a clinical isolate of AIEC. In these models, mice are colonized sub-clinically with AIEC and later exposed to secondary infection stimuli with either S. Typhimurium or Citrobacter rodentium, modeling what might occur in healthy AIEC-colonized individuals exposed to acute infectious gastroenteritis. S. Typhimurium is a prominent enteric pathogen linked to food poisoning in humans that triggers a strong inflammation in the gut,63 while C. rodentium is a murine pathogen that recapitulates the gastroenteritis mediated by enteropathogenic E. coli in humans.64 Using these models, we showed that AIEC-colonized mice exposed to acute infectious gastroenteritis have a significantly worsened outcome compared to AIEC-naïve animals exposed to the same infection stimuli.65 To our surprise, disease activity was not driven by the secondary pathogen but rather by AIEC in the post-infectious period when the host had largely constrained the secondary pathogen. Acute bacterial gastroenteritis induced an AIEC bloom in mucosal tissue that was associated with host damage. Importantly, rendering AIEC susceptible to host defense peptides, or quelling the inflammation induced by the secondary pathogen prevented AIEC blooms and mitigated the disease activity, indicating that AIEC is a tractable disease modifier following acute infectious gastroenteritis.
Other environmental factors can instigate gut inflammation that might be relevant to AIEC colonization dynamics. For example, dysbiosis triggered by antibiotics or a “Western diet” high in sugar and fat was shown to cause a reduction in short-chain fatty acid (SCFA) levels in the intestine.66-68 SCFA can mediate anti-inflammatory functions in the gut via the activation of regulatory T cells,69 and the reduction of oxygen availability.69 Antibiotic treatment was shown to increase the inflammatory tone of the gut mucosa in mice, characterized by increased infiltration of neutrophils and inflammatory monocytes in the lumen. The converging view is that the induction of inflammation in mice with either antibiotics,62 Western diet,66 or acute infectious gastroenteritis65 causes expansion of resident AIEC that is linked to worsening levels of gut pathology, leading to the suggestion that inflammation provides AIEC with a selective advantage in the gut (Fig. 1).
Figure 1.
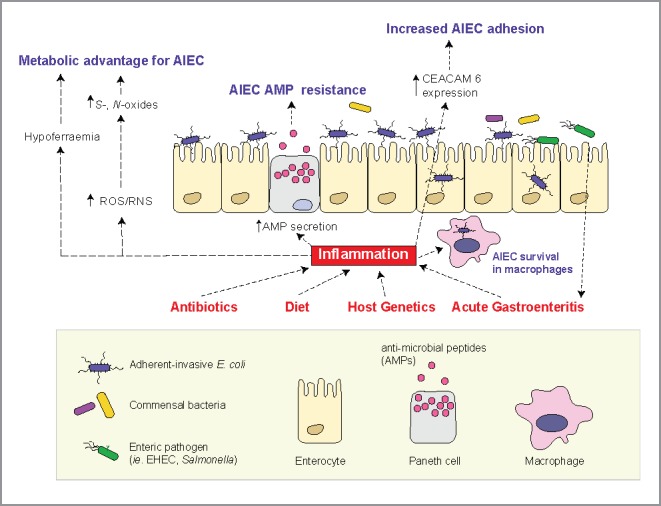
A schematic diagram that summarizes some of the inflammation-mediated changes in the gut and their influence on AIEC colonization. Gut inflammation can be triggered by multiple factors like host genetics, antibiotic administration, diet, and acute gastroenteritis. The resulting proinflammatory environment mediates pronounced changes in the human gut, which includes hypoferremia, the generation of alternative electron acceptors, increased secretion of antimicrobial peptides, and overexpression of CEACAM6 surface receptors by intestinal epithelia. During their evolution, AIEC strains have gained traits that confer on them a competitive advantage in the inflamed gut. This includes the ability to utilize S- and N- oxides as alternative electron acceptors, an abundance of iron acquisition genes, resistance to antimicrobial peptides and the ability to bind to CEACAM6 through a modified FimH protein.
In addition to the overt pathological changes triggered by inflammatory reactions at mucosal sites, inflammation can change nutrient availability in the gut. For example, hypoferremia is a hallmark of gut inflammation, and acts as a host defense mechanism against invading pathogens by limiting iron availability.70 Genes involved in iron utilization such as siderophores are enriched in AIEC compared to E. coli strains from other pathotypes.71 Siderophores are also commonly found in extraintestinal E. coli species such as UPEC where it is an important virulence factor. Since siderophores, like aerobactin, provide a strong fitness advantage in hypoferremic conditions,72 this may suggest that AIEC emerge under selection in an iron-poor environment. Similar to the closely related UPEC and APEC, aerobactin was found to be important for AIEC virulence.17 The production of reactive oxygen and nitrogen species by neutrophils is another important outcome of gut inflammation.68 Upon release into the gut lumen, reactive oxygen and nitrogen species can react with organic sulphides and tertiary amines to generate S- and N- oxides that can be used as alternative electron acceptors by some facultative anaerobes. It is conceivable that nutritional changes associated with inflammation might create a selective niche for AIEC that brings about their pathogenic potential, or perhaps renders hosts more susceptible to de novo colonization through loss of some facet of colonization resistance, a host state that we are just beginning to understand with granularity.73 More studies are required to investigate the metabolic behavior of AIEC during gut inflammation, which might provide insights to the evolutionary trajectories that shaped this pathovar.
Concluding remarks
While a microbial basis for Crohn's pathogenesis is well founded, it is currently unclear how microbes influence, and are influenced by, the inflammatory environment in the gut. The role that environmental risk factors play in disease expression is also not fully understood, however the interactions created along a genes-environment-microbe axis hold the key to unlock future preventions and therapies. Emerging data from multiple studies place microbes at the epicentre of CD pathogenesis, with members of the emergent AIEC pathovar being possible disease modifiers. Evidence that the host environment influences AIEC evolution, colonization, and disease potential makes it a fascinating case study in host-pathogen interactions. For example, if gut inflammation plays a fundamental role in evoking the pathogenic character of AIEC, then many questions emerge. How does host genotype affect AIEC behavior in vivo? Do anti-inflammatory medicines used in CD treatments have secondary effects on AIEC colonization dynamics that improve disease outcome? How does AIEC influence disease potential of the microbial community in which it resides? Are individuals with gut inflammation more susceptible to host-to-host transmission of AIEC? Phenotypic changes in the gut induced by inflammatory reactions might provide the selective niche for the evolution of AIEC strains from different phylogroups and explain the lack of a common ancestor. To mitigate the burden of CD, a primary goal of research centers on the interactions between genes, the environment, and intestinal microbes using robust preclinical models. More studies investigating AIEC pathogenesis in the context of host genetics and environmental risk factors linked to CD will be informative towards this goal. Finally, as evidence mounts against AIEC as a tractable disease modifier, understanding the provenance of AIEC, their movement through the environment, and their transmission dynamics from host-to-host is likely to yield major public health dividends.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Research on AIEC in the Coombes laboratory is funded by grants from the Canadian Institutes of Health Research and Crohn's and Colitis Canada. A.O. is supported by a Canada Graduate Scholarship from the Canadian Institutes of Health Research. B.K.C. is the Canada Research Chair in Infectious Disease Pathogenesis.
References
- [1].Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterol. 2017;152:313-21e2. doi: 10.1053/j.gastro.2016.10.020. [DOI] [PubMed] [Google Scholar]
- [2].Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn's disease. Lancet. 2017;389:1741-55. doi: 10.1016/S0140-6736(16)31711-1. PMID:27914655 [DOI] [PubMed] [Google Scholar]
- [3].Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298-306. doi: 10.1038/nature10208. PMID:21677746 [DOI] [PubMed] [Google Scholar]
- [4].Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al.. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118-25. doi: 10.1038/ng.717. PMID:21102463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu TC, Stappenbeck TS. Genetics and pathogenesis of inflammatory bowel disease. Annu Rev Pathol. 2016;11:127-48. doi: 10.1146/annurev-pathol-012615-044152. PMID:26907531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Korzenik JR, Fuchs CS, Willett WC, Richter JM, Chan AT. A prospective study of long-term intake of dietary fiber and risk of Crohn's disease and ulcerative colitis. Gastroenterol. 2013;145:970-7. doi: 10.1053/j.gastro.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc. 2006;81:1462-71. doi: 10.4065/81.11.1462. PMID:17120402 [DOI] [PubMed] [Google Scholar]
- [8].Ungaro R, Bernstein CN, Gearry R, Hviid A, Kolho KL, Kronman MP, Shaw S, Van Kruiningen H Colombel JF, Atreja A.. Antibiotics associated with increased risk of new-onset Crohn's disease but not ulcerative colitis: a meta-analysis. Am J Gastroenterol. 2014;109:1728-38. doi: 10.1038/ajg.2014.246. PMID:25223575 [DOI] [PubMed] [Google Scholar]
- [9].Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015;31:69-75. doi: 10.1097/MOG.0000000000000139. PMID:25394236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al.. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382-92. doi: 10.1016/j.chom.2014.02.005. PMID:24629344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterol. 2014;146:1489-99. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al.. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. PMID:23013615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780-5. doi: 10.1073/pnas.0706625104. PMID:17699621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Oberc A, Coombes BK. Convergence of external Crohn's disease risk factors on intestinal bacteria. Front Immunol. 2015;6:558. doi: 10.3389/fimmu.2015.00558. PMID:26579131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Forbes JD, Van Domselaar G Bernstein CN. The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol. 2016;7:1081. doi: 10.3389/fmicb.2016.01081. PMID:27462309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterol 1998;115:1405-13. doi: 10.1016/S0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- [17].Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, Ziebell K, Torres AG, Karmali MA, Coombes BK. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics. 2010;11:667. doi: 10.1186/1471-2164-11-667. PMID:21108814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barnich N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli and Crohn's disease. Curr Opin Gastroenterol. 2007;23:16-20. doi: 10.1097/MOG.0b013e3280105a38. PMID:17133079 [DOI] [PubMed] [Google Scholar]
- [19].Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterol. 2011;140:1720-28. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- [20].Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterol. 2004;127:412-21. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- [21].Kotlowski R, Bernstein CN, Sepehri S, Krause DO. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut. 2007;56:669-75. doi: 10.1136/gut.2006.099796. PMID:17028128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].O'Brien CL, Bringer MA, Holt KE, Gordon DM, Dubois AL, Barnich N, Darfeuille-Michaud A, Pavli P. Comparative genomics of Crohn's disease-associated adherent-invasive Escherichia coli. Gut. 2016. 66:1382-1389. doi: 10.1136/gutjnl-2015-311059. PMID:27196580 [DOI] [PubMed] [Google Scholar]
- [23].Negroni A, Costanzo M, Vitali R, Superti F, Bertuccini L, Tinari A, Minelli F, Di Nardo G, Nuti F, Pierdomenico M, et al.. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:913-24. doi: 10.1002/ibd.21899. PMID:21994005 [DOI] [PubMed] [Google Scholar]
- [24].Conte MP, Longhi C, Marazzato M, Conte AL, Aleandri M, Lepanto MS, Zagaglia C, Nicoletti M, Aloi M, Totino V, et al.. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn's disease patients: phenotypic and genetic pathogenic features. BMC Res Notes. 2014;7:748. doi: 10.1186/1756-0500-7-748. PMID:25338542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterol. 2004;127:80-93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- [26].Croxen MA, Finlay BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol. 2010;8:26-38. PMID:19966814 [DOI] [PubMed] [Google Scholar]
- [27].Coombes BK, Gilmour MW, Goodman CD. The evolution of virulence in non-O157 shiga toxin-producing Escherichia coli. Front Microbiol. 2011;2:90. doi: 10.3389/fmicb.2011.00090. PMID:21833329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Coombes BK, Wickham ME, Mascarenhas M, Gruenheid S, Finlay BB, Karmali MA. Molecular analysis as an aid to assess the public health risk of non-O157 Shiga toxin-producing Escherichia coli strains. Appl Environ Microbiol. 2008;74:2153-60. doi: 10.1128/AEM.02566-07. PMID:18245257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Desilets M, Deng X, Rao C, Ensminger AW, Krause DO, Sherman PM, Gray-Owen SD. Genome-based definition of an inflammatory bowel disease-associated adherent-invasive Escherichia coli pathovar. Inflamm Bowel Dis. 2016;22:1-12. doi: 10.1097/MIB.0000000000000574. PMID:26444104 [DOI] [PubMed] [Google Scholar]
- [30].Krause DO, Little AC, Dowd SE, Bernstein CN. Complete genome sequence of adherent invasive Escherichia coli UM146 isolated from ileal Crohn's disease biopsy tissue. J Bacteriol. 2011;193:583. doi: 10.1128/JB.01290-10. PMID:21075930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reid SD, Herbelin CJ, Bumbaugh AC, Selander RK, Whittam TS. Parallel evolution of virulence in pathogenic Escherichia coli. Nature. 2000;406:64-7. doi: 10.1038/35017546. PMID:10894541 [DOI] [PubMed] [Google Scholar]
- [32].Schreiber HLT, Conover MS, Chou WC, Hibbing ME, Manson AL, Dodson KW, Hannan TJ, Roberts PL, Stapleton AE, Hooton TM, et al.. Bacterial virulence phenotypes of Escherichia coli and host susceptibility determine risk for urinary tract infections. Sci Transl Med. 2017;9. pii: eaaf1283. doi: 10.1126/scitranslmed.aaf1283. PMID:28330863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zdziarski J, Brzuszkiewicz E, Wullt B, Liesegang H, Biran D, Voigt B, Grönberg-Hernandez J, Ragnarsdottir B, Hecker M, Ron EZ, et al.. Host imprints on bacterial genomes–rapid, divergent evolution in individual patients. PLoS Pathog. 2010;6. e1001078. doi: 10.1371/journal.ppat.1001078. PMID:20865122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alvarez Dorta D, Chalopin T, Sivignon A, de Ruyck J, Dumych TI, Bilyy RO, Deniaud D, Barnich N, Bouckaert J, Gouin SG. Physiochemical tuning of potent Escherichia coli anti-adhesives by microencapsulation and methylene homologation. ChemMedChem. 2017;12:986-98. doi: 10.1002/cmdc.201700061. PMID:28257558 [DOI] [PubMed] [Google Scholar]
- [35].Alvarez Dorta D, Sivignon A, Chalopin T, Dumych TI, Roos G, Bilyy RO, Deniaud D, Krammer EM, de Ruyck J, Lensink MF, et al.. The antiadhesive strategy in Crohn's disease: orally active mannosides to decolonize pathogenic Escherichia coli from the gut. Chembiochem. 2016;17:936-52. doi: 10.1002/cbic.201600018. PMID:26946458 [DOI] [PubMed] [Google Scholar]
- [36].Sivignon A, Yan X, Alvarez Dorta D, Bonnet R, Bouckaert J, Fleury E, Bernard J, Gouin SG, Darfeuille-Michaud A, Barnich N. Development of heptylmannoside-based glycoconjugate antiadhesive compounds against Adherent-Invasive Escherichia coli bacteria associated with Crohn's disease. mBio. 2015;6:e01298-15. doi: 10.1128/mBio.01298-15. PMID:26578673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brument S, Sivignon A, Dumych TI, Moreau N, Roos G, Guerardel Y, Chalopin T, Deniaud D, Bilyy RO, Darfeuille-Michaud A, et al.. Thiazolylaminomannosides as potent antiadhesives of type 1 piliated Escherichia coli isolated from Crohn's disease patients. J Med Chem. 2013;56:5395-406. doi: 10.1021/jm400723n. PMID:23795713 [DOI] [PubMed] [Google Scholar]
- [38].Mydock-McGrane LK, Hannan TJ, Janetka JW. Rational design strategies for FimH antagonists: new drugs on the horizon for urinary tract infection and Crohn's disease. Expert Opin Drug Dis. 2017;12:711-31. doi: 10.1080/17460441.2017.1331216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mydock-McGrane LK, Cusumano ZT, Janetka JW. Mannose-derived FimH antagonists: a promising anti-virulence therapeutic strategy for urinary tract infections and Crohn's disease. Expert Opin Ther Patents. 2016;26:175-97. doi: 10.1517/13543776.2016.1131266. [DOI] [PubMed] [Google Scholar]
- [40].Low D, Tran HT, Lee IA, Dreux N, Kamba A, Reinecker HC, Darfeuille-Michaud A, Barnich N, Mizoguchi E. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterol. 2013;145:602-12 e9. doi: 10.1053/j.gastro.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chassaing B, Rolhion N, de Vallee A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Söderholm JD, Hugot JP, Colombel JF, et al.. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer's patches via long polar fimbriae. J Clin Invest. 2011;121:966-75. doi: 10.1172/JCI44632. PMID:21339647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn's disease. Mol Microbiol. 2003;48:781-94. doi: 10.1046/j.1365-2958.2003.03468.x. PMID:12694621 [DOI] [PubMed] [Google Scholar]
- [43].Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, et al.. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007;117:1566-74. doi: 10.1172/JCI30504. PMID:17525800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dreux N, Denizot J, Martinez-Medina M, Mellmann A, Billig M, Kisiela D, Chattopadhyay S, Sokurenko E, Neut C, Gower-Rousseau C, et al.. Point mutations in FimH adhesin of Crohn's disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog. 2013;9:e1003141. doi: 10.1371/journal.ppat.1003141. PMID:23358328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rolhion N, Barnich N, Bringer MA, Glasser AL, Ranc J, Hebuterne X, Hofman P, Darfeuille-Michaud A. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut. 2010;59:1355-62. doi: 10.1136/gut.2010.207456. PMID:20587550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577-94. doi: 10.1053/j.gastro.2007.11.059. PMID:18242222 [DOI] [PubMed] [Google Scholar]
- [47].Vazeille E, Chassaing B, Buisson A, Dubois A, de Vallee A, Billard E, Neut C, Bommelaer G, Colombel JF, Barnich N, et al.. GipA factor supports colonization of Peyer's patches by Crohn's disease-associated Escherichia coli. Inflamm Bowel Dis. 2016;22:68-81. doi: 10.1097/MIB.0000000000000609. PMID:26512715 [DOI] [PubMed] [Google Scholar]
- [48].Stanley TL, Ellermeier CD, Slauch JM. Tissue-specific gene expression identifies a gene in the lysogenic phage Gifsy-1 that affects Salmonella enterica serovar typhimurium survival in Peyer's patches. J Bacteriol. 2000;182:4406-13. doi: 10.1128/JB.182.16.4406-4413.2000. PMID:10913072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gibold L, Garenaux E, Dalmasso G, Gallucci C, Cia D, Mottet-Auselo B, Faïs T, Darfeuille-Michaud A, Nguyen HT, Barnich N, et al.. The Vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn's disease-associated Escherichia coli. Cell Microbiol. 2016;18:617-31. doi: 10.1111/cmi.12539. PMID:26499863 [DOI] [PubMed] [Google Scholar]
- [50].Jarry A, Cremet L, Caroff N, Bou-Hanna C, Mussini JM, Reynaud A, Servin AL, Mosnier JF, Liévin-Le Moal V, Laboisse CL. Subversion of human intestinal mucosa innate immunity by a Crohn's disease-associated E. coli. Mucosal Immunol. 2015;8:572-81. doi: 10.1038/mi.2014.89. PMID:25269707 [DOI] [PubMed] [Google Scholar]
- [51].Cieza RJ, Hu J, Ross BN, Sbrana E, Torres AG. The IbeA invasin of adherent-invasive Escherichia coli mediates interaction with intestinal epithelia and macrophages. Infect Immun. 2015;83:1904-18. doi: 10.1128/IAI.03003-14. PMID:25712929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ho BT, Dong TG, Mekalanos JJ. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe. 2014;15:9-21. doi: 10.1016/j.chom.2013.11.008. PMID:24332978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen H, Yang D, Han F, Tan J, Zhang L, Xiao J, Zhang Y, Liu Q. The Bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the Ca2+-dependent MAPK-Jnk pathway. Cell Host Microbe. 2017;21:47-58. doi: 10.1016/j.chom.2016.12.004. PMID:28081443 [DOI] [PubMed] [Google Scholar]
- [54].Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, Whitney JC, Radey MC, Peterson SB, Ruhland BR, et al.. Secreted effectors encoded within and outside of the Francisella Pathogenicity Island promote intramacrophage growth. Cell Host Microbe. 2016;20:573-83. doi: 10.1016/j.chom.2016.10.008. PMID:27832588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sana TG, Baumann C, Merdes A, Soscia C, Rattei T, Hachani A, Jones C, Bennett KL, Filloux A, Superti-Furga G, et al.. Internalization of Pseudomonas aeruginosa strain PAO1 into epithelial cells is promoted by interaction of a T6SS effector with the microtubule network. mBio. 2015;6:e00712. doi: 10.1128/mBio.00712-15. PMID:26037124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356-68. doi: 10.1038/nrmicro2546. PMID:21423246 [DOI] [PubMed] [Google Scholar]
- [57].McPhee JB, Small CL, Reid-Yu SA, Brannon JR, Le Moual H Coombes BK. Host defense peptide resistance contributes to colonization and maximal intestinal pathology by Crohn's disease-associated adherent-invasive Escherichia coli. Infect Immun. 2014;82:3383-93. doi: 10.1128/IAI.01888-14. PMID:24866805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Porter CK, Tribble DR, Aliaga PA, Halvorson HA, Riddle MS. Infectious gastroenteritis and risk of developing inflammatory bowel disease. Gastroenterol. 2008;135:781-6. doi: 10.1053/j.gastro.2008.05.081. [DOI] [PubMed] [Google Scholar]
- [59].Garcia Rodriguez LA, Ruigomez A, Panes J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterol. 2006;130:1588-94. doi: 10.1053/j.gastro.2006.02.004. [DOI] [PubMed] [Google Scholar]
- [60].Gradel KO, Nielsen HL, Schonheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after Salmonella or Campylobacter gastroenteritis. Gastroenterol. 2009;137:495-501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- [61].Hajishengallis G, Lamont RJ. Dancing with the stars: how choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016;24:477-89. doi: 10.1016/j.tim.2016.02.010. PMID:26968354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Small CL, Reid-Yu SA, McPhee JB, Coombes BK. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Comm. 2013;4:1957. doi: 10.1038/ncomms2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol. 2015;13:191-205. doi: 10.1038/nrmicro3420. PMID:25749450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. Citrobacter rodentium: Infection, inflammation and the microbiota. Nat Rev Microbiol. 2014;12:612-23. doi: 10.1038/nrmicro3315. PMID:25088150 [DOI] [PubMed] [Google Scholar]
- [65].Small CL, Xing L, McPhee JB, Law HT, Coombes BK. Acute infectious gastroenteritis potentiates a Crohn's disease pathobiont to fuel ongoing inflammation in the post-infectious period. PLoS Pathogens. 2016;12(10):e1005907. doi: 10.1371/journal.ppat.1005907. PMID:27711220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Agus A, Denizot J, Thevenot J, Martinez-Medina M, Massier S, Sauvanet P, Bernalier-Donadille A, Denis S, Hofman P, Bonnet R, et al.. Western diet induces a shift in microbiota composition enhancing susceptibility to adherent-invasive E. coli infection and intestinal inflammation. Sci Rep. 2016;6:19032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Garner CD, Antonopoulos DA, Wagner B, Duhamel GE, Keresztes I, Ross DA, Young VB, Altier C. Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar typhimurium murine model of infection. Infect Immun. 2009;77:2691-702. doi: 10.1128/IAI.01570-08. PMID:19433544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Winter SE, Lopez CA, Baumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013;14:319-27. doi: 10.1038/embor.2013.27. PMID:23478337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-73. doi: 10.1126/science.1241165. PMID:23828891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500-10. doi: 10.1038/nri3863. PMID:26160612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al.. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis. 2014;20:1919-32. doi: 10.1097/MIB.0000000000000183. PMID:25230163 [DOI] [PubMed] [Google Scholar]
- [72].Gao Q, Wang X, Xu H, Xu Y, Ling J, Zhang D, Gao S, Liu X. Roles of iron acquisition systems in virulence of extraintestinal pathogenic Escherichia coli: salmochelin and aerobactin contribute more to virulence than heme in a chicken infection model. BMC Microbiol. 2012;12:143. doi: 10.1186/1471-2180-12-143. PMID:22817680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Olsan EE, Byndloss MX, Faber F, Rivera-Chavez F, Tsolis RM, Baumler AJ. Colonization resistance: The deconvolution of a complex trait. J Biol Chem. 2017;292:8577-81. doi: 10.1074/jbc.R116.752295. PMID:28389556 [DOI] [PMC free article] [PubMed] [Google Scholar]