

Abstract
Tumor suppressor/transcription factor p53 is mutated in over 50% of all cancers. Some mutant p53 proteins have not only lost tumor suppressor activities but they also gain oncogenic functions (GOF). One of the most frequently expressed GOF p53 mutants is Arg175His (p53R175H) with well‐documented roles in cancer development and progression. Plakoglobin is a cell adhesion and signaling protein and a paralog of β‐catenin. Unlike β‐catenin that has oncogenic function through its role in the Wnt pathway, plakoglobin generally acts as a tumor/metastasis suppressor. We have shown that plakoglobin interacted with wild type and a number of p53 mutants in various carcinoma cell lines. Plakoglobin and mutant p53 interacted with the promoter and regulated the expression of several p53 target genes. Furthermore, plakoglobin interactions with p53 mutants restored their tumor suppressor/metastasis activities in vitro. GOF p53 mutants induce accumulation and oncogenic activation of β‐catenin. Previously, we showed that one mechanism by which plakoglobin may suppress tumorigenesis is by sequestering β‐catenin's oncogenic activity. Here, we examined the effects of p53R175H expression on β‐catenin accumulation and transcriptional activation and their modifications by plakoglobin coexpression. We showed that p53R175H expression in plakoglobin null cells increased total and nuclear levels of β‐catenin and its transcriptional activity. Coexpression of plakoglobin in these cells promoted β‐catenin's proteasomal degradation, and decreased its nuclear levels and transactivation. Wnt/β‐catenin targets, c‐MYC and S100A4 were upregulated in p53R175H cells and were downregulated when plakoglobin was coexpressed. Plakoglobin‐p53R175H cells also showed significant reduction in their migration and invasion in vitro.
Keywords: β‐catenin, invasion, p53, p53R175H, plakoglobin
1. INTRODUCTION
p53 is a sequence‐specific transcription factor with tumor and metastasis suppressor activities.1, 2 It plays pivotal roles in the regulation of the cell cycle, DNA repair, senescence, apoptosis, and metabolism by responding to various cellular stress signals such as DNA damage, hypoxia, mitotic stress, oncogenic signaling etc.3, 4 As a transcription factor, p53 downregulates the expression of genes involved in tumor development and cancer progression.5, 6, 7 p53 is mutated or lost in over half of all cancers and in more than 80% of metastatic tumors.3, 8 In addition to the loss/partial loss of tumor suppressor activities, some p53 mutants also gain oncogenic functions (GOF) that contribute to tumor cell growth, aggressiveness, metastasis and drug resistance.9 p53 inactivation can result from genetic alterations, decreased stability, defective post‐translational modifications and interaction with intracellular partners.10
There are over 30 000 somatic mutations in TP53, including missense, nonsense, deletions, frameshifts and temperature sensitive.11 Most of these changes occur within the DNA‐binding domain with more than 75% single missense mutations, 40% of which are represented by 6 hot spot mutations (Arg175, Gly245, Arg248, Arg249, Arg273 and Arg282) that are highly frequent in tumors of different origins.11 Hot spot mutations are further classified into 2 groups: contact mutations (Arg248, Arg273) inhibit direct interaction between p53 and DNA leading to a loss of sequence‐specific transactivation, whereas structural mutations (Arg175, Gly245, Arg249, and Arg282) alter local or global conformation of p53 causing indirect loss of DNA binding.12 Among the hot spot structural mutations, Arg175His (R175H), is the most frequent GOF p53 mutant13, 14 that increases cancer cell proliferation, migration and invasion by deregulating different signaling pathways involved in tumorigenesis and metastasis.
p53 functions are regulated by post‐translational modifications and protein‐protein interactions.15 We have identified plakoglobin (γ‐catenin) as an endogenous interacting partner of wild type as well as a number of most frequent mutant p53 (mp53) in various carcinoma cell lines of different origins and have shown that its interaction with mp53 restores mp53 tumor suppressor activities in vitro.16, 17, 18, 19 Plakoglobin is an armadillo protein family member and is a paralog of β‐catenin with similar dual cell‐cell adhesion and signaling activities.20, 21 However, unlike β‐catenin that acts as an oncogene through its interaction with the transcription factors T‐cell factor/lymphoid enhancer‐binding factor (TCF/LEF), and activation of Wnt signaling pathway,22, 23 plakoglobin generally acts as a tumor/metastasis suppressor.20, 21, 24, 25, 26, 27 We have shown that plakoglobin can act as a tumor/metastasis suppressor by at least 3 mechanisms: regulation of stability and subcellular localization of growth regulating molecules,19, 25, 28 interaction with transcription factors involved in the regulation of cell growth and metastasis16, 17, 18, 19, 25 and sequestration of β‐catenin oncogenic activities;29 see also.30, 31, 32, 33, 34
p53 GOF mutations can induce aberrant accumulation and increased transcriptional activation of β‐catenin in cancer cells.35, 36, 37 In the absence of Wnt, excess cytoplasmic β‐catenin is degraded by phosphorylation and proteasomal degradation.38, 39, 40 Upon Wnt activation, β‐catenin is stabilized and translocates into the nucleus where it binds to TCF/LEF and activates the expression of Wnt targets including cyclin D1, c‐Myc, MMPs, S100A4, and survivin etc.38, 39, 40, 41, 42, 43 This triggers an epithelial to mesenchymal phenotypic transition, cell proliferation, migration, invasion and metastasis.41, 43 β‐Catenin is also degraded through its ubiquitination by Siah‐1, an E3 ubiquitin ligase that enhances β‐catenin's proteasomal degradation independent of the canonical Wnt signaling pathway.44, 45
In the present study, our goal was to assess the effects of GOF p53R175H mutant (herein referred to as p53R175H) alone or together with plakoglobin on excess β‐catenin accumulation and its transcriptional activation. To this end, plakoglobin‐deficient and p53 null H1299 cells were transfected with wild‐type p53 (herein referred to as p53) or p53R175H with or without plakoglobin. p53R175H‐expressing H1299 cells showed significantly higher levels of total and nuclear β‐catenin relative to the p53‐expressing transfectants. H1299 cells expressing plakoglobin or coexpressing plakoglobin and p53 or p53R175H had significantly lower levels of total and nuclear β‐catenin. Plakoglobin and β‐catenin interacted with TCF‐4 and expression of plakoglobin decreased the β‐catenin/TCF interaction. p53R175H cells showed a significant increase in β‐catenin/TCF luciferase reporter activity, whereas coexpression of plakoglobin in these cells significantly decreased the reporter activity. β‐Catenin target genes, c‐MYC and S100A4 were upregulated in p53R175H cells and were significantly downregulated when plakoglobin was coexpressed. p53R175H expression also increased the in vitro migration and invasion of H1299 cells, which were significantly reduced when plakoglobin was coexpressed.
2. MATERIALS AND METHODS
2.1. Cell lines and culture conditions
H1299, the non‐small‐cell lung carcinoma cells have been described18 and were grown in minimum essential medium (MEM) supplemented with 10% FBS, and 1% penicillin‐streptomycin‐kanamycin (PSK) antibiotics. SW620 colon carcinoma cells were grown in Leibovitz's L‐15 medium supplemented with 2 mmol/L l‐glutamine, 10% FBS and 1% PSK.
2.2. Plasmid construction and transfection
Hemagglutinin (HA)‐tagged p53 has been described previously.18, 46 The pcDNA3.1/hygro‐plakoglobin construct was generated using the previously described FLAG‐tagged plakoglobin as a template.29 The p53R175H expression construct was a gift from Dr Giovanni Blandino.47
H1299 cells were cultured in 60‐mm dishes and transfected at 60% confluency with 9 μg DNA using calcium phosphate. Twenty hours after transfection, cells were rinsed with media and allowed to recover for 24 hours in complete MEM. Stable transfectants were selected by placing cultures in media containing 500 μg/mL Hygromycin B (plakoglobin transfectants) or 400 μg/mL G418 (p53 R175H transfectants) or both (double transfectants) for 2‐3 weeks. Resistant clones were screened for p53 and plakoglobin expression by immunofluorescence (IF) and immunoblot assays and maintained in media containing 350 μg/mL Hygromycin B or 200 μg/mL G418 or both. Positive clones were subcultured by limiting dilution. Both parental and multiple single cell isolated clones were tested for plakoglobin and p53 expression using various assays and the results are presented for 1 representative clone.
2.3. Cell fractionation, preparation of cell extracts and western blot analysis
Total cellular proteins were extracted by solubilizing confluent 100‐mm cultures in SDS sample buffer (10 mmol/L Tris‐HCl pH 6.8, 2% [w/v] SDS, 50 mmol/L DTT, 2 mmol/L EDTA, 0.5 mmol/L PMSF, 1 mmol/L NaF, 1 mmol/L Na3VO4). Equal amounts of total cellular proteins were separated by SDS‐PAGE and transferred onto nitrocellulose membranes (Bio‐Rad). Membranes were incubated in specific primary antibodies overnight at 4°C followed by the appropriate secondary antibodies at room temperature (Table 1). Membranes were scanned using an Odyssey CLx infrared imaging system.
Table 1.
Antibodies and their respective dilutions in specific assays
| Species | Assay | Source/Catalog number | |||
|---|---|---|---|---|---|
| WB | IP | IF | |||
| Primary antibodies | |||||
| p53‐ DO‐1 | Mouse | 1:1000 | 1:100 | ‐ | Santa Cruz Biotechnology/sc‐126 |
| Plakoglobin (γ‐catenin) | Mouse | 1:1000 | 1:100 | 1:100 | BD Transduction Laboratories/610254 |
| β‐catenin | Mouse | 1:1000 | 1:100 | 1:100 | Sigma Aldrich/C‐7207 |
| β‐catenin (nuclear) | Mouse | 1:1000 | ‐ | ‐ | Abcam/ab 19451‐50 |
| TCF‐4 | Mouse | 1:500 | 1:100 | ‐ | Upstate Biotechnology/05‐511 |
| β‐actin | Mouse | 1:1000 | ‐ | ‐ | Santa Cruz Biotechnology/sc‐47778 |
| Lamin B1 | Rabbit | 1:1000 | ‐ | ‐ | Abcam/ab 16048 |
| Secondary antibodies | |||||
| Anti‐mouse, light chain IgG | Goat | 1:20 000 | ‐ | ‐ | Jackson ImmunoResearch/115‐625‐174 |
| Anti‐rabbit, light chain IgG | Goat | 1:20 000 | ‐ | ‐ | Jackson ImmunoResearch/211‐652‐171 |
| Alexa Fluor 488 | Mouse | ‐ | ‐ | 1:2000 | Molecular Probes Biotechnology/A11029 |
| Alexa Fluor 546 | Rabbit | ‐ | ‐ | 1:3000 | Molecular Probes Biotechnology/A11035 |
IF, Immunofluorescence; IP, Immunoprecipitation; WB, western blot; ‐, Not applicable.
Nuclear fractions were prepared with Thermo Fisher Scientific (Waltham, MA, USA) NE‐PER Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer's protocol. Purity of nuclear fractions was verified by immunoblotting with nuclear lamin antibodies (Table 1).
2.4. Immunoprecipitation (IP)
Cultures (100 mm) were washed with cold PBS containing 1 mmol/L NaF, Na3VO4 and CaCl2 and extracted in 1 mL lysis buffer (20 mmol/L Tris‐HCl pH 7.5, 150 mmol/L NaCl, 1% NP‐40, 0.1% sodium deoxycholate, 100 mmol/L NaF,18 and protease inhibitor cocktail (Roche Diagnostics) for 30 minutes at 4°C on a rocker. Cells were then scraped and centrifuged at 48 000 g for 10 minutes. Supernatants were divided into equal aliquots and processed for immunoprecipitation with p53, plakoglobin and β‐catenin antibodies (Table 1) and 40 μL protein G agarose beads (Pierce Biotechnology) overnight at 4°C. To ensure complete depletion, immunoprecipitates were centrifuged at 14 000 g for 2 minutes and supernatants were separated and processed for a second immunoprecipitation for 3 hours. Beads from the 2 immunoprecipitations were combined and washed 3 times with the lysis buffer. Immune complexes were solubilized in 60 μL SDS sample buffer, separated by SDS‐PAGE and processed for western blot (WB) as described above.
2.5. Immunofluorescence and confocal microscopy
Immunofluorescence and confocal microscopy were carried out as described in detail previously.48 Briefly, confluent cultures of various cell lines were established on glass coverslips and rinsed with cold PBS containing 1 mmol/L each of NaF, Na3VO4 and CaCl2. Cells were fixed with 3.7% formaldehyde in PBS for 20 minutes and extracted with cytoskeleton extraction buffer (CSK; 50 mmol/L NaCl, 300 mmol/L sucrose, 10 mmol/L PIPES pH 6.8, 3 mmol/L MgCl2, 0.5% Triton X‐100, 1.2 mmol/L PMSF, and 1 mg/mL DNase and RNase) for 10 minutes. Coverslips were blocked with 4.0% goat serum and 50 mmol/L NH4Cl in PBS containing 0.2% BSA for 1 hour. Coverslips were then incubated in the specific primary antibodies for 1 hour followed by the species‐specific secondary antibodies for 30 minutes at concentrations indicated in Table 1. Nuclei were stained with DAPI (1:2000). Coverslips were mounted in elvanol containing paraphenylene diamine (PPD, 0.2% [w/v]) and viewed using a 63 × objective lens of a Zeiss confocal microscope.
2.6. RNA isolation, RT‐PCR and real‐time PCR
Total RNA was isolated from 100‐mm cultures with Trizol reagent (Invitrogen‐Thermo Fisher Scientific), treated with DNase I and reverse‐transcribed with RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
For real‐time PCR, SYBR Green Master Mix (Thermo Fisher Scientific) and specific forward and reverse primers for MYC, S100A4 and ACTB (β‐actin) (Table 2) were used according to the manufacturer's instructions.
Table 2.
Oligos/primer sequences used for RT‐qPCR
| Construct | Primers | Size (nt) | |
|---|---|---|---|
| c‐MYC | Forward | 5′‐CAGCTGCTTAGACGCTGGATT‐3′ | 21 |
| Reverse | 5′‐GTAGAAATACGGCTGCACCGA‐3′ | 21 | |
| S100A4 | Forward | 5′‐GATGAGCAACTTGGACAGCAA‐3′ | 21 |
| Reverse | 5′‐CTGGGCTGCTTATCTGGGAAG‐3′ | 21 | |
| ACTB | PrimePCR SYBR Green Assay ACTB Human, Cat No. 10025636 | ||
2.7. Proteasome inhibition assay
Replicate 100 mm cultures remained untreated or were treated with 1 μmol/L proteasome inhibitor MG132 (Sigma Chemical Co., St Louis, MO, USA) for 16 hours. Untreated and treated cells were then lysed and total cell lysates were used for western blot with β‐catenin antibodies as described above.
2.8. Luciferase reporter assay
To measure β‐catenin‐driven transactivation, confluent 35 mm cultures of parental H1299 cells and H1299 transfectants were cotransfected with 5 μg pTOPFLASH plasmid49 and 3 μg Renilla luciferase plasmid (pRL‐TK) serving as a control for transfection efficiency.50 Luciferase activities were measured 48 hours later and normalized to Renilla activities. Each experiment was repeated 4 times and the mean and standard errors were calculated.
2.9. In vitro migration and invasion assays
Migration and invasion assays were carried out using Transwell inserts (BD Biosciences, Franklin Lakes, NJ, USA) as described in detail previously.18 The migrated/invaded cells were counted in 5 random fields for each membrane using the NIH ImageJ Cell Counter program. Numbers for each cell line were averaged and normalized to those of the parental untransfected cells and histograms constructed. Histograms represent the average ± SD of 3‐6 independent assays for each cell line.
2.10. Statistical analysis
Values are presented as means ± SD. Statistical differences between groups were assessed by Student's t tests. P‐value <.05 was considered significant.
3. RESULTS
3.1. Plakoglobin interacted with p53R175H and decreased β‐catenin protein levels
We first validated the expression of plakoglobin, p53 and p53R175H in single and double H1299 transfectants by processing total cell extracts from all transfectants for western blots with plakoglobin and p53 antibodies (Figure 1A).
Figure 1.
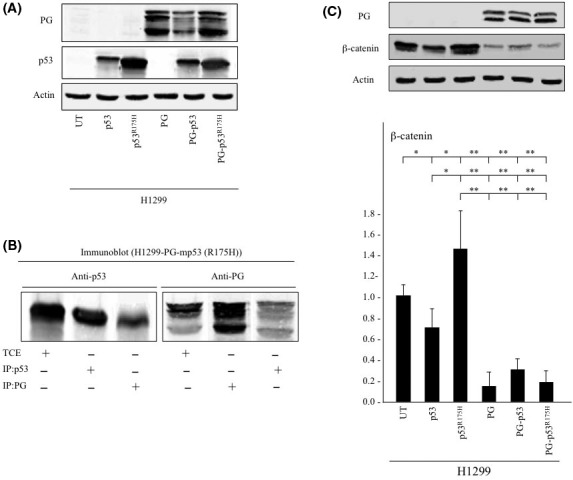
Plakoglobin (PG) interacts with the mutant p53R175H and decreases β‐catenin levels. A, Total cell extracts from untransfected H1299 cells and H1299 cells transfected with p53, p53R175H, PG, PG and p53 (PG‐p53) or PG and p53R175H (PG‐p53R175H) were processed for immunoblot using plakoglobin and p53 antibodies. β‐Actin levels in the same extracts were used as an internal control. B, Total cell extracts (TCE) from H1299 cells coexpressing plakoglobin and p53R175H were processed for reciprocal and sequential coimmunoprecipitation (Co‐IP) and immunoblotting (IB) using p53 and plakoglobin antibodies. C, Total cell extracts from parental H1299 cells and H1299 transfectants expressing p53 or p53R175H with or without plakoglobin were processed for immunoblot using plakoglobin and β‐catenin antibodies. β‐Actin levels were used as loading control. Histograms represent the average ± SD of 4 separate experiments. All values are normalized to H1299 cells. UT, untransfected. *P < .05 **P < .001
Previously, we have shown that plakoglobin interacted with p53 and several mp53 proteins using different carcinoma cell lines.16, 17, 18, 19 To verify plakoglobin interaction with p53R175H, we processed H1299 transfectants coexpressing plakoglobin and p53R175H for reciprocal coimmunoprecipitation (Co‐IP) and immunoblotting (IB) with plakoglobin and p53 antibodies (Figure 1B). Co‐Immunoprecipitation the double transfectants total cell extracts with p53 antibodies precipitated p53 (Figure 1B, lane 2; IP: p53, IB: p53) and coprecipitated plakoglobin (PG) (Figure 1B, lane 6, IP: p53, IB: PG). Reciprocal Co‐IP using PG antibodies confirmed this finding, as PG antibodies pulled down total cellular PG (Figure 1B lane 5, IP: PG, IB: PG) and coprecipitated p53 (Figure 1B, lane 3, IP: PG, IB: p53) from cells coexpressing both proteins.
Figure 1C shows plakoglobin and β‐catenin protein expression in H1299 transfectants expressing p53 or p53R175H with or without plakoglobin. Relative to H1299 cells, β‐catenin levels were decreased in p53 expressing H1299 cells (Figure 1C, β‐catenin, lane 2), whereas it was increased when p53R175H was expressed (Figure 1C, β‐catenin, lane 3). Plakoglobin expression significantly reduced β‐catenin levels in parental as well as in p53 or p53R175H‐expressing H1299 transfectants (Figure 1C, β‐catenin, lanes 4‐6).
3.2. Expression of plakoglobin decreased β‐catenin protein levels by promoting its proteasomal degradation
Western blot analysis of the nuclear extracts from parental H1299 cells and H1299 transfectants was used to examine the cytoplasmic and nuclear levels of plakoglobin and β‐catenin (Figure 2A,B). The highest level of nuclear and cytoplasmic β‐catenin was detected in H1299‐p53R175H transfectants (Figure 2A,B, β‐catenin [N, C] lane 3). In contrast, significantly lower amounts of β‐catenin were detected in parental as well as in p53 or p53R175H cells when plakoglobin was coexpressed (Figure 2A,B, β‐catenin [N] and [C] lanes 4‐6).
Figure 2.
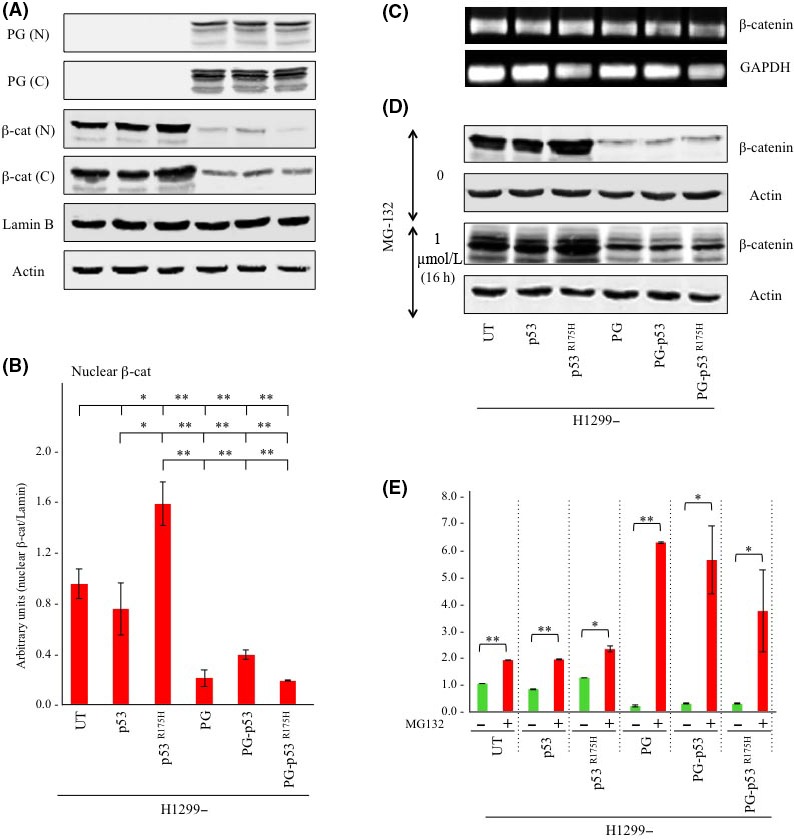
Plakoglobin (PG) expression decreases cytoplasmic and nuclear β‐catenin levels by promoting its proteasomal degradation in H1299‐p53R175H cells. A, Equal amounts of nuclear (N) and cytoplasmic (C) extracts from parental H1299 cells and H1299 transfectants were processed for immunoblot using plakoglobin and β‐catenin antibodies. Lamin B and actin levels were probed from the nuclear and cytoplasmic extracts of different transfectants, respectively, to confirm equal loadings. B, Nuclear β‐catenin blots in (A) were quantitated by NIH ImageJ software. Histograms represent the average ± SD of 4 experiments. All values were normalized to untransfected H1299 cells. *P < .05; **P < .001. C, Cultures of H1299 cells and H1299 transfectants were processed for RT‐PCR using β‐catenin and GAPDH (control) primers as described in Materials and Methods. D, Replicate cultures of H1299 cells and H1299 transfectants remained untreated or were treated with 1 μmol/L MG132 for 16 h. Total cell extracts from the untreated and treated cells were processed for immunoblot using β‐catenin antibodies. Blots were also probed with β‐actin antibodies to confirm equal protein loadings. E, Blots in (C) were quantitated and histograms were generated that represent the average ± SD of 4 separate experiments. All values were normalized to the untransfected and untreated cells. *P < .05; **P < .001. UT, untransfected; β‐cat, β‐catenin
Examination of β‐catenin's mRNA levels in various transfectants showed no significant differences (Figure 2C) suggesting that changes in β‐catenin levels in plakoglobin‐expressing cells occur post‐transcriptionally. This led us to assess whether downregulation of β‐catenin is due to increased degradation upon plakoglobin expression. β‐Catenin degradation can occur through 2 pathways: glycogen synthase kinase 3 beta (GSK3β) and/or Siah1 mediated. GSK3β proteasomal degradation is the primary regulator of cellular β‐catenin levels and requires β‐catenin's ubiquitination. To be ubiquitinated, β‐catenin has to be phosphorylated on serine 33 and 37 residues by GSK3β, which enables the protein to bind to E3 ubiquitin ligase TrCP1 (β‐TrCP) and marks it for proteasomal degradation.51, 52 The second degradation pathway is mediated by Siah1 and is independent of GSK3β. Siah1 is a target of p53 and E2F1 and is minimally expressed in H1299 cells,53, 54 suggesting that decreased β‐catenin levels in plakoglobin‐expressing H1299 cells is likely as a result of the increased proteasomal degradation by GSK3β. To address this possibility, we used MG132, which inhibits 20S proteasome activity and degradation of ubiquitinated proteins.29 Replicate cultures of H1299 cells and H1299 transfectants remained untreated or were treated with 1 μmol/L MG132 for 16 hours and processed for western blot using β‐catenin antibodies. As shown in Figure 2D, β‐catenin was detected as multiple bands in MG132‐treated cultures, which was consistent with the inhibition of degradation of the ubiquitinated protein. Quantitation of β‐catenin protein levels in untreated and treated cell lines showed that in untreated cultures, plakoglobin expression increased β‐catenin degradation by ~5.5‐, 3‐ and 4.6‐fold in H1299, H1299‐p53 and H1299‐p53R175H cells, respectively (Figure 2E). MG132 treatment decreased β‐catenin degradation by >2‐fold in the absence of plakoglobin, whereas this reduction was significantly higher (up to 7‐fold) in plakoglobin‐expressing transfectants (Figure 2E). These results suggested that plakoglobin decreased β‐catenin levels by promoting its proteasomal degradation.
We also examined the subcellular distribution of β‐catenin in H1299 cells and H1299 transfectants by confocal double immunofluorescence microscopy (Figure 3). Confluent cultures of various cell lines were fixed with formaldehyde, extracted with CSK buffer and processed for double immunofluorescence staining with plakoglobin and β‐catenin antibodies.48 There was no detectable plakoglobin staining in H1299, H1299‐p53 and H1299‐p53R175H cells, whereas it was distributed throughout the cytoplasm and at the membrane in H1299‐PG, ‐PG‐p53 and ‐PG‐p53R175H transfectants. β‐Catenin was expressed in all cell lines, although with different intensity. Relative to H1299 cells, β‐catenin staining was significantly reduced in H1299‐p53 transfectants, whereas it was significantly increased in H1299‐p53R175H cells. Plakoglobin co/expression dramatically reduced β‐catenin's staining (nuclear and cytoplasmic) in all transfectants, particularly in H1299‐p53R175H cells (Figure 3, H1299‐PG‐p53R175H). These results were consistent with the western blot studies and further confirmed that plakoglobin expression reduced β‐catenin levels in both cytoplasmic and nuclear pools. Of note, we did not detect significant changes in the morphology of plakoglobin‐expressing transfectants relative to parental cells as they both showed an epithelioid phenotype. H1299 cells are E‐cadherin deficient and express N‐cadherin,55 which similar to E‐cadherin, interacts with β‐catenin and plakoglobin leading to the appearance of an epithelioid morphology in both parental cells and transfectants.
Figure 3.
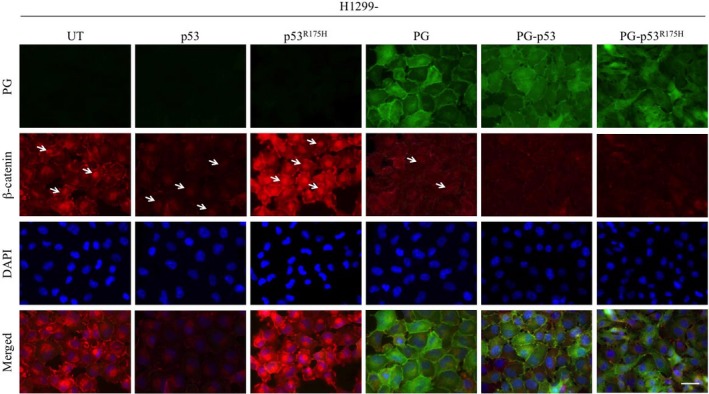
Subcellular localization of plakoglobin (PG) and β‐catenin in H1299 transfectants. H1299 cells and H1299 transfectants were grown to confluency on coverslips, fixed with formaldehyde and permeabilized with CSK buffer as described in Materials and Methods. Coverslips were processed for confocal microscopy using plakoglobin (green) and β‐catenin (red) antibodies. Nuclei were stained with DAPI (blue). Arrows indicate nuclei with β‐catenin staining. Bar, 25 μmol/L. UT, untransfected
3.3. Plakoglobin expression decreased β‐catenin interaction with TCF‐4, reduced β‐catenin/TCF‐4 reporter activity and downregulated target gene expression
So far, the results showed that plakoglobin expression decreased the nuclear pool of β‐catenin, which may influence the interaction between β‐catenin and its cognate transcription factor TCF. Therefore, we used Co‐IP experiments to examine the interactions between plakoglobin and β‐catenin with TCF in parental H1299 cells and H1299 transfectants. In Figure 4A, equal amounts of total cellular protein from parental H1299 cells and H1299 transfectants were processed for sequential Co‐IP and immunoblotting with nuclear β‐catenin, plakoglobin and TCF antibodies. TCF was coprecipitated with β‐catenin in H1299, H1299‐p53 and H1299‐p53R175H cells and its level was significantly lower in H1299‐p53 transfectants (Figure 4A, IP: β‐cat, IB: TCF). Interestingly, very little/no TCF was detected in β‐catenin precipitates from the plakoglobin‐expressing transfectants. In these cells, TCF was only detected in association with plakoglobin (Figure 4A, H1299‐PG, ‐p53, ‐p53R175H; IP: PG, IB: TCF). Consistent with the earlier results, nuclear β‐catenin levels were decreased in p53 and in all plakoglobin‐expressing transfectants (H1299‐p53, ‐PG, ‐PG‐p53, ‐PG‐p53R175H) relative to parental H1299 cells (Figure 4A; TCE, β‐catenin). In contrast, TCF level was not notably different among various cell lines (Figure 4A; TCE, IB: TCF). These results indicated that plakoglobin coexpression significantly reduced the interactions between β‐catenin and TCF in H1299, H1299‐p53 and H1299‐p53R175H transfectants.
Figure 4.
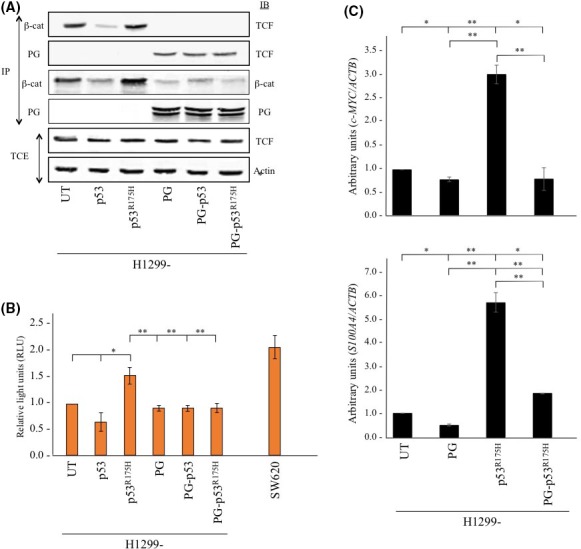
Plakoglobin (PG) expression reduces β‐catenin interaction with T‐cell factor 4 (TCF‐4), decreases β‐catenin/TCF‐4 reporter activity and target gene expression. A, Equal amounts of total cellular extracts (TCE) from H1299 cells and H1299 transfectants were processed for sequential immunoprecipitation (IP) and immunoblotting (IB) using plakoglobin, nuclear β‐catenin (β‐cat) and TCF‐4 (TCF) antibodies. Equal loadings were confirmed by processing total cell extracts from all cell lines for immunoblotting using β‐actin antibodies. B, Untransfected H1299 cells and H1299 transfectants were cotransfected with pTOPFLASH β‐catenin/TCF reporter construct and p‐RL‐TK Renilla reporter plasmid and luciferase activities were measured 48 h post‐transfection. Levels of luciferase activities from pTOPFLASH were normalized to those of Renilla plasmid. Four independent experiments were carried out and the results were normalized to H1299 parental cells. SW620 cells were included as a positive control. UT, untransfected. *P < .05, **P < .001. C, Total cellular RNA was extracted from untransfected H1299 cells and H1299 transfectants, reverse transcribed and processed for real‐time PCR for C‐MYC (top) and S100A4 (bottom) using specific primers (Table 2). Expression levels were first normalized to the amount of ACTB in the same cell line and then to H1299 untransfected cells. Histograms were constructed based on the average ± SD. UT, untransfected. *P < .05, **P < .001
We next assessed whether decreased nuclear β‐catenin/TCF association was reflected in β‐catenin‐dependent TCF reporter activity (Figure 4B). Parental H1299 cells and H1299 transfectants were transiently transfected with pTOPFLASH and pRL‐TK Renilla reporter constructs and luciferase activities were measured in all cell lines. As a positive control, SW620, a colon carcinoma cell line expressing mutant APC and signaling‐competent β‐catenin56 was included in these studies (Figure 4B). Luciferase activities of all cell lines were normalized to that of parental H1299 cells. Results showed no significant differences in luciferase activity among H1299, H1299‐p53 and H1299‐PG cells (Figure 4B). In contrast, relative to parental H1299 cells, H1299‐p53R175H transfectants showed over 60% higher luciferase activity (Figure 4B), which was significantly reduced when plakoglobin was coexpressed in these cells (Figure 4B, H1299‐PG‐p53R175H).
Based on these observations, we reasoned that decreased β‐catenin/TCF transactivation should result in decreased expression of their target genes. Specifically, we focused on c‐MYC and S100A4, 2 β‐catenin/TCF target genes that are known to participate in tumorigenesis and metastasis.41, 42, 43, 57, 58, 59 RT‐qPCR experiments showed that the levels of c‐MYC and S100A4 mRNA were significantly increased in H1299‐p53R175H transfectants compared to H1299 cells (Figure 4C). Coexpression of plakoglobin in these cells (H1299‐PG‐p53R175H) led to over 3‐and 5‐fold decrease in c‐MYC and S100A4 mRNA levels, respectively (Figure 4C). Together, the results of the experiments in Figure 4 suggested that plakoglobin expression in p53R175H cells reduced β‐catenin/TCF association and the activation of at least 2 of their target genes involved in tumorigenesis and metastasis.
3.4. Plakoglobin expression decreased migratory and invasive properties of p53R175H‐expressing H1299 cells
To assess the biological significance of decreased β‐catenin/TCF transactivation, we examined the in vitro migration and invasion of parental H1299 cells and H1299 transfectants.
As shown in Figure 5A, p53 and plakoglobin expression decreased migration by 45% and 34% relative to H1299 cells, respectively, whereas coexpression of both proteins reduced migration by 73% (Figure 5A, migration; H1299‐p53, H1299‐PG and H1299‐PG‐p53; also see18). In contrast, expression of p53R175H increased the migration of H1299 cells by ~20% (Figure 5A, migration, H1299‐ p53R175H), which was reduced by >40% when plakoglobin was coexpressed (Figure 5A, H1299‐PG‐p53R175H).
Figure 5.
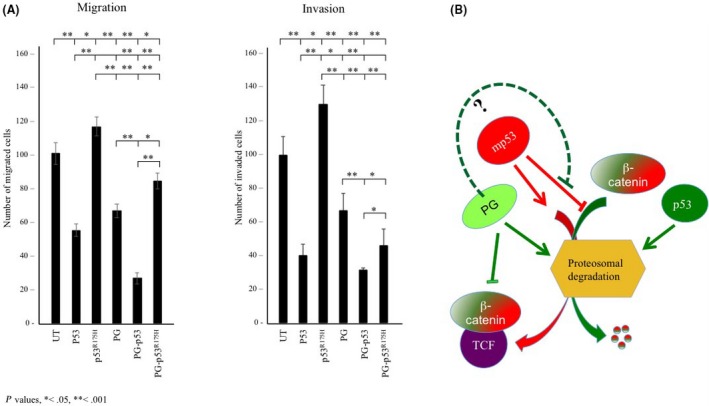
A, Plakoglobin (PG) reduced in vitro migration and invasion of p53R175H‐expressing H1299 cells. H1299 and H1299 transfectants were processed for 24‐h Transwell migration and invasion assays as described in Materials and Methods. Number of migrated/invaded cells in 5 random fields were counted using ImageJ cell counter program. Four separate experiments were carried out for each cell line and the histograms represent the average ± SD of the number of the migrated/invaded cells for each cell line. All values were normalized to H1299 untransfected cells. UT, untransfected. *P < .05, **P < .001. B, Hypothetical model for restoration of tumor‐suppressor activity of mutant p53 by plakoglobin. p53 enhances β‐catenin's proteasomal degradation by increasing the assembly of the destruction complex and/or inducing the expression of Siah‐1, whereas mp53 inhibit β‐catenin degradation (see text for details). This would lead to increased stability and translocation of β‐catenin to the nucleus and transactivation of target genes in mp53‐expressing cells. Based on the current study and our previous findings, plakoglobin may counteract the oncogenic function of mutant p53 by increasing β‐catenin's proteasomal degradation and by restoring tumor‐suppressor activities of mutant p53 through enabling their interaction with promoters of wild‐type p53 target genes16, 17 or modifying mutant p53 target gene expression.16 TCF, T‐cell factor
Similarly, the invasiveness of H1299‐p53 and H1299‐PG cells was decreased by 60% and 33%, respectively, whereas the invasiveness of H1299‐PG‐p53 cells was decreased by 68% relative to parental H1299 cells (Figure 5A, invasion; H1299‐p53, H1299‐PG and H1299‐PG‐p53, also see18). In contrast, p53R175H expression in H1299 cells increased their invasiveness by 30%, and the coexpression of plakoglobin in these cells reduced their invasiveness by >60% (Figure 5A, invasion, H1299‐p53R175H and H1299‐PG‐p53R175H). These results indicated that plakoglobin acted synergistically with p53 to decrease migration and invasion and significantly reduced the migration and invasion‐promoting effects of p53R175H.
4. DISCUSSION
p53R175H is one of the most common hot spot mutations that are frequently expressed in many cancers.13, 14 p53R175H expression has been shown to increase genomic instability,60 induce oncogenic miRNAs expression61, 62 and promote cancer stem cell population expansion,63 epithelial to mesenchymal transition,62, 63, 64, 65, 66 and drug resistance.67, 68, 69 p53R175H mice models show tumor formation and metastasis characteristics of the inherited Li‐Fraumeni syndrome, the disease that is associated with germline mutations in the TP53 gene.70, 71
In the present study, we used invasive and metastatic H1299 cells with the activated Wnt/β‐catenin pathway.72 This cell line is plakoglobin deficient and p53 null and has been extensively used to assess the function of p53 and p53 mutants. We showed that the expression of p53R175H increased β‐catenin levels, its interaction with TCF and activation of c‐MYC and S100A4, 2 known β‐catenin/TCF target genes.41, 42, 43, 57, 58, 59 Increased β‐catenin levels and activation were concurrent with increased migration and invasion of p53R175H‐expressing cells. We further showed that the oncogenic effects of p53R175H were counteracted by the coexpression of plakoglobin in these cells. Plakoglobin interacted with p53R175H and reduced β‐catenin level, its interaction with TCF and the expression of c‐MYC and S100A4. These changes were concurrent with decreased migration and invasion of these transfectants and are consistent with the previously reported activated Wnt/β‐catenin pathway in these cells.72
β‐Catenin is the main downstream effector of the canonical Wnt signaling pathway.38, 39, 40, 41, 42, 43, 44, 45 In the absence of Wnt signal, Axin/APC/GSK3β/CKI forms the destruction complex that recruits and phosphorylates excessive cytoplasmic β‐catenin, which is subsequently ubiquitinated and degraded by the proteasome pathway.38, 39, 40 In the presence of Wnt, the destruction complex is inactivated and the stabilized β‐catenin translocates into the nucleus, binds to TCF/LEF and induces the expression of Wnt target genes involved in tumorigenesis and metastasis.38, 39 β‐Catenin can also be activated independent of the Wnt signal. Mutations that interfere with β‐catenin's interaction with the components of the destruction complex or with the phosphorylation of its N‐terminal serine/threonine residues (S33, S37, S45, T41) required for its degradation also activate β‐catenin in the absence of the Wnt signal.73, 74
In agreement with previous reports, we showed that wild‐type p53 expression in H1299 cells reduced the total and nuclear β‐catenin levels and its transcriptional activity. There are several mechanisms by which p53 reduces β‐catenin protein levels and activation. p53 interacts with and activates GSK3β and/or accelerates the movement of the scaffolding protein, Axin, into the destruction complex, both of which lead to increased phosphorylation of β‐catenin and its subsequent degradation.75, 76 p53 also inhibits the activity of CK2, which phosphorylates and protects β‐catenin from proteasomal degradation.77 Furthermore, p53 can inhibit the Wnt pathway by inducing the expression of the Wnt antagonizer, Dickkopf‐1, the E3 ubiquitin ligase Siah1 that mediates the degradation of β‐catenin independent of the GSK3β and miR‐34 tumor suppressor, which inhibits the expression of several components of the Wnt pathway.44, 45, 78, 79, 80 In contrast, GOF mutant p53 have been shown to inhibit β‐catenin degradation and promote its oncogenic activities and suppress miR‐34 expression.35, 36, 37, 44, 45, 79, 81 Here, we showed that p53R175H expression significantly reduced β‐catenin degradation through the proteasome pathway. p53R175H‐expressing cells had increased total and nuclear β‐catenin and β‐catenin/TCF reporter activity and showed upregulation of the Wnt target genes.41, 42, 43, 57, 58, 59
Previously, we have shown that plakoglobin interacted with both wild type and a number of mutant p53 proteins and this interaction was direct (data not shown) and mediated by the DNA‐binding domain of p53 and the C‐terminal transactivation domain of plakoglobin.16, 17, 18, 19 We showed that plakoglobin and p53 associated with promoters of a number of p53 target genes including tumor suppressors SFN (14‐3‐3δ) and NME1 and the oncogenic genome organizer SATB1.16, 17, 18, 19 Furthermore, plakoglobin expression in plakoglobin‐deficient and mp53‐expressing cells reduced growth, migration and invasion of these cells in vitro.16, 17, 19 Plakoglobin has also been shown to regulate the expression of HAI‐1 and to reduce migration in a p53‐dependent way in NSCLC cells.27 Coimmunoprecipitation experiments indicated that plakoglobin interacted with p53.R175H Furthermore, expression of plakoglobin in p53R175H cells promoted β‐catenin's proteasomal degradation and significantly reduced its total and nuclear levels, as had been reported previously.82 Plakoglobin expression also reduced β‐catenin/TCF‐4 interaction, and the expression of c‐MYC and S100A4. These observations are also supported by our previous microarray studies, which identified p44 and p56 of the 26S proteasome and S100A4 as transcripts that were upregulated and downregulated, respectively, in plakoglobin‐expressing cells relative to their plakoglobin‐deficient and mutant p53‐expressing parental cells.29 S100A4, a recently identified target of β‐catenin/TCF83 was shown to be an early factor in EMT and its elevated level in various carcinoma cells and cancers was correlated with poor prognosis.84, 85
Apart from increasing β‐catenin's proteasomal degradation, plakoglobin may inhibit β‐catenin's transcriptional activity. β‐Catenin and plakoglobin interact with 2 sequential and non‐overlapping domains in the N‐terminus of TCF.32 However, whereas binding of the β‐catenin/TCF complex to DNA is highly efficient; plakoglobin/TCF binding to DNA is inefficient with significantly weaker transcriptional activities.86, 87, 88 Our Co‐IP studies showed significant reduction in β‐catenin‐TCF association in plakoglobin‐expressing cells. These results further support the decreased c‐Myc and S100A4 expression in H1299‐PG‐p53R175H transfectants and are consistent with our previous observations in another mutant p53‐expressing carcinoma cell line.29 Studies from other groups have also shown that transcriptional activity of β‐catenin downstream of Wnt signaling was significantly reduced upon increased accumulation of plakoglobin in the nucleus.32, 33 Plakoglobin also repressed Wnt/β‐catenin signaling and target gene expression (DICER and AXIN2) through its association with the transcription factor SOX4 and inhibition of β‐catenin‐SOX4 interaction.26
In addition to upregulating the Wnt pathway, p53R175H has been shown to activate other signaling pathways including EGFR/PI3K/AKT, TGF‐β and c‐Met, leading to enhanced migratory and invasiveness of cancer cells.89, 90, 91, 92, 93 Our results also showed increased migration and invasion of H1299 cells expressing p53R175H and their significant decrease when plakoglobin was coexpressed in these cells. Although the effects of mp53 on enhancing migratory and invasive properties of cancer cells have been studied extensively, the effects of plakoglobin on hindering the in vitro metastatic features of p53R175H is novel and has not been previously reported.
In conclusion, our data suggest that plakoglobin promoted β‐catenin's proteasomal degradation and reduced its transcriptional activation independent of p53 status. Furthermore, its coexpression with p53R175H clearly counteracted the gain‐of‐function activities of this mutant, which is mediated, at least in part, by activating the oncogenic function of β‐catenin. These observations together with our previous studies suggest that plakoglobin may counteract oncogenic functions of mutant p53 by at least 2 different mechanisms: plakoglobin augments β‐catenin proteasomal degradation and reduces Wnt pathway activation, and it associates with mutant p53 and may either interfere with the expression of mutant p53 target genes and/or enable them to interact with and regulate wild‐type p53 target genes (Figure 5B). The latter possibility is supported by our previous studies that have shown activation of p53 target genes in plakoglobin‐deficient and mutant p53‐expressing cell lines upon plakoglobin expression as well as our microarray experiments that have identified a number of growth/metastasis inhibiting and oncogenic promoting targets that are up‐ and downregulated, respectively, in mutant p53‐expressing cells when plakoglobin is expressed.16, 17 Overall, these results suggest that plakoglobin may act as a tumor/metastasis suppressor protein in mutant p53‐expressing cells by downregulating the Wnt/β‐catenin axis and oncogenic activation of mutant p53, 2 pathways that are known to be frequently dysregulated in many cancers. These findings provide insight into the possibility of developing therapeutic drugs that can mimic plakoglobin to concurrently inhibit the oncogenic effects of β‐catenin and restore wild‐type tumor suppressor activities of mutant p53 in cancer.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
We would like to thank Drs G. Blandino and S. Di Agostino for the p53R175H construct. This work was supported by the Canadian Breast Cancer Foundation Prairies/NWT Chapter (MP) and the generous supporters of the Lois Hole Hospital for Women through the Women and Children's Health Research institute (M.A.). M.A. is the recipient of the Cathy and Harold Roozen Entrance Scholarship, Dr. Herbert Meltzer Memorial Fellowship) and Andrew Stewart Memorial Graduate Prize.
Alaee M, Nool K, Pasdar M. Plakoglobin restores tumor suppressor activity of p53R175H mutant by sequestering the oncogenic potential of β‐catenin. Cancer Sci. 2018;109:1876–1888. https://doi.org/10.1111/cas.13612
Funding information
Cathy and Harold Roozen Entrance Scholarship (MA) (Grant/Award Number: NA), Canadian Breast Cancer Foundation Prairies/NWT Chapter (MP) (Grant/Award Number: NA), Lois Hole Hospital for Women through the Women and Children's Health Research Institute (MA) (Grant/Award Number: NA), Dr. Herbert Meltzer Memorial Fellowship and Andrew Stewart Memorial Graduate Prize (MA) (Grant/Award Number: NA)
REFERENCES
- 1. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393‐405. [DOI] [PubMed] [Google Scholar]
- 2. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15‐16. [DOI] [PubMed] [Google Scholar]
- 3. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl). 2011;89:237‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self‐renewing divisions in mammary stem cells. Cell. 2009;138:1083‐1095. [DOI] [PubMed] [Google Scholar]
- 6. Golubovskaya VM, Cance W. Focal adhesion kinase and p53 signal transduction pathways in cancer. Front Biosci (Landmark Ed). 2010;15:901‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mukhopadhyay UK, Mak AS. p53: is the guardian of the genome also a suppressor of cell invasion? Cell Cycle. 2009;8:2481. [DOI] [PubMed] [Google Scholar]
- 8. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2‐8. [DOI] [PubMed] [Google Scholar]
- 9. Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild‐type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011;18:2‐11. [DOI] [PubMed] [Google Scholar]
- 10. Collavin L, Lunardi A, Del Sal G. p53‐family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901‐911. [DOI] [PubMed] [Google Scholar]
- 11. http://p53.free.fr/.
- 12. Mello SS, Attardi LD. Not all p53 gain‐of‐function mutants are created equal. Cell Death Differ. 2013;20:855‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oren M, Rotter V. Mutant p53 gain‐of‐function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Freed‐Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268‐1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aktary Z, Kulak S, Mackey J, Jahroudi N, Pasdar M. Plakoglobin interacts with the transcription factor p53 and regulates the expression of 14‐3‐3σ. J Cell Sci. 2013;126:3031‐3042. [DOI] [PubMed] [Google Scholar]
- 17. Aktary Z, Pasdar M. Plakoglobin represses SATB1 expression and decreases in vitro proliferation, migration and invasion. PLoS One. 2013;8:e78388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alaee M, Padda A, Mehrabani V, Churchill L, Pasdar M. The physical interaction of p53 and plakoglobin is necessary for their synergistic inhibition of migration and invasion. Oncotarget. 2016;7:26898‐26915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alaee M, Danesh G, Pasdar M. Plakoglobin reduces the in vitro growth, migration and invasion of ovarian cancer cells expressing N‐Cadherin and Mutant p53. PLoS One. 2016;11:e0154323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aktary Z, Pasdar M. Plakoglobin: role in tumorigenesis and metastasis. Int J Cell Biol. 2012;2012:189521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aktary Z, Alaee M, Pasdar M. Beyond cell‐cell adhesion: plakoglobin and the regulation of tumorigenesis and metastasis. Oncotarget. 2017;8:32270‐32291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller RK, Hong JY, Muñoz WA, McCrea PD. Beta‐catenin versus the other armadillo catenins: assessing our current view of canonical Wnt signaling. Prog Mol Biol Transl Sci. 2013;116:387‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim W, Kim M, Jho EH. Wnt/β‐catenin signalling: from plasma membrane to nucleus. Biochem J. 2013;450:9‐21. [DOI] [PubMed] [Google Scholar]
- 24. Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. The vertebrate adhesive junction proteins beta‐catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol. 1992;118:681‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aktary Z, Chapman K, Lam L, et al. Plakoglobin interacts with and increases the protein levels of metastasis suppressor Nm23‐H2 and regulates the expression of Nm23‐H1. Oncogene. 2010;29:2118‐2129. [DOI] [PubMed] [Google Scholar]
- 26. Lai YH, Cheng J, Cheng D, et al. SOX4 interacts with plakoglobin in a Wnt3a‐dependent manner in prostate cancer cells. BMC Cell Biol. 2011;12:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sechler M, Borowicz S, Van Scoyk M, et al. Novel role for γ‐catenin in the regulation of cancer cell migration via the induction of hepatocyte growth factor activator inhibitor type 1 (HAI‐1). J Biol Chem. 2015;290:15610‐15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lam L, Aktary Z, Bishay M, et al. Regulation of subcellular distribution and oncogenic potential of nucleophosmin by plakoglobin. Oncogenesis. 2012;1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L, Chapman K, Hu X, Wong A, Pasdar M. Modulation of the oncogenic potential of beta‐catenin by the subcellular distribution of plakoglobin. Mol Carcinog. 2007;46:824‐838. [DOI] [PubMed] [Google Scholar]
- 30. Zhurinsky J, Shtutman M, Ben‐Ze'ev A. Plakoglobin and beta‐catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000;113:3127‐3139. [DOI] [PubMed] [Google Scholar]
- 31. Williams BO, Barish GD, Klymkowsky MW, Varmus HE. A comparative evaluation of beta‐catenin and plakoglobin signaling activity. Oncogene. 2000;19:5720‐5728. [DOI] [PubMed] [Google Scholar]
- 32. Miravet S, Piedra J, Miro F, Itarte E, Garcia de Herreros A, Dunach M. The transcriptional factor Tcf‐4 contains different binding sites for beta‐catenin and plakoglobin. J Biol Chem. 2002;277:1884‐1891. [DOI] [PubMed] [Google Scholar]
- 33. Chen YJ, Lee LY, Chao YK, et al. DSG3 facilitates cancer cell growth and invasion through the DSG3‐plakoglobin‐TCF/LEF‐Myc/cyclin D1/MMP signaling pathway. PLoS One. 2013;8:e64088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lombardi R, da Graca Cabreira‐Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 2011;109:1342‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cagatay T, Ozturk M. P53 mutation as a source of aberrant beta‐catenin accumulation in cancer cells. Oncogene. 2002;21:7971‐7980. [DOI] [PubMed] [Google Scholar]
- 36. Sadot E, Geiger B, Oren M, Ben‐Ze'ev A. Down‐regulation of beta‐catenin by activated p53. Mol Cell Biol. 2001;21:6768‐6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prowald A, Cronauer MV, von Klot C, et al. Modulation of beta‐catenin‐mediated TCF‐signalling in prostate cancer cell lines by wild‐type and mutant p53. Prostate. 2007;67:1751‐1760. [DOI] [PubMed] [Google Scholar]
- 38. Clevers H, Nusse R. Wnt/β‐catenin signaling and disease. Cell. 2012;149:1192‐1205. [DOI] [PubMed] [Google Scholar]
- 39. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767‐779. [DOI] [PubMed] [Google Scholar]
- 40. Kretzschmar K, Clevers H. Wnt/β‐catenin signaling in adult mammalian epithelial stem cells. Dev Biol. 2017;428:273‐282. [DOI] [PubMed] [Google Scholar]
- 41. Tarabykina S, Griffiths TR, Tulchinsky E, Mellon JK, Bronstein IB, Kriajevska M. Metastasis‐associated protein S100A4: spotlight on its role in cell migration. Curr Cancer Drug Targets. 2007;7:217‐228. [DOI] [PubMed] [Google Scholar]
- 42. Stein U, Arlt F, Smith J, et al. Intervening in β‐catenin signaling by sulindac inhibits S100A4‐dependent colon cancer metastasis. Neoplasia. 2011;13:131‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sack U, Walther W, Scudiero D, et al. S100A4‐induced cell motility and metastasis is restricted by the Wnt/β‐catenin pathway inhibitor calcimycin in colon cancer cells. Mol Biol Cell. 2011;22:3344‐3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J, Stevens J, Rote CA, et al. Siah‐1 mediates a novel beta‐catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001;7:927‐936. [DOI] [PubMed] [Google Scholar]
- 45. Matsuzawa SI, Reed JC. Siah‐1, SIP, and Ebi collaborate in a novel pathway for beta‐catenin degradation linked to p53 responses. Mol Cell. 2001;7:915‐926. [DOI] [PubMed] [Google Scholar]
- 46. Abou Zeinab R, Wu H, Sergi C, Leng RP. Residues 240‐250 in the C‐terminus of the Pirh2 protein complement the function of the RING domain in self‐ubiquitination of the Pirh2 protein. PLoS One. 2013;8:e82803. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47. Valenti F, Ganci F, Fontemaggi G, et al. Gain of function mutant p53 proteins cooperate with E2F4 to transcriptionally downregulate RAD17 and BRCA1 gene expression. Oncotarget. 2015;6:5547‐5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pasdar M, Nelson WJ. Kinetics of desmosome assembly in Madin‐Darby canine kidney epithelial cells: temporal and spatial regulation of desmoplakin organization and stabilization upon cell‐cell contact. II. Morphological analysis. J Cell Biol. 1988;106:687‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Molenaar M, van de Wetering M, Oosterwegel M, et al. XTcf‐3 transcription factor mediates beta‐catenin‐induced axis formation in Xenopus embryos. Cell. 1996;86:391‐399. [DOI] [PubMed] [Google Scholar]
- 50. Baker JM, Boyce FM. High‐throughput functional screening using a homemade dual‐glow luciferase assay. J Vis Exp. 2014; https://doi.org/10.3791/50282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stamos JL, Weis WI. The β‐catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5:a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Çelen İ, Ross KE, Arighi CN, Wu CH. Bioinformatics knowledge map for analysis of beta‐catenin function in cancer. PLoS One. 2015;10:e0141773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fiucci G, Beaucourt S, Duflaut D, et al. Siah‐1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah‐1b promoter. Proc Natl Acad Sci USA. 2004;101:3510‐3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie W, Jin L, Mei Y, Wu M. E2F1 represses beta‐catenin/TCF activity by direct up‐regulation of Siah1. J Cell Mol Med. 2009;13:1719‐1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pallier K, Cessot A, Côté JF, et al. TWIST1 a new determinant of epithelial to mesenchymal transition in EGFR mutated lung adenocarcinoma. PLoS One. 2012;7:e29954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta‐catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA. 1997;94:10330‐10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yochum GS, Cleland R, Goodman RH. A genome‐wide screen for beta‐catenin binding sites identifies a downstream enhancer element that controls c‐Myc gene expression. Mol Cell Biol. 2008;28:7368‐7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Herbst A, Jurinovic V, Krebs S, et al. Comprehensive analysis of β‐catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β‐catenin signaling. BMC Genom. 2014;15:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rennoll S, Yochum G. Regulation of MYC gene expression by aberrant Wnt/β‐catenin signaling in colorectal cancer. World J Biol Chem. 2015;6:290‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Samassekou O, Bastien N, Lichtensztejn D, Yan J, Mai S, Drouin R. Different TP53 mutations are associated with specific chromosomal rearrangements, telomere length changes, and remodeling of the nuclear architecture of telomeres. Genes Chromosom Cancer. 2014;53:934‐950. [DOI] [PubMed] [Google Scholar]
- 61. Donzelli S, Fontemaggi G, Fazi F, et al. MicroRNA‐128‐2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012;19:1038‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masciarelli S, Fontemaggi G, Di Agostino S, et al. Gain‐of‐function mutant p53 downregulates miR‐223 contributing to chemoresistance of cultured tumor cells. Oncogene. 2014;33:1601‐1608. [DOI] [PubMed] [Google Scholar]
- 63. Lu X, Liu DP, Xu Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene. 2013;32:2900‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rieber M, Strasberg Rieber M. DN‐R175H p53 mutation is more effective than p53 interference in inducing epithelial disorganization and activation of proliferation signals in human carcinoma cells: role of E‐cadherin. Int J Cancer. 2009;125:1604‐1612. [DOI] [PubMed] [Google Scholar]
- 65. Kogan‐Sakin I, Tabach Y, Buganim Y, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial‐mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011;18:271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Y, Yan W, Chen X. Mutant p53 disrupts MCF‐10A cell polarity in three‐dimensional culture via epithelial‐to‐mesenchymal transitions. J Biol Chem. 2011;286:16218‐16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsang WP, Chau SP, Fung KP, Kong SK, Kwok TT. Modulation of multidrug resistance‐associated protein 1 (MRP1) by p53 mutant in Saos‐2 cells. Cancer Chemother Pharmacol. 2003;51:161‐166. [DOI] [PubMed] [Google Scholar]
- 68. Tsang WP, Ho FY, Fung KP, Kong SK, Kwok TT. p53‐R175H mutant gains new function in regulation of doxorubicin‐induced apoptosis. Int J Cancer. 2005;114:331‐336. [DOI] [PubMed] [Google Scholar]
- 69. Capponcelli S, Pedrini E, Cerone MA, et al. Evaluation of the molecular mechanisms involved in the gain of function of a Li‐Fraumeni TP53 mutation. Hum Mutat. 2005;26:94‐103. [DOI] [PubMed] [Google Scholar]
- 70. Lang GA, Iwakuma T, Suh YA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li‐Fraumeni syndrome. Cell. 2004;119:861‐872. [DOI] [PubMed] [Google Scholar]
- 71. Liu G, McDonnell TJ, Montes de Oca Luna R, et al. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci USA 2000;97:4174‐4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Qu J, Li M, An J, et al. MicroRNA‐33b inhibits lung adenocarcinoma cell growth, invasion, and epithelial‐mesenchymal transition by suppressing Wnt/β‐catenin/ZEB1 signaling. Int J Oncol. 2015;47:2141‐2152. [DOI] [PubMed] [Google Scholar]
- 73. Cui J, Zhou X, Liu Y, Tang Z, Romeih M. Wnt signaling in hepatocellular carcinoma: analysis of mutation and expression of beta‐catenin, T‐cell factor‐4 and glycogen synthase kinase 3‐beta genes. J Gastroenterol Hepatol. 2003;18:280‐287. [DOI] [PubMed] [Google Scholar]
- 74. Provost E, Yamamoto Y, Lizardi I, et al. Functional correlates of mutations in beta‐catenin exon 3 phosphorylation sites. J Biol Chem. 2003;278:31781‐31789. [DOI] [PubMed] [Google Scholar]
- 75. Watcharasit P, Bijur GN, Zmijewski JW, et al. Direct, activating interaction between glycogen synthase kinase‐3beta and p53 after DNA damage. Proc Natl Acad Sci USA. 2002;99:7951‐7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Levina E, Oren M, Ben‐Ze'ev A. Downregulation of beta‐catenin by p53 involves changes in the rate of beta‐catenin phosphorylation and Axin dynamics. Oncogene. 2004;23:4444‐4453. [DOI] [PubMed] [Google Scholar]
- 77. Schuster N, Götz C, Faust M, et al. Wild‐type p53 inhibits protein kinase CK2 activity. J Cell Biochem. 2001;81:172‐183. [DOI] [PubMed] [Google Scholar]
- 78. Wang J, Shou J, Chen X. Dickkopf‐1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene. 2000;19:1843‐1848. [DOI] [PubMed] [Google Scholar]
- 79. Iwai A, Marusawa H, Matsuzawa S, et al. Siah‐1L, a novel transcript variant belonging to the human Siah family of proteins, regulates beta‐catenin activity in a p53‐dependent manner. Oncogene. 2004;23:7593‐7600. [DOI] [PubMed] [Google Scholar]
- 80. Hosain SB, Khiste SK, Uddin MB, et al. Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial‐mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget. 2016;7:60575‐60592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kasinski AL, Slack FJ. miRNA‐34 prevents cancer initiation and progression in a therapeutically resistant K‐ras and p53‐induced mouse model of lung adenocarcinoma. Cancer Res. 2012;72:5576‐5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Salomon D, Sacco PA, Roy SG, et al. Regulation of beta‐catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin‐proteasome system. J Cell Biol. 1997;139:1325‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stein U, Arlt F, Walther W, et al. The metastasis‐associated gene S100A4 is a novel target of beta‐catenin/T‐cell factor signaling in colon cancer. Gastroenterology. 2006;131:1486‐1500. [DOI] [PubMed] [Google Scholar]
- 84. Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial‐mesenchymal transformation. Am J Physiol. 1997;273:F563‐F574. [DOI] [PubMed] [Google Scholar]
- 85. Dahlmann M, Kobelt D, Walther W, Mudduluru G, Stein U. S100A4 in Cancer metastasis: Wnt signaling‐driven interventions for metastasis restriction. Cancers (Basel). 2016;8:E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Simcha I, Shtutman M, Salomon D, et al. Differential nuclear translocation and transactivation potential of beta‐catenin and plakoglobin. J Cell Biol. 1998;141:1433‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim YM, Ma H, Oehler VG, et al. The gamma catenin/CBP complex maintains survivin transcription in β‐catenin deficient/depleted cancer cells. Curr Cancer Drug Targets. 2011;11:213‐225. [DOI] [PubMed] [Google Scholar]
- 88. Maeda O, Usami N, Kondo M, et al. Plakoglobin (gamma‐catenin) has TCF/LEF family‐dependent transcriptional activity in beta‐catenin‐deficient cell line. Oncogene. 2004;23:964‐972. [DOI] [PubMed] [Google Scholar]
- 89. Boudreau HE, Casterline BW, Burke DJ, Leto TL. Wild‐type and mutant p53 differentially regulate NADPH oxidase 4 in TGF‐β‐mediated migration of human lung and breast epithelial cells. Br J Cancer. 2014;110:2569‐2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dong P, Xu Z, Jia N, Li D, Feng Y. Elevated expression of p53 gain‐of‐function mutation R175H in endometrial cancer cells can increase the migratory and invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol Cancer. 2009;8:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gurtner A, Starace G, Norelli G, Piaggio G, Sacchi A, Bossi G. Mutant p53‐induced up‐regulation of mitogen‐activated protein kinase kinase 3 contributes to gain of function. J Biol Chem. 2010;285:14160‐14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Grugan KD, Vega ME, Wong GS, et al. A common p53 mutation (R175H) activates c‐Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol Ther. 2013;14:853‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim NH, Kim HS, Kim NG, et al. p53 and microRNA‐34 are suppressors of canonical Wnt signaling. Sci Signal. 2011;4:ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]