

Abstract
Background
Sarcopenia is the age‐related degeneration characterized with the decline of skeletal muscle mass, strength, and function. The imbalance of protein synthesis and degradation which jeopardizes immune, hormone regulation, and muscle‐motor neuron connection is the main cause of sarcopenia. There is limited knowledge regarding molecular mechanism of sarcopenia. As the endoplasmic reticulum is the control centre of the protein syntheses and degradation, we hypothesized that endoplasmic reticulum stress and unfolded protein response (UPR) play an important in the development of sarcopenia. Understanding the sarcopenia molecular mechanisms may benefit the therapeutic diagnosis and treatment in the future.
Methods
Mouse myoblast C2C12 cells are exposed to designated time and concentration of indoxyl sulfate (IS), a uremic toxin of chronic kidney disease. The proliferation, differentiation, and the expression of atrogin 1 are examined. The protein and mRNA expression of IS treated‐C2C12 cells are inspected to distinguish the role of ER stress and oxidative stress underlying the sarcopenia.
Results
Indoxyl sulfate inhibits myoblast differentiation. We demonstrate that as the number of multi‐nuclei myotube decreased, the differentiation markers including myoD, myoG, and myosin heavy chain are also suppressed. Indoxyl sulfate inhibits myoblast proliferation and induces the myotubular atrophy marker atrogin‐1 protein expression. Indoxyl sulfate stimulates eIF2α phosphorylation and XBP1 mRNA splicing in UPR. Interestingly, the oxidative stress is related to eIF2α phosphorylation but not XBP1 mRNA splicing. The eIF2α phosphorylation triggered by IS reduces myoD, myoG, and myosin heavy chain protein expression, which represents the anti‐myogenic modulation on the early differentiation event. The XBP1 mRNA splicing induced by IS, however, is considered the adaptive response to restore the myogenic differentiation.
Conclusions
Our studies indicated that the ER stress and UPR modulation are critical in the chronic kidney disease uremic toxin‐accumulated sarcopenia model. We believe that UPR‐related signals showed great potential in clinical application.
Keywords: Chronic kidney disease, Indoxyl sulfate, ER stress, Unfolded protein response, Myogenesis
Introduction
Sarcopenia is the progressive loss of muscle mass and strength that comes with aging.1 The causes of sarcopenia include decreased sex hormones, increased proinflammatory cytokines, neurodegeneration, and so on.2, 3, 4, 5 In cellular and molecular levels, the imbalance of protein synthesis and degradation is the most characteristic feature of sarcopenia. Skeletal muscle differentiation (myogenesis) is crucial for muscle tissue formation and regeneration. Myogenic regulatory factors (MRFs) are the master regulators for myogenic precursor cells, and the myoblast terminal differentiation. Myf5 and MyoD were suggested to be the early MRF, while MRF4 and myogenin (myoG) were the late differentiation MRF.6, 7, 8, 9, 10 Thus, MyoD, MyoG, and myosin heavy chain (MyHC) expression were applied to monitor myogenesis.11 In addition, Akt activation enhances the expression of muscle specific proteins and elicits muscle differentiation.12 The AKT–mTOR–p70S6K anabolic cascade promotes protein synthesis and muscle growth.13 Akt also inactivates FoxO1, which induces muscle atrophy by transcriptional regulation, through protein phosphorylation.14, 15, 16 Furthermore, myostatin negatively regulates myogenesis through myoD down‐regulation17 and Akt/TORC1/p70S6K signalling counteraction.18, 19
Similar to what has been observed in sarcopenia, advanced chronic kidney disease (CKD) patients show protein‐energy wasting with the presentation of skeletal muscle atrophy.20, 21 Indoxyl sulfate (IS), a protein‐bound uremic toxin, is elevated in the micromilieu of CKD patients. By the action of intestinal bacteria, dietary tryptophan is metabolized to indole, which is absorbed, metabolized, and sulfonated to IS in the liver.22, 23 The failure of renal excretion causes IS accumulation, which causes numerous uremic symptoms and promotes premature aging.24, 25, 26, 27 Accordingly, it is considered that CKD is a premature aging syndrome and uremic milieu provides a good model to study the aging process.
The endoplasmic reticulum (ER) is an organelle that functions to maintain protein quality control.28 ER functional disturbance causes ER stress and leads to accumulation of unfolded or misfolded proteins, thereby trigging the unfolded protein response (UPR) to alleviate cellular stress and re‐establish homeostasis.29 Protein kinase RNA‐activated (PKR)‐like ER kinase (PERK), inositol‐requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6) are three ER transmembrane sensor proteins, which participate in the UPR.30 Activation of PERK results in the translational attenuation through phosphorylation of the eukaryotic translation initiation factor‐2α (eIF2α), controlling the quantity of proteins in the ER lumen.31 ATF6, which is a transcription factor, induces the expression of ER chaperones to assist protein folding.32, 33 IRE1 is a dual protein kinase/ribonuclease. IRE1‐mediated unconventional XBP1 mRNA splicing in the cytoplasm to generate a mature mRNA, which encodes splicing XBP1 (XBP1s), and then enforces expression of ER chaperones and components of ER‐associated protein degradation.34, 35, 36 Recently, studies imply decline of chaperones and folding enzymes compromise proper protein folding and the adaptive response of the UPR with age.37 In addition, in comparison with young mice, lower ER stress tolerance has been observed in old mice as revealed by elevated UPR signalling.38, 39 These references highlight the significant role of UPR in age‐related disease. Given that the sarcopenia is an age‐related disease, in the present study, we use a uremic milieu premature aging model to investigate the molecular mechanisms, especially UPR, involved in sarcopenia progression.
Materials and methods
Culture of C2C12 mouse myoblasts and myogenic differentiation
Cells were obtained from Bioresource Collection and Research Center (Hsinchu City, Taiwan) and cultured in growth medium (GM) consisting of Dulbecco's modified Eagle's medium (ThermoFisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (ThermoFisher Scientific) at 37°C in a 5% CO2‐humidified environment. For myogenic differentiation, C2C12 myoblasts were placed in differentiation medium (DM) consisting of an equal mixture of two serum‐free media Nutrient Mixture F‐12 K Ham medium (ThermoFisher Scientific) and MCDB201 (Sigma‐Aldrich, St. Louis, MO), along with 2% horse serum (ThermoFisher Scientific) to induce differentiation.
Reagents and antibodies
Indoxyl sulfate (IS), N‐Acetyl‐l‐cysteine (NAC), 2′,7′‐dichlorofluorescin diacetate (DCF‐DA), and salubrinal were purchased from Sigma‐Aldrich. The following antibodies were used: Phospho‐eIF2α (Ser51) (9721S) and Phospho AKT (Ser473) (9271S) from Cell Signalling Technology (Danvers, MA); eIF2α (D‐3) and MYH (H‐300) from Santa Cruz Biotechnology (Dallas, TX); BiP (610978) from BD Biosciences; myoG (GTX63352), GAPDH (GTX100118), and β‐actin (GTX109639) from Genetex (Hsinchu City, Taiwan). Atrogin 1 (ab168372) from Abcam (Cambridge, MA); myoD (5.8A) (NB100‐56511) from Novus Biologicals (Littleton CO), and Myc (CSB‐MA000041M0m) from Cusabio (China). AKT1 (C20) was kindly provided by Dr. Jim‐Tong Horng of Chang Gung University, Taiwan. Anti‐mouse IgG (H + L) and anti‐rabbit IgG (H + L) antibodies were purchased from Genetex. siRNA against XBP1 (siXBP1:CCUUGUAGUUGAGAACCAGGAGUUA) and scrambled siRNA (siCtrl: medium GC of Stealth negative control duplex) were purchased from ThermoFisher Scientific and transfected into cells with Lipofectamine(R) 2000 reagent (ThermoFisher Scientific), according to the manufacturer's protocol. The Smart Quant Green master mix for real‐time quantitative PCR assay was purchased from Protech (Taipei, Taiwan).
Plasmid construction
The full length of eIF2a S52D mutant was amplified by PCR from the cDNA prepared from C2C12 cells using following primers: 5′‐ATAAAGCTTATGCCGGGGCTAAGTTGT‐3′, 5′‐ATATCTAGAGCATCTTCAGCTTTGGCTTCCATTTC‐3′, 5′‐ AGTGAATTAGATAGACGACGTATCC‐3′, and 5′‐GGATACGTCGTCTATCTAATTCACT‐3. The product was inserted into the HindIII and XbaI sites of pcDNA3.1(+)/myc‐His B (ThermoFisher Scientific).
Assessment of cellular proliferation and cell death
Cell proliferation assays were conducted in 96‐well plates. C2C12 myoblasts were seeded at 5 × 103 cells per well in GM for 16 h and then treated with IS (0–1 mM). After 48 h incubation, cell proliferation was determined using an MTS assay of CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) as described by the manufacturer. The data were collected with SpectraMax 190 Microplate Reader (Molecular Devices, Sunnyvale, CA). Trypan blue exclusion assay was used to assess cell death following IS treatment. C2C12 myoblasts (1 × 105 cells per well) were seeded in 6‐well plates in GM for 16 h and then treated with IS (0–1 mM). After 48 h of incubation, cells were washed with PBS and trypsinized with 0.25% trypsin–EDTA (ThermoFisher Scientific). The cells were resuspended in 0.4% trypan blue solution (ThermoFisher Scientific) and counted using a haemocytometer. Each sample was run in triplicate.
Morphological analysis
After 3 or 4 days of cell culture in differentiation media, the mature C2C12 myotubes were determined morphologically by analysis of multinucleated myotube formation with haematoxylin and eosin (H&E) staining. The myotube formation was examined under a Leica DMi8 inverted microscope equipped with Leica DFC7000 T CCD Microscope Camera (Wetzlar, Germany). Random five separate views at magnification of ×100 were captured and quantified. The proportion of 2–5, 5–10, and >10 nuclear myotubes per number of all myotubes was shown.
Immunoblotting
Cells were lysed in RIPA lysis buffer (Cell Signalling Technology) containing a protease inhibitor cocktail (Roche, Basel, Switzerland). Lysates were incubated for 10 min on ice and then centrifuged at 14 000 rpm for 10 min at 4°C. The supernatants were collected, and the protein concentrations were measured by the Coomassie Protein Assay Reagent (ThermoFisher Scientific). Equal amounts of proteins (30–50 μg) were subjected to SDS‐PAGE. Following electrophoresis, the proteins were electrotransferred to the polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membrane was blocked with TBST (0.2% Tween 20 (vol/vol)) containing 5% nonfat milk for 1 h. Membranes were immunoblotted with specific primary antibodies followed by horseradish peroxidase (HRP)‐conjugated secondary antibodies, and developed with Immobilon Western HRP Chemiluminescent Substrate (Millipore). The chemiluminescent image was captured with BioSpectrum 810 Imaging System (UVP, Upland, CA).
RNA extraction, reverse transcription, RT‐PCR, and quantitative real‐time PCR
DNase‐treated total RNA was extracted using GENEzol™ TriRNA Pure Kit (Geneaid, New Taipei City, Taiwan). One microgram of RNA was reverse transcribed with iScript reverse transcription (RT) supermix (Bio‐Rad, Hercules, CA). The resulting cDNA products were amplified with specific primer pairs to detect mRNA abundance using a StepOnePlus real‐time PCR system (ThermoFisher Scientific). The relative expression of the target genes was calculated using the comparative threshold cycle (C T) method (ΔΔC T). Primers used for real‐time PCRs are as follows: myoG forward primer 5′‐ACGAAACCATGCCCAACTGA‐3′ and myoG reverse primer‐5′ CCACTTAAAAGCCCCCTGCT‐3′; myoD forward primer 5′‐TGAATGAGGCCTTCGAGACG‐3′ and myoD reverse primer 5′‐ACCTTCGATGTAGCGGATGG‐3′; MyHC forward primer 5′‐GAAGCGAGGCACAAAATGTGA‐3′ and MyHC reverse primer 5′‐TTGCTTGCAAAGGAACTTGGG‐3′; BiP forward primer 5′‐AGAAACTCCGGCGTGAGGTAGA‐3′ and BiP reverse primer 5′‐TTCCTGGACAGGCTTCATGGTAG‐3′; GAPDH forward primer 5′‐TGACTCCACTCACGGCAAAT‐3′ and GAPDH reverse primer 5′‐GTCTCGCTCCTGGAAGATGG‐3′. XBP1u and XBP1s mRNA were monitored by semi‐quantitative PCR using the following primers: XBP1 forward primer 5′‐GTCTGCTGAGTCCGCA‐3′ and XBP1 reverse primer 5′‐TCCTTCTGGGTAGACCTCTGGGAG‐3′. GAPDH forward primer 5′‐TGACTCCACTCACGGCAAAT‐3′ and GAPDH reverse primer 5′‐GGCATGGACTGTGGTCATGAG‐3′.
Intracellular ROS detection
DCF‐DA was used to detect intracellular reactive oxygen species (ROS) production. Cultured C2C12 cells were treated by IS with or without 30 min of pre‐incubation with NAC. After 48 h, cells were treated with 25 μM DCF‐DA for 30 min at 37°C, and fluorescence was then analysed using flow cytometry BD LSRII (BD Bioscience).
Statistics
Results are presented as the mean ± SEM. The significance of difference was determined by the Student's two‐tailed t test. A P‐value of <0.05 was considered to be significant.
Results
Indoxyl sulfate treatment inhibits myoblast differentiation in a dose‐dependent manner
We first assessed the effects of IS during the myogenic differentiation process. As shown in Figure 1A, addition of IS (0, 0.1, 0.5, and 1 mM) 2 days before induction of differentiation, to mimic the uremic milieu, markedly inhibited the formation of matured myotubes after 4 days of DM exposure. In total, 6 days of IS exposure significantly suppressed myoblast differentiation in a dose‐dependent manner. Moreover, to keep an eye on myoblast fusion, numbers of nuclei per myotube were assessed. As shown in Figure 1B, IS treatment reduced the percentage of myotube with 5 to 10 nuclei, and none of the myotube had the nuclei more than 10 when the concentration of IS raised up to 1 mM. These data demonstrated that IS treatment dose‐dependently inhibits myoblasts differentiation. We further examined the expression of differentiation markers myoG and MyHC. Compared with non‐treated cells, IS‐treated cells exhibited lower levels of myoG and MyHC at both mRNA and protein levels (Figure 1C‐F).
Figure 1.

IS impairs C2C12 myogenic differentiation (MD) in a dose‐dependent manner. (A) H&E staining of C2C12 myotubes under IS treatment, scale bars = 200 μm. The arrows indicate the fused myoblasts. (B) The number of nuclei per myotube at 4 days of differentiation were counted. The values were then classified in three categories and divided by the total number of myotubes in a field. The data were expressed as mean ± SED from three independent experiments. (C, E, and F) Protein expression of myogenin and MyHC expression in differentiated myoblasts treated with or without IS (1 mM) are examined by western blot and quantified. (D) The relative myogenin and MyHC mRNA expression levels are determined by real‐time PCR. Data represent means ± SEM for three independent experiments. *P < 0.05, ** P < 0.01, and *** P < 0.001, as compared with untreated control.
IS inhibits myoblast proliferation and induces atrogin 1 expression
Impaired proliferation of myoblasts and enhanced myotube atrophy would precede loss of muscle mass. We thus performed MTS assay and trypan blue exclusion assay to investigate whether IS treatment diminishes proliferation and induces cell death. C2C12 myoblast cells were seeded in GM. After plating down for 24 hours, cells were treated with indicated concentration of IS (0–1 mM). The results showed that IS had growth inhibitory effect (Figure 2A) but had no sign of cell death on myoblast (Figure 2B) after 48 hours exposure. These results confirm that the inhibitory effects of IS on myogenic differentiation are not due to a nonspecific cytotoxic effect. The ubiquitin‐proteasome system is the degradation system involved in myotube atrophy. Atrogin 1 is an important enzyme in ubiquitin‐mediated proteolysis; therefore, we treated 4‐days differentiated C2C12 myotubes with or without 1 mM IS for additional 2 days and cell extracts made were analysed by immunoblot using anti‐atrogin 1 antibody. IS treatment up‐regulates atrogin 1 protein level and down‐regulates MyHC, a substrate of muscle‐specific E3 ubiquitin ligase under atrophy condition40 (Figure 2C‐E). Enoki et al. demonstrated that atrogin 1 expression is induced by continuous IS exposure throughout myotube differentiation.41 In our study, however, IS can trigger the atrogin 1 expression even at time point near the completion of the differentiation.
Figure 2.
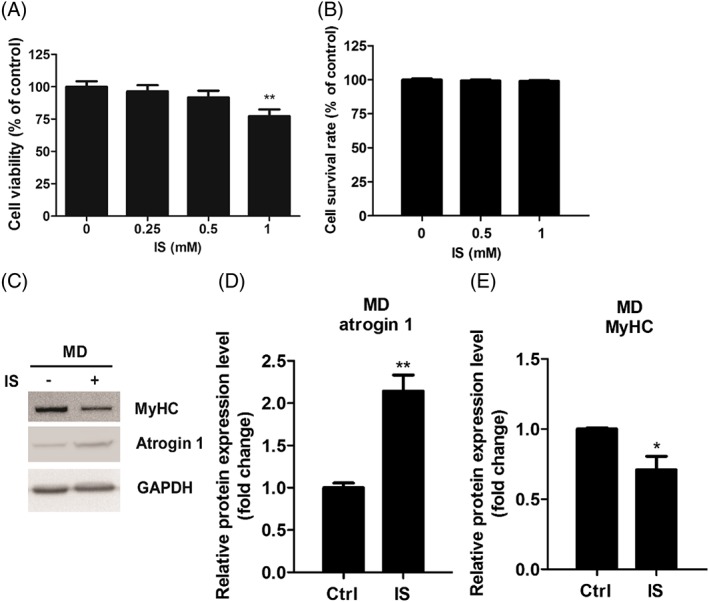
IS treatment inhibits myoblast proliferation and enhances atrogin 1 expression in differentiated myoblasts. (A) C2C12 myoblasts were incubated with growth medium containing indicated concentrations of IS for 48 h. Cell viability was determined via MTS assay. (B) Myoblast apoptotic potential are demonstrated by trypan blue exclusion assay. (C–E) Atrogin 1 and MyHC expression in differentiated myoblasts treated with or without IS (1 mM) are determined by western blot and quantified. Data represent means ± SEM for three independent experiments. *P < 0.05 and ** P < 0.01, as compared with untreated control.
IS down‐regulates promyogenic AKT signalling and myoD expression
Accordingly, myoD is believed to be an important regulator of gene expression in myogenic initiation, and up‐regulation of MyoD during differentiation process is required for terminal differentiation.6 Therefore, we examined the changes in myoD expression level during myoblast differentiation in IS‐treated cells. As shown in Figure 3A and 3B, IS exposure suppressed myoD protein expression. Furthermore, previous study indicated that myoD expressed in the proliferating myoblasts at the beginning of myogenic differentiation, which mediates cell cycle withdrawal, and then promoting myogenic differentiation.42 To determine whether IS treatment decreases myoD expression, C2C12 myoblasts were cultured in GM and examined for the expression of myoD after 2 days of IS incubation. Our results showed that the amount of myoD protein decreased significantly in IS‐treated myoblast compared with mock‐treated control (Figure 3C and 3D). AKT activation favours myoblast differentiation and is positively correlated with myoD, MyoG, and consequent MyHC protein expression.12, 43 We thus hypothesized that the myogenesis inhibitory activity of IS might rely on modulating AKT activity. We examined the changes in AKT‐phosphorylation levels in myoblasts and in DM‐incubated cells with or without IS treatment. We found that IS treatment significantly reduces phosphor‐AKT expression (Figure 3E‐H). These observations showed that IS treatment inhibits AKT‐phosphorylation and downstream myoD expression.
Figure 3.
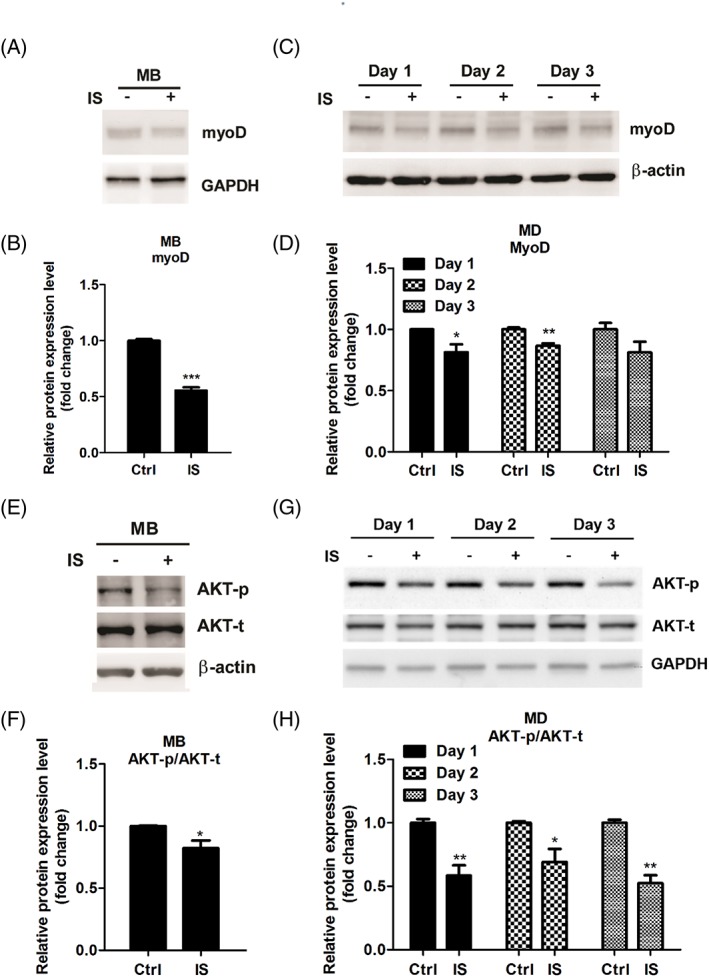
IS treatment represses myoD expression and Akt phosphorylation during myoblast (MB) proliferation and myogenic differentiation (MD). (A and B) Protein expression of myoD in myoblsts treated with or without IS (1 mM) for 48 h is examined by western blot and quantified. (C and D) pPotein expression of myoD in differentiated myoblasts treated with or without IS (1 mM) at the indicated time points by western blot and quantified. (E–F) Akt expression and phosphorylation in myoblasts treated with or without IS (1 mM) for 48 h are determined by western blot and quantified. (G–H) Akt expression and phosphorylation during myoblasts differentiation treated with or without IS (1 mM) at the indicated time point are determined by western blot and quantified. Data represent means ± SEM for three independent experiments. *P < 0.05, ** P < 0.01, and *** P < 0.001, as compared with untreated control.
Indoxyl sulfate treatment elicits unfolded protein response signalling in C2C12 cells
ER stress response has been implicated in aging and age‐related disease,37, 44 we then questioned whether exposure of IS induces ER stress in C2C12 cells. We first assessed UPR signalling in undifferentiated myoblasts. Results showed that BiP mRNA expression level and phosphorylation of eIF2α increased after 2 days exposure of IS (Figure 4A‐C). To investigate whether IS induces IRE1–XBP1 pathway activation by splicing of XBP1 mRNA, we performed semi‐quantitative PCR to measure XBP1u and XBP1s mRNA levels. It can be observed that the levels of either XBP1u or XBP1s mRNA were not altered by IS treatment (Figure 4D and 4E). Next, we explored whether IS exposure modulates ER stress during differentiation. Similar to what has been observed in IS‐treated myoblast, IS treatment enhances BiP mRNA expression and eIF2α phosphorylation (Figure 4F‐H). Notably, IS treatment significantly induces XBP1s mRNA expression compared with non‐treated condition during myogenesis (Figure 4I and 4J).
Figure 4.
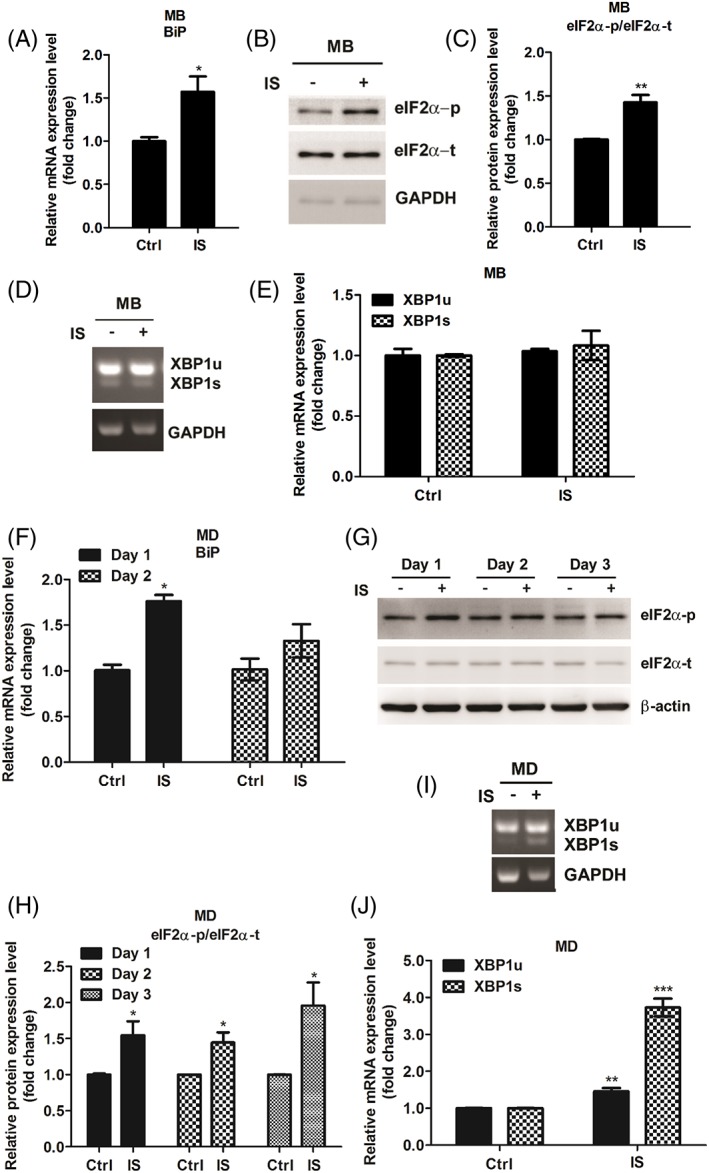
IS modulates myoblast and myotube UPR signalling pathways. (A–C) BiP mRNA expression level and phosphorylation of eIF2α are examined in C2C12 myoblasts (MB) after 2 days exposure of IS (1 mM). (D and E) The levels of both XBP1u or XBP1s mRNA were determined by semi‐quantitative PCR. (F–H) BiP mRNA expression level and phosphorylation of eIF2α increased are examined in IS‐treated well myogenic differentiation (MD) cells. (I and J) The level of XBP1u and XBP1s mRNA expression is determined in well MD cells with or without IS (1 mM) for 48 h. The data were expressed as mean ± SED from three independent experiments. *P < 0.05, ** P < 0.01, and *** P < 0.001, as compared with untreated control.
Divergent effects of phospho‐eIF2α and XBP1 on myoblast differentiation
Given that up‐regulation of phospho‐eIF2α is an early event in IS‐induced suppression of myogenesis (Figure 4B and 4C), we treated cells with salubrinal, an eIF2α dephosphorylation inhibitor,45, 46, 47 to investigate the effect of eIF2α phosphorylation on myogenesis. Figure 5A and 5B showed an increase of eIF2α phosphorylation in the salubrinal treatment condition which confirms the effectiveness of salubrinal. Altering eIF2α phosphorylation level influences myogenesis as demonstrated by decreased myoD and phospho‐AKT expression in salubrinal‐treated myoblasts. In addition, salubrinal treatment also down‐regulated the expressions of myoG, MyHC, and phospho‐AKT in DM‐incubated cells (Figure 5C and 5D). In agreement, the suppressor role of phospho‐eIF2α was further supported by overexpression of mouse S52D phospho‐mimetic eIF2α mutant, which leads to suppress myogenesis with reduction of myogenic markers in myoblasts and in DM‐incubated cells (Figure 5E‐H). Furthermore, we used XBP1‐targeted siRNA to investigate the role of XBP1 on IS‐suppressed myogenesis. Control siRNA or XBP1 siRNA‐transfected myoblasts were treated with IS for 24 h in GM, followed by 2 days of IS exposure in the differentiation condition. We found that decreased expression of MyHC but not myoG were detected in XBP1‐deficient cells, indicating XBP1 may be related to the late differentiation phase (Figure 6A and 6B). In addition, depletion of XBP1 presents decreased phospho‐AKT expression level (Figure 6C and 6D). To further access the importance of XBP1 in the regulation of late phase myogenic differentiation, the 2 days differentiated C2C12 cells were shifted to GM and transfected with siXBP1. After 24 h, the medium was removed and replaced with DM for 2 days as illustrated in Figure 6E. Phase contrast photomicroscopy revealed XBP1‐deficient cells fuse to form short myotubes compared with cells transfected with scrambled siRNA (Figure 6F). These data showed that XBP1 is responsible for mediating late phase myogenic differentiation probably through regulation of MyHC expression. Collectively, our results demonstrated the pro‐ and anti‐myogenesis roles of XBP1 and phospho‐eIF2α, respectively, in different stages of myoblast differentiation.
Figure 5.
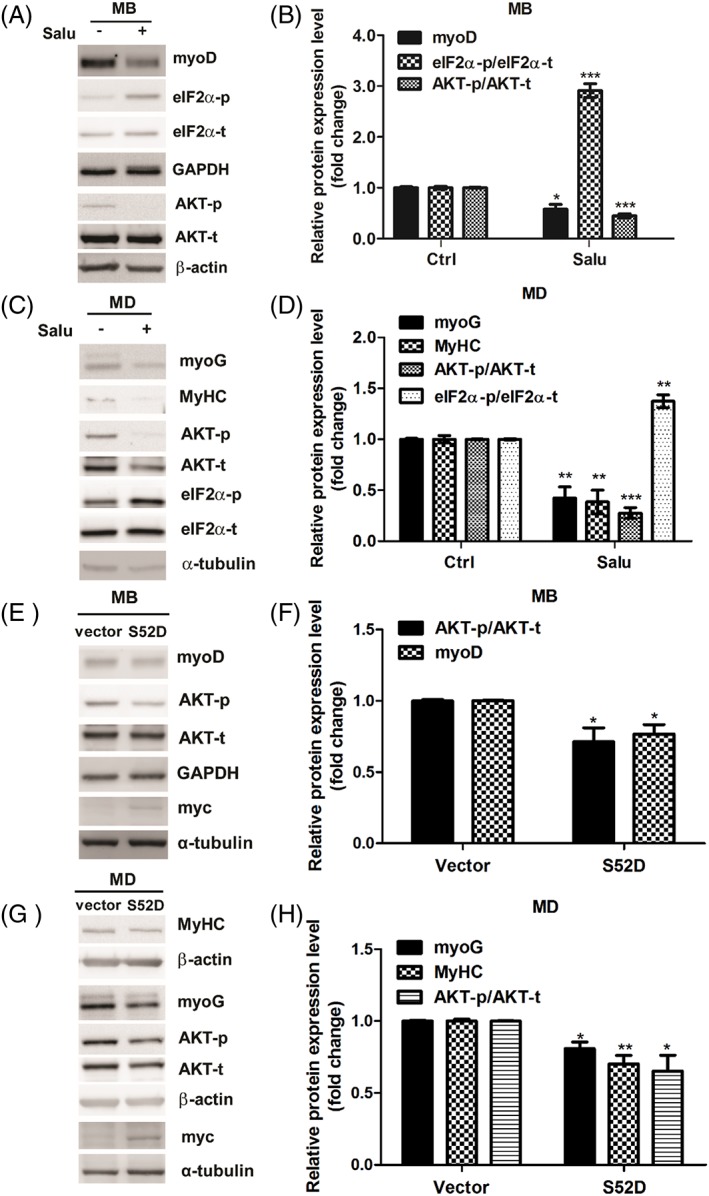
Salubrinal, an eIF2α dephosphorylation inhibitor, deregulates C2C12 myogenic differentiation (MD). (A and B) Myoblasts (MB) were treated with or without 20 μM salubrinal for 48 h. MyoD, phospho‐eIF2α, and phospho‐AKT expressions are examined with western blot and quantified. (C and D) Myoblasts under 48 h differentiation are simultaneously exposed with or without 20 μM salubrinal. MyoG, MyHC, phospho‐AKT, and phospho‐ eIF2α expression level are determined with western blot and quantified. (E and F) Myoblasts are overexpressed with or without mouse S52D phosphomimetic eIF2α mutant for 48 h. The expression of eIF2α S52D was detected with anti‐myc antibody. MyoD and phospho‐AKT expressions are examined with western blot and quantified. (G and H) myoblasts under 48 h differentiation are simultaneously transfected with or without S52D mutant. The expression of eIF2α S52D was detected with anti‐myc antibody. MyoG, MyHC, and phospho‐AKT expression level are determined with western blot and quantified.
Figure 6.
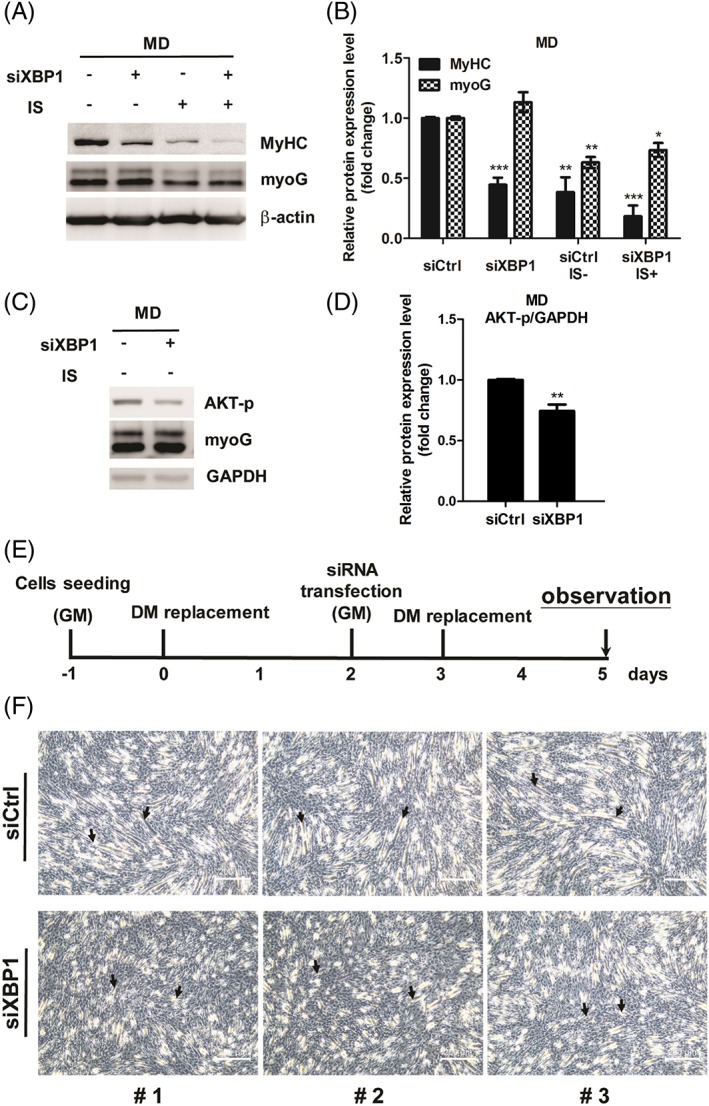
Promyogenic role of XBP1 in myoblast differentiation (MD). (A–D) The expression of MyHC, myoG, and phospho‐AKT in XBP1‐dificent cells is detected by western bot and quantified. (E) Diagram illustrates timeline of experiment. (F) Phase contrast photomicroscopy revealed differentiated morphology in XBP1‐deficient myoblasts compared with ordinary myoblasts. The arrows indicate the fusion of myoblasts. Random views of three independent experiments at a magnification of ×100 were shown, scale bars = 200 μm.
Involvement of ROS in IS‐modulated eIF2α phosphorylation and antimyogenesis
IS induces ROS in several cell lines.48, 49, 50 To investigate whether ROS are involved in IS‐induced suppression of myogenesis, C2C12 myoblasts were preincubated with or without ROS scavanger NAC for 30 min and then treated with IS for 2 days. Then cells were washed with PBS and maintained in DM or NAC‐containing DM for 30 min followed by IS treatment as illustrated in Figure 7A. Here, we showed that NAC treatment effectively decreases IS‐induced ROS levels, which confirms antioxidant activity of NAC (Figure 7B). By the end of 4 days of incubation in DM, H&E staining results reveal that NAC treatment reverses IS‐induced defect in myotube formation (Figure 7C). The results presented in Figure 4 suggested that eIF2α phosphorylation involved in IS‐mediated myogenesis suppression. According to the interaction between ROS and ER stress has been implicated in many research fields, we examined the changes of eIF2α phosphorylation induced by IS with or without NAC. Results showed NAC treatment reverses IS‐induced up‐regulation of phospho‐eIF2α (Figure 7D and 7E). Consistently, the inhibitory effect of IS on myogenesis was reversed by NAC treatment as demonstrated by increased myoG and MyHC expression as well as activation of AKT (Figure 7F and 7G); however, there is no change in IS‐induced splicing of XBP1 mRNA (Figure 7H and 7I), indicating eIF2α phosphorylation is selectively activated by ROS upon IS treatment. In addition, we found that increased atrogin 1 protein level could be reversed by NAC (Figure 7J and 7K). These results showed that ROS‐eIF2α axis is important for the anti‐myogenesis activity of IS.
Figure 7.
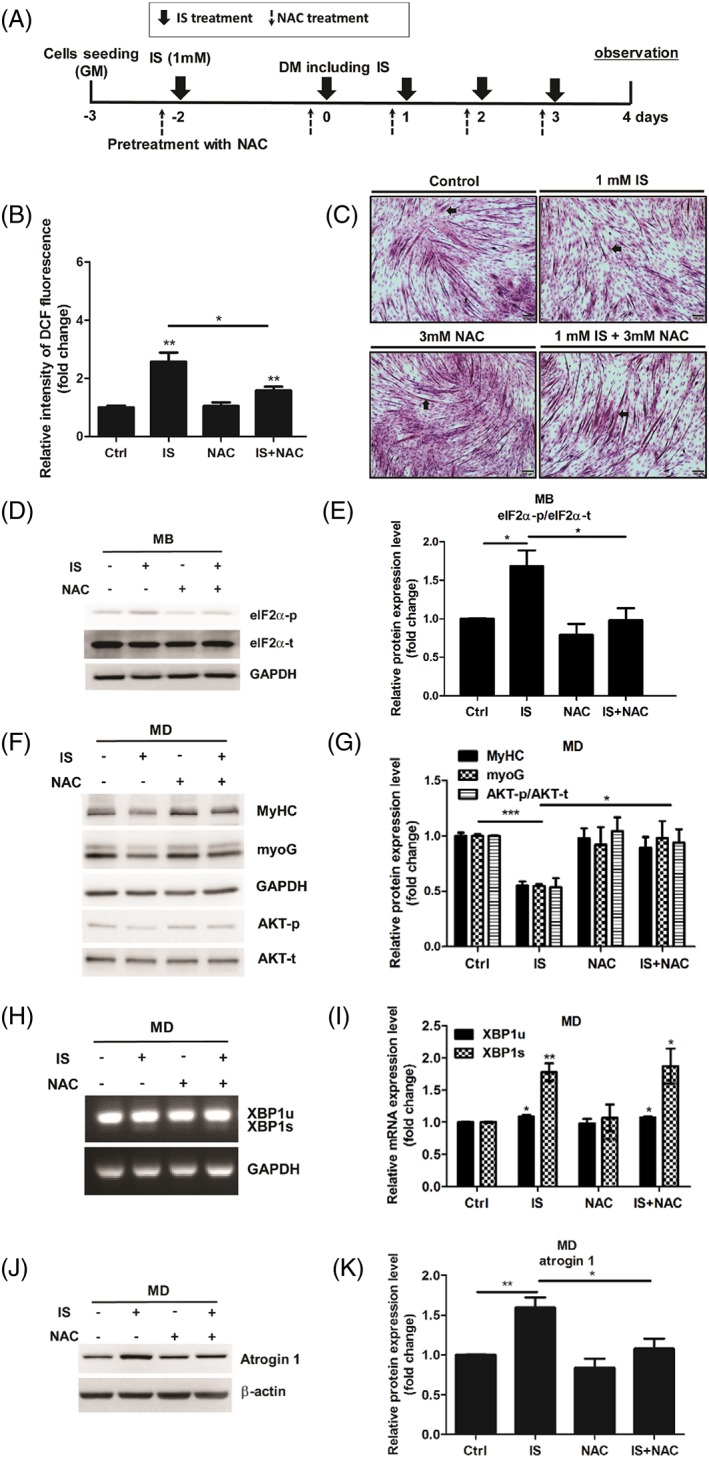
NAC treatment reverses IS‐induced defect in myogenic differentiation (MD). (A) Diagram illustrates timeline of experiment. (B) Flow cytometric quantitation of DCF fluorescence. (C) H&E staining of C2C12 myotubes under NAC (3 mM) or IS (1 mM) treatment, scale bars = 100 μm. The arrows indicate the fused myotubes. (D and E) myoblasts (MB) were treated with NAC (3 mM) or IS (1 mM) for 48 h. Phospho‐eIF2α expression was examined with western blot and quantified. (f‐k) Myoblasts under 48 h differentiation are simultaneously exposed with NAC (3 mM) or IS (1 mM). MyHC, myoG, and phospho‐AKT expression were determined with western blot and quantified (F and G). XBP1u and XBP1s mRNA expression are examined by agarose gel electrophoresis (H and I). Atrogin 1 expression are measured by western blot and quantified (J and K). The data were expressed as mean ± SED from three independent experiments. *P < 0.05, ** P < 0.01, and *** P < 0.001, as compared with untreated control.
Discussion
Using a uremic milieu premature aging model, we demonstrated that UPR signalling pathways engage myogenesis regulation (Figure 8). Since IS‐induced myoblast differentiation defect was identical to the effect of salubrinal treatment or overexpression of phosphomimetic eIF2α S52D, we concluded that IS treatment causes eIF2α phosphorylation followed by decreased phospho‐AKT, reduced myoD expression, and impaired myogenesis. Moreover, phase contrast photomicroscopy and the expression of differentiation markers revealed that depletion of XBP1 causes abnormal myotube formation, which indicates IS‐induced XBP1 serves as an adaptive response. We further demonstrated that ROS accumulation triggers eIF2α phosphorylation, and antioxidant NAC reverses the eIF2α‐mediated AKT dephosphorylation in IS‐treated myoblasts. However, as the crosstalk between oxidative stress and ER stress have been raised, it is possible that the protective activity of NAC is afforded by means of reducing the overall level of ROS and ER stress directly.
Figure 8.
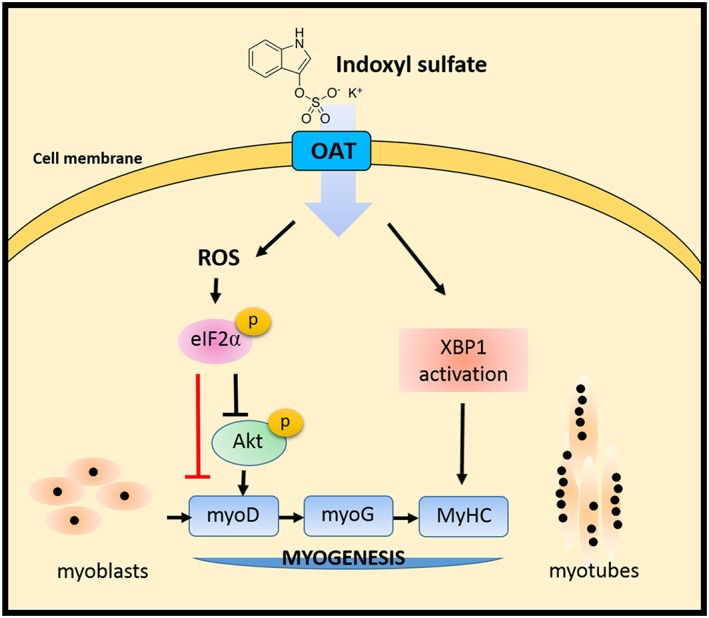
Model of IS‐induced defect in myoblast differentiation. The pro‐ and anti‐myogenesis roles of XBP1 and phospho‐eIF2α in different stages of myogenesis are pointed out. Notably, the ROS‐eIF2α axis is important for the antimyogenesis activity of IS. The red line indicates translational attenuation caused by phosphorylation of eIF2α probably results in decreased myoD expression. Overall, our research demonstrates the pivotal role of UPR signalling in age‐related muscle loss and sarcopenia.
UPR has been shown to participate in modulating myogenesis.51, 52, 53 Selective activation of ATF6 in apoptotic myoblasts rather than lived cells is associated with muscle development probably through induction of caspase‐12 cascade to remove vulnerable cells, which in turn benefits myogenic progression of the rest surviving cells.51 Recently, studies in PERK/eIF2α arm of the UPR indicated that eIF2α phosphorylation is persistent in muscle stem cell to maintain the quiescence and self‐renewal while eIF2α dephosphorylation initiate myogenic differentiation.52, 53 Accordingly, these observations lead in considering phospho‐eIF2α plays as a molecular switch between cell quiescence and differentiation. The eIF2α phosphorylation declines during myoblast differentiation,52 and sustained eIF2α phosphorylation blocks myogenesis under pathological conditions such as uremic milieu or aging (Figures 4 and 5). It presently remains unresolved how phospho‐eIF2α manipulates IS‐suppressed myogenesis. Presumably, phosphorylation of eIF2α attenuates global protein synthesis, and decreased muscle protein synthesis is associated with both myoblast dysfunction and myotube atrophy. However, phospho‐eIF2α paradoxically selective translates transcripts containing upstream open reading frames within their 5′ untranslated region commonly referred to as the integrated stress response.54 Therefore, it will be intriguing to determine whether integrated stress response target genes act as myogenic suppressors in future study.
Salubrinal, an eIF2α dephosphorylation inhibitor, will increase the phosphorylation of eIF2α as we have already known, but what is its exact effect on the ER stress is still controversial. One landmark study by Yuan JY's group mentioned that salubrinal acts as an ER stress inhibitor.45 However, the same group stated that salubrinal induced a marked eIF2α phosphorylation, and potentiated ER stress in β‐islet cells.47 Because of the controversial role of salubrinal, we designed the experiment to investigate the influence of salubrinal in our system. We pre‐treated C2C12 cells with salubrinal followed by ER stress inducer tunicamycin exposure, and we found salubrinal cannot attenuate the tunicamycin‐induced BiP and XBP1s mRNA up‐regulation, but potentiate the induction of CHOP, a downstream of ER stress effector (Supplementary Figure S1). In addition to the earlier findings, our recent works also demonstrated that salubrinal potentiated cisplatin‐induced tubular cells death.55 Taken together, we suggested that salubrinal potentiated ER stress‐induced adverse effect in C2C12 differentiation.
Each UPR or Akt signalling pathway has a specific role in cellular development, differentiation, and cell fate determination. Studies focused on the interplay between UPR and AKT signalling, however, are rare. AKT regulates PERK/eIF2α pathway controlling cell fates under pathological condition such as tumour and hypoxia.56, 57 Inhibition of Akt synergistically sensitizes acute lymphoblastic leukaemia cells to 2‐deoxy‐D‐glucose and down‐regulates expression of UPR factors BiP, IRE1, and phospho‐eIF2α. In addition, ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy.58 Interestingly, XBP1s forms complex with PI3K regulatory subunits p85α and p85β, which facilitates nuclear translocation of XBP1s and downstream genes expression.59, 60 In the present study, we found that eIF2α phosphorylation and depletion of XBP1 cause dephosphorylation of AKT. To our knowledge, this is the first work that shows both eIF2α and XBP1 control activation of AKT, thereby regulating myogenic progression. The underlying mechanisms, however, await further investigation.
XBP1s induces expression of genes involved in maintaining ER homeostasis including protein folding, modification, trafficking and ER‐associated protein degradation. Recently, it was shown that XBP1s regulates glucose homeostasis via FoxO1 interaction and subsequent FoxO1 proteosomal degradation.61 Short myotube formation in XBP1‐deficient condition suggested a crucial role for XBP1 in the late phase of myogenesis. The molecular mechanisms under XBP1 regulation in myotube formation remain unclear. XBP1s, as a transcription factor, possibly induce myogenic proteins expression. Alternatively, regarding XBP1 is required for AKT phosphorylation, XBP1 may protect cells from atrophy through AKT‐FoxO1 signalling pathway. It is also possible that XBP1 prevents myotube atrophy through its inhibitory interaction with FoxO1. In any case, it is interesting to study further the exact role of XBP1 in myogenesis and other related disease.
In the present study, myogenesis is repressed by high (1 mM) and lower (0.5 mM) concentration of IS (Figure 1A and 1B) while only high concentration shows inhibitory effect in myoblast proliferation (Figure 2A). As proliferation precedes myotube differentiation, we then chose 1 mM IS to do the subsequent experiments. In CKD patients, IS concentration achieves nearly 53 ± 21 mg/L (0.25 ± 0.1 mM)62, 63 which is one third of our treatment. Because uremic patients prolongedly and continually exposed to pathological IS concentration, higher IS concentration would be rational in in‐vitro shorten exposure cell culture study. The same concept is also mentioned in another study.27
Given that depression of protein synthesis and increase in degradation lead to muscle loss, studies targeting repressed protein synthesis or protein degradation as therapeutic strategies are emerging topics.64, 65 Urolithin B, an ellagic acid metabolite, up‐regulates protein synthesis and inhibits ubiquitin‐proteasome‐mediated protein degradation in myotube. The mRNA levels of ubiquitin ligases atrogin 1 and MuRF1 are decreased under Urolithin B treatment.64 In addition, compound EMBL ID#704946 has been identified as an inhibitor of MuRF1 in the context of protein expression, E3 ligase activity, and MuRF1‐titin complexation. Intriguingly, EMBL ID#704946 not only attenuates skeletal muscle atrophy but also presents protective effect against cardiac cachexia.65 Sarcopenia during cancer and aging share many common features.66, 67 Inspired by our results, it is possible that UPRs also play regulatory roles in cancer cachexia or anticancer treatment‐induced cardiac wasting.68 Further investigations are needed for verification.
Conclusions
Our studies indicated that the ER stress and UPR modulation are critical in the CKD uremic toxin‐accumulated sarcopenia. These findings improve our understating of the role of ER stress and uremic toxin, IS, in myogenesis, and provides new insight for developing therapies to improve myofiber formation in aging and uremic sarcopenia.
Conflicts of Interest
The authors declare no competing financial interests.
Supporting information
Fig S1. Salubrinal cannot attenuate tunicamycin‐induced ER stress. C2C12 myoblasts were pre‐treated with or without 20 μM salubrinal for 1 h, followed by 2.5 μg/mL ER stress inducer Tunicamycin (Sigma‐Aldrich) treatment for 8 h. The mRNA levels of (a) BiP, CHOP, and (b and c) XBP1 were examined and quantified. The increased CHOP mRNA level confirms the effectiveness of salubrinal. The data were expressed as mean ± SED from three independent experiments. ** P < 0.01 and *** P < 0.001, as compared with untreated control.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.69
We thank the Second Core Laboratory of the Department of Medical Research in the National Taiwan University Hospital for equipment and facility support.
This work was supported by grants from the Taiwan Ministry of Science and Technology (MOST‐104‐2314‐B‐002‐126‐MY3), the National Taiwan University Hospital (NTUH‐104‐S2810 and NTUH‐105‐S3180), NTUH‐TVGH Joint Research Program (VN104‐05), and E‐Da Hospital‐NTUH Joint Research Program (106‐EDN1).
Jheng, J.‐R. , Chen, Y.‐S. , Ao, U. I. , Chan, D.‐C. , Huang, J.‐W. , Hung, K.‐Y. , Tarng, D.‐C. , and Chiang, C.‐K. (2018) The double‐edged sword of endoplasmic reticulum stress in uremic sarcopenia through myogenesis perturbation. Journal of Cachexia, Sarcopenia and Muscle, 9: 570–584. doi: 10.1002/jcsm.12288.
References
- 1. Metter EJ, Conwit R, Tobin J, Fozard JL. Age‐associated loss of power and strength in the upper extremities in women and men. J Gerontol A Biol Sci Med Sci 1997;52:B267–B276. [DOI] [PubMed] [Google Scholar]
- 2. Maggio M, Lauretani F, Ceda GP. Sex hormones and sarcopenia in older persons. Curr Opin Clin Nutr Metab Care 2013;16:3–13. [DOI] [PubMed] [Google Scholar]
- 3. Messier V, Rabasa‐Lhoret R, Barbat‐Artigas S, Elisha B, Karelis AD, Aubertin‐Leheudre M. Menopause and sarcopenia: a potential role for sex hormones. Maturitas 2011;68:331–336. [DOI] [PubMed] [Google Scholar]
- 4. Frost RA, Lang CH. Protein kinase B/Akt: a nexus of growth factor and cytokine signaling in determining muscle mass. J Appl Physiol 2007;103:378–387. [DOI] [PubMed] [Google Scholar]
- 5. Rygiel KA, Picard M, Turnbull DM. The ageing neuromuscular system and sarcopenia: a mitochondrial perspective. J Physiol 2016;594:4499–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo K, Wang J, Andres V, Smith RC, Walsh K. MyoD‐induced expression of p21 inhibits cyclin‐dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol 1995;15:3823–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martelli F, Cenciarelli C, Santarelli G, Polikar B, Felsani A, Caruso M. MyoD induces retinoblastoma gene expression during myogenic differentiation. Oncogene 1994;9:3579–3590. [PubMed] [Google Scholar]
- 8. Zhang W, Behringer RR, Olson EN. Inactivation of the myogenic bHLH gene MRF4 results in up‐regulation of myogenin and rib anomalies. Genes Dev 1995;9:1388–1399. [DOI] [PubMed] [Google Scholar]
- 9. Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993;364:501–506. [DOI] [PubMed] [Google Scholar]
- 10. Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up‐regulation of the myogenic HLH gene Myf‐5 and results in apparently normal muscle development. Cell 1992;71:383–390. [DOI] [PubMed] [Google Scholar]
- 11. Miller JB. Myogenic programs of mouse muscle cell lines: expression of myosin heavy chain isoforms, MyoD1, and myogenin. J Cell Biol 1990;111:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang BH, Aoki M, Zheng JZ, Li J, Vogt PK. Myogenic signaling of phosphatidylinositol 3‐kinase requires the serine‐threonine kinase Akt/protein kinase B. Proc Natl Acad Sci U S A 1999;96:2077–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drummond MJ, Dreyer HC, Fry CS, Glynn EL, Rasmussen BB. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J Appl Physiol 2009;106:1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Machida S, Spangenburg EE, Booth FW. Forkhead transcription factor FoxO1 transduces insulin‐like growth factor's signal to p27Kip1 in primary skeletal muscle satellite cells. J Cell Physiol 2003;196:523–531. [DOI] [PubMed] [Google Scholar]
- 16. Briata P, Lin WJ, Giovarelli M, Pasero M, Chou CF, Trabucchi M, et al. PI3K/AKT signaling determines a dynamic switch between distinct KSRP functions favoring skeletal myogenesis. Cell Death Differ 2012;19:478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down‐regulating MyoD expression. J Biol Chem 2002;277:49831–49840. [DOI] [PubMed] [Google Scholar]
- 18. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 2009;296:C1258–C1270. [DOI] [PubMed] [Google Scholar]
- 19. Matsakas A, Mouisel E, Amthor H, Patel K. Myostatin knockout mice increase oxidative muscle phenotype as an adaptive response to exercise. J Muscle Res Cell Motil 2010;31:111–125. [DOI] [PubMed] [Google Scholar]
- 20. Kooman JP, Kotanko P, Schols AM, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol 2014;10:732–742. [DOI] [PubMed] [Google Scholar]
- 21. Stenvinkel P, Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis 2013;62:339–351. [DOI] [PubMed] [Google Scholar]
- 22. Niwa T. Uremic toxicity of indoxyl sulfate. Nagoya J Med Sci 2010;72:1–11. [PMC free article] [PubMed] [Google Scholar]
- 23. Banoglu E, Jha GG, King RS. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur J Drug Metab Pharmacokinet 2001;26:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deguchi T, Ohtsuki S, Otagiri M, Takanaga H, Asaba H, Mori S, et al. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int 2002;61:1760–1768. [DOI] [PubMed] [Google Scholar]
- 25. Ohtsuki S, Asaba H, Takanaga H, Deguchi T, Hosoya K, Otagiri M, et al. Role of blood‐brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: its involvement in neurotransmitter metabolite clearance from the brain. J Neurochem 2002;83:57–66. [DOI] [PubMed] [Google Scholar]
- 26. Shimizu H, Yisireyili M, Nishijima F, Niwa T. Stat3 contributes to indoxyl sulfate‐induced inflammatory and fibrotic gene expression and cellular senescence. Am J Nephrol 2012;36:184–189. [DOI] [PubMed] [Google Scholar]
- 27. Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, et al. Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 2017;8:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hurtley SM, Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 1989;5:277–307. [DOI] [PubMed] [Google Scholar]
- 29. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012;13:89–102. [DOI] [PubMed] [Google Scholar]
- 30. Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000;101:451–454. [DOI] [PubMed] [Google Scholar]
- 31. Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, Krause GS. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J Neurochem 2005;94:1235–1242. [DOI] [PubMed] [Google Scholar]
- 32. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000;6:1355–1364. [DOI] [PubMed] [Google Scholar]
- 33. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis‐acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose‐regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 1998;273:33741–33749. [DOI] [PubMed] [Google Scholar]
- 34. Lee AH, Iwakoshi NN, Glimcher LH. XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci U S A 2010;107:16113–16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001;107:881–891. [DOI] [PubMed] [Google Scholar]
- 37. Naidoo N. ER and aging―protein folding and the ER stress response. Ageing Res Rev 2009;8:150–159. [DOI] [PubMed] [Google Scholar]
- 38. Ghosh AK, Mau T, O'Brien M, Garg S, Yung R. Impaired autophagy activity is linked to elevated ER‐stress and inflammation in aging adipose tissue. Aging (Albany NY) 2016;8:2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Segev Y, Michaelson DM, Rosenblum K. ApoE epsilon4 is associated with eIF2alpha phosphorylation and impaired learning in young mice. Neurobiol Aging 2013;34:863–872. [DOI] [PubMed] [Google Scholar]
- 40. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Enoki Y, Watanabe H, Arake R, Sugimoto R, Imafuku T, Tominaga Y, et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress‐mediated expression of myostatin and atrogin‐1. Sci Rep 2016;6 https://doi.org/10.1038/srep32084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol Cell Biol 1999;19:5073–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu Q, Wu Z. The insulin‐like growth factor‐phosphatidylinositol 3‐kinase‐Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma‐derived RD cells. J Biol Chem 2000;275:36750–36757. [DOI] [PubMed] [Google Scholar]
- 44. Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum―new mechanisms in kidney disease. Nat Rev Nephrol 2014;10:369–378. [DOI] [PubMed] [Google Scholar]
- 45. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005;307:935–939. [DOI] [PubMed] [Google Scholar]
- 46. Teng Y, Gao M, Wang J, Kong Q, Hua H, Luo T, et al. Inhibition of eIF2alpha dephosphorylation enhances TRAIL‐induced apoptosis in hepatoma cells. Cell Death Dis 2014;5 https://doi.org/10.1038/cddis.2014.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid‐induced endoplasmic reticulum stress and causes pancreatic beta‐cell dysfunction and apoptosis. J Biol Chem 2007;282:3989–3997. [DOI] [PubMed] [Google Scholar]
- 48. Yang K, Xu X, Nie L, Xiao T, Guan X, He T, et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol Lett 2015;234:110–119. [DOI] [PubMed] [Google Scholar]
- 49. Dou L, Jourde‐Chiche N, Faure V, Cerini C, Berland Y, Dignat‐George F, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost 2007;5:1302–1308. [DOI] [PubMed] [Google Scholar]
- 50. Shimizu H, Saito S, Higashiyama Y, Nishijima F, Niwa T. CREB, NF‐kappaB, and NADPH oxidase coordinately upregulate indoxyl sulfate‐induced angiotensinogen expression in proximal tubular cells. Am J Physiol Cell Physiol 2013;304:C685–C692. [DOI] [PubMed] [Google Scholar]
- 51. Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 2005;169:555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, et al. Phosphorylation of eIF2alpha is a translational control mechanism regulating muscle stem cell quiescence and self‐renewal. Cell Stem Cell 2016;18:79–90. [DOI] [PubMed] [Google Scholar]
- 53. Xiong G, Hindi SM, Mann AK, Gallot YS, Bohnert KR, Cavener DR, et al. The PERK arm of the unfolded protein response regulates satellite cell‐mediated skeletal muscle regeneration. Elife 2017;6:https://doi.org/10.7554/eLife.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Translational RD. control in the endoplasmic reticulum stress response. J Clin Invest 2002;110:1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu SH, Wu CT, Huang KH, Wang CC, Guan SS, Chen LP, et al. C/EBP homologous protein (CHOP) deficiency ameliorates renal fibrosis in unilateral ureteral obstructive kidney disease. Oncotarget 2016;7:21900–21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mounir Z, Krishnamoorthy JL, Wang S, Papadopoulou B, Campbell S, Muller WJ, et al. Akt determines cell fate through inhibition of the PERK‐eIF2alpha phosphorylation pathway. Sci Signal 2011;4 https://doi.org/10.1126/scisignal.2001630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blaustein M, Perez‐Munizaga D, Sanchez MA, Urrutia C, Grande A, Risso G, et al. Modulation of the Akt pathway reveals a novel link with PERK/eIF2alpha, which is relevant during hypoxia. PLoS One 2013;8 https://doi.org/10.1371/journal.pone.0069668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010;6:239–247. [DOI] [PubMed] [Google Scholar]
- 59. Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, et al. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP‐1 and increase its nuclear translocation. Nat Med 2010;16:429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3‐kinase increases the nuclear accumulation of X‐box‐binding protein‐1 to modulate the unfolded protein response. Nat Med 2010;16:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, et al. Regulation of glucose homeostasis through a XBP‐1‐FoxO1 interaction. Nat Med 2011;17:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med 1994;124:96–104. [PubMed] [Google Scholar]
- 63. Kim HY, Yoo TH, Hwang Y, Lee GH, Kim B, Jang J, et al. Indoxyl sulfate (IS)‐mediated immune dysfunction provokes endothelial damage in patients with end‐stage renal disease (ESRD). Sci Rep 2017;7 https://doi.org/10.1038/s41598-017-03130-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodriguez J, Pierre N, Naslain D, Bontemps F, Ferreira D, Priem F, et al. Urolithin B, a newly identified regulator of skeletal muscle mass. J Cachexia Sarcopenia Muscle 2017;8:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, et al. Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle 2017;8:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Argiles JM, Busquets S, Felipe A, Lopez‐Soriano FJ. Muscle wasting in cancer and ageing: cachexia versus sarcopenia. Adv Gerontol 2006;18:39–54. [PubMed] [Google Scholar]
- 67. Argiles JM, Busquets S, Felipe A, Lopez‐Soriano FJ. Molecular mechanisms involved in muscle wasting in cancer and ageing: cachexia versus sarcopenia. Int J Biochem Cell Biol 2005;37:1084–1104. [DOI] [PubMed] [Google Scholar]
- 68. Ishida J, Saitoh M, Springer J. Is cardiac wasting accompanied by skeletal muscle loss in breast cancer patients receiving anticancer treatment? J Cachexia Sarcopenia Muscle 2017;8:851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Salubrinal cannot attenuate tunicamycin‐induced ER stress. C2C12 myoblasts were pre‐treated with or without 20 μM salubrinal for 1 h, followed by 2.5 μg/mL ER stress inducer Tunicamycin (Sigma‐Aldrich) treatment for 8 h. The mRNA levels of (a) BiP, CHOP, and (b and c) XBP1 were examined and quantified. The increased CHOP mRNA level confirms the effectiveness of salubrinal. The data were expressed as mean ± SED from three independent experiments. ** P < 0.01 and *** P < 0.001, as compared with untreated control.