

Summary
We previously piloted the concept of a Connectivity Map (CMap), whereby genes, drugs and disease states are connected by virtue of common gene-expression signatures. Here, we report more than a 1,000-fold scale-up of the CMap as part of the NIH LINCS Consortium, made possible by a new, low-cost, high throughput reduced representation expression profiling method that we term L1000. We show that L1000 is highly reproducible, comparable to RNA sequencing, and suitable for computational inference of the expression levels of 81% of non-measured transcripts. We further show that the expanded CMap can be used to discover mechanism of action of small molecules, functionally annotate genetic variants of disease genes, and inform clinical trials. The 1.3 million L1000 profiles described here, as well as tools for their analysis, are available at https://clue.io.
Keywords: Functional genomics, gene expression profiling, chemical biology
Graphical abstract
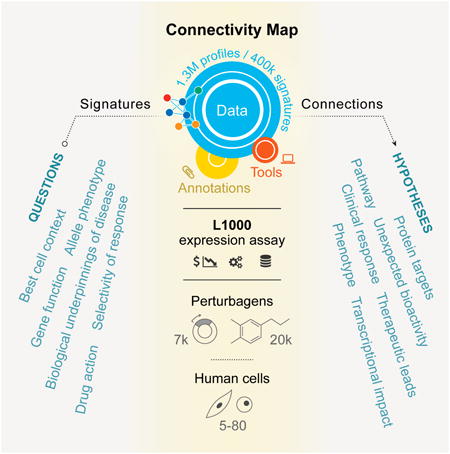
The next generation Connectivity Map, a large-scale compendium of functional perturbations in cultured human cells coupled to a gene expression read-out, facilitates the discovery of connections between genes, drugs, and diseases.
Introduction
The sequencing of the human genome provided the parts list of life, and this in turn has led to an explosion of new insights into the genetic basis of disease. Genome-wide association studies have identified risk-associated loci for major diseases, and the sequencing of human tumors has similarly identified the somatic mutations that underlie many types of cancer. The research community has benefitted from these genomic resources by being able to readily look up sequence variants in large-scale compendia of genomic variation. Such look-up tables of biology have transformed how modern research is done.
A challenge, however, is that a parts list and its association with disease is generally not sufficient to establish causality, or to provide mechanistic and circuit-level biological insights. Truly understanding cellular function requires perturbing the system – modulating the expression of a gene of interest, and monitoring the downstream consequences. Unfortunately, large-scale compendia of the cellular effects of genetic perturbation have yet to be established as a community resource.
Similarly, there has been no method to systematically determine the cellular effects of chemical compounds. For example, it would be desirable to be able to query a functional look-up table to discover unexpected activities of a compound – such activities often being discovered only late in the drug development process, resulting in side effects that limit clinical use.
Hughes and colleagues were the first to suggest that such inference of function could be gleaned from a compendium of gene expression profiles (Hughes et al., 2000). Working in yeast, they showed that genetic variants and pharmacologic treatment could be related by virtue of common gene expression signatures they induce.
We subsequently hypothesized that the compendium concept could be extended to human signatures representing genetic or pharmacologic perturbation. Signatures that proved to be similar might thus represent previously unrecognized connections (e.g., between two proteins operating in the same pathway, between a small molecule and its protein target, or between two small molecules of similar function but structural dissimilarity). Such a catalog of connections could thus serve as a functional look-up table of the human genome, and we termed this the Connectivity Map (Lamb et al., 2006).
We piloted the Connectivity Map (CMap) concept by treating cells with 164 drugs and tool compounds, and then performing mRNA expression profiling using Affymetrix microarrays, thereby generating a public resource with over 18,000 users. Examples of CMap use include the anthelmintic drug parbendazole as an inducer of osteoclast differentiation (Brum et al., 2015), celastrol as a leptin sensitizer (Liu et al., 2015), compounds targeting COX2 and ADRA2A as potential diabetes treatments (Zhang et al., 2015), small molecules that mitigate skeletal muscular atrophy (Dyle et al., 2014) and spinal muscular atrophy (Farooq et al., 2009), and new therapeutic hypotheses for the treatment of inflammatory bowel disease (Dudley et al., 2011) and cancer (Singh et al., 2016); (Muthuswami et al., 2013; Wang et al., 2008); (Schnell et al., 2015); (Fortney et al., 2015; Wang et al., 2011); (Churchman et al., 2015); (Rosenbluth et al., 2008); (Saito et al., 2009); (Stockwell et al., 2012).
Despite the popularity of the pilot Connectivity Map pilot dataset, its small scale limits its utility. With only 164 drug perturbations in only 3 cancer cell lines, the database lacks the necessary richness of a truly genome-scale resource. Missing is a diversity of chemical perturbations, genetic perturbations as well as a diversity of cell types. Unfortunately, the high cost of commercial gene expression microarrays and even RNA sequencing precludes such a genome-scale Connectivity Map. We therefore describe here a new approach to gene expression profiling based on a reduced representation of the transcriptome. This method, which we call L1000, is high-throughput and low-cost, and is thus well-suited to a large-scale Connectivity Map. We report here the first 1,319,138 L1000 profiles as part of the NIH LINCS initiative.
Results
Reduced representation of transcriptome
We hypothesized that it might be possible to capture at low cost any cellular state by measuring a reduced representation of the transcriptome. To explore this, we analyzed 12,031 Affymetrix HGU133A expression profiles in the Gene Expression Omnibus (GEO). We used these to identify the optimal number of informative transcripts, which we term ‘landmark’ transcripts, k. If k was too small, too much information might be lost, whereas if k was too large, sufficient cost reduction compared to the entire transcriptome might be not be achieved. This analysis showed that 1,000 landmarks were sufficient to recover 82% of the information in the full transcriptome (see STAR Methods). The selection of the 1,000 landmarks was done using a data-driven approach rather than selecting transcripts based on prior biological knowledge, as detailed in STAR Methods.
L1000 assay platform
To measure the 1,000 landmark transcripts, we adapted a method involving ligation-mediated amplification (LMA) followed by capture of the amplification products on fluorescently-addressed microspheres (Peck et al., 2006). We extended this method to a 1,000-plex reaction (Figure 1A; protocols at clue.io/sop-L1000.pdf). Briefly, cells growing in 384-well plates were lysed and mRNA transcripts captured on oligo-dT-coated plates. cDNAs were synthesized and subjected to LMA using locus-specific oligonucleotides harboring a unique 24-mer barcode sequence and a 5′ biotin label. The biotinylated LMA products were detected by hybridization to polystyrene microspheres (beads) of distinct fluorescent color, each coupled to an oligonucleotide complementary to a barcode, and then stained with streptavidin-phycoerythrin. Thus, each bead was analyzed both for its color (denoting landmark identity) and fluorescence intensity of the phycoerythrin signal (denoting landmark abundance). Because only 500 bead colors are commercially available, we devised a strategy that allows two transcripts to be identified by a single bead color (STAR Methods and Figure 1B). 955 shRNAs targeting landmark transcripts were used to empirically validate L1000 probes (Figure 1C). The final assay, which we call L1000, contains 1,058 probes for 978 landmark transcripts and 80 control transcripts (Table S2) chosen for their invariant expression across cell states (see STAR Methods). The reagent cost of the L1000 assay is approximately $2.
Figure 1. L1000 gene expression platform implementation and validation.
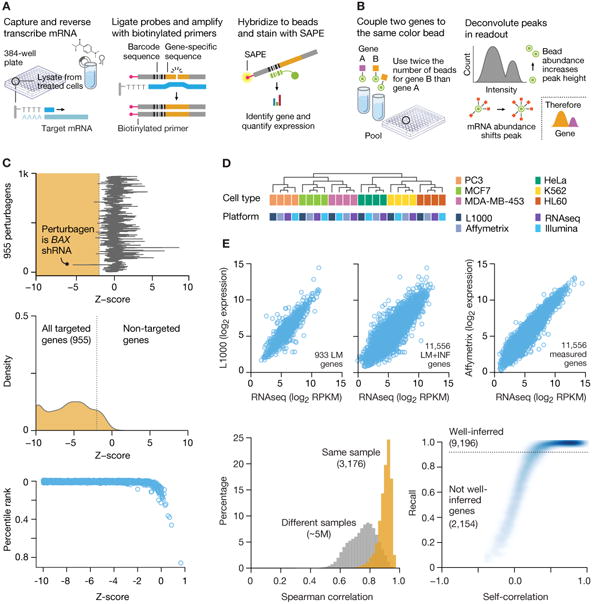
A. Overview of ligation-mediated amplification. Cells are treated in 384-well plates, lysed and mRNA captured on oligo-dT plates. mRNA is reverse-transcribed and oligonucleotide probes designed with transcript-specific, 24-mer unique barcode and universal primer sequences annealed to the cDNA, ligated and PCR-amplified using biotinylated primers. PCR product is hybridized to optically addressed polystyrene microspheres, where each bead is coupled to an oligonucleotide complementary to a landmark gene's barcode. Transcript abundance is quantified by fluorescence using a Luminex FlexMap 3D scanner.
B. Deconvoluting 1,000 landmark genes using 500 bead colors. Each bead is analyzed for its bead color (denoting landmark gene identity) and phycoerythrin intensity (denoting transcript abundance). Aliquots of the same bead color, separately coupled to two different gene barcodes, are combined in a ratio of 2:1. A distribution of fluorescent intensities reveals two peaks (partitioned by k-means clustering), the larger peak designating the landmark for which double number of beads were used.
C. Validation of L1000 probes using shRNA knockdown. MCF7 and PC3 cells transduced with shRNAs targeting 955 landmark genes. Differential expression values (z-scores) were computed for each landmark and the percentile rank of expression z-scores in the experiment in which it was targeted relative to all other experiments was computed. 841/955 genes (88%) rank in the top 1% of all experiments and 907/955 (95%)rank in the top 5%. Top panel: z-score of BAX gene in every experiment. Middle panel: Z-score distribution from all targeted (orange) and non-targeted (white) genes. Distribution from the targeted set is significantly lower than non-targeted (p value <10-16). Bottom panel: Scatter of percentile rank versus expression z-score for 955 targeted genes.
D. Comparison of L1000 with other platforms. Samples of RNA from 6 human cancer cell lines were profiled on L1000, Affymetrix GeneChip HG-U133 Plus 2.0 Array, Illumina Human HT-12 v4 Expression BeadChip Array, and mRNA-seq (Illumina Hi-Seq).
E. Comparison of L1000 with RNA-seq and Affymetrix using patient-derived samples. RNA samples from 3,176 tissue specimens profiled on L1000 and RNA-seq, and a subset on Affymetrix microarrays. Top panels: Scatter plots of L1000 expression versus RNA-seq in landmark (left, Spearman correlation of 0.86) and landmark plus inferred (middle, Spearman correlation of 0.91) expression for a single sample. Bottom left:Spearman correlation distribution for L1000 vs RNA-seq of landmark genes for the same sample (orange) and different samples (gray), across all 3,176 patient samples. Bottom right: All L1000 inferred genes were subject to recall analysis by comparison with their RNA-seq measured equivalents. Scatter plot shows R versus cross-platform correlation for all inferred genes. 9,196 of 11,350 (81%) have an R in the 95th percentile (dotted line).
Having chosen the L1000 landmark transcripts using an entirely data-driven approach optimized for maximal information rather than on biological function, we asked whether the landmarks were enriched in any particular functional class (e.g., transcription factors). We computed hypergeometric overlap statistics and found no substantial enrichment for any particular protein class (see STAR Methods). Similarly, we found no evidence of developmental lineage bias based on an analysis of landmark expression patterns across 30 tissue types (Figure S1B).
L1000 reproducibility
Technical replicates of 6 cancer cell lines in which aliquots of the same RNA sample were subjected to replicate L1000 profiling (12 replicates in each of 3 batches, yielding 36 replicates per cell line) showed that for 88% of all pairwise comparisons of replicates, Spearman correlation was >0.9, suggesting low sample-to-sample variability (Figure S1C). Furthermore, intra-batch variation (median pairwise correlation 0.97) was comparable to inter-batch variation (median pairwise correlation 0.95), indicating high technical reproducibility.
Comparison of L1000 to RNA-seq
RNA sequencing (RNA-seq) has become the standard for gene expression profiling, and thus we sought to benchmark L1000 against it. We note that while RNA-seq is attractive given its unbiased nature, it suffers from inability to detect non-abundant transcripts without deep sequencing that results in higher costs. The L1000 platform is hybridization-based, thus making the detection of non-abundant transcripts feasible. As an initial assessment of cross-platform performance, mRNA samples from 6 cell lines were profiled on L1000, Affymetrix U133A and Illumina BeadChip arrays, and by RNA-seq. Hierarchical clustering of these data grouped samples by cell type, not measurement platform (Figure 1D and 1E, upper panel).
To more extensively compare L1000 to RNA-seq, we analyzed 3,176 samples (previously sequenced by the GTEx Consortium (The GTEx Consortium, 2015)) profiled on both platforms. This analysis showed that cross-platform similarity was high (median self-correlation 0.84), with a right-shifted distribution compared to non-self correlations (Figure 1E, lower panel left). Recall analysis similarly showed that 98% of samples had a sample recall > 99% (indicating 99th percentile) (see STAR Methods). Taken together, these results indicate a strong degree of similarity of profiles across L1000 and RNA-seq platforms.
Inferring gene expression from L1000 landmarks
Using 8,555 RNA-seq samples (Dataset DSGTEx-rnaseq) as an independent test set, we used landmark transcript measurements to infer the remainder of the transcriptome. As a test of inference accuracy, we analyzed gene-level recall (Rgene) for each of the inferred genes and assessed performance by comparing the result to a null distribution of correlations between all inferred transcripts and all measured transcripts. This analysis showed that inference was accurate (defined as Rgene > 0.95) for 9,196 of the 11,350 inferred genes (81%). When combined with the 978 measured landmarks, the L1000 platform thus measures or infers with high fidelity 83% of transcripts, but yields poor inference for 17% (Figure 1E, lower panel right and Table S3). Inferences for these 17% were therefore not used in any of the analyses that follow.
Generation of the first million L1000 Connectivity Map profiles
Having validated L1000, we set out to expand on the CMap pilot dataset in several dimensions. First, we increased the small molecule perturbations from 164 drugs to 19,811 small molecule drugs, tool compounds and screening library compounds including those with clinical utility, known mechanism of action, or nomination from the NIH Molecular Libraries Program. Each compound was profiled in triplicate, either at 6 or 24 hours following treatment.
Second, we expanded in the dimension of genetic perturbation by knocking down and overexpressing 5,075 genes selected on the basis of their association with human disease or membership in biological pathways. Each genetic perturbation was profiled in triplicate, 96 hours after infection. For overexpression studies, a single cDNA clone was used, whereas three distinct shRNAs targeting each gene were profiled.
Third, we expanded in the dimension of cell lines. Well-annotated genetic and small molecule perturbagens were profiled in a core set of 9 cell lines, yielding a reference dataset we refer to as Touchstone v1. Uncharacterized small molecules without known mechanism of action (MOA) were profiled variably across 3 to 77 cell lines, yielding a dataset we refer as Discovery v1 (Table S4).
In total, we generated 1,319,138 L1000 profiles from 42,080 perturbagens (19,811 small molecule compounds, 18,493 shRNAs, 3,462 cDNAs, and 314 biologics), corresponding to 25,200 biological entities (19,811 compounds, shRNA and/or cDNA against 5,075 genes, and 314 biologics) for a total of 473,647 signatures (consolidating replicates), representing over a 1,000-fold increase over the CMap pilot dataset. We term this first release of an L1000-based compendium CMap-L1000v1 (Figure 2A). All data, at multiple levels of pre-processing are available via GEO (accession GSE92742 and pre-processing code via GitHub), and for easier use via the CLUE analysis environment (https://clue.io; see below and Figure 2B).
Figure 2. L1000 dataset coverage, signature generation, and data access.
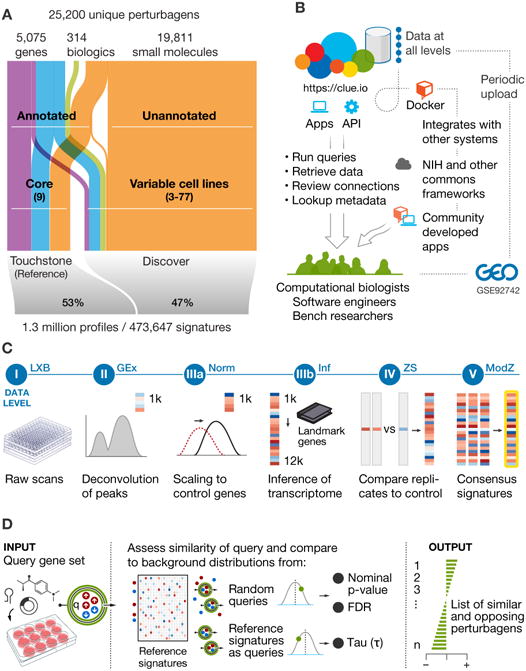
A. Classification of data in CMap-L1000v1. The 25,200 unique perturbagens correspond to 19,811 compounds, shRNA and/or cDNA targeting 5,075 genes, and 314 biologics. Annotated perturbagens profiled systematically across 9 core cell lines comprise the reference or Touchstone portion of the dataset, while the unannotated reagents make up the Discover portion.
B. Modes of access to analysis tools and data. The clue.io software platform enables computational biologists, bench scientists, and software engineers to leverage CMap by offering web applications for analysis, and APIs and docker containers for code and data access.
C. Signature generation and data levels. I) Raw bead count and fluorescence intensity measured by Luminex scanners II) Deconvoluted data to assign expression levels to two transcripts measured on the same bead IIIa) Normalization to adjust for non-biological variation IIIb) Inferred expression of 12,328 genes from measurement of 978 landmarks IV) Differential expression values V) Signatures representing collapse of replicate profiles.
D. Schematic of query analysis. Query is specified by sets of up- and down-regulated genes. Similarities between the query and all signatures in CMap are computed. Normalized similarities are converted to a p-value and FDR, by comparison with a compendium of random queries, and to τ via comparison with reference signature queries. Perturbagens are then sorted by τ to generate most similar and opposing perturbagens.
CMap query methodology
The connectivity workflow involves interrogating the CMap database of signatures with a query (a set of differentially expressed genes representing a biological state of interest). Each of the signatures in the database represents a weighted average across the 3 biological replicate perturbations (see STAR Methods). This moderated z-score procedure serves to mitigate the effects of uncorrelated or outlier replicates (Figure 2C). The similarity of the query to each of the CMap signatures is computed, thus yielding a rank ordered list of the 473,647 signatures in the CMap-L1000v1 dataset. However, simply sorting by degree of similarity can be misleading because it does not address issues such as magnitude of gene expression change or specificity of observed connections.
We therefore developed a Connectivity Score (Figure 2D) that provides three measures of confidence: 1) a nominal p-value derived by comparing the similarity between the query and reference signature, using the Kolmogorov-Smirnov enrichment statistic (Subramanian et al., 2005), to a null distribution of random queries; 2) a false discovery rate (FDR) that adjusts the p-value to account for multiple hypothesis testing; and 3) Tau (τ), which compares an observed enrichment score to all others in the database (see STAR Methods).
These Connectivity Score metrics constitute a statistical framework that provides a holistic quantification of the relationship between a query and a perturbagen, as opposed to merely sorting by degree of similarity. Additionally, while the Connectivity Scores are generated on each cell type individually, we summarize those scores across all profiled cell types and thus provide a measure of robustness. Importantly, this analytical approach is platform-independent, allowing users to create query signatures from any gene expression platform.
Feasibility of querying a million-profile compendium
We next tested the CMap for its ability to produce biologically meaningful connections. While our analysis of replicate measurements demonstrated that L1000 is robust, it is conceivable that as the size of the dataset increased, so might biological and technical noise, thereby obscuring real signal. To address this, we compiled 7,578 perturbational signatures from public sources from which we identified 1,143 perturbational profiles (across multiple expression platforms; Table S5)–that matched a CMap-L1000v1 perturbagen, and were therefore eligible for Recall analysis. For each query, we assessed whether it connected to its equivalent in CMap-L1000v1 at a high level of confidence (defined as NP <=0.05, FDR <= 0.25 and |τ| >= 90). 909/1,143 queries (80%) exhibited the expected connectivity. We note that the inference of expression values from landmarks was essential to recovering connections. 20% of connections were lost when the analysis was restricted to landmarks only. Furthermore, 48 query signatures contained zero landmark transcripts and were therefore not analyzable without inference of the remainder of the transcriptome.
Discovering off-target effects of shRNAs
The scope of the L1000 dataset provides an unprecedented opportunity to examine the biological effects of shRNAs, in particular, their off-target effects. We analyzed 13,187 shRNAs targeting 3,799 genes across 9 cell lines, and compared each pair of shRNA-induced L1000 profiles, comparing similarity between shRNAs targeting the same gene (“shared gene”) and shRNAs targeting different genes but sharing the 2-8 nucleotide seed sequence known to contribute to off-target effects (“shared seed”) (Jackson et al., 2003). Figure 3A shows that shared gene similarity is only slightly greater than random. In contrast, shared seed pairs were dramatically more similar compared to the null distribution, indicating that the magnitude of off-target effects of shRNAs substantially exceeds the magnitude of their on-target effect. We reasoned that while on-target gene expression effects of different shRNAs targeting the same gene should be the same, their off-target effects should not. We therefore developed an algorithm to produce a Consensus Gene Signature (CGS) that reflects the consistent (and therefore on-target) gene expression effects of shRNAs and used the CGS output for all analyses that follow. The CGS method and its validation are described in detail elsewhere (Smith et al., 2017) .
Figure 3. Analysis of genetic loss of function perturbations.
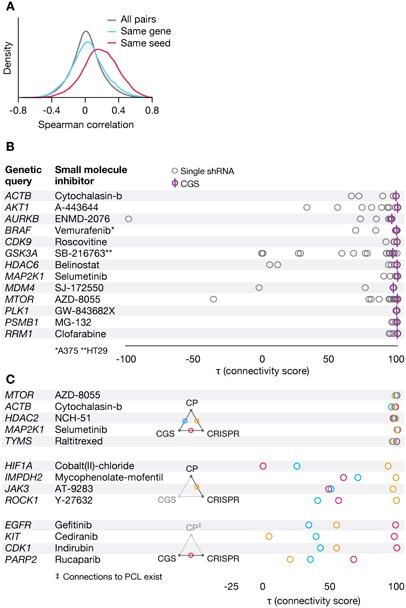
A. Off-target effects of shRNAs. Distributions of Spearman correlations between signatures of 12,961 shRNAs in A549 cells targeting the same gene but different seed sequences (blue), targeting different genes but the same seed (red) and all pairs of shRNAs (gray). Distribution was randomly down-sampled to 10 million points. All pairwise comparisons of distributions were significantly different (adjusted p < 10e-7 by Kruskal-Wallis test followed by Dunn test with Benjamini-Hochberg correction).
B. Consensus Gene Signature (CGS) improves on-target signal. A consensus gene signature (CGS) is computed from a weighted average of signatures of independent shRNAs targeting the same gene. Connectivity to annotated small molecules targeting each gene (horizontal axis) is markedly improved by CGS over individual shRNAs (with τ closer to 100), suggesting that the CGS procedure mitigates the seed effect inherent to individual shRNAs and enhances on-target signal. Connections are shown summarized across cell lines unless otherwise indicated.
C. CRISPR knockout augments compound-target analysis. Top: Consistency between Loss of Function (LoF) signatures from CRISPR and CGS enhances confidence in connectivity to small molecules (CP). Middle: CRISPR-based LoF recovers some connections to small molecules missed by CGS. Bottom: Lack of compound-target connectivity, despite consistency between LoF reagents and validated compound signature suggests non-equivalency of genetic and pharmacological agent-derived signatures.
Characterizing small molecule function
A theoretical feature of a large-scale CMap is the ability to determine mechanism of action (MOA) of a small molecule, based simply on similarity to profiles of genetic perturbagens or compounds of known function. We first determined whether known MOAs could be recovered by the CMap. This is challenging, however, because the definitive list of protein targets (and their associated pathways) of small molecule drugs is unknown. Nevertheless, we used multiple resources to associate 1,902 compounds to protein targets and associated pathway members profiled in the CMap. This led to 58,820 expected relationships that could plausibly be recovered in the CMap (see STAR Methods) (Corsello et al., 2017). We then sought to recover those relationships from among the approximately 160 million pairwise relationships (connections) that could be assessed across CMap.
For each compound, we computed the true positive rate (i.e., recovery of expected relationships). We refer to these expected relationships as expected pairs. To estimate the false positive rate, we counted the connections between compounds and genetic or pharmacologic perturbagens annotated as having a relationship with a different small molecule in the dataset. We refer to such relationships as null pairs. We then plotted the true positive rate against the false positive rate at various thresholds of statistical significance, thereby generating an ROC curve from which an AUC could be calculated. An AUC >0.6 is typically regarded as signifying a positive signal. At that cut-off, an average of 45% of expected relationships were recovered in any one of the 9 cell lines tested (range 29%-58%). This number rose to 63% when Connectivity Scores were summarized across all 9 lines (see STAR Methods, Table S6).
Defining perturbagen classes (PCLs)
A challenge in CMap interpretation is that the analysis returns a rank-ordered list of connections, leaving the user to extract biological meaning from the list. We reasoned that while any given member of an MOA class would likely have a multitude of targets, integrating signatures across several examples of an MOA class would sharpen the on-target signal, while diminishing off-target effects. We codified this by identifying compounds that share MOA and by identifying genetic perturbagens belonging to the same gene family or were targeted by the same compounds. These perturbagen classes (PCLs) were then further refined by excluding compounds that failed to connect with their cognate class members based on L1000 connectivity analysis (see STAR Methods and Figure 4A). This yielded 171 high confidence PCLs (Table S7).
Figure 4. Reference perturbagen classes for CMap discovery.
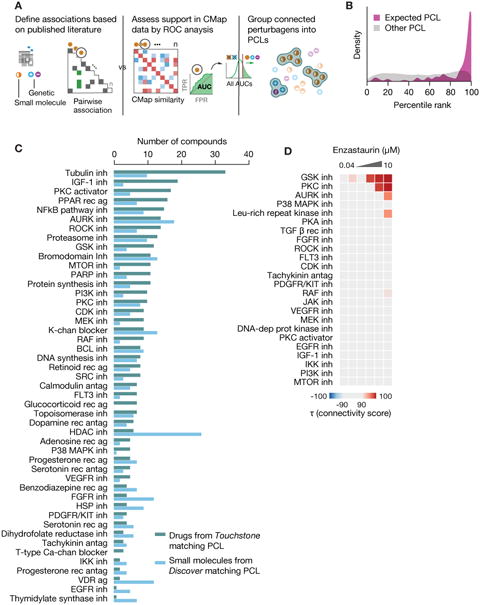
A. Process for defining Perturbagen Classes (PCLs). Left: Annotations gathered from literature sources to construct pairwise association matrix between perturbagens based on shared descriptors such as MoA, target gene and pathway membership. Middle: Each perturbagen is subject to ROC analysis to determine whether it recovers expected connections. Right: Remaining members are grouped based on shared annotations and assessed for intra-group connectivity of CMap signatures. Groups sufficiently interconnected are retained as PCLs.
B. PCL validation. 137 compounds with known activities corresponding to one or more of 54 PCLs, but not used in PCL construction, were profiled across multiple cell types. Histogram shows rank of each expected PCL connection for the compounds (purple) versus the rank of all unexpected PCL connections (grey). The expected PCL distribution is significantly right-shifted (one-sided p < 2.2e-16 via two-sample KS test).
C. Using PCLs for discovery. 3,333 known drugs and 2,418 unannotated but transcriptionally active compounds were subject to PCL analysis. Count of strong and selective connections to validated PCLs byknown drugs (teal) and unannotated compounds (blue). Abbreviations: inh. inhibitor, ag. agonist, rec.receptor, antag. antagonist, and chan. channel.
D. Detecting multiple drug activities using PCLs. The PKC inhibitor enzastaurin was profiled in CMap across multiple doses. Connectivity to each established kinase inhibitor PCL is shown in the heatmap. Strong dose-responsive connections were observed to PKC and GSK3 inhibitor PCLs.
To test the hypothesis that PCLs would increase confidence in biological interpretation, we profiled 137 test compounds known to share a mechanism with one or more of 54 small molecule PCLs, but which were not used in the construction of the PCL. For 41/54 classes (76%), the test compounds connected to their designated PCL in multiple cell types (Figure 4B). For an additional 7/54 (13%), a selective connection was observed in a single cell type. The remaining 6/54 (11%) did not connect at a threshold of τ >90.
We next performed PCL connectivity analysis on 3,333 drugs and 2,418 unannotated compounds and observed a variety of strong, selective connections to PCLs. Importantly, many drugs showed strong PCL connections to mechanisms other than those for which the drugs were developed, representing potential off-target or secondary effects (Figure 4C and Figure S3A). 132 drugs (3.9%) had such off-target connections (see STAR Methods). For example, compounds showing connectivity to the protein kinase C (PKC) inhibitor PCL were often also strongly connected to the GSK3 inhibitor PCL. 44 such dually connected compounds were found (τ>=95, selectivity >=0.85), including the PKC inhibitor enzastaurin which showed dose-responsive connectivity to both PKC and GSK3 inhibitor classes (τGSK=99.79 τPKC=99.47, selectivity=0.88) (Figure 4D). Interestingly, synergy between compounds targeting these pathways has been reported (Rovedo et al., 2011), and the biochemical profiling confirms that enzastaurin is indeed also a potent GSK3 inhibitor with a KD of 8 nM (Davis et al., 2011).
In the future, splitting PCLs to reflect subclasses with distinct patterns of selectivity may be possible. For example, the histone deacetylase (HDAC) inhibitor PCL class currently has 20 members, each with varying selectivity against the 13 HDAC proteins. Clustering the L1000 gene expression data revealed clear substructure within the PCL, with pan-HDAC-inhibitory compounds forming a distinct cluster, and compounds selective for either HDAC6 or HDAC1,3 and 8 forming distinct clusters (Figure 5A).
Figure 5. Characterizing known and unexpected activities of small molecules.
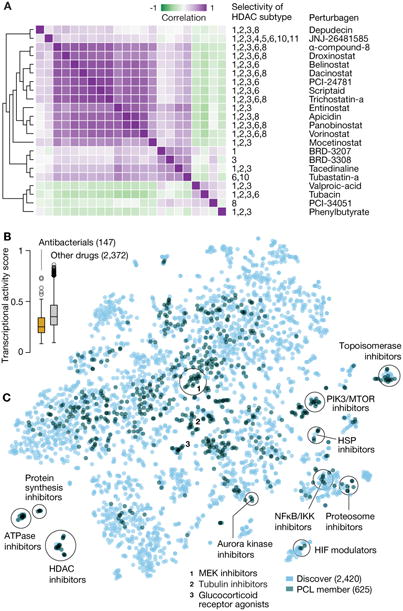
A. HDAC inhibitor PCL substructure. Hierarchical clustering of pairwise connectivities of the HDAC inhibitor PCL members reveals substructure within the class. Pan-HDAC inhibitors cluster together, distinct from more isoform-selective compounds.
B. Antibacterials exhibit lower transcriptional activity than other drugs. Distributions of the maximum TASper compound for 147 antibacterials and 2,372 known drugs in CMap-TS. The antibacterials' TAS distribution is significantly lower (p < 3e-11) than that of other drugs.
C. Comparison of unannotated compounds with known drugs. t-SNE projection of the signatures of 2,418unannotated but transcriptionally active compounds (blue) with PCL members (teal). Some unannotated compounds occupy regions not covered by drugs, presenting opportunities for novel chemical development.
Cellular context
To study the effect of cellular context on perturbational responses, we compared the signatures of 2,429 drugs across 9 cancer cell lines. On average, 38% of compounds scored as transcriptionally active in any single cell type (range 28%-45%) and 92% of small molecule drugs scored as active in at least one cell line. Of 1,399 (58%) compounds active in at least 3 cell lines, 26% (corresponding to 15% of all compounds) produced highly similar signatures across the entire panel, whereas the remainder were active in only 1 or 2 cell lines or produced a diversity of cellular signatures (see STAR Methods and Figure S2B, S2C).
As might be expected, connections with support across multiple cell types tended to target core cellular processes (e.g., ribosomal function, proteasome complex), whereas compounds with reproducibly cell-type-selective patterns of connectivity tended to target more specialized mechanisms. For example, connectivity between multiple glucocorticoid receptor agonists was strongest in those cell types in which the glucocorticoid receptor was expressed (Figure S2D, upper panel). Connectivity between multiple PPARG agonists was greatest in HT29 and PC3, the two core cell lines with the highest baseline expression of PPARG (Figure S2D, lower panel). Similarly, the connection between androgen receptor (AR) knockdown and the AR antagonist nilutamide was strongest in the AR-expressing cell line VCAP (Figure S2E). We also note that the naturally occurring genetic diversity can be informative. For example, connections between genetic perturbation of the MAP kinase pathway and small molecule inhibitors of RAF or MEK kinases were strongest in the cell lines that harbor BRAF V600E kinase-activating mutations (Figure S2E).
Identifying bioactive subsets of small molecule screening libraries
It is now possible to create large numbers of structurally diverse small molecule compounds. However, many compounds fail to engage specific protein targets or to even enter living cells. We asked whether an L1000 profile could serve as a sensor for biological activity. If so, screening chemical libraries with L1000 might enable rapid elimination of compounds lacking obvious activity and help prioritize others for subsequent cell-based screening. Consistent with our earlier studies (Wawer et al., 2014), we found that whereas 2,232/2,429 (92%) established drugs yielded a strong L1000 transcriptional response (defined as Transcriptional Activity Score (TAS) >0.2; see STAR Methods), only 2,418/16,527 (15%) un-optimized compounds had high TAS scores. We note, however, that compounds with cell-type selective bioactivity might be missed by this approach.
Interestingly, the TAS-low drugs were enriched in antimicrobial agents that would not be expected to target human proteins (Figure 5B). An exception to this was the antimicrobial triclosan, which yielded a high TAS score, consistent with its having effects in mammalian cells. The safety of triclosan has recently been questioned (Dinwiddie et al., 2014; Yueh et al., 2014).
Discovery of MOA of unannotated small molecules
Having demonstrated the ability to recover MOA from optimized drugs and tool compounds, we next asked whether CMap could identify the MOA of previously uncharacterized compounds. Projection of TAS-high compounds in two dimensions shows that many uncharacterized compounds cluster with existing PCLs (Figure 5C). We focused on novel kinase inhibitors simply because of the availability of methods for validating CMap predictions. For example, our analysis indicated that the unannotated compound BRD-2751 showed strong connectivity to the Rho-associated protein kinase (ROCK) PCL, suggesting that it might in fact be a ROCK inhibitor. To test this hypothesis, we subjected the compound to kinome-wide binding measurements (using the Kinomescan assay) and found that precisely as predicted, the compound has a KD of 56 nM against ROCK1 (Figure 6A). We note that while the compound had not been previously reported to be a ROCK1 inhibitor, its chemical structure is reminiscent of canonical ROCK inhibitory compounds. As another example, several compounds (BRD-5161, BRD-5657, and BRD-9186) were predicted to function as MTOR and/or PI3 kinase inhibitors. Kinomescan dose-response profiling confirmed that the three compounds were indeed MTOR/PI3K inhibitors, spanning a range of potencies and selectivities (Figure S3B).
Figure 6. Kinase inhibitor discovery using reference transcriptional signatures.
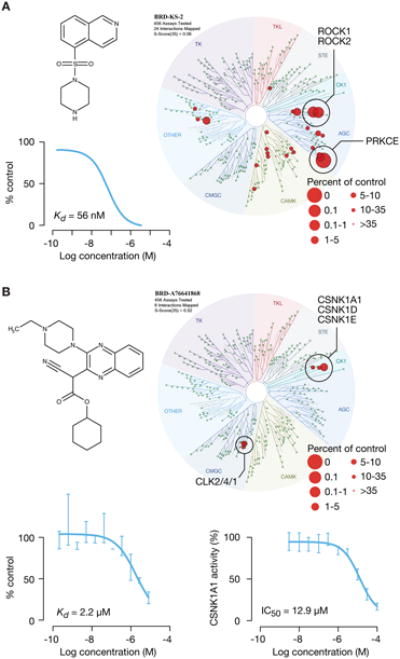
A. Discovery of ROCK1/ROCK2 inhibitor. Top left panel: chemical structure of BRD-2751, predicted to be aROCK inhibitor. Right: TREEspot selectivity profile of Kinomescan binding assay confirmed compound binding to ROCK1/ROCK2. Bottom left: Dose response testing by Kinomescan showed ROCK1 KD of 56 nM.
B. Discovery of novel CSNK1A1 inhibitor. Top left panel: The chemical structure of BRD-1868. Top right:TREEspot image of Kinomescan binding assay performed with BRD-1868 at 10 uM demonstrated inhibition of6/456 kinases tested including CSNK1A1. Bottom left: CSNK1A1 binding by BRD-1868 confirmed by Kinomescan, with Kd 2.2 uM. Bottom right: BRD-1868 inhibits phosphorylation of peptide substrate byCSNK1A1, with IC50 12.9 uM. Error bars indicate standard deviation between technical replicates.
Discovery of a selective CSNK1A1 inhibitor
We next asked whether we could use the CMap to discover a compound with a particular activity – in this case, Casein Kinase 1A1 (CSNK1A1). CSNK1A1 is a serine-threonine kinase that was reported as an essential gene in certain subtypes of myelodysplastic syndrome and acute myeloid leukemia, and also has been shown to be targeted for degradation by the drug lenalidomide, which is particularly effective in MDS patients with chromosome 5q deletion (the locus of the CSNK1A1 gene) (Järås et al., 2014; Krönke et al., 2015; Schneider et al., 2014). Furthermore, CSNK1A1 has been reported as a mediator of drug resistance to EGFR inhibitors in lung cancer (Lantermann et al., 2015). Unfortunately, potent and selective CSNK1A1 small molecule inhibitors have yet to be reported.
As CSNK1A1 was among the 3,799 genes subjected to shRNA-mediated knock-down, we used the CMap to generate a signature of CSNK1A1 loss of function. We then queried all compounds in the database against this signature to identify perturbations that phenocopied CSNK1A1 loss. One unannotated compound, BRD-1868, showed strong connectivity to CSNK1A1 knockdown in two cell types. This suggested that BRD-1868 might function as a novel CSNK1A1 inhibitor. To test this hypothesis, we subjected the compound to kinase specificity profiling, testing its ability to bind to 456 kinases using the Kinomescan assay. This confirmed BRD-1868's ability to bind CSNK1A1 with high specificity and modest potency (KD 2.2 uM). Follow-up enzymatic assays confirmed that BRD-1868 not only binds CSNK1A1, but also inhibits its enzymatic activity (Figure 6B), making it an strong candidate for further chemical optimization. Most importantly, the result highlights the power of the L1000 Connectivity Map as a starting point for drug discovery – even in the absence of prior examples of the drug class.
Using L1000 data to assess allele function
The preceding analyses focused primarily on using CMap to annotate chemical compounds. We next asked whether a similar strategy could be used to annotate the function of an allelic series of genes. Building on our prior results (Berger et al., 2016), we sought to determine whether the CMap could distinguish the downstream consequences of overexpression of cDNAs harboring particular somatic mutations observed in human tumors. For example, the ubiquitin ligase FBXW7 is a negative regulator of MYC protein expression. As expected, CMap showed that overexpression of wild-type FBXW7 strongly connected to knock-down of MYC. In addition, overexpression of 6 cancer-associated alleles (I347M, V464E, R465C, R465H, A502V, and R505C) all lost this connection to MYC loss-of-function, whereas 4 other alleles retained connectivity to MYC knockdown (Figure 7A, lower panel). Examination of the substrate-bound FBXW7 crystal structure (Hao et al., 2007) indicated that the mutations predicted by the CMap to be damaging map to the substrate-recognition pocket, whereas the non-damaging alleles do not (Figure 7A, upper panel).
Figure 7. Assessing impact of allelic variants and drug response in clinical trials.
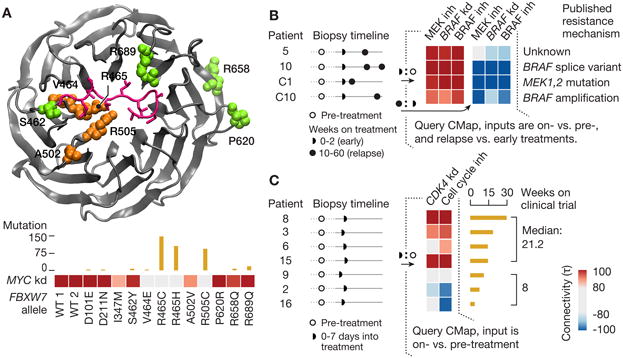
A. Predicting LoF alleles. Clinically-observed FBXW7 alleles were overexpressed and L1000 profiles obtained. Protein structure shows residues in question. Wild-type FBXW7 connects strongly to MYC shRNA, which is a known target (heat map). Mutations at residues adjacent to the substrate recognition site lose the MYC connection. τ values are summarized across multiple cell types. Bar plot above heat map indicates incidence of each mutation in COSMIC database.
B. Interpreting drug resistance. Transcriptional profiles of pre-treatment, early on-treatment, and relapse tumor biopsies obtained from clinical trials of BRAF and MEK inhibitors. Queries from on-treatment versus pre-treatment biopsies exhibited connectivity to pharmacologic inhibition of BRAF or MEK as well as BRAF shRNA in A375 cells, reflecting target engagement in vivo (left 3 columns in heat map). MAP kinase signaling was re-activated, as indicated by a strong negative connection to the same CMap signature in the subset of relapse biopsies with known MAP kinase pathway-related resistance mutations (right 3 columns of heat map).
C. Predicting therapeutic efficacy. Transcriptional profiles of pre-treatment and on-treatment biopsies from clinical trial of PHA-793887. Differential expression between the two time points yielded variable connectivity to negative regulators of cell cycle. Patients with strong positive connectivity to cell cycle inhibition signatures remained on trial for a median of 21 weeks; patients with negative connections remained on study for only 8 weeks.
The CMap similarly predicted the functional impact of the tumor suppressor KEAP1. Nineteen alleles of KEAP1 were subjected to L1000 profiling. Whereas over-expression of wild-type KEAP1 showed the expected CMap connection to knock-down of its transcriptional target NFE2L2, multiple alleles of KEAP1 lacked the NFE2L2 connection, suggesting that these were KEAP1 loss-of-function alleles. A subset of these alleles were recently functionally characterized and reported to result in loss of KEAP1 function, as predicted by the CMap analysis (Hast et al., 2014) (Figure S3C, left panel). A similar phenomenon was observed with alleles of the phosphatase PTEN, which negatively regulates PI3K activity. Whereas overexpression of wild-type PTEN showed connectivity to signatures of PI3K inhibitors, such connectivity was lost with PTEN mutations at residues M35 (mutated in Cowden's syndrome), G127 (important for active site conformation) and G129 (required for phosphatase activity) (Figure S3C, right panel).
Using CMap to interpret clinical trial results
The CMap has been developed to support research, not routine clinical care. However, we hypothesized that there might be potential to inform clinical investigation. Toward that end, we analyzed two oncology clinical trials in which tumor samples were obtained before and after treatment.
In the first study, 21 patients with melanoma were treated with the RAF inhibitors dabrafenib or vemurafenib and 9 patients were treated with dabrafenib plus the MEK inhibitor trametinib (Carlino et al., 2013; Long et al., 2014). Biopsies were obtained prior to treatment and at the time of relapse, and on-treatment biopsies were taken in four patients. The authors performed expression profiling on the Illumina beadchip platform (GSE50509, GSE61992). Comparing four on-treatment biopsies to the pre-treatment biopsies, we observed strong positive connectivity to multiple signatures of MAP kinase inhibition, consistent with drug-induced silencing of the MAP kinase pathway. Analysis at the time of relapse showed that several patients showed strong negative connectivity to these same CMap perturbations, suggestive of reactivation of the MAP kinase pathway – a known mechanism of drug resistance in melanoma (Wagle et al., 2014). One of those patients (patient 10) had a MAP kinase-activating BRAF splice variant, consistent with the CMap results (Figure 7B). Pathway reactivation was also detected in a resistant tumor with MAP2K1 mutation (patient C1) and in a resistant tumor with BRAF amplification (patient C10).
In the second study, patients with solid tumors were treated with the pan-CDK inhibitor PHA-793887 in a phase I clinical trial. Seven patients from that trial were subjected to gene expression profiling of biopsies pre-treatment and on-treatment using Agilent microarrays (Locatelli et al., 2010; Massard et al., 2011). For each patient, the on-treatment expression profile was compared to their pre-treatment profile and the difference used as a signature to query the the CMap. This analysis showed an association between duration of therapy (a proxy for clinical benefit) and connectivity to the overexpression of key negative regulators of the cell cycle such as CDKN1A and CDKN2A. Strong connectivity was also observed to knock-down of the cyclin-dependent kinase CDK4 – one of the targets of the drug (Figure 7C). Interestingly, the patients with rapidly progressive disease showed anti-correlation to this cell cycle inhibition signature, possibly reflective of a feedback mechanism to reactivate the cell cycle in the face of CDK4 inhibition. These results, while reflecting only a small number of patients, are encouraging. First, they suggest that while PHA-793887 may be a pan-CDK inhibitor, inhibition of CDK4 may be the most clinically relevant. Second, on-treatment biopsy coupled to Connectivity Map analysis may prove useful as an early molecular readout of target engagement in patients.
Accessing Connectivity Map data
All of the CMap data described in this report are available without restriction–to the research community including commercial entities. To enhance accessibility and utility, we developed a number of computational-visualization tools that enable users to interact with data at multiple levels (from raw to processed to normalized data), using methods optimized for technical and non-technical users (e.g., restful Application Programming Interfaces (APIs) for computational biologists and software engineers, and web applications for biologists). The most efficient method of accessing the data and tools is via the secure, cloud-based computing environment that we termed CLUE (Connectivity Map Linked User Environment), at https://clue.io. To enable computational researchers to reproduce our findings exactly, code is available at GitHub, and the entire preprocessing workflow is available as a container in the AWS Docker registry. Raw data are also available for download from GEO (accession GSE92742), but users will find it more efficient to interact with the data using CLUE.
Discussion
This study demonstrates the feasibility of a large-scale compendium of functional perturbations coupled to an information-rich gene expression read-out. By making L1000 expression profiling inexpensive, scale up became tractable. The L1000 platform has certain attributes and limitations worth considering. Because L1000 is hybridization-based, it is possible to monitor the expression of non-abundant transcripts. While such rare transcripts (e.g., encoding transcription factors) can also be detected by RNA-seq, high depth of sequence coverage is needed, and this can become cost-prohibitive. Nevertheless, as sequencing costs drop, RNA-sequencing-based approaches such as Perturb-Seq (Dixit et al., 2016) should be considered.
We chose the ∼1,000 landmark transcripts in an unbiased manner, based on their orthogonal expression patterns. Alternative probe-selection methods, however, have been proposed (Donner et al., 2012). Whether alternative sets of 1,000 transcripts would improve the ability to discover connections (the primary goal of CMap) remains to be established.
The ability to infer the expression of genes not directly measured in the L1000 assay was also explored. We found that a simple ordinary least squares model predicted the expression of 81% of non-measured transcripts. We also note that while our inference method was successful in the cell types tested, it is conceivable that it might perform less well in cell types dissimilar to those used to train the model.
The 1,319,138 L1000 profiles reported here represent 42,080 genetic and small molecule perturbations profiled across a variable number of cell types. To our knowledge, this far exceeds any other publicly available resource of cellular perturbation. An important question, however, is the extent to which this CMap resource can be used to discover important biological connections (e.g., to inform MOA of compounds, to discover pathway membership of gene products, or to connect disease states to pathways and small molecules).
For example, we used the annotation of protein targets of small molecule drugs and tool compounds to determine whether such targets could be recovered from the CMap. Our analysis showed that the CMap results were highly enriched in the correct targets for up to 63% of small molecules tested. While this result is encouraging, 37% of compounds showed no evidence of connection to their expected targets. Failure to recover such connections could be explained by many factors including i) incomplete inhibition of the target by the compound, ii) off-target effects of compounds and genetic perturbations, iii) missing information in the L1000 read-out, iv) incorrect literature-based annotations of compounds, v) biological differences between small molecule inhibition of specific aspects of protein function (e.g., enzymatic inhibition) compared to complete loss of function (e.g., scaffolding functions) induced by shRNA-mediated knock-down, and vi) the existence of previously unrecognized bona fide connections that effectively penalize the known connections – particularly if the novel connections are stronger than the expected ones.
Perhaps the most interesting use of CMap is to functionally annotate previously uncharacterized small molecules. For example, we discovered a novel inhibitor of the casein kinase CSNK1A1 – a newly emerging protein essential for survival of certain myeloid malignancies and also implicated in EGFR inhibitor resistance. The compound, BRD-1868, was discovered entirely through computational analysis; no laboratory experiments were needed to generate the CSNK1A1 inhibitory hypothesis. We note that this discovery underscores the value of having a large-scale compendium of genetic and pharmacologic perturbations.
Our analysis of 18,493 shRNA profiles showed that the off-target effects of shRNAs far exceed their on-target effects, consistent with recent reports (Tsherniak et al., 2017). However, the generation of a Consensus Gene Signature (CGS) that identifies gene expression changes common to multiple shRNAs targeting the same gene substantially improved the ability to discover on-target connections by minimizing off-target effects. Nevertheless, the CGS procedure is imperfect, and some off-target effects likely remain. Preliminary studies of CRISPR/Cas9-mediated gene knock-out suggest that genome editing approaches may recover some genetic connections to small molecules that were missed by RNA interference-based perturbation. Two caveats bear mentioning. First, we and others have shown that CRISPR/Cas9-based genome editing results in non-specific toxicity directly proportional to the number of cuts to the genome (Aguirre et al., 2016). The extent to which such non-specific effects can be computationally corrected in the context of CMap analysis remains to be determined. This is particularly relevant when performing genetic perturbations in cancer cell lines that often harbor copy number alterations. Second, it remains to be determined whether complete gene knock-out (via CRISPR) or partial knock-down (via shRNA) better phenocopies the effect of a small molecule.
Our analysis across multiple cell types revealed that some perturbations yielded universal signatures, whereas 43% of compounds yielded cell-type selective gene expression signatures. The fact that many compounds yield a universal signature regardless of cell type also has important implications. Specifically, the value of continuing to profile such compounds across a large number of cell lines is probably low. Future iterations of the CMap might therefore benefit from an adaptive experimental design whereby the selection of future cell lines is chosen based on the performance of an initial set.
Importantly, the CMap concept is not restricted to mRNA expression. Other groups are generating proteomic or high-content imaging readouts following perturbation (Litichevskiy et al., 2017); (Rohban et al., 2017) consistent with early reports of feasibility of annotating compounds based on cellular consequences (Seiler et al., 2008).
Biomedical research in the 21st century reflects a dramatic increase in the sheer amount of data available for analysis, and a commensurate need for increasingly sophisticated computational tools. In the past, researchers would download genomic datasets to their own computers, and run computational analyses locally. In the era of big data, however, it is advantageous to bring computation to the data. While the present 1.3 million L1000 profiles are not too large to download, making the data available to users on the cloud will increase computational efficiency. We have therefore created a cloud-based data storage and analysis system called CLUE. In the CLUE environment, users can access all publicly available CMap data, append their private data, and access a collection of user-friendly analysis apps designed for intuitive use by experimental biologists. Computational biologists can access data using data APIs at clue.io/api. We note that additional L1000 analytical tools developed by others are available through the LINCS data coordinating center at lincsproject.org.
A future, comprehensive CMap might expand in multiple dimensions. First, the number of small molecules profiled would increase to include much larger collections. Second, the genetic perturbations would include allelic series of important disease-associated genes. Third, future iterations of the CMap should explore new cell types including patient-derived iPS cells and genome-edited isogenic cell lines. Fourth, future expansion should include different types of perturbational read-outs (e.g., high content imaging, limited proteomic profiling). An important goal for the years ahead should be to establish which of these alternative data types are most complementary to transcriptional profiling.
As with all large-scale community resources, the full potential of the Connectivity Map will only be realized with time. Whether it proves most useful for elucidating small molecule mechanism of action, for providing functional readouts of genetic variants, or for generating new therapeutic hypotheses remains to be seen. Such emerging utility should guide the further expansion of a future CMap.
Star Methods
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Todd Golub (golub@broadinstitute.org).
Experimental Model and Subject Details
Cell lines
Cell lines were obtained from ATCC (https://www.atcc.org/) unless otherwise indicated. Information on tissue, tumor type and optimal growth conditions, (for normal growth and or profiling) were obtained from the CCLE project (https://portals.broadinstitute.org/ccle) and the protocols including the number of cells employed for these L1000 experiments are described at https://clue.io/sop-cell.pdf.
Method Details
Dataset for Landmark selection (DSGEO)
We assembled a large, diverse collection of 12,063 gene expression samples profiled on Affymetrix HG-U133A microarrays from the Gene Expression Omnibus (GEO) (Edgar et al., 2002). These data were used to identify the subset of universally informative transcripts to be measured, which we term ‘Landmark Genes’ (Dataset DSGEO).
Selecting landmark transcripts
As DSGEO contains a non-uniform representation of various aspects of biology (for example certain tumor types such as breast and lung cancer were disproportionately represented), we applied Principal Component Analysis (PCA) as a dimensionality reduction procedure to minimize bias toward any particular lineage or cellular state. In this reduced eigenspace of 386 components (which explained 90% of the variance), cluster analysis was performed to identify tight clusters of commonly co-regulated transcripts. We applied an iterative peel-off procedure to select the centroids (Tseng and Wong, 2005). Specifically, at each iterative step in the tight clustering process, the k-means algorithm with K ranging 20-100 was applied repeatedly on 100 independent random subsamples each comprising 75% of the original data. This procedure yielded a consensus matrix that contained the proportion of trials a pair of genes were in the same cluster. Thresholding the consensus matrix yielded sets of genes that co-clustered in more than 80% of the trials. The genes belonging to the stable clusters were noted, excluded from the data and the procedure was repeated to identify additional clusters. Because high-dimensional data is challenging to partition into definitive clusters, the advantage of this approach is that gene-gene clusters are derived through the tendency of genes to be grouped together under repeated resampling and hence are more robust to the initialization and cluster size thresholds. Transcripts nominated as landmarks through this process were then tested empirically to assess ability to measure levels accurately in the L1000 assay as described in “Probe and primer design for the L1000 assay” and experimental validation as described in the L1000 reproducibility sections below.
Evaluating performance of reduced representations of the transcriptome
To simulate performance of measuring a subset of the transcriptome, we asked what number of landmarks (k) would optimally recover the observed connections seen in the pilot Connectivity Map dataset based on Affymetrix arrays (Dataset DSCMAP-AFFX). Specifically, prior work indicated that 25 query signatures yielded robust and expected connections to small molecules in the CMap pilot dataset (Table S1). We therefore used those 25 signatures to query the inferred DSCMAP-AFFX dataset for various values of k, counting how often we recovered the connections observed in the original dataset at a comparable rank based on the Kolmogorov-Smirnov statistic. At values of k ranging from 100-10,000, we generated an imputed version of DSCMAP-AFFX using OLS regression (trained on samples from DSGEO) with the k landmarks as the independent variables, queried it with the benchmark signatures, and assessed the percentage of connections that were recovered.
Baseline expression of landmark genes across a diversity of tissue types
Our procedure for selecting Landmark Genes was data-driven and the simulations presented above indicate that both the landmark and inferred genes capture relevant information about cell state. However, given a new state, any inference algorithm will only work if a fair number of the landmark genes are expressed in that state. We examined expression across lineage using the Genotype Tissue Expression (GTEx) RNA-seq dataset (DSGTEx-RNA-seq) of 3,176 patient-derived expression profiles from 30 different tissue types (Figure S1B). We quantified the expression levels of the landmark genes reported in the dataset and observed that at a RPKM threshold of 1 at least 86% of Landmark Genes are expressed in each of the 3,176 samples (with an average of 92% expressed in each sample), and that range of expression is similar across tissue types.
Functional enrichment analysis of landmark gene content
Our data-driven procedure suggested genes to include as landmarks based on analysis of the 12,063 sample compendium DSGEO. We then asked if genes suggested by this data driven approach were enriched in particular known biological pathways or categories.
For every landmark gene we accessed from NCBI entrez its current gene description and family assignment. We also annotated every landmark gene with the pathway (as defined in MSigDB) in which it is thought to function (when available). Finally, we looked up its biological/molecular category from Gene Ontology (GO). These annotations were analyzed for functional enrichment to ask if the landmarks, when considered as a set, are dominated by a few functions or if on the whole they map to many different functions. For example, at one extreme the transcriptionally active genes could belong to basic regulatory processes (e.g transcription factors).
To do this analysis we intersected the 978 landmarks with a database of 1,533 gene sets compiled in Gene Ontology using the hypergeometric statistic (gene to GO gene ontology, conditional test for over-representation). We used the R Bioconductor package GOstats (v2.36.0) and the ontology from GO.db (v3.2.2). The results show that while some categories are enriched (e.g ATP binding, nucleoside/nucleotide activity, transcription factor binding, kinase regulator activity) the percentage of the 978 genes that are in any such set is small. While we did observe a number of classes to be enriched in the landmark genes, these categories tend to be generic (e.g. enzyme binding, protein kinase binding, catalytic activity, ATP binding) and/or contain only a small fraction of the landmark genes (e.g. protein kinase binding, which contains 84 of 978 landmarks). Taken together, we did not find any particular functional category dominating the list of landmarks chosen.
Probe and primer design for the L1000 assay
Each transcript of interest was targeted with an upstream and downstream probe pair. Upstream and downstream probes were each designed with a 20nt gene specific region (40nt contiguous sequence per probe pair), a unique identifying barcode, and a universal primer site. The gene specific sequences were blasted against the human genome to verify that each is unique to the targeted gene of interest, as described in the steps below. In addition to gene specific sequence, upstream probes contained a T7 primer site, and a 24-nucleotide (nt) barcode, and downstream probes, which were 5′ phosphorylated, contained the T3 primer site. Barcode sequences are shown in supplementary Table S2. Probes were synthesized by IDT (Integrated DNA Technologies).
We followed an iterative process of probe design followed by empirical probe validation, as follows, until we achieved ∼1,000 landmark genes with a validated probe.
Landmark genes proposed based on computational analysis.
-
For each gene, select a 40 base sequence using the following design principles, then split into two 20-mers
a. Empirical probe design rules:- the region must be contiguous with no gaps
- must be 3′ biased to minimize RNA degradation
- choose regions with few repeats to minimize cross-reactivity
-
Perform computational sequence QC by aligning against human reference genome (assembly HG19) using BLAT (Kent, 2002)
Ensure a perfect alignment to intended gene's reference sequence
Check for non-specific alignment of the probe sequence to other genes
If either checks (a) or (b) fail, then redesign the probe sequence
-
Build upstream and downstream probes using T7 and T3 primer sites and FlexMAP tag
T7 primer site 5′ TAA TAC GAC TCA CTA TAG GG 3′
T3 primer site 5′ TCC CTT TAG TGA GGG TTA AT 3′
Uni-bio-T7 5′/5Bio/TAA TAC GAC TCA CTA TAG GG 3′
Uni-T3 5′ ATT AAC CCT CAC TAA AGG GA 3′
Cell lysate preparation
Cells were cultured in appropriate media and 40 μl was transferred into each well of a 384-well clear bottom, tissue culture treated plate with an automatic liquid handler. Plates were incubated at 37°C, 5% CO2. Cells were either treated with chemical or genetic perturbations, the details of which are reported in section 6 below. For cell lysis, media was removed from the wells without disturbing the cells and 25 μl/well of TCL Lysis Buffer (Qiagen) was added. Plates were sealed with adherent foil seals and incubated at room temperature for 30 minutes prior to storage at -80°C.
Coupling barcodes to Luminex beads
To detect gene-specific sequences, Luminex beads were coupled to DNA barcodes complementary to each barcode used in our collection of probes. Because Luminex produces 500 distinct bead colors and the L1000 set consists of 978 genes, 2 barcodes were coupled to beads of each color (see below); this was done in separate batches - one barcode per batch - and then the pairs were mixed in a 2:1 ratio prior to use. Luminex magnetic beads were added in 500 μl aliquots to each well of 96 deep-well plates. Beads were pelleted and resuspended in 62.5 μl binding buffer (0.1 M 2- [N-morpholino]ethanesulfonic acid; pH 4.5), to which was added 100 pmol capture barcode. 6.25 μl of freshly prepared 10 mg/ml aqueous solution of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Pierce, Milwaukee, WI, USA) was added to each well followed by incubation at room temperature in the dark for 30 minutes. This step was repeated and then 180 μl 0.02% Tween-20 was added. Beads were pelleted and washed in 0.1% SDS in TE, pH 8.0 buffer. Beads were stored in TE in the dark at 4°C for up to one month. Mixtures of beads were freshly prepared in 1.5× TMAC buffer (4.5 mol/l tetramethylammonium chloride, 0.15% N-lauryl sarcosine, 75 mmol/l tris-HCl [pH 8.0], and 6 mmol/l EDTA [pH 8.0]).
Ligation-mediated amplification
For mRNA capture, 20 μl lysate was transferred to Turbocapture (Qiagen) plates coated with oligo dT. Following a 60-minute incubation at room temperature, unbound lysate was removed by inverting the plates onto a highly absorbent towel followed by centrifugation at 1000 rpm for one minute. First-strand cDNA was prepared from the mRNA by adding 5 μl master mix consisting of # units M-MLV reverse transcriptase and # umol/l of each dNTP. Plates were incubated at 37°C for 90 minutes. Probes were annealed to the first-strand cDNA using 5ul Probe Anneal master mix, which contains 100 femtomole of each probe in 1× Taq ligase buffer. Denaturation was accomplished by incubating the plates at 95°C for 2 minutes and then decreasing the temperature from 70°C to 40°C over a 6-hour period. Plates were then inverted onto an absorbent towel and spun at 1,000 RPM for 1 minute to remove unbound probe.
To ligate juxtaposed probe pairs, 5 μl mix containing 2.5 units Taq DNA ligase in ligase buffer was added, plates were sealed, and incubation proceeded at 45°C for 1 hour followed by 65°C for 10 minutes. The plate wells were emptied as described above, and the resulting amplification templates were subject to PCR using T3 and 5′-biotinylated T7 universal primers. PCR was initiated by adding 15 μl master mix, containing 1.5 umole of each primer, 2.4 nmol of each dNTP, and 4.8e-4 units of HotStarTaq in reaction buffer. Plates were sealed and loaded into a Thermo Electron MBS 384 Satellite Thermal Cycler. Initial denaturation was performed at 95°C for 15 minutes, and then the plates were subjected to 29 cycles as follows, one minute per step: 92°C(denature), 60°C (anneal), 72°C (elongation). The resulting amplicons were gene-specific, barcoded, and biotinylated.
Hybridization of amplicon to bead
Because a sequence complementary to the barcode on each probe has also been coupled to a Luminex bead, the amplicons (and hence the gene-specific sequence) can be identified by hybridization to the beads. A volume of 5 μl of PCR amplicon was transferred to a well containing 30 μl of L1000 bead mix (∼ 100 beads/region/well). The plate was sealed and incubated at 95°C for 2 minutes to denature the DNA. Incubation continued at 45°C for 18 hrs. Beads were pelleted, washed, and stained with 20 μl of 10 ng/ul streptavidin R-phycoerythrin conjugate (Molecular Probes) in 1× TMAC buffer (3 mol/l tetramethylammonium chloride, 0.1% N-lauryl sarcosine, 50 mmol/l tris-HCl [pH 8.0], 4 mmol/l EDTA [pH 8.0]) at 45°C for ten minutes.
Tag Duo dual detection and peak deconvolution
The Luminex FlexMap 3D platform is capable of detecting 500 different bead colors while the L1000 assay needs to measure ∼1,000 mRNA transcripts. One option would be to read these in 2 different detection sets, each of 500. However, that would introduce inevitable batch effects and also reduce detection throughput by half.
Therefore it was provident to devise a strategy that allows two different transcripts to be identified by a single bead color. The 978 landmarks were divided into pairs and barcodes representing each gene were coupled to beads of the same color (one gene per bead). Genes coupled to the same bead type (color) in two separate batches were combined in a ratio of 2:1 prior to use. When the beads are hybridized with the sample templates and analyzed by the Luminex scanner, two values are obtained from each bead: one indicating the color of the bead and the other indicating the intensity of the signal, which is a reflection of the expression of the gene. Identification of the bead color associates the intensity to the correct gene pair and signal intensity provides a measure of the abundance of the transcripts of the two genes. Deconvolution of the composite fluorescent intensity signal into its component gene expression values is done computationally as described below. To make it easier to resolve the peaks, rather than pairing genes at random, during design of the L1000 system, we optimized pairing of genes to maximize the average difference in their expression levels across the training GEO compendium.
Detection
Hybridization of amplicon to complimentary barcodes was detected using a Luminex FlexMap 3D flow cytometer, which detects both bead color (i.e., transcript identity) and the biotin label on the probe (i.e. transcript abundance; as measured by the phycoerythrin channel). Analysis was done using a sample volume of 40 μl.
Invariant genes as controls for data QC and normalization
We developed a set of internal controls to assess quality, to provide real-time feedback during the scanning process, and to use in normalization. Importantly, rather than using a single “housekeeping” gene (e.g. GAPDH), we adopted an approach that utilizes control values across the entire spectrum of gene expression. We adapted the approach described in the Illumina BeadChip studio (Illumina Inc., 2007) by defining a set of genes that are rank invariant across all samples. To identify these genes, we analyzed human gene expression profiles from DSGEO and selected genes whose expression is relatively invariant (coefficient of variation < 10%) across a variety of tissue types and experimental conditions. To further minimize the variance, rather than picking single genes as invariants, we grouped the genes into 10 sets of 8 genes each based on their level of expression across all samples. The 10 gene sets were ordered by increasing levels of expression, with the first level corresponding to genes with the lowest expression and the tenth level to genes most highly expressed. Because these gene sets exhibit a consistent expression pattern, they can be used to adjust the data for non-biological variation. Importantly, in addition to being useful for data normalization, the invariant genes provide a simple quality check in real time as detection occurs, which is valuable in a high-throughput process.
Quantification and Statistical Analysis
L1000 reproducibility using reference mRNA
Samples of purified total RNA from six human cancer cell lines, purchased from Life Technologies, were subjected to L1000 profiling. L1000 expression profiles were generated for six cell lines in 3 independent LMA batches, each with 12 technical replicates, for a total of 216 total profiles (6 cell line × 12 replicates × 3 batches = 216). Within each cell line, we computed the Spearman correlation between all pairwise combinations of replicates (data level 3, see below), excluding the comparison of each replicate to itself. Three examples of paired comparisons and the full spectrum of correlations are shown in Figure S1C. We then computed the median correlation between each replicate and all others, yielding 36 values per cell line; and finally summarized using the median of medians so as to derive one value per cell line. These analyses showed that in general the L1000 assay has very high technical reproducibility.
L1000 reproducibility using reference mRNA and cross platform analysis
Samples of purified total RNA from six human cancer cell lines were purchased from Life Technologies. One gene-expression profile per sample was generated using the Affymetrix GeneChip HG-U133 Plus 2.0 Array, the Illumina Human HT-12 v4 Expression BeadChip Array and mRNA-seq (Illumina Hi-Seq) by Expression Analysis, a genomics contract research organization. The L1000 samples were profiled in multiple replicates. Data were normalized within platform (level 3, see below for details). For each cell line, we selected the L1000 replicate with highest technical quality (by LISS goodness of fit, see below) for comparison with the other three platforms. We then performed ComBat batch correction to adjust for cross-platform differences (Johnson et al., 2007), and subjected the data to hierarchical clustering in the space of the 952 genes commonly measured by all four platforms. We observe that the data cluster by cell line and not by platform, suggesting that the cross-platform differences are smaller than the biological differences between cell lines.
L1000 reproducibility using shRNAs
The fidelity of L1000 depends on being able to quantify endogenous levels of intended landmark genes accurately and specifically. In synthesizing landmark gene-specific oligonucleotide probes we followed several computational procedures that maximized matches to the target DNA sequence while minimizing non-specific hybridization. However, as sequence-based QC methods are imperfect and measurement of a transcript might degrade in a multiplexed gene assay (e.g due to cross hybridization), we designed an experiment to empirically confirm probe performance.
To assess the specificity of L1000 landmark probe measurements, we procured shRNAs that target landmark genes from The RNAi Consortium (TRC). We restricted this experiment to shRNAs that had been validated to down-regulate their intended target through RT-PCR assays conducted by TRC. We plated MCF7 and PC3 cells onto 384-well plates and used standard arrayed lentiviral protocols to infect the cells with these shRNAs, each of which targets a specific landmark gene, and then profiled the cells by L1000.
The resulting L1000 signature was used to calculate the targeted landmark gene down-regulation and rank relative to all other shRNAs in the experiment. For each gene in each sample, we computed differential expression values (z-scores) by comparing the gene's expression value in the given sample to that same gene's expression values in all other samples in the cohort and then collapsed replicate samples (DSLM-KD). The resulting dataset contains, as columns, an individual shRNA targeting a landmark gene performed in either MCF7 or the PC3 cancer cell line. Rows are replicate-collapsed z-scores (level 5, see below) of all measured landmark genes.
A probe designed against a landmark gene was progressed if its z-score when targeted by an shRNA was -2.0 or lower. When the initial probe design showed non-specific reactivity, failed to correlate with reference mRNA standards or failed to register adequate knockdown, we redesigned the probe sequence and retested. After a few cycles of iteration between design and empirical testing, we were able to show that 846 of the 955 targeted landmark genes (89%) were down-regulated by at least one targeting shRNA (z-scores less than -2). However, a low z-score doesn't in itself imply specificity—for example, a sample corrupted by dead cells might have yielded low mRNA across the board, leading to many genes with low z-scores. To guard against nonspecific reduction of z-scores, we compared the distribution of targeted gene z-scores to non-targeted gene z-scores and observed that the former was significantly left-shifted, indicating that the observed down-regulation is largely specific to the targeted genes (Figure 1C, middle panel). For each targeted gene, we computed the rank of its z-score in the experiment in which it was targeted relative to all other experiments in the dataset where it was not targeted. We observe that 841 of 955 genes (88%) rank in the top 1% and 907 of 955 (95%) rank in the top 5% (Figure 1C, bottom panel). These results indicate that the large majority of L1000 probes are specifically measured.
Definition of Recall
An absolute measure of similarity (e.g. Spearman correlation) between samples or genes does not in itself convey how uncommon that similarity is. Hence, in addition to computing the similarity (sim) between designated samples or genes, it is also useful to compare this similarity value to a reference distribution of similarity values (SIMnull), which can aid in interpretation of sim. To that end, we compute recall (R) as the fraction of SIMnull that is lower than sim. High R values correspond to unusually high values of sim. Thus, R provides an assessment of how well a particular pair of samples or genes match each other relative to an appropriate null.
L1000 comparison to RNA-seq
We sought to compare expression profiles generated using L1000 with those generated using Affymetrix and RNA-seq, the most widely employed platforms for gene expression profiling. In conjunction with the NIH's Genotype Tissue Expression (GTEx) project (http://commonfund.nih.gov/GTEx/index), we profiled 3,176 samples on L1000 and obtained from GTEx the RNA-seq (Illumina TrueSeq RNA sequencing) data for different aliquots of these same samples (DSGEO-RNA-seq and DSGEO-L1000). The data were quantile normalized independently by platform (level 3, see below) and then batch-corrected using the ComBat algorithm, an empirical Bayes-based method commonly used to remove batch effects across gene expression datasets (Johnson et al., 2007).
A small subset of these samples were also profiled on Affymetrix, and Figure 1E, top panel, shows comparisons of the platforms with each other for a single such sample. We observe that L1000 measurements and inferred expression values are as similar with RNA-seq as RNA-seq is with Affymetrix.
To more thoroughly compare L1000 to RNA-seq, we then computed sample self-correlations (using Spearman rank correlation) for the 3,176 samples in the space of the 970 genes directly measured by both platforms. There are 8 L1000 landmark genes that were not included in the DSGEO-RNA-seq. Level 3 L1000 data were used, and the GTEx RNA-seq data were quantile normalized, log2 scaled 1+RPKM values. We then computed sample self-correlations for the 3,176 samples and the median sample self-correlation was 0.84, with a notably right-shifted distribution relative to non-self correlations (Figure 1E, lower panel left). We also measured sample Recall (Rsample, see STAR Methods), wherein a given L1000 profile is forced to compete with all other RNA-seq profiles in order to find its RNA-seq counterpart. This analysis yielded 3,103/3,176 samples (98%) with a Rsample > 0.99 (indicating 99th percentile) and all but 5 (99.84%) had a Rsample > 0.95 (Figure S1D).
Identifying well inferred genes
We sought to assess the inference quality of the 12,232 features corresponding to inferred-only genes in DSGEO-OLS. For this test, we used a compendium of 8,555 RNA-seq profiles, generated as part of the GTEx project. We applied the DSGEO-OLS inference model on DSGTEx-RNA-seq-lmonly which resulted in DSGTEx-RNA-seq-INF. To assess inference performance, we computed the correlation of every inferred feature in DSGTEx-rnase-INF to its corresponding gene in DSGTEx-RNA-seq. We then analyzed these data to identify genes with statistically significant inferred to measured correlation, as these genes represent the most reliable inference predictions. To generate a null distribution of correlations, we computed the correlation between every inferred probeset in DSGTEx-RNA-seq-INF and every non-matched gene in DSGTEx-RNA-seq. We then computed p-values for every inferred gene by computing the percentage of the null distribution with higher correlation than the given inferred gene. We observed that 9,196 of the 11,350 inferred genes (81%) correlated with p-value less than or equal to 0.05. This set of 9,196 inferred genes, plus the 978 landmarks, are referred to as the Best Inferred Genes (BING) and are presented in Table S3.
Gene space summary
The L1000 assay directly measures 978 genes and infers 11,350 more, for a total of 12,328 genes. Of the 11,350 inferred genes, 9,196 are considered well inferred, based on the analysis described above. All datasets are provided in the full 12,328 gene space. Table S3 indicates which genes are measured or well-inferred.
Data preprocessing
The L1000 automated data processing pipeline captures raw data from Luminex scanners as it is generated, deconvolutes 978 transcripts from only 500 Luminex bead colors, normalizes the data based on 80 invariant control genes, infers the expression of the non-measured transcripts, determines differentially expressed genes following a perturbation compared to controls, and generates composite signatures across biological replicates.
Level 1 - Raw (LXB)
Level 1 data comprises the bead identity and raw fluorescent intensity (FI) values measured for every bead detected by the Luminex scanner. The FI is proportional to the amount of amplicon bound to the bead, and hence also proportional to the transcript abundance of the genes that particular bead is interrogating.
Level 2 - Deconvolute (GEX)
The raw FI values associated with each bead color are analyzed in a peak deconvolution step to associate the expression levels with the appropriate genes. This step is necessary because each bead color is associated with two genes rather than one. To facilitate the analysis, separate bead batches that identify each gene are mixed in a 2:1 ratio for use in the assay. To deconvolute the composite expression signal into two values and associate them with the appropriate genes, we construct a histogram of FI values. This yields a distribution that generally consists of two peaks, a larger one that designates expression of the gene for which a larger proportion of beads are present, and a smaller peak representing the other gene. Using the k-means clustering algorithm, the distribution is partitioned into two distinct clusters, such that the ratio of cluster membership is as close as possible to 2:1, and the median expression value for each cluster is then assigned as the expression value of the appropriate gene.
Level 3 - Normalization (NORM)
In order to reduce artifacts (non-biological sample variation) from the data, we use a rescaling procedure called L1000 Invariant Set Scaling, or LISS, involving 80 control transcripts (8 each at 10 levels of low to high expression) that we empirically found to be invariant in expression across the DSGEO. The 80 genes are used to construct a calibration curve for each sample. Each curve is computed using the median expression of the 8 invariant genes at each of the 10 pre-defined invariant levels. We then loess-smooth the data and fit the following power law function using non-linear least squares regression:
where x is the unsealed data and a, b, and c are constants estimated empirically. The entire sample is then rescaled using the obtained model. LISS therefore serves as a method to both adjust for technical variation and to convert between measured Luminex intensity and more traditional Affymetrix log2-expression values.
After applying LISS, we standardize the shape of the expression profile distributions on each plate by applying quantile normalization, or QNORM. This is done by first sorting each profile by expression level, and then normalizing the data by setting the highest-ranking value in each profile to the median of all the highest ranking values, the next highest value to the median of the next highest values, and so on down to the data for the lowest expression level.
Normalization yields the expression values of the 978 landmark genes. To obtain expression values for all the remaining genes in the transcriptome, we assume that an unmeasured gene x can be predicted from the measured landmark genes li via linear regression:
where the wi constitute the model weights and have been estimated using DSGEO. These weights are provided in the dataset DSGEO-OLS. Repeating this procedure for all unmeasured genes gives predicted measurements of all 12,328 genes reported (measured plus inferred) by the L1000 assay.
Level 4 - Differential Expression (ZSPC)
To obtain a measure of relative gene expression, we use a robust z-scoring procedure to generate differential expression values from normalized profiles. We compute the differential expression of gene x in the ith sample on the plate as
where X is the vector of normalized gene expression of gene x across all samples on the plate, MAD is the median absolute deviation of X, and the factor of 1.4826 makes the denominator a consistent estimator of scale for normally distributed data.
Level 5 - Replicate-consensus signatures (MODZ)
L1000 experiments are typically done in 3 or more biological replicates. We derive a consensus replicate signature by applying the moderated z-score (MODZ) procedure as follows. First, a pairwise Spearman correlation matrix is computed between the replicate signatures in the space of landmark genes with trivial self-correlations being ignored (set to 0). Then, weights for each replicate are computed as the sum of its correlations to the other replicates, normalized such that all weights sum to 1. Finally, the consensus signature is given by the linear combination of the replicate signatures with the coefficients set to the weights. This procedure serves to mitigate the effects of uncorrelated or outlier replicates, and can be thought of as a ‘de-noised’ representation of the given experiment's transcriptional consequences.
Identifying batches in CMap data
Because the CMap resource was generated over a number of years, it is inherently comprised of multiple smaller batches of data. The most predominant form of batch is the 384 well plate in which each experiment was performed. Samples on a given physical plate were processed together in the lab (i.e cell plating, treatments, amplification and detection). To mitigate plate-level effects, normalization and differential expression are computed within individual plates (see data levels 3 and 4 above). Each profile is labelled with the name of the plate and the individual well in which the experiment was done. These fields are named ‘rna_plate’ and ‘rna_well’, respectively, in the provided sample metadata.
Query methodology
The fundamental unit of CMap analysis is the query. A query (q) consists of a set of genes corresponding to any biological state of interest. Each gene in the query carries a sign indicating whether it is up-regulated or down-regulated. Thus each query yields a pair of mutually exclusive gene lists (qup, qdown). The query is compared to each signature in the CMap reference database (Touchstone) using the similarity metric described below to assess connectivity viz. the degree to which the up-regulated query genes (qup) appear toward the top of the rank-ordered signature and the down-regulated query genes (qdown) appear toward the bottom of the signature (positive connectivity) or vice-versa (negative connectivity). The result of a query is a rank ordered list of CMap signatures ordered by their connectivity scores.
Computing similarities - Weighted Connectivity Score (WTCS)
The weighted connectivity score (WTCS) represents a non-parametric, similarity measure based on the weighted Kolmogorov-Smirnov enrichment statistic (ES) described previously (Subramanian et al., 2005). WTCS is a composite, bi-directional version of ES. For a given query gene set pair (qup, qdown) and a reference signature r, WTCS is computed as follows:
Where ESup is the enrichment of qup in r and ESdown is the enrichment of qdown in r. WTCS ranges between -1 and 1. It will be positive for signatures that are positively related and negative for those that are inversely related, and near zero for signatures that are unrelated. A null (0) score is assigned for cases when both ESup and ESdown are the same sign.
Normalization of Connectivity Scores
To allow for comparison of connectivity scores across cell types and perturbation types, the scores are normalized to account for global differences in connectivity that might occur across these covariates. Given a vector of WTCS values w resulting from a query, we normalize the values within each cell line and perturbagen type to obtain normalized connectivity scores (NCS) as follows:
where NCSC,t, wC,t, and are the normalized connectivity scores, raw weighted connectivity scores, and signed means of the raw weighted connectivity scores (the mean of positive and negative values evaluated separately) within the subset of Touchstone signatures corresponding to cell line c and perturbagen type t, respectively.
Overall, this procedure is similar to that used in Gene Set Enrichment Analysis, with the addition of bidirectional gene sets (i.e up and down) as queries.
Connectivity Map Score
Tau (τ) compares an observed enrichment score to all others in a reference database. In principle, τ can be computed by comparison to scores from any database of reference signatures, and the most common approach is to generate a null distribution by random permutation. However, a more stringent test that avoids having to make assumptions regarding the complex correlation structure of gene expression data is to use a compendium of diverse, biologically relevant perturbational signatures, such as those in CMap-LlOOOvl, as it is these reference signatures against which any novel connection must compete. Thus, query results are scored with τ as a standardized measure ranging from -100 to 100; a τ of 90 indicates that only 10% of reference perturbations showed stronger connectivity to the query. Because the reference is fixed, τ can be used to compare results across queries - a connection with a significant p-value and FDR but low τ would suggest a highly promiscuous relationship whose connections are not unique.
Calculating τ
While meaningful comparisons can be made between the NCS values of reference signatures with respect to query q, it is also useful to assess if the connectivity between q and a particular signature r is significantly different from that observed between r and other queries. This is done by comparing each observed NCS value ncsq,r between the query q and a reference signature r to a distribution of NCS values representing the similarities between a reference compendium of queries (Qref) and r. This procedure results in a standardized measure we refer to as Tau (τ) that ranges from -100 to +100 and represents the percentage of queries in Qref with a lower |NCS| than |ncsq,r|, adjusted to retain the sign of ncsq,r:
where ncsq,r is the normalized connectivity score for signature r w.r.t query q, ncsi,r is the normalized connectivity score for signature r relative to the i-th query in Qref and N is the number of queries in Qref Our standard practice is that Qref be comprised of queries obtained from exemplar signatures of Touchstone perturbagens that match the cell line and perturbation type of signature r. In principle any arbitrary compendium of gene sets (as long as they are large enough) could be used.
Summarization Across Cell Lines
When examining query results, it is often convenient to obtain a perturbagen-centric measure of connectivity that summarizes the results observed in individual cell types. This can be particularly helpful when searching for connections that persist across cell lines or when one is unsure which cell line to examine. Given a vector of normalized connectivity scores for perturbagen p, relative to query q, across all cell lines in which p was profiled, a cell-summarized connectivity score is obtained using a maximum quantile statistic:
where ncsp,c is a vector of normalized connectivity scores for perturbagen p, relative to query q, across all cell lines in which p was profiled, and Qhi and Qlo are upper and lower quantiles respectively. This procedure compares the Qhi and Qlo quantiles of ncsp,c and retains whichever is of higher absolute magnitude. Thus, maximum quantile is more sensitive to signal in a subset of the cell lines than measures of central tendency such as mean or median. In the analyses presented here, we used Qhi = 67, Qlo = 33
Off-target effects of shRNAs
In an effort to mitigate the strong off-target effects of shRNAs, we developed an algorithm to produce a Consensus Gene Signature (CGS) that reflects the consistent (and therefore on-target) gene expression effects of shRNAs. To generate a consensus gene signature (CGS), we first create a pairwise Spearman correlation matrix between all shRNA signatures targeting the same gene, explicitly setting self-correlations to 0. Each shRNA signature is then assigned a weight given by the sum of its correlations to the other signatures, with the weights normalized to sum to 1. The CGS is computed as the linear combination of the shRNA signatures, with coefficients set to the weights.
Assessing recovery of expected connections
To assess the degree to which each perturbagen profiled in L1000 recovered its expected connections to other perturbagens in Touchstone we leveraged annotations compiled from various sources. First, the annotations were used to construct a pairwise binary association matrix for all perturbagens in Touchstone. A pair of perturbagens were considered to be associated if they shared at least one type of annotation. For example, a pair of small-molecules were associated if they shared the same MoA. Similarly a compound and a genetic perturbagen could be associated if they shared the same gene target. We retained 1,902 small-molecule, 994 genetic over-expression, and 1,634 CGS perturbagens after excluding those that had too few (<10) or too many (>3,000) connection pairs. Then for each perturbagen p, we partitioned all associated perturbagen-pairs into a collection of expected connection pairs (Ep) whose members were associated with p and and a collection of background pairs (BP) whose members were not associated with p. Finally, ROC analysis was performed wherein the connectivities between members of Ep were compared to that between members of Bp at different threshold values for connectivity t ranging from (0, 100). At each threshold we computed true positive rates (TPR) as the fraction of Ep that were connected, and false positive rates (FPR) as the fraction of Bp that were connected, thereby generating an ROC curve from which an AUC was derived.
To further explore the known relationship between a compound's gene expression signature and cell line genotype, we profiled the MDM2 inhibitor AMG-232 in a panel of ten MCF10A isogenic cell lines. We observed AMG-232 had a dramatic reduction in TAS only in the cell line in which TP53, which is negatively regulated by MDM2, was homozygously deleted compared to the other 9 cell lines which were all TP53 wild-type. This result may indicate the utility of a more general screening approach by which the potential target(s) of a compound could be identified by generating L1000 profiles across a diversity of genetic backgrounds.
Defining perturbational classes
In order to define perturbational classes we first obtained annotations for as many Touchstone perturbagens as possible. For compounds, mechanism of action and gene target annotations were collated from multiple sources (Corsello et al., 2017). For genes, family and pathway annotations were obtained from HGNC as of July 2016. Annotations for both compounds and genes were manually regularized. We next grouped perturbagens by shared annotation to generate candidate classes. For example, all compounds that share the same mechanism of action were assigned to the same class.
For each perturbagen member of a candidate class, we assessed whether it sufficiently recovered its expected connections to other perturbagens in at least one cell line via ROC analysis (more detail below). The class definition was refined to include only those members that passed this criterion. Finally, the classes were assessed for sufficient interconnectivity. We required that classes had at least 3 members and exhibited a median pairwise τ of at least 80 in one or more cell lines. Those classes that passed this filter were codified into perturbagen classes (PCLs). This process resulted in 171 PCLs (92 compound, 60 LoF, and 17 GoF classes) corresponding to 930 unique perturbagens. PCLs range in size from 3 to 44 members, with an average size of 5.8 members. PCLs were required to contain only perturbagens of the same type and although perturbagens were allowed to belong to more than one PCL, most PCLs are completely distinct, with a median pairwise overlap of zero members. 95% of PCL members belong to just one PCL.
The majority of PCLs show strong inter-member connectivity in multiple cell types with 132 PCLs (77%) having a cell-summarized median pairwise τ >= 80. 24 PCLs (14%) had significantly stronger connectivity in a particular cell type than in cell-summarized mode, indicating that for these PCLs the connectivity was driven by cell context. Some examples include PPAR receptor agonists in HT29 and PC3 cell lines and estrogen-receptor agonists and antagonists in MCF7.
Compound PCLs were also assessed for structural similarity. The 2D structural similarity of all pairwise combinations of compounds within each PCL was measured using Tanimoto coefficient calculated from binary fingerprints, which were obtained from SMILES strings representing structures of the compounds in PCLs. SMILES strings were converted to binary fingerprints using the Open Babel implementation of the Daylight fingerprint standard (O'Boyle et al., 2011). We found that the vast majority of PCLs were structurally diverse. All but one PCL had a median pairwise Tanimoto below 0.8. Detailed information on all PCLs is available in Supplementary Table S7.
Computing Connectivity to PCLs
Connectivity of a query to PCLs is computed using the same approach described earlier for summarization of connectivities to a perturbagen across cell lines. Given a vector of normalized connectivity scores for the members of a PCL p, relative to query q, in a given cell line, we apply the maximum quantile procedure to obtain a summarized NCS value (NCSPCL). We then compute a PCL-level τ from NCSPCL by comparison to a reference distribution comprised of PCL-aggregated scores corresponding to the Qref queries described above.
This ensures that t is always computed relative to an equivalent background distribution and keeps it on a scale comparable to that of individual perturbagens.
Selectivity of PCL Connections
We define the PCL selectivity s of a query q as the fraction of PCLs whose connectivity to q is less than a given threshold τth. The fewer the number of PCLs connected to by q, the higher its selectivity.
Where sq is the PCL selectivity of query q, N is the number of PCLs, τi is the connectivity of q to the ith PCL and τth is the connectivity threshold.
PCL validation
In order to test the accuracy of PCL connections, we profiled 137 holdout compounds known to share a mechanism with one or more of 54 small-molecule PCLs, but which were not used in the construction of the PCL itself. We subjected the resulting signatures to connectivity analyses as described above and observed that for 41/54 classes (76%), the test compounds connected to their designated PCL in multiple cell types (Figure 4B). For an additional 7/54 (13%), a selective connection was observed in a single cell type. The remaining 6/54 (11%) did not reconnect at a threshold of t >90. Thus, 48 of the 54 assessed PCLs (89%) were considered validated in that they successfully connected to their corresponding holdout compound(s).
Unexpected Connections Between Drugs and PCLs
We assessed whether a validated PCL had strong, selective, but unexpected connections to 3,333 annotated small molecule compounds. To focus on unexpected connections, we identified all compounds that connected to a validated PCL at τ ≥ 98 of which the given compound was not a member and whose members' gene targets did not overlap with the given compounds' gene targets. For each compound, we computed its PCL selectivity as the fraction of the 171 PCLs to which it failed to connect with τ ≥ 90 and considered only compounds with selectivity of at least 0.9. We identified 225 novel connections between drugs and validated PCLs, corresponding to 132 drugs (3.9% of total assessed). We applied the same analysis to 2,418 unannotated but transcriptionally active compounds and identified 194 strong, selective connections corresponding to 111 compounds (4.6% of total assessed).
HDAC Inhibitor PCL Clustering
We performed hierarchical clustering on the 22 members of the HDAC inhibitor PCL in the space of their pairwise connectivities to each other across 9 cell lines using spearman correlation as the similarity metric with complete linkage. Hierarchical clustering of pairwise connectivities of the HDAC inhibitor PCL members reveals substructure within the class. The pan-HDAC inhibitors generally cluster together, distinct from the more isoform-selective compounds, suggesting that gene expression can be used to further stratify compounds within the same class.
Cellular context
A common question with respect to perturbational signatures is the extent to which they are consistent across different cellular contexts. To investigate this, we first restricted our analysis to the cell lines in which each perturbagen gave a signature whose transcriptional activity score (TAS) was greater than 95% of that of negative controls and considered only perturbagens that had high-TAS signatures in at least three cell lines. Using these thresholds, we analyzed 1,399 of 2,429 compounds, 1,088 of 2,160 cDNAs, and 3,926 of 13,187 shRNAs.
We then computed the pairwise similarity (using WTCS) between signatures of the same perturbagen in different cell lines, yielding an N × N matrix of similarity values, where N is the number of cell lines in which the perturbagen gave a high-TAS signature. Next, we computed the median WTCS between each cell line and all others, yielding a vector of N median WTCS values (WTCSmed). We then computed the median of medians (MoM) and range of WTCSmed, yielding WTCSMoM and WTCSrange, metrics which indicate the aggregate similarity and the variability thereof between signatures of the same perturbagen in different cell lines. Perturbagens that give a single signature across multiple cell types should have high WTCSMoM and low WTCSrange values, respectively. To estimate significance, we computed WTCSMoM and WTCSrange for 1,000 random combinations of N high-TAS signatures for values of N between 3 and 9. For a perturbagen to be considered as giving a single signature, we required that its WTCSMoM be greater than 95% and its WTCSrange be less than 95% of its size-matched null.
Using these thresholds, we found that 26% of compounds, 8% of cDNAs, and 34% of shRNAs gave a single signature across multiple cell lines. The comparatively larger proportion of shRNAs that give a single signature may be attributed to the higher transcriptional activity of shRNAs. We observe that about 36% of genes with at least 3 high-TAS shRNAs have at least 50% of those shRNAs flagged as single-signature reagents. This is not notably different from the 34% rate at which shRNAs give single signatures in general, suggesting that whether or not an shRNA gives a single signature is more dependent on the shRNA itself (and possibly its off-target effects) than it is on the specific gene the shRNA is targeting. cDNAs least frequently give a common signature. This was somewhat unexpected, given that they have a similar transcriptional impact as compounds, and may be due to their relative lack of off-target effects. Having fewer off-target effects may result in a signature that predominantly contains the effect of over-expressing a single gene, which may be quite different depending on the gene and cell context.
Amongst those perturbagens that were identified as having a single signature, we observed many that target core biological processes such as heat shock response, cell cycle, and HDAC and topoisomerase inhibition, among others. These results suggest that the transcriptional response to perturbing each of these fundamental pathways is conserved across cell contexts.
We also observed a number of classes of perturbagens whose members tended to give multiple unique signatures. For example, 23 of 32 EGFR inhibitors were identified as having multiple signatures and 31 of 34 serotonin receptor antagonists gave multiple signatures, one extreme example being pindolol, whose signature in HCC515 was strongly dissimilar to its signature in other cell lines. These results suggest that the transcriptional response to perturbing these and other pathways may be context- and/or reagent-dependent.
Neuronal cell line comparison
To extend this analysis to include specialized primary cell types, we considered 768 compounds that had been profiled in both neural progenitor cells (NPC) and differentiated neurons (NEU) as well as the 9 core cancer cell lines. For each compound, we computed the similarity, using WTCS, between all pairwise combinations of cell lines and converted to τ using the pairwise similarities between all 768 compounds in all 11 cell lines as reference (Qref). We observed that 189 of the 768 compounds (25%) connected with τ ≥ 90 when comparing NPC to NEU. For each pairwise combination of the 11 cell lines (NPC, NEU + 9 core) we computed the fraction of the 189 compounds that self-connected above 90. We then computed the average fraction that self-connected when considering NPC to cancer (34%), NEU to cancer (25%) and cancer to cancer (50%). This suggests that the neuronal lines are more different from the cancer lines than the cancer lines are from each other, at least in the space of these 189 compounds. Therefore, expanding the cell line set into neuronal cell types may be beneficial.
Replicate Correlation (CC)
Each L1000 experiment consists of multiple biological replicates. To derive an aggregate measure of replicate reproducibility, we compute the 75th quantile of the Spearman correlations between all pairwise combinations of replicate level 4 profiles for a given experiment.
Signature Strength (SS)
We compute signature strength (SS) as the number of differentially expressed genes within a signature; that is, the number of landmark genes with absolute z-score greater than or equal to 2. The z-scores are adjusted to offset shrinkage of z-scores that occurs with increasing number of replicates. This allows SS values derived from signatures of different numbers of replicates to be compared with each other
Where z, nrep are a vector of moderated z-scores and the number of replicates respectively
Transcriptional Activity Score (TAS)
The transcriptional activity score (TAS) is computed as the geometric mean of SS and CC for a signature. TAS is scaled by the square root of the number of landmark genes (978) so the final score ranges between 0 and 1.
Where SS and CC are the signature strength and replicate correlation for the given signature, respectively.
Analysis of unannotated small-molecule screening libraries
We began with a collection of 16,527 unannotated small molecules for which we had generated L1000 profiles. These compounds were derived from a variety of sources, including the Broad Institute's diversity oriented synthesis (DOS) library and the NIH's Molecular Libraries Probe Production Centers Network (MLPCN). We focused on the 2,418 compounds whose 75th quantile of TAS was as least 0.2 and whose signatures had a median pairwise WTCS of at least 0.3 across cell lines, indicating a robust transcriptional response in at least a subset of cell lines. We termed these compounds Discovery, and attempted to assign functional annotations via comparison with annotated drugs and genes in the L1000 Touchstone (reference) part of the data.
To obtain a high-level view of these Discovery compounds relative to known drugs, we ran t-SNE analysis on the Discovery signatures and those of every compound belonging to a PCL. t-SNE is a non-linear dimensionality reduction and visualization technique that attempts to preserve local-structure from high-dimensional datasets ensuring that samples that are similar in the high dimensional space are plotted close together in the embedding (Maaten and Hinton, 2008). t-SNE was run on consensus signatures across cell types for each perturbagen in landmark space, with initial dimensions set to 50 and a perplexity of 30.
In addition, we performed query analysis on these compounds' signatures to derive their connectivities to Touchstone perturbagens and PCLs. We found that 111 Discovery compounds had strong and selective connections to PCLs (τ >= 98; PCL specificity >= 0.9).
Data and Software Availability
The data generated in this study are publicly available, at multiple levels of pre-processing, via GEO (accession GSE92742 and pre-processing code via GitHub https://github.com/cmap/cmapM), and for easier use via the CLUE analysis environment at https://clue.io. In particular, a web app that allows users to input gene sets from their study of interest so as to find matching conditions in the CMap database is available at https://clue.io/l1000-query
Additional Resources
Detailed protocols for the L1000 assay are provided at https://clue.io/sop-L1000.pdf. In addition, several documents in the connectopedia knowledgebase https://clue.io/connectopedia provide details to users on how to access utilize the dataset and tools. The website http://lincsproject.org provides information about the umbrella LINCS consortium, including the various metadata standards.
Supplementary Material
Figure S1. Properties and technical validation of Landmark genes, Related to Figure 1, A. Evaluating dimensionality reduction. Simulation showing the mean percentage (+/- SEM) of 33 benchmark connections recovered from an inferred CMap pilot dataset as a function of number of landmark genes used to train the inference model, indicating that around 1000 landmarks were sufficient to recover 82% of expected connections
B. Landmark gene expression. Distributions of baseline expression of landmark genes across the 30 tissue types present in dataset DSGTEx-RNA-seq. The majority of landmark genes are expressed in each tissue and the distributions of expression are similar across tissues, suggesting that landmarks are not over-optimized to a particular cell context.
C. Technical replicate reproducibility. Top: Scatter plots of 3 technical replicates for a sample of commercial MCF7 RNA. Bottom: Distributions of median Spearman correlations between technical replicates of commercial RNA samples from 6 cell lines (36 replicates per cell line). Nearly all correlations exceed 0.90, suggesting low sample-to-sample variability.
D. Comparison with RNA-seq. Scatter plot of recall (R) vs. cross-platform Spearman correlation for 3,176 patient-derived samples in the space of of 970 landmark genes profiled on RNA-seq and L1000. 3,103/3,176 samples (98%) had an R > 0.99 (indicating that their cross-platform self-correlations exceeded 99 percent of background different-sample correlations, see STAR Methods) and all but 5 samples (99.84%) had an R > 0.95, suggesting high similarity between the two platforms.
Figure S2. Characteristics of perturbational signatures, Related to STAR Methods, A. MDM2 inhibitor AMG-232 shows TP53-dependent transcriptional response. Left: Correlation matrix of AMG-232's signatures derived from isogenic MCF10A breast epithelial cell lines that were either TP53 wild-type or homozygously deleted at 6-point dose [0.04-10 uM range]. Signatures from a background of TP53homozygous deletion lose correlation with signatures from TP53 wild type background. Right: TAS of AMG-232signature is notably lower in the presence of TP53 homozygous deletion compared to wild type.
B. Cell context and transcriptional activity. The bars show the percentage of 2,429 small molecule compounds that produced high TAS signatures (defined as TAS ≥ 0.2) in a specified number of cell lines. The line represents cumulative percentages and indicates that 92% of compounds produce a high-TAS signature in at least one cell line.
C. Multiplicity of signatures. The 2,429 drugs profiled in the Touchstone dataset have differing degrees of transcriptional activities across the panel of 9 core cell lines. The majority of the compounds are active in 3 or more cell lines with 364 (15%) producing a single concordant signature per molecule and 1,045 (43%) giving rise to multiple signatures, suggesting that transcriptional response is dependent on cell context. Only 194 (8%) of the compounds gave no measurable response in any of the cell lines tested.
D. Context-specific interconnectivity. The bar plot shows the baseline expression of the target gene across the9 core cell lines. The thumbnail heatmaps show the inter-connectivity of members of the compound class that targets the indicated gene. Top: NR3C1 / glucocorticoid receptor antagonists (n=44). Bottom: PPARG / PPARG agonists (n=16). In both cases, the PCL interconnectivity is highest in the cell lines in which the target is most highly expressed.
E. Context-dependent connection between compounds and their gene targets. The raster plot shows the connectivity between the indicated gene knockdown and its targeting compounds, where each circle is a cell line. Filled circles indicate cell lines in which the compound-gene connection is expected, either due to mutation in (BRAF, MAPK genes) or high expression of (all others) the target gene. The strong connections are enriched for the expected cell lines, suggesting that CMap connectivities can be indicative of context specificity.
Figure S3. Experimental follow-up of CMap hypotheses and functional annotation of allelic variants, Related to Figure 4, A. Confirmation of predicted bromodomain inhibitors. TREEspot selectivity profile indicating the inhibitory activity of droxinostat, BRD-1103, and XMD11-85H against the 32 bromodomains tested by the BROMOmax assay. All three compounds were active against at least one bromodomain. Images generated using TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation.
B. Confirmation of predicted PI3K/MTOR inhibitors. Chemical structures and affinities (kD) between three predicted PI3K/MTOR inhibitors and AKT1, MTOR, and 4 PI3K isoforms, as measured by the Kinomescan assay. All three compounds show at least micromolar affinity for a subset of the predicted targets, with two compounds showing nanomolar affinities.
C. CMap connectivities correspond to loss of function mutations. Left: Heat map showing the connectivity between the over-expression signatures of various KEAP1 alleles to the knockdown of NFE2L2 in A549 cells. Right: Heat map showing the connectivity between the over-expression signatures of various PTEN alleles and the PI3K and MTOR inhibitor classes. Cell line is indicated by the colored bar below the heat map. For bothKEAP1 and PTEN, mutations at known active residues important for protein function result in loss of expected connectivity.
Table S1. Queries, Related to STAR Methods
Table S2. L1000 Probes, Related to Figure 1
Table S3. Inferred Genes and Accuracy, Related to STAR Methods
Table S4. CMap-L1000v1 Content, Related to Figure 2
Table S5. External Query Results, Related to STAR Methods
Table S6. AUC Results, Related to Figure 4
Table S7. PCL Statistics, Related to Figure 4
Highlights.
A new gene expression profiling method, L1000, dramatically lowers cost
The Connectivity Map now includes 1.3 million publicly accessible L1000 profiles
Facilitates discovery of small-molecule mechanism and annotation of genetic variants
The work establishes feasibility and utility of a truly comprehensive Connectivity Map
Acknowledgments
We thank Broad Compound Management and Genetic Perturbation Platform, Harvard Medical School LINCS Center, LINCS collaborators, P. Tamayo, J. Bradner and G. Shapiro for helpful discussions. We thank Luminex Corporation for support with the FlexMap 3D system, and Qiagen for assistance with TurboCapture kits. Supported in part by NIH grants 5U54HG006093 (T.R.G. and A.S.), U54HG008699 (T.R.G. and A.S.) 5U01HG008699 (T.R.G. and A.S), CA009172 (S.M.C.), TR001100 (S.M.C.), and the Conquer Cancer Foundation of ASCO Young Investigator Award (S.M.C.). J.L and W.R.B are shareholders and employees of Genometry, Inc. A.S, R.N, D.D.P, and X.L are shareholders of Genometry, Inc.
Footnotes
Author Contributions: Conceptualization, A.S., R.N., S.M.C., D.D.P., T.E.N., J.L., and T.R.G.; Methodology, A.S., R.N., S.M.C., D.D.P., T.E.N., D.W., I.S., L.H., N.S.G., W.R., J.G.D., and T.R.G.; Software, R.N., T.E.N., J.G., J.K.A., D.L.L., M.K., C.T., and C.F.; Validation, R.N., S.M.C., D.D.P., T.E.N., X.L., and J.F.D.; Formal analysis, A.S., R.N., S.M.C., T.E.N., J.G., D.W., I.S., M.O., O.M.E., C.F., L.H., R.H., P.G., and J.A.B.; Investigation, S.M.C., D.D.P., X.L., J.F.D., M.D., B.J., D.N., M.B., F.P., A.H.B., D.D, S.A.J., N.J.L., X.W., W.Z., W.R., and X.W.; Resources, F.P., A.H.B., A.S., A.N.B., A.V., D.T., K.A., N.S.G., P.A.C., S.S., X.W., W.Z., S.J.H., L.V.R., J.S.B., S.L.S., J.A.B., and D.E.R.; Data Curation, R.N., S.M.C., T.E.N., J.E.H., Z.L., A.L., and J.A.B.; Writing – Original draft, A.S., R.N., S.M.C., D.D.P., T.E.N., J.E.H., J.A.B., B.W., and T.R.G.; Writing – Review & Editing, A.S., R.N., S.M.C., T.E.N., J.E.H., A.H.B., A.S., D.T., J.L., P.A.C., X.W., S.J.H., L.V.R., J.G.D., J.A.B., D.E.R., B.W., and T.R.G.; Visualization, S.M.C., A.A.T., M.K., and B.W.; Supervision, A.S., D.D.P., J.G.D., B.W., and T.R.G.; Project Administration, A.S., J.R., and T.R.G.; Funding Acquisition, A.S., and T.R.G.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M, Hogstrom L, et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. 2016;30:214–228. doi: 10.1016/j.ccell.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum AM, van de Peppel J, van der Leije CS, Schreuders-Koedam M, Eijken M, van der Eerden BCJ, van Leeuwen JPTM. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc Natl Acad Sci U S A. 2015;112:12711–12716. doi: 10.1073/pnas.1501597112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlino MS, Gowrishankar K, Saunders CAB, Pupo GM, Snoyman S, Zhang XD, Saw R, Becker TM, Kefford RF, Long GV, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12:1332–1342. doi: 10.1158/1535-7163.MCT-13-0011. [DOI] [PubMed] [Google Scholar]
- Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, Payne-Turner D, Althoff MJ, Song G, Chen SC, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell. 2015;28:343–356. doi: 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Dinwiddie MT, Terry PD, Chen J. Recent evidence regarding triclosan and cancer risk. Int J Environ Res Public Health. 2014;11:2209–2217. doi: 10.3390/ijerph110202209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016;167:1853–1866e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner Y, Feng T, Benoist C, Koller D. Imputing gene expression from selectively reduced probe sets. Nat Methods. 2012;9:1120–1125. doi: 10.1038/nmeth.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational Repositioning of the Anticonvulsant Topiramate for Inflammatory Bowel Disease. Sci Transl Med. 2011;3:96ra76–ra96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, Bullard SA, Dierdorff JM, Adams CM. Systems-based Discovery of Tomatidine as a Natural Small Molecule Inhibitor of Skeletal Muscle Atrophy. J Biol Chem. 2014;289:14913–14924. doi: 10.1074/jbc.M114.556241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A. p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet. 2009;18:4035–4045. doi: 10.1093/hmg/ddp352. [DOI] [PubMed] [Google Scholar]
- Fortney K, Griesman J, Kotlyar M, Pastrello C, Angeli M, Sound-Tsao M, Jurisica I. Prioritizing Therapeutics for Lung Cancer: An Integrative Meta-analysis of Cancer Gene Signatures and Chemogenomic Data. PLoS Comput Biol. 2015;11:e1004068–17. doi: 10.1371/journal.pcbi.1004068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, Walter V, Zheng N, Hayes DN, Major MB. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014;74:808–817. doi: 10.1158/0008-5472.CAN-13-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai HY, He Y, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Illumina Inc. BeadStudio Normalization Algorithms for Gene Expression Data. 2007 Illumina Technical Bulletin Pub. No.470-2007-005. [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Järås M, Miller PG, Chu LP, Puram RV, Fink EC, Schneider RK, Al-Shahrour F, Peña P, Breyfogle LJ, Hartwell KA, et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J Exp Med. 2014;211:605–612. doi: 10.1084/jem.20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lantermann AB, Chen D, McCutcheon K, Hoffman G, Frias E, Ruddy D, Rakiec D, Korn J, McAllister G, Stegmeier F, et al. Inhibition of Casein Kinase 1 Alpha Prevents Acquired Drug Resistance to Erlotinib in EGFR-Mutant Non-Small Cell Lung Cancer. Cancer Res. 2015;75:4937–4948. doi: 10.1158/0008-5472.CAN-15-1113. [DOI] [PubMed] [Google Scholar]
- Litichevskiy L, Peckner R, Abelin JG, Asiedu JK, Creech AL, Davis JF, Davison D, Dunning CM, Egertson JD, Egri S, et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. 2017 doi: 10.1016/j.cels.2018.03.012. bioRxiv https://doi.org/10.1101/185918. [DOI] [PMC free article] [PubMed]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli G, Bosotti R, Ciomei M, Brasca MG, Calogero R, Mercurio C, Fiorentini F, Bertolotti M, Scacheri E, Scaburri A, et al. Transcriptional Analysis of an E2F Gene Signature as a Biomarker of Activity of the Cyclin-Dependent Kinase Inhibitor PHA-793887 in Tumor and Skin Biopsies from a Phase I Clinical Study. Mol Cancer Ther. 2010;9:1265–1273. doi: 10.1158/1535-7163.MCT-09-1163. [DOI] [PubMed] [Google Scholar]
- Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- Maaten L van der, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- Massard C, Soria JC, Anthoney DA, Proctor A, Scaburri A, Pacciarini MA, Laffranchi B, Pellizzoni C, Kroemer G, Armand JP, et al. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle. 2011;10:963–970. doi: 10.4161/cc.10.6.15075. [DOI] [PubMed] [Google Scholar]
- Muthuswami M, Ramesh V, Banerjee S, Viveka Thangaraj S, Periasamy J, Bhaskar Rao D, Barnabas GD, Raghavan S, Ganesan K. Breast Tumors with Elevated Expression of 1q Candidate Genes Confer Poor Clinical Outcome and Sensitivity to Ras/PI3K Inhibition. PLoS One. 2013;8:e77553–16. doi: 10.1371/journal.pone.0077553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J. A method for high-throughput gene expression signature analysis. Genome Biol. 2006;7:R61. doi: 10.1186/gb-2006-7-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohban MH, Singh S, Wu X, Berthet JB, Bray MA, Shrestha Y, Varelas X, Boehm JS, Carpenter AE. Systematic morphological profiling of human gene and allele function via Cell Painting. Elife. 2017;6 doi: 10.7554/eLife.24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth JM, Mays DJ, Pino MF, Tang LJ, Pietenpol JA. A Gene Signature-Based Approach Identifies mTOR as a Regulator of p73. Mol Cell Biol. 2008;28:5951–5964. doi: 10.1128/MCB.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovedo MA, Krett NL, Rosen ST. Inhibition of glycogen synthase kinase-3 increases the cytotoxicity of enzastaurin. J Invest Dermatol. 2011;131:1442–1449. doi: 10.1038/jid.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-ya K, Tsuruo T, Tomida A. Chemical Genomics Identifies the Unfolded Protein Response as a Target for Selective Cancer Cell Killing during Glucose Deprivation. Cancer Res. 2009;69:4225–4234. doi: 10.1158/0008-5472.CAN-08-2689. [DOI] [PubMed] [Google Scholar]
- Schneider RK, Ademà V, Heckl D, Järås M, Mallo M, Lord AM, Chu LP, McConkey ME, Kramann R, Mullally A, et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell. 2014;26:509–520. doi: 10.1016/j.ccr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell SA, Ambesi-Impiombato A, Sanchez-Martin M, Belver L, Xu L, Qin Y, Kageyama R, Ferrando AA. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood. 2015;125:2806–2814. doi: 10.1182/blood-2014-10-608448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, Brudz S, Sullivan JP, Muhlich J, Serrano M, et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008;36:D351–D359. doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AR, Joshi S, Zulcic M, Alcaraz M, Garlich JR, Morales GA, Cho YJ, Bao L, Levy ML, Newbury R, et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS One. 2016;11:e0150836–22. doi: 10.1371/journal.pone.0150836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I, Greenside P, Wadden D, Tirosh I, Natoli T, Narayan R, Root DE, Golub TR, Subramanian A, Doench JG. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. 2017 doi: 10.1371/journal.pbio.2003213. bioRxiv https://doi.org/10.1101/147504. [DOI] [PMC free article] [PubMed]
- Stockwell SR, Platt G, Barrie SE, Zoumpoulidou G, te Poele RH, Aherne GW, Wilson SC, Sheldrake P, McDonald E, Venet M, et al. Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS One. 2012;7:e28568–16. doi: 10.1371/journal.pone.0028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Wong WH. Tight clustering: a resampling-based approach for identifying stable and tight patterns in data. Biometrics. 2005;61:10–16. doi: 10.1111/j.0006-341X.2005.031032.x. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–576e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Ye Y, Yang X, Liao H, Zhao C, Liang S. Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma. PLoS One. 2011;6:e14573–e14577. doi: 10.1371/journal.pone.0014573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, Pandiella A, Arteaga CL. Transforming Growth Factor Engages TACE and ErbB3 To Activate Phosphatidylinositol-3 Kinase/Akt in ErbB2-Overexpressing Breast Cancer and Desensitizes Cells to Trastuzumab. Mol Cell Biol. 2008;28:5605–5620. doi: 10.1128/MCB.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer MJ, Li K, Gustafsdottir SM, Ljosa V, Bodycombe NE, Marton MA, Sokolnicki KL, Bray MA, Kemp MM, Winchester E, et al. Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc Natl Acad Sci U S A. 2014;111:10911–10916. doi: 10.1073/pnas.1410933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueh MF, Taniguchi K, Chen S, Evans RM, Hammock BD, Karin M, Tukey RH. The commonly used antimicrobial additive triclosan is a liver tumor promoter. Proc Natl Acad Sci U S A. 2014;111:17200–17205. doi: 10.1073/pnas.1419119111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Luo H, Xi Z, Rogaeva E. Drug Repositioning for Diabetes Based on “Omics” Data Mining. PLoS One. 2015;10:e0126082–13. doi: 10.1371/journal.pone.0126082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Properties and technical validation of Landmark genes, Related to Figure 1, A. Evaluating dimensionality reduction. Simulation showing the mean percentage (+/- SEM) of 33 benchmark connections recovered from an inferred CMap pilot dataset as a function of number of landmark genes used to train the inference model, indicating that around 1000 landmarks were sufficient to recover 82% of expected connections
B. Landmark gene expression. Distributions of baseline expression of landmark genes across the 30 tissue types present in dataset DSGTEx-RNA-seq. The majority of landmark genes are expressed in each tissue and the distributions of expression are similar across tissues, suggesting that landmarks are not over-optimized to a particular cell context.
C. Technical replicate reproducibility. Top: Scatter plots of 3 technical replicates for a sample of commercial MCF7 RNA. Bottom: Distributions of median Spearman correlations between technical replicates of commercial RNA samples from 6 cell lines (36 replicates per cell line). Nearly all correlations exceed 0.90, suggesting low sample-to-sample variability.
D. Comparison with RNA-seq. Scatter plot of recall (R) vs. cross-platform Spearman correlation for 3,176 patient-derived samples in the space of of 970 landmark genes profiled on RNA-seq and L1000. 3,103/3,176 samples (98%) had an R > 0.99 (indicating that their cross-platform self-correlations exceeded 99 percent of background different-sample correlations, see STAR Methods) and all but 5 samples (99.84%) had an R > 0.95, suggesting high similarity between the two platforms.
Figure S2. Characteristics of perturbational signatures, Related to STAR Methods, A. MDM2 inhibitor AMG-232 shows TP53-dependent transcriptional response. Left: Correlation matrix of AMG-232's signatures derived from isogenic MCF10A breast epithelial cell lines that were either TP53 wild-type or homozygously deleted at 6-point dose [0.04-10 uM range]. Signatures from a background of TP53homozygous deletion lose correlation with signatures from TP53 wild type background. Right: TAS of AMG-232signature is notably lower in the presence of TP53 homozygous deletion compared to wild type.
B. Cell context and transcriptional activity. The bars show the percentage of 2,429 small molecule compounds that produced high TAS signatures (defined as TAS ≥ 0.2) in a specified number of cell lines. The line represents cumulative percentages and indicates that 92% of compounds produce a high-TAS signature in at least one cell line.
C. Multiplicity of signatures. The 2,429 drugs profiled in the Touchstone dataset have differing degrees of transcriptional activities across the panel of 9 core cell lines. The majority of the compounds are active in 3 or more cell lines with 364 (15%) producing a single concordant signature per molecule and 1,045 (43%) giving rise to multiple signatures, suggesting that transcriptional response is dependent on cell context. Only 194 (8%) of the compounds gave no measurable response in any of the cell lines tested.
D. Context-specific interconnectivity. The bar plot shows the baseline expression of the target gene across the9 core cell lines. The thumbnail heatmaps show the inter-connectivity of members of the compound class that targets the indicated gene. Top: NR3C1 / glucocorticoid receptor antagonists (n=44). Bottom: PPARG / PPARG agonists (n=16). In both cases, the PCL interconnectivity is highest in the cell lines in which the target is most highly expressed.
E. Context-dependent connection between compounds and their gene targets. The raster plot shows the connectivity between the indicated gene knockdown and its targeting compounds, where each circle is a cell line. Filled circles indicate cell lines in which the compound-gene connection is expected, either due to mutation in (BRAF, MAPK genes) or high expression of (all others) the target gene. The strong connections are enriched for the expected cell lines, suggesting that CMap connectivities can be indicative of context specificity.
Figure S3. Experimental follow-up of CMap hypotheses and functional annotation of allelic variants, Related to Figure 4, A. Confirmation of predicted bromodomain inhibitors. TREEspot selectivity profile indicating the inhibitory activity of droxinostat, BRD-1103, and XMD11-85H against the 32 bromodomains tested by the BROMOmax assay. All three compounds were active against at least one bromodomain. Images generated using TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation.
B. Confirmation of predicted PI3K/MTOR inhibitors. Chemical structures and affinities (kD) between three predicted PI3K/MTOR inhibitors and AKT1, MTOR, and 4 PI3K isoforms, as measured by the Kinomescan assay. All three compounds show at least micromolar affinity for a subset of the predicted targets, with two compounds showing nanomolar affinities.
C. CMap connectivities correspond to loss of function mutations. Left: Heat map showing the connectivity between the over-expression signatures of various KEAP1 alleles to the knockdown of NFE2L2 in A549 cells. Right: Heat map showing the connectivity between the over-expression signatures of various PTEN alleles and the PI3K and MTOR inhibitor classes. Cell line is indicated by the colored bar below the heat map. For bothKEAP1 and PTEN, mutations at known active residues important for protein function result in loss of expected connectivity.
Table S1. Queries, Related to STAR Methods
Table S2. L1000 Probes, Related to Figure 1
Table S3. Inferred Genes and Accuracy, Related to STAR Methods
Table S4. CMap-L1000v1 Content, Related to Figure 2
Table S5. External Query Results, Related to STAR Methods
Table S6. AUC Results, Related to Figure 4
Table S7. PCL Statistics, Related to Figure 4