

Abstract
Alpha-Synuclein (α-Syn) is an important protein in the pathogenesis of Parkinson disease (PD) as it accumulates as fibrillar inclusions in affected brain regions including dopaminergic neurons in the substantia nigra. Elevated levels of α-Syn seem to be crucial in mediating its toxicity. Thus, detailed information regarding the regulatory mechanism of α-Syn expression in several layers such as transcription, post-transcription and post-translation is needed in order to devise therapeutic interventions for PD. Previously, we reported that expression of α-Syn is repressed by microRNA-7 (miR-7) through its effect on the 3′-untranslated region (UTR) of α-Syn mRNA. Here, we show that miR-7 also accelerates the clearance of α-Syn and its aggregates by promoting autophagy in differentiated ReNcell VM cells. Further, miR-7 facilitates the degradation of pre-formed fibrils of α-Syn transported from outside the cells. This additional mechanism for reducing α-Syn levels show miR-7 to be an important molecular target for PD and other alpha-synucleinopathies.
Keywords: alpha-Synuclein, Autophagy, MicroRNA-7, Parkinson disease
Introduction
Parkinson disease (PD) is a common neurodegenerative disorder that affects 2–3% of the population over age 65 [21]. It is clinically characterized by disabling motor abnormalities including bradykinesia, tremor, rigidity, and poor balance. These impairments result from the progressive loss of dopaminergic neurons in the substantia nigra pars compacta. Loss of dopaminergic neurons in the substantia nigra and the intracellular inclusions containing aggregated forms of alpha-Synuclein (α-Syn) are the neuropathological hallmarks of PD. Although inherited forms of PD only represent 5–10% of all cases, investigations on these have provided clues to the PD pathogenesis. To date, more than ten distinct genes including α-Syn, parkin, dj-1, pink1 and lrrk2 have been identified to cause PD when they are mutated [28]. Among these, α-Syn is thought to be a crucial player in the pathogenesis of PD based on genetic, pathological, and cellular/molecular lines of evidence [8, 16, 19]. In addition to point mutations linked to dominantly inherited forms of PD [15, 22, 33], growing evidence suggests that high levels of α-Syn are harmful to dopaminergic neurons [5, 17]. Therefore, strategies that downregulate α-Syn levels or its aggregates are attractive targets for therapeutic intervention. Notably, microRNAs (miRs) have been recognized as critical post-transcriptional regulators of gene expression with vital roles in various cellular pathways. miRs are a class of endogenous 21–25 base-long single-stranded RNAs, which inhibit gene expression by binding to their target sequences mostly in the 3′ untranslated region (UTR) of mRNAs [10]. Growing evidence has suggested that miR dysfunction contributes to the pathogenesis of neurodegenerative disorders including PD [12]. Previously, we and others reported that expression of α-Syn is repressed by miR-7 through its effect on the 3′-UTR of α-Syn mRNA [7, 11, 24].
Autophagy is responsible for eliminating aggregated proteins via their sequestration into double-membrane vesicles called autophagosomes and their subsequent degradation by lysosomal proteases after autophagosome/lysosome fusion [23, 25]. Further, it is known that α-Syn aggregates are degraded by autophagy [31]. Autophagy is a crucial cellular process and impairment of autophagy can lead to neurodegenerative disorders including PD [1, 2, 30]. The capacity of autophagy in removing toxic protein aggregates offers strategies for future drug development for neurodegenerative disorders characterized by protein misfolding and aggregation [18, 26]. Here, we report that miR-7 accelerates the clearance of α-Syn and its aggregates by promoting autophagy, in addition to repressing α-Syn expression through acting on the 3′-UTR.
Materials and Methods
Cell culture and transfection
Human neural progenitor cell line ReNcell VM (EMD Millipore) were cultured in DMEM: F12 growth medium supplemented with 1X B27 (Life Technologies), 1X glutamax (Gibco), 10 U/ml heparin (Sigma-Aldrich), 50 μg/ml Gentamycin (Gibco), 20 ng/ml basic fibroblast growth factor 2 (bFGF-2, Sigma-Aldrich) and 20 ng/ml epidermal growth factor (EGF, Sigma-Aldrich). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2. ReNcell VM can be differentiated into neuron-like cells by two different protocols. For standard differentiation (std), cells were seeded at 30,000 cells/cm2 in a laminin-coated plate and cultured to confluency in growth medium over a 3–4 day period. Differentiation was initiated by omitting growth factors. Alternatively, ReNcell VM cells were differentiated into dopaminergic neurons by the pre-aggregation differentiation (pre-agg) protocol requiring dibutyryl-cAMP and GDNF [6]. Transfections were performed using Lipofectamine RNAiMax (Invitrogen) with pre-miR-SC (scrambled) (Ambion), pre-miR-7 (Ambion).
Viral vectors and transduction
HEK293T cells were used for generation of lentivirus as reported previously [4]. Lentiviral vector pLemiR (Open Biosystems, GE healthcare) containing human pri-miR-7–2 gene as well as red fluorescent protein (RFP) gene as a bicistronic transcript was used for generating lenti-miR-7 virus. Concentrated virus (1 × 1010 TU/ml) was used to transduce differentiated ReNcell VM cells at a multiplicity of infection (MOI) of 10. Adeno-associated virus 2 (AAV2) containing α-Syn gene (1.5 × 1013 viral genomes (vg)/ml) or eGFP gene (8.1 × 1012 vg/ml) were obtained from the vector core of University of North Carolina. These concentrated AAV2 virus were used to transduce differentiated ReNcell VM cells at an MOI of 1 × 105 vg per cell.
Western blot analysis
Cells were rinsed with ice-cold phosphate-buffered saline (PBS) and lysed in PBS containing 2% sodium dodecyl sulfate (SDS) with protease cocktail and phosphatase inhibitors (Roche). Cell lysates were sonicated for 30 sec and protein concentrations were quantified using BCA Protein Assay Reagent (Thermo Scientific). Cell lysates were analyzed by Western blotting as reported previously [13] using anti-α-Syn (BD Biosciences, Cat# 610786), anti-GFP (Santa Cruz Biotech., Cat# sc-8334), anti-LC3B (Cell Signaling, Cat# 4108), and anti-β-actin (Sigma-Aldrich, Cat# A5316), followed by incubation with horseradish peroxidase-conjugated anti-mouse antibody or anti-rabbit antibody. Band intensity was measured using Image J (NIH).
Immunocytochemistry
To stain α-Syn, cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 5% donkey serum in PBS. Cells were then incubated with anti-α-Syn (SYN-1, BD Biosciences, Cat# 610786) diluted in PBS containing 1% bovine serum albumin (BSA), overnight at 4°C. After washing with PBS, cells were incubated with Alexa Fluor 488-conjugated anti-mouse IgG (Jackson Immunoresearch) diluted in PBS containing 1% BSA for 1h. For nuclear staining, cells were incubated with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) in PBS for 1 min. After washing with PBS, cells were analyzed under a fluorescence microscope (Axiovert 2000, Carl Zeiss). The number of cells containing α-Syn aggregate(s) were counted in 7 different microscopic fields containing 10–20 lentivirus-transduced cells. To stain LC3 puncta, cells transfected with pre-miRs for 48h were fixed and permeabilized, and incubated with anti-LC3B (Cell Signaling Technology, Cat# 4108) overnight at 4°C. Subsequently, cells were incubated with rhodamine red-conjugated anti-rabbit IgG. The number of cells containing LC3 puncta were counted in 7 different microscopic fields including 10–20 cells, and the percentage of cells containing LC3 puncta was calculated. In addition, the number of LC3 puncta in a cell was counted from 20 LC3 puncta-containing cells.
Preparation of pre-formed fibril (pff) of α-Syn
Full-length α-Syn cDNA was cloned into pGEX-4T-1 (GE Healthcare) as a GST fusion. GST-α-Syn recombinant protein was purified using glutathione Sepharose column, and α-Syn was isolated after cleaving between GST and α-Syn by thrombin. α-Syn pff was prepared as previously reported [29].
Statistical analysis
Quantitative data are presented as means ± SEM or means ± SD and statistical significance between two groups was analyzed by student’s t-test. Level of significance was set at p < 0.05.
Results
We previously reported that miR-7, which is highly expressed in the tyrosine hydroxylase (TH)-positive nigral neurons, represses α-Syn expression by targeting the 3′-UTR of its mRNA [11]. Because strategies targeting the removal of preformed α-Syn aggregates are thought to be viable options in PD therapeutics, we sought to investigate whether miR-7 facilitates the degradation of preformed α-Syn aggregates. For this, we employed a human neural progenitor cell line, ReNcell VM, which can be differentiated into neuron-like cells [3, 4, 6]. Cells can be differentiated by two different protocols, but we chose the standard differentiation method because endogenous α-Syn expression was negligible (Fig. 1A), where the majority of α-Syn was expressed from the transduction of AAV2-α-Syn virus. After 7 days of differentiation, cells were infected with AAV2-α-Syn, and followed by the transduction of lentivirus encoding miR-7 or scramble sequence (SC) (Fig. 1B). We found that overexpression of miR-7 in differentiated ReNcell VM cells using lenti-miR-7 virus (Dharmacon) resulted in a dramatic decrease in the level of monomeric and high molecular weight (HMW) forms of α-Syn expressed from AAV2-α-Syn (Fig. 1C). In contrast, miR-7 overexpression was not able to decrease the level of eGFP, which is expressed from AAV2-eGFP, the same vector as AAV2-α-Syn (Fig. 1C). This result suggests that the decrease of α-Syn upon miR-7 overexpression is not due to the inhibition of de novo expression of α-Syn. As we utilized AAV2-α-Syn that lacks the 3′-UTR in this experiment, we also suggest that overexpression of miR-7 can decrease α-Syn levels without targeting the 3′-UTR of the α-Syn mRNA. In particular, immunocytochemistry studies showed that α-Syn aggregates are significantly reduced by overexpression of miR-7 in differentiated ReNcell VM cells (Fig. 2A). While a 64 ± 5.2% (mean ± SEM) of cells contained α-Syn inclusion(s) as indicated by arrowheads in control cells (lenti-miR-SC), overexpression of miR-7 significantly reduced the percentage of cells having aggregates to 28 ± 4.6% (Fig. 2B). As autophagy is known to clear the protein aggregates [23, 25], we tempted to determine whether miR-7 overexpression leads to the degradation of α-Syn and its aggregates by promoting autophagy. We found that the decrease of α-Syn is prevented by the addition of lysosome inhibitors, NH4Cl/leupeptin (AC/LP) (Fig. 3A). Further, miR-7 expression increased the conversion of LC3-I to LC3-II (lower band of LC3B), which indicates increased autophagosome formation (Fig. 3B). Addition of lysosome inhibitors further increased the level of LC3-II, suggesting that the increase of LC3-II upon miR-7 overexpression is due to an increase of autophagy influx. In addition, we found that transfection of miR-7 significantly increased both the percentage of cells having LC3 puncta and the number of LC3 puncta in a cell compared to miR-SC-transfected cells (Fig. 3C and 3D). Therefore, these results suggest that miR-7 promotes the clearance of α-Syn and its aggregates by promoting autophagy. Next, we investigated whether overexpression of miR-7 facilitates the degradation of α-Syn aggregates transported from outside the cell. Exposure of preformed-fibril (pff) of α-Syn to differentiated ReNcell VN cells generates intracellular a-Syn aggregates, and these α-Syn aggregates are significantly reduced by overexpression of miR-7 (Fig. 4A). While a 33 ± 5% (mean ± SEM) of cells contained α-Syn inclusion(s) as indicated by arrowheads in control cells (lenti-miR-SC), overexpression of miR-7 significantly reduced the percentage of cells having aggregates to 17 ± 1% (Fig. 4B). In addition, Western blot analysis showed that exposure of α-Syn pff to differentiated ReNcell VN cells generates HMW as well as monomeric α-Syn, and the levels of these forms of α-Syn were also decreased by overexpression of miR-7 (Fig. 4C). Further, this decrease of α-Syn is prevented by the addition of NH4Cl/leupeptin, but not by lactacystin, a proteasome inhibitor, corroborating our conclusion that miR-7 overexpression facilitates the degradation of α-Syn/its aggregates by promoting autophagy.
Fig. 1.
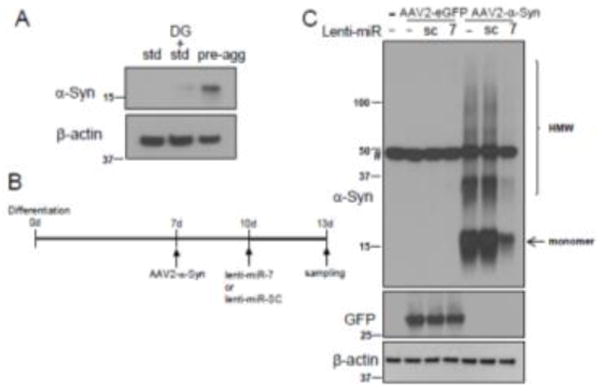
miR-7 decreases the levels of monomeric and HMW forms of α-Syn. (A) α-Syn expression is negligible in the neuron-like cells differentiated from ReNcell VM cells with standard protocol. ReNcell VM cells were differentiated for 13 days as indicated, and then these cells were subjected to Western blot analyses to detect the expression of α-Syn, and β-actin. Std, pre-agg and DG denote standard differentiation, preaggregation differentiation and dibutyryl-cAMP/GDNF, respectively. (B) Time-course showing differentiation and viral transductions. (C) Cells transduced as indicated were subjected to Western blot analyses to detect the expression of α-Syn, GFP and β-actin. HMW indicates high molecular weight complex. # denotes the nonspecific band. These results are representative of three separate experiments.
Fig. 2.
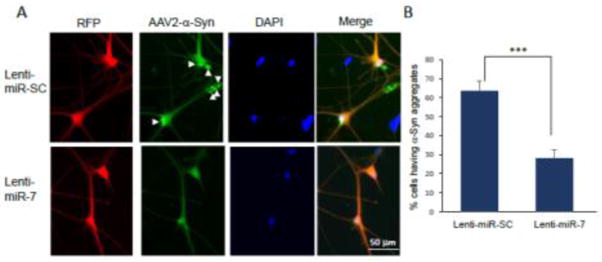
Immunocytochemistry showing the decrease of α-Syn aggregates in differentiated ReNcell VM cells with miR-7 overexpression. (A) A representative figure showing miR-7-induced decrease of α-Syn aggregates. Lentivirus-infected cells are detected as red fluorescence and α-Syn expression is detected as green fluorescence. α-Syn aggregates are indicated as white arrowheads. Scale bar = 50 μm. (B) Cells having α-Syn aggregate(s) were counted in 7 randomly-selected fields comprising 10–20 RFP-positive cells (lentivirus-transduced cells). The data represent means ± SEM. *** p < 0.001. These results are representative of three separate experiments.
Fig. 3.
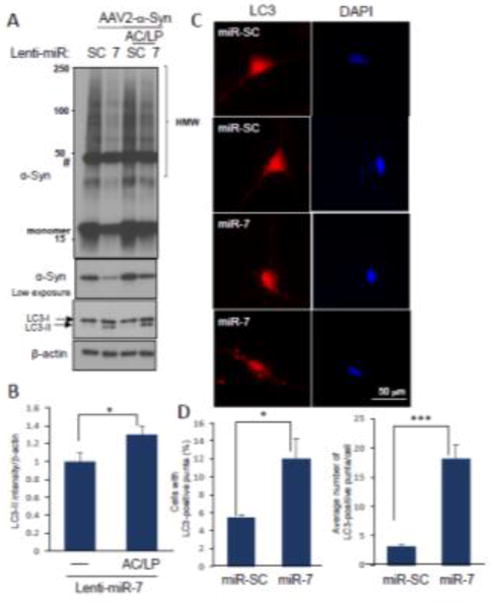
miR-7 decreases the levels of α-Syn and its aggregates through promoting autophagy. (A) Differentiated ReNcell VM cells transduced as indicated were further treated with AC (NH4Cl (10 mM)) and LP (leupeptin (100 μM)) for 12 h. Cells were subjected to Western blot analyses to detect α-Syn, LC3B and β-actin. (B) The LC3B-II band is used to measure autophagy flux. Intensity of the LC3B-II band was normalized to that of the β-actin band. The data represent means ± SD. * p < 0.05. (C) Representative figures showing that transfection of pre-miR-7 leads to an increase in the formation of LC3 puncta (red color). Two pictures are provided for each experimental group. (D) Quantitative data showing the percentage of cells having LC3 puncta and the number of LC3 puncta in a cell. The number of cells containing LC3 puncta were counted in 7 different microscopic fields including 10–20 cells for each sample and the percentage of cells having LC3 puncta was calculated. The number of LC3 puncta in a cell was counted with 20 cells containing LC3 puncta. The data represent means ± SEM. * p < 0.05. *** p < 0.001. These results are representative of three separate experiments.
Fig. 4.
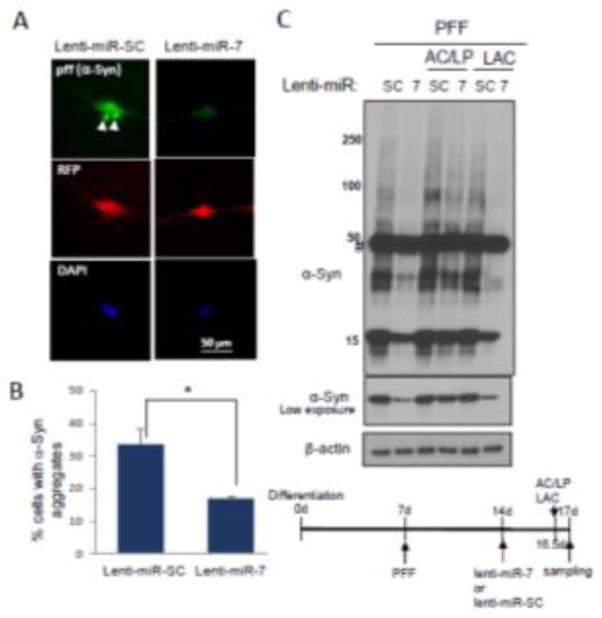
miR-7 decreases the levels of intracellular α-Syn transported from the outside as pff. Differentiated ReNcell VM cells were treated with pff (100ng). After 7 days, these cells were transduced with either lenti-miR-SC or lenti-miR-7 for another 3 days. (A) Representative figures showing miR-7-induced decrease of α-Syn aggregates (pff). Lentivirus-infected cells are detected as red fluorescence and α-Syn expression is shown as green fluorescence. α-Syn aggregates are indicated as white arrowheads. (B) Cells having α-Syn aggregate(s) were counted in 7 randomly selected fields comprising 10–20 RFP-positive cells (lentivirus-transduced cells). The data represent means ± SEM. * p < 0.01. (C) During the period of lentiviral transduction, cells were further treated with AC (NH4Cl (10 mM)), LP (leupeptin (100 μM)) or LAC (lactacystin (5 μM)) for 12 h before harvest. Cells were subjected to Western blot analyses to detect α-Syn and β-actin. Time-course showing differentiation, PFF exposure, viral transductions and chemical treatments is presented. These results are representative of three separate experiments.
Discussion
In the present study, we have found that overexpression of miR-7 leads to an increased degradation of α-Syn and its aggregates by promoting autophagy. In our experimental conditions, human α-Syn was overexpressed by AAV2-α-Syn infection or by pff introduction in differentiated ReNcell VM cells. In both conditions, overexpression of miR-7 significantly reduced the level of α-Syn and its aggregates, as shown by immunocytochemistry and Western blot analyses. In addition to finding that miR-7 increases the formation of LC3 puncta, the observation that the miR-7-mediated decrease in α-Syn expression was prevented by lysosome inhibitors, supports our conclusion.
We previously reported that miR-7 represses α-Syn expression by targeting the 3′-UTR of its mRNA. Therefore, we believe that miR-7 could decrease the level of α-Syn by two mechanisms: one is by repressing α-Syn expression by targeting the 3′-UTR of its mRNA, and the other is by facilitating the degradation of α-Syn protein/aggregates by promoting autophagy. This latter effect appears to be particularly important in clearing the α-Syn aggregates transported from outside the cell and in preventing further seeding and intracellular aggregation formation, thereby protecting against aggregate propagation. These two effects on lowering a-Syn level demonstrate miR-7 to be an excellent molecular target for potential disease-modifying therapeutics.
Although the precise mechanism behind our observation is currently unknown, we suggest that it could involve the miR-7-mediated reduction of negative factors for autophagy. Among the reported targets of miR-7, epidermal growth factor receptor (EGFR), an oncogenic receptor tyrosine kinase, reportedly regulates autophagy [32]. Active EGFR inhibits the autophagy pathway by inhibiting Beclin1 activity by catalyzing tyrosine phosphorylation. Thus, it is possible to speculate that miR-7-mediated repression of EGFR expression results in increased autophagy, and subsequently in the degradation of α-Syn/its aggregates. Specifically, Gefitinib (trade name Iressa), a chemical inhibitor of EGFR tyrosine kinase [27], can be tested for its ability to facilitate the degradation of α-Syn/aggregates in the cellular system. Gefitinib is a drug used for certain breast and lung cancers [27]. Similarly, c-Abl tyrosine kinase inhibitor, Nilotinib, a drug for acute myelogenous leukemia, was shown to protect cells from DA neuron death in MPTP-intoxicated mouse model [14], and has further shown early promise in patients with Parkinson’s disease with dementia and Lewy body dementia [20]. In addition, it was reported that mTOR mRNA is targeted by miR-7 [9]. Inhibition of mTOR activity is well known to promote autophagy, thus it is possible to speculate that miR-7-mediated decreases in mTOR expression leads to stimulation of autophagy and subsequently increased degradation of α-Syn. Successful identification of miR-7 targets involved in promoting autophagy might provide a clue into therapeutic avenues for pharmacological interventions of EGFR or mTOR using Gefinitib or Torin, respectively.
In summary, the present study provides evidence that miR-7 facilitates the degradation of α-Syn and its aggregates by promoting autophagy. In addition to repressing α-Syn expression by targeting the 3′-UTR of its mRNA, this effect of miR-7 on autophagy proves that miR-7 is an attractive target for therapeutic intervention for PD.
Highlights.
miR-7 decreases α-Syn expression independent of targeting 3′-UTR of its mRNA.
miR-7 facilitates the degradation of α-Syn and its aggregates by promoting autophagy.
miR-7 accelerates the degradation of pre-formed fibrils of α-Syn transported from outside the cells.
Acknowledgments
This work was supported by the National Institute of Health NS70898 (EJ) and partly NS095003 (EJ). Authors thank Aleta Murphy for critical reading of this manuscript.
Abbreviations
- α-Syn
alpha-Synuclein
- bFGF
basic fibroblast growth factor
- DMEM
Dulbecco’s modified Eagle’s medium
- EGF
epidermal growth factor
- miR-7
microRNA-7
- miR-SC
scrambled microRNA control
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson disease
- RFP
red fluorescent protein
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alegre-Abarrategui J, Wade-Martins R. Parkinson disease, LRRK2 and the endocytic-autophagic pathway. Autophagy. 2009;5:1208–1210. doi: 10.4161/auto.5.8.9894. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaudhuri AD, Kabaria S, Choi DC, Mouradian MM, Junn E. MicroRNA-7 Promotes Glycolysis to Protect against 1-Methyl-4-phenylpyridinium-induced Cell Death. J Biol Chem. 2015;290:12425–12434. doi: 10.1074/jbc.M114.625962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi DC, Chae YJ, Kabaria S, Chaudhuri AD, Jain MR, Li H, Mouradian MM, Junn E. MicroRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J Neurosci. 2014;34:12725–12737. doi: 10.1523/JNEUROSCI.0985-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eriksen JL, Dawson TM, Dickson DW, Petrucelli L. Caught in the act: alpha-synuclein is the culprit in Parkinson’s disease. Neuron. 2003;40:453–456. doi: 10.1016/s0896-6273(03)00684-6. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y, Xue JL, Shen Q, Chen J, Tian L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology. 2012;55:1852–1862. doi: 10.1002/hep.25576. [DOI] [PubMed] [Google Scholar]
- 10.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 11.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep. 2014;4:4874. doi: 10.1038/srep04874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 16.Lim KL, Dawson VL, Dawson TM. The cast of molecular characters in Parkinson’s disease: felons, conspirators, and suspects. Ann N Y Acad Sci. 2003;991:80–92. doi: 10.1111/j.1749-6632.2003.tb07465.x. [DOI] [PubMed] [Google Scholar]
- 17.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven CC. Genetic Epidemiology of Parkinson’s Disease, Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 18.Moors TE, Hoozemans JJ, Ingrassia A, Beccari T, Parnetti L, Chartier-Harlin MC, van de Berg WD. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol Neurodegener. 2017;12:11. doi: 10.1186/s13024-017-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mouradian MM. Recent advances in the genetics and pathogenesis of Parkinson disease. Neurology. 2002;58:179–185. doi: 10.1212/wnl.58.2.179. [DOI] [PubMed] [Google Scholar]
- 20.Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, Wilmarth BM, Howard H, Dunn C, Carlson A, Lawler A, Rogers SL, Falconer RA, Ahn J, Li Z, Moussa C. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J Parkinsons Dis. 2016;6:503–517. doi: 10.3233/JPD-160867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 22.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 23.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 24.Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nature communications. 2012;3:1084. doi: 10.1038/ncomms2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 26.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 27.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 28.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec No 2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 29.Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B, Abraham N, Gao G, Yang Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl Neurodegener. 2016;5:19. doi: 10.1186/s40035-016-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 32.Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–1284. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]