

Abstract
Aims
To test whether circulating neurotoxic autoantibodies increase in adult type 2 diabetes mellitus with Parkinson’s disease (PD) or dementia. To identify the G-protein coupled receptor on neuroblastoma cells mediating neural inhibitory effects in diabetic Parkinson’s disease plasma autoantibodies. To determine the mechanism of accelerated neuroblastoma cell death and acute neurite retraction induced by diabetic Parkinson’s disease and dementia autoantibodies.
Methods
Protein-A eluates from plasma of twelve older adult male diabetic patients having Parkinson’s disease (n=10) or dementia (n=2), and eight age-matched control diabetic patients were tested for ability to cause accelerated N2A neuroblastoma cell death and acute neurite retraction. Specific antagonists of G protein coupled receptors belonging to the G alpha q subfamily of heterotrimetric G-proteins, the phospholipase C/inositol triphosphate/Ca2+ pathway, or the RhoA/Rho kinase pathway were tested for ability to block diabetic Parkinson’s disease/dementia autoantibody-induced neurite retraction or N2A accelerated cell loss. Sequential Liposorber LA-15 dextran sulfate cellulose/protein-A affinity chromatography was used to obtain highly-purified fractions of diabetic Parkinson’s disease autoantibodies.
Results
Mean accelerated neuroblastoma cell loss induced by diabetic Parkinson’s disease or dementia autoantibodies significantly exceeded (P = 0.001) the level of N2A cell loss induced by an identical concentration of the diabetic autoantibodies in control patients without these two co-morbid neurodegenerative disorders. Co-incubation of diabetic Parkinson’s disease and dementia autoantibodies with two-hundred nanomolar concentrations of M100907, a highly selective 5-HT2AR antagonist, completely prevented autoantibody-induced accelerated N2A cell loss and neurite retraction. A higher concentration (500 nM-10μM) of alpha-1 adrenergic, angiotensin II type 1, or endothelin A receptor antagonists did not substantially inhibit autoantibody-induced neuroblastoma cell death or prevent neurite retraction. Antagonists of the inositol triphosphate receptor (2-APB, 50μM), the intracellular calcium chelator (BAPTA-AM, 30 μM) and Y27632 (10 μM), a selective RhoA/Rho kinase inhibitor, each completely blocked acute neurite retraction induced by sixty nanomolar concentrations of diabetic Parkinson’s disease autoantibodies. Co-incubation with 2-APB (1–2 μM) for 8 hours’ prevented autoantibody-induced N2A cell loss. The highly-purified fraction obtained after Liposorber LA/protein-A affinity chromatography in hypertriglyceridemic diabetic dementia and Parkinson’s disease plasmas had apparent MWs > 30 kD, and displayed enhanced N2A toxicity requiring substantially higher concentrations of 5-HT2AR antagonists (M100907, ketanserin, spiperone) to effectively neutralize.
Conclusion
These data suggest increased autoantibodies in older adult diabetes with Parkinson’s disease or dementia cause accelerated neuron loss via the 5-hydroxytryptamine 2 receptor coupled to inositol triphosphate receptor-mediated cytosolic Ca2+ release.
Keywords: 5-HT2A receptor, diabetes, autoantibodies, Parkinson’s disease, dementia
Introduction
Parkinson’s disease is a progressive movement disorder characterized by the selective loss of dopaminergic neurons in the substantia nigra region of the midbrain. Sporadic Parkinson’s disease (PD) which comprises the vast majority of cases is thought to arise through complex gene-environment interactions [1]. Although the motor symptoms in Parkinson’s disease (tremor, rigidity, bradykinesia, and postural instability) can be lessened by medications and surgery, the disease itself is incurable. Non-motor symptoms in PD (including altered sense of smell, depression, cognitive dysfunction, and constipation) may precede the onset of motor symptoms by years indicative of a systemic neurodegenerative disorder [2]. The current lack of specific biomarker(s), however, has hampered drug development targeted at an early stage in the neurodegenerative disease process.
In a recent population-based study, adult type 2 diabetes was associated with a substantially increased (1.85-fold) hazard ratio for incident Parkinson’s disease [3]. Yet the underlying mechanism for the association between type 2 diabetes and Parkinson’s disease is unknown. We previously reported increased plasma autoantibodies in adult type 2 diabetes suffering with co-morbid depression [4]. The autoantibodies caused accelerated N2A neuroblastoma cell death via activation of the phospholipase C/inositol triphosphate receptor/calcium (PLC/IP3R/Ca2+) signaling pathway [4]. Diabetic depression autoantibody neurotoxicity could be prevented (in vitro) by co-incubating cells with specific antagonists of the 5-HT2A receptor [4]. Since the 5HT-2AR is concentrated on neurons in specific brain regions (anterior olfactory nucleus, transentorhinal cortex, substantia nigra, brainstem, hippocampus, medial prefrontal cortex) [5] affected by PD- or Alzheimers’type- neurodegeneration [6], in the present study we tested a hypothesis that circulating agonist, 5-HT2A receptor autoantibodies increase (and may contribute to pathophysiology) in older adult type 2 diabetes suffering with co-morbid Parkinson’s disease and/or dementia.
Patients and Methods
Patients
All twenty-three patients provided informed consent for the Investigational Review Board-approved study and were enrolled consecutively from the Diabetes and Endocrinology clinics at Veterans Affairs New Jersey Healthcare System (VANJHCS) between August 2017 and March 2018. In order to be included in the study patients needed to have been previously evaluated and followed up in the Neurology clinics at the VANJHCS and have a clinical diagnosis of Parkinson’s disease with type 2 diabetes mellitus (n=10), dementia with type 2 diabetes (n=2), Parkinson’s disease with pre-diabetes (n=3), or type 2 diabetes without Parkinson’s disease or dementia (n=8). The baseline clinical characteristics in the twenty study participants having diabetes with or without Parkinson’s disease or dementia are shown in Table 1. Three age-matched patients with Parkinson’s disease, but lacking overt type 2 diabetes (not shown in Table 1) were enrolled at the end of the study to test for the presence of factors similar to those found in diabetic patients having Parkinson’s disease.
Table 1.
Baseline characteristics in study patients
| Parkinson’s disease, dementia (N =12) | Control (N =8) | P-value^,* | |
|---|---|---|---|
| Age (years) | 70.8 ±5.6 | 70.4 ± 7.5 | 0.96 |
| BMI (kg/m2) | 30.5 ± 3.8 | 40.1±6.4 | 0.002 |
| HbA1c (%) | 7.8 ± 1.4 | 8.7 ± 1.6 | 0.29 |
| Diabetes duration (years) | 15.8 ± 10.3 | 20.0 ± 9.4 | 0.44 |
| Nephropathy (yes/no) | (7/5) | (4/4) | 1 |
| Depression (yes/no) | (4/8) | (2/6) | 1 |
| Painful neuropathy (yes/no) | (5/7) | (1/7) | 0.33 |
| Atrial fibrillation (yes/no) | (3/9) | (4/4) | 0.36 |
| Cancer (yes/no) | (6/6) | (1/7) | 0.16 |
T-test for continuous variable;
Fischer’s exact test for dichotomous variables BMI- body mass index; HbA1c- glycosylated hemoglobin.
Subgroups
Parkinson’s Disease
All patients with a diagnosis of Parkinson’s disease had been evaluated by a movement disorder specialist in the Department of Neurology at the Veterans Affairs New Jersey Health Care System, East Orange, New Jersey. The clinical diagnosis of Parkinson’s disease was based on the presence of two of the classic motor symptoms (bradykinesia, tremor, rigidity, postural instability) and a positive response to L-dopa therapy. Eight of ten patients were at an early stage in the PD disease process, i.e. modified Hoehn and Yahr stages 1–2 [7]. All PD patients were ambulatory, but two of ten patients had experienced frequent falls secondary to postural instability. At least four of the ten PD patients had complained of memory difficulty, and one additional patient (Pt 4) without complaint of memory impairment had undergone CT imaging (after a fall) which demonstrated moderate global cerebral volume loss.
Depression And Other Co-Morbidities
All patients having a diagnosis of major depressive disorder were evaluated by psychiatry staff at the Veterans Affairs New Jersey Health Care System as previously reported [4]. Painful diabetic neuropathy is defined according to previously reported criteria [8]. Diabetic nephropathy is defined as persistent micro-or macro-albuminuria, urinary albumin excretion > 300 mg/g creatinine or urinary protein excretion > 500 mg/g creatinine.
Diabetic Dementia (DMT), Parkinson’s Disease Plus Dementia (PDD) Or PD
Patient 1
A 62-year-old Hispanic male with a thirty- year history of type 2 diabetes complicated by mild retinopathy, painful peripheral neuropathy, recurrent major depression, type 2b hyperlipidemia, suspicion of glaucoma, prior transient ischemic attack, and mild neurocognitive disorder progressing to dementia. The patient had undergone radical prostatectomy to treat prostate cancer a few years prior to the onset of memory problems. MRI of the brain to evaluate progressive memory loss showed mild central volume loss, mild chronic per ventricular white matter ischemic changes with patchy scattered areas of prolonged T2 signal within the deep white matter posterior hemispheres.
Patient 2
A 77-year-old African-American man with a 17-year history of type 2 diabetes complicated by nephropathy, hyperparathyroidism, refractory hypertension, blindness secondary to primary open angle glaucoma, cerebrovascular accident and dementia. Labs were remarkable for proteinuria and type 2b hyperlipidemia. CT scan of the brain to evaluate memory loss showed mild, global volume loss with microvascular angiopathy and lacunar infarcts.
Patient 3
Parkinson’s disease plus dementia (PDD): A 70-year-old Caucasian male with diabetes, hypertension, cardiovascular disease, suspicion of glaucoma, depression, mild diabetic retinopathy, proteinuria and a history of chronic lymphocytic leukemia (CLL).
Patient 4
A 73-year-old African-American male with diabetes, hypertension, atherosclerotic heart disease, hyperlipidemia, a trial fibrillation, depression, obstructive sleep apnea, prostate cancer, and Parkinson’s disease. CT scan of the brain after a fall revealed moderate, global volume loss with micro vascular angiopathy and lacunar infarcts.
Blood drawing
Blood drawing was performed in the morning in fasted participants. An exception was one of two blood sample(s) obtained from Patient 1, for the sequential Liposorber/Protein-A affinity purification of autoantibodies from dyslipidemic plasma, in which a non-fasting morning sample was obtained approximately two hours after a meal.
Protein-A Affinity Chromatography
Protein-A affinity chromatography was carried out as previously reported [4].
Liposorber-LA-15 dextran sulfate cellulose chromatography
Liposorber LA-15 and sequential Liposorber-LA/protein-A chromatography were performed as previously reported [9]. The Liposorber LA-15, LDL Adsorption Column was kindly provided by Maasaki Fukunishi (Kaneka Pharma America, LLC, Newark, CA)
Mouse N2 A Neuroblastoma cells
Mouse N2a cells were kindly provided by Dr. Janet Alder, Dr. Smita Thakker-Varia and Shavonne Teng (Department of Neuroscience, Rutgers/Robert Wood Johnson Medical School, Piscataway, New Jersey) and were cultured as previously described [4].
Mouse HL1 Atrial Cardiomyocytes
Mouse HL1 cells were kindly provided by Dr. Zui Pan (University of Texas, Arlington, College of Nursing and Health Innovation, Dallas, TX) and were cultured as previously reported [10].
N2A Neurite retraction assays
N2A neurite retraction assays were performed as previously described [4].
N2A cell survival assays
N2a cell survival assays were carried out as previously reported [4].
Chemicals
M100907, ketanserin, spiperone, SB204741, bosentan, losartan, prazosin, BAPTA-AM, 2-APB, and Y27632 were obtained from Sigma Chemical Co. (St Louis, MO). All other chemicals and reagents were analytical grade.
Protein determinations
Protein concentrations were determined by a bicinchoninic acid protein assay kit (Pierce Chemical Co., Rockford, IL).
Statistics
All data are the mean ± SD (Tables 1–4) or ± SEM (Figure 1–6) as indicated. Comparisons were made by Student’s t-test for a continuous variable with a significance level, p=0.05; or by Fischer’s exact test or χ2 test for dichotomous variables (Tables 1,4).
Table 4.
Baseline characteristics in Parkinson’s disease (PD) subsets according to differential response in protein-A eluates to heat treatment.
| PD subset 1 (N=6) | PD subset 2 (N=6) | P-value** | |
|---|---|---|---|
| Age (years) | 67.5 ± 5.9 | 72.7 ± 2.8 | 0.11 |
| BMI (kg/m2) | 31.6 ± 2.4 | 31.4 ± 4.2 | 0.94 |
| Diab Duration (years) | 21.7 ± 11.4 | 9.2 ± 3.8 | 0.4 |
| HbA1c (%) | 8.7 ± 1.2 | 7.8 ± 1.7 | 0.35 |
| Prostate cancer (yes/no) | (1/5) | (5/1)* | 0.02^ |
Chronic lymphocytic leukemia (n=1), bladder cancer (n=1), prostate cancer (n=3);
T-test;
χ2 test
Figure 1.

Diabetes Parkinson’s disease (n=10) or dementia (n=2) auto antibodies (60 nM) caused significant N2a neuroblastoma cell loss after 24 hrs incubation compared to identical concentration of autoantibodies in the protein-A eluate of plasma in diabetic patients without neurodegenerative disorder (control EL). B) Neuroblastoma cell loss induced by (60 nM) concentration of diabetic PD (n=5) or dementia (n=1) auto antibodies was completely prevented by co incubation with 200 nM concentration of M100907, a highly selective 5-HT2A receptor antagonist. N2A cells were incubated for 24 hours at 37 degrees. Cell number was determined as described in Methods. Results are mean ± SEM.
Figure 6.

Heating (65 deg C x 30 mins) caused gain in N2A neurotoxicity in a subset of six diabetic patients with co-morbid PD (n=5) or dementia (n=1) (A). There was no effect of heating on N2A toxicity in protein-A eluates from two diabetic control patients without neurodegenerative disease co morbidity (B). Heating caused a modest gain in N2A proliferation (n=3) or a loss in N2A toxicity in a second subset of six diabetic patients with co-morbid PD (n=5) or dementia (n=1) (C).
Results
Clinical Characteristics In Study Patients
The baseline clinical characteristics in the study patients are shown in Table 1. Patients with neurodegenerative disease (n=10 Parkinson’s disease, N= 2 diabetic dementia) did not different significantly in their mean: age, baseline HbA1c or known diabetes duration from control diabetic patients without PD or dementia. Parkinson’s disease and dementia patients had significantly lower mean body mass index (BMI) compared to control diabetic patients without PD or dementia (Table 1).
Effect Of Protein A Eluates On N2A Cell Survival
Mean N2A neuroblastoma cell survival in the protein-A eluate fraction of plasma (6–9 μg/mL) was significantly reduced in patients with Parkinson’s disease and dementia (N=12) compared to diabetic patients without Parkinson’s disease or dementia (N=8) (Figure 1, P = 0.001). In all six PD and dementia protein A eluates tested (n=5 PD; n=1 DMT), co-incubation with 200 nM concentrations of the highly selective 5-HT2AR antagonist M100907 completely protected N2A cells against accelerated cell death caused by the Parkinson’s disease and dementia autoantibodies (P =0.001; Figure 1B).
Effect of Parkinson’s Disease or Dementia Autoantibodies on N2a Cell Morphology
Forty-sixty nano molar concentrations of Parkinsons’ disease autoantibodies (n=2 patients) or a five-fold lower concentration of diabetic dementia autoantibodies (n=1 Patient 1) caused acute N2A cell contraction and 75–87% mean N2A neurite length-shortening after 20 minutes’ exposure time (Figure 2A). After 10 minutes’ incubation time, 40–60 nM concentrations of Parkinsons’ disease autoantibodies (n=3) caused significant (mean 58%) N2A neurite shortening compared to neurite length in untreated N2a cells (P < 0.01; Fig 2B). The PD autoantibody-induced neurite retraction (n=3 patients) was nearly completely prevented by co-incubating N2A cells with 200 nanomolar concentrations of the selective 5-HT2A receptor antagonist, M100907 (P < 0.01; Figure 2B). The 5-HT2AR antagonists ketanserin and spiperone (200 nM concentrations) each substantially protected (60–90%) against PD autoantibody-induced N2a neurite- shortening (Table 3). A higher concentration (1–10 μM) of three different GPCR antagonists belonging to the Gαq subclass of GPCR receptors (i.e. bosentan, losartan or prazosin, specific for the endothelin1, the angiotensin II, type 1, or the alpha 1 adrenergic receptor, respectively had no significant protective effect (0–20%) against PD autoantibody-induced N2a neurite length- shortening (Table 2).
Figure 2.
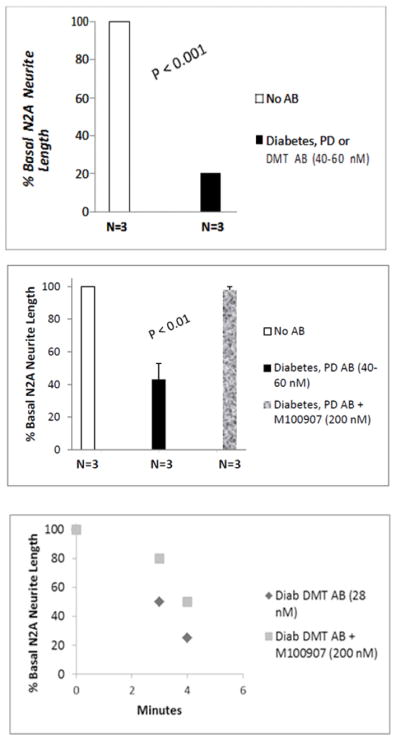
Diabetic Parkinson’s disease (n=2) or dementia autoantibodies (n=1) caused significant suppression of N2A cell neurite extension compared to control cells (A); Mean N2A neurite length suppression induced by three different diabetic PD autoantibodies was nearly completed prevented by co-incubation with 200 nM M100907, a highly selective 5-HT2A receptor antagonist (B). Potent, time-dependent suppression of N2A neurite outgrowth by diabetic dementia (Pt 1) autoantibodies was partially prevented by co-incubation with M100907(C).
Table 3.
Effect of IP3R/Ca2+ pathway or RhoA/Rho kinase inhibitors on diabetic PD autoantibody-induced N2A neurite retraction
| Treatment | [Conc] | % Diabetic PD autoantibody neurite retraction |
|---|---|---|
| Diab PD AB | 60 nM | 100% ± 0% |
| 2-APB (IP3R inhibitor) | 50 μM | 0% ± 0% |
| BAPTA-AM (Ca2+chelator) | 30 μM | 0% ± 0% |
| Y27632 (ROCK inhibitor) | 10 μM | −8% ± 2% |
Results are expressed as the mean± SD of two experiments.
Table 2.
Effect of GPCR receptor antagonists on diabetic Parkinson’s disease autoantibody-induced neurite retraction.
| Antagonist | [Conc] | GPRC | %Neutralization of Diabetic PD Autoantibody Neurite Retraction |
|---|---|---|---|
| M100907 | 200 nM | 5-HT2A | 95% |
| Spiperone | 200 nM | 5-HT2A | 90% |
| Ketanserin | 200 nM | 5-HT2A | 60% |
| SB 204741 | 1 μM | 5-HT2B | 10% |
| Losartan | 10 μM | AT-1R | 20% |
| Bosentan | 10 μM | ET1-R | 15% |
| Prazosin | 2 μM | A1-AR | 0% |
Results are expressed as mean of two determinations (which varied by < 15%) and were calculated as 1-[neurite length in presence of antagonist + diab PD autoantibodies (~60 nM) divided by (neurite length in presence of diab PD autoantibodies alone)] x 100.
High Potency Neurotoxicity In Diabetic Dementia Autoantibodies
Twenty-eight nanomolar concentrations of the diabetic dementia (Pt 1) autoantibodies caused rapid onset, substantial collapse in proximally-located N2A neurites, i.e. (50% length-shortening after 3 minutes; and 75% shortening after 4 minutes incubation time)(solid diamonds, Figure 2C). The diabetic dementia (Pt 1) autoantibody-induced neurite retraction was only partially blocked by co-incubation with 200 nanomolar concentrations of M100907 (solid squares, Figure 2C) suggesting increased potency of diabetic DMT compared to the diabetic PD autoantibodies.
Mechanism Of Neurite Retraction Induced By Diabetic PD Autoantibodies
Pre-incubation (for 5–10 minutes) with the indicated concentrations of the IP3 receptor antagonist (2-APB) or the calcium chelator (BAPTA-AM) each completed prevented neurite retraction induced by 60 nanomolar concentrations of diabetic PD autoantibodies (Table 3). Pre-incubation with 10 μM concentrations of Y27632, a selective RhoA/Rho kinase inhibitor, completely prevented neurite retraction induced by 60 nanomolar concentrations of the diabetic PD autoantibodies (Table 3). These data suggest that PD autoantibodies cause neurite retraction via 5-HT2A receptor activation positively coupled to IP3R/Ca2+ release as well as activation of RhoA/ROCK downstream signaling as previously reported [4].
Purification Of Neurotoxic Diabetic PD,& Dementia Plasma Autoantibodies Using Liposorber
Liposorber apheresis is an FDA-approved method for removing Apo-lipoprotein B-100- bearing low-density and very- low- density lipoprotein (LDL, vLDL) particles from the circulation in patients with familial hypercholesterolemia [11]. Highly potent neurotoxic autoantibodies were previously reported in an adult type 1 diabetes patient suffering with recurrent small vessel stroke, severe retinopathy, and dementia [9]. A highly- purified fraction was previously obtained from hypertriglyceridemic diabetic dementia plasma using a simple two-step purification procedure consisting of Liposorber affinity chromatography (LIPOSORBER LA-15) followed by protein-A affinity chromatography. We next subjected small aliquots of diabetic dementia (Pt-1) or Parkinson’s disease plus dementia (PDD; Pt 3) plasma (0.15 mL) to dextran sulfate cellulose chromatography (LIPOSORBER LA-15) followed by protein A-affinity chromatography.
Roughly 90% of the starting protein was recovered in the Liposorber flow- through fraction (Figure 3A); ~ 5% was recovered in the Liposorber eluate fraction (Figure 3A). The protein- A eluate fraction (0.5 μg/mL IgG) caused significantly greater N2A cell loss compared to either the Liposorber eluate (P < 0.01;Figure 3A) or the protein-A flow-through fractions (P < 0.05; Figure 3A). The highly-purified diabetic DMT autoantibodies caused dose-dependent inhibition of N2A neurite extension (Figure 3B): half-maximal suppression of neurite outgrowth occurred at an 8-fold lower concentrations of the autoantibodies (3.5 nM) than in the corresponding protein-A eluate fraction(s) obtained directly from starting plasma (28 nM, Figure. 2C). Diabetic dementia and PDD autoantibodies caused dose-dependent, three-fold greater loss of N2A cells than identical concentrations (10 nM) of the DMT Liposorber eluate fraction (Figure 3C).
Figure 3.

Neuroblastoma cell toxicity was present in the purified fraction eluting from protein A following Liposorber/protein-A sequential chromatography, but was lacking in the other fractions (A). The highly purified protein-A eluate fraction of diabetic dementia plasma caused significantly greater dose-dependent suppression of N2A neurite outgrowth compared to the Liposorber eluate fraction from the same patient (B). Dose-dependent N2A cell toxicity in the protein-A eluates from diabetic dementia (Pt 1) and PDD (Pt 3) significantly exceeded toxicity in the corresponding Liposorber DMT eluate fraction (C). ** P < 0.01; * P < 0.05.
Neutralization Of Highly-Purified Neurotoxic PDD, DMT Autoantibodies By 5-HT2AR Antagonists
Accelerated N2A cell loss caused by three nanomolar concentrations of the highly-purified diabetic DMT (Pt 1) and PDD (Pt 3) autoantibodies was nearly completed blocked by co-incubation with 500 nanomolar concentrations of each of three different 5-HT2A receptor antagonists: M100907 (Figure. 4A); spiperone (Figure 4B); and ketanserin (Figure. 4C). M100907, spiperone and ketanserin (500 nM) alone had no significant effect on N2A cell survival (not shown in Figure 4).
Figure 4.

The neuroblastoma cell toxicity in the highly-purified Liposorber/protein-A eluates of diabetic dementia and Parkinson’s disease with dementia was significantly blocked by co-incubation with 500 nM concentrations of A) M100907, B) spiperone, or C) ketanserin.
Apparent MW Of Highly-Purified Neurotoxic Diabetic DMT AutoAntibodies
The highly purified protein-A eluate fraction obtained after sequential Liposorber/Protein A chromatography in Patients 1 and 3 plasmas was dialyzed on a 30 kilodalton (kD) MW cutoff membrane. Ten nanomolar concentrations of the concentrated (> 30 kD MW) retentate fraction caused significantly accelerated N2A cell loss (~50%) after 24 hours’ incubation. Much less N2A cell loss (13%) occurred after 24 hours incubation with an identical concentration of the (< 30 kD) MW flow-through fraction (Fig 5A). These data suggest peak neurotoxicity eluting from protein- A had an apparent MW in the range of Ig heavy chains or intact IgG (50, 150 kD).
Figure 5.

Peak neurotoxicity in the highly purified diabetic dementia autoantibodies was retained following dialysis on a 30 kD MW cutoff membrane (A). The N2A neurotoxicity in PD and diabetic dementia auto antibodies was completed prevented by co-incubation with 1–2 μM concentrations of the IP3R blocker 2-APB(B). Mean N2A neurotoxicity in autoantibodies from five different diabetic PD or dementia patients was not significantly affected by co-incubation with Losartan, bosentan or prazosin (C).
N2A cell loss induced by the (> 30 kD MW), Pt 1 diabetic dementia autoantibodies (3nM) could be substantially decreased (by 55%) following co-incubation with 200 nanomolar concentrations of the selective 5-HT2AR antagonist M100907 (data not shown in Figure 5).
Mechanism and pharmacologic specificity of PD autoantibody-induced N2A cell loss
Co-incubating ~ 60 nanomolar concentrations of diabetic dementia (Pt 2) or diabetic PD (Pt 4) autoantibodies with 1–2 μM concentrations of the inositol triphosphate receptor blocker 2-APB (for 8 hours at 37 degrees C) significantly protected against N2A cell loss (P< 0.01; Figure 5B). 2-APB alone (1–2 μM) had no significant effect on N2A survival (Figure 5B). Co-incubation of diabetic PD or diabetic DMT autoantibodies (n=5 different patients) together with losartan (2.5 μM), bosentan (2.5μM), or prazosin (0.5 μM) did not significantly prevent autoantibody-induced N2a cell loss (Figure 5C). Losartan (2.5 μM), bosentan (2.5 μM) or prazosin (0.5 μM) alone each had no significant effect on N2A cell survival (not shown in Figure 5).
Heat Stability Of Neurotoxicity In Parkinson’s Disease Protein- A Eluates
Protein-misfolding disorders are characterized by aggregation in disordered protein fragments which can assume a highly-stable, β-rich structural conformation [12]. Heat treatment causes IgG aggregation and denaturation of highly-ordered protein structure. We next tested the heat stability of neurotoxicity in protein-A eluates from Parkinson’s disease (n=10) or dementia (n=2) patients. In a subset of six patients (five PD and one dementia, i.e. Neurodegenerative Diseases subset 1, Table 4) heating the protein A eluate (65 degrees x 30 mins) caused a gain in inhibitory toxicity (i.e. lower percent N2A cell survival) compared to the unheated protein-A eluate fraction (54 ± 17% vs 84 ± 5%; P =0.005; Figure 6A) consistent with aggregation in a heat-stable, toxic Ig fragment. In two diabetic control patients without PD, heating the protein-A eluates had no significant effect on basal N2A cell proliferation (n =2; Figure 6B). In a second subset of six patients (five of whom suffered with co-morbid cancer, i.e. Neurodegenerative Diseases subset 2, Table 4) heat treatment of the protein-A eluates either eliminated basal neurotoxicity (n=3; Figure 6C) or caused a modest increase in mean N2A cell proliferation (n=3; Figure 6C) consistent with denaturation of IgG tertiary structure or promotion of a stimulatory IgG immune complex. Taken together, these data suggest molecular heterogeneity in diabetic PD protein-A eluates some of which (i.e. Neurodegenerative Diseases subset 2, Table 4) may have contained immune complexes as previously reported in uro-epithelial cancers [13] and in chronic lymphocytic leukemia (CLL) [14].
Effect Of Diabetic PD Autoantibodies In Mouse HL1 Atrial Cardiomyocytes
Hypertension, diabetic dyslipidemia and atrial fibrillation are associated with cerebral hypoperfusion, and accelerated cognitive decline through complex, and poorly understood mechanisms [15]. Since the 5-HT2A receptor is expressed on smooth muscle cells in the gut, heart and vasculature where it mediates 5-HT-induced-contraction [16] and -vasoconstriction [17], we next tested whether HL1 mouse atrial cardiomyocytes, a type of vascular smooth muscle cell, responds to diabetic PD protein-A eluates. Sixty nanomolar concentrations of the Pt 4, diabetic PD autoantibodies caused sustained contraction in dissociated HL1 mouse atrial cardiomyocytes in vitro within 5 minutes of exposure (Fig 7). Pre-incubation of HL1 cells (for five minutes) with (500 nM) concentrations of M100907, a highly selective 5-HT2AR antagonist, prevented the Pt 4, diabetic PD autoantibody-induced HL1 cell contraction (not shown in Figure 7). Similar results were obtained with the protein-A eluate fraction from another diabetic PD patients’ plasma.
Figure 7.

Diabetic PD autoantibodies (60 nM) caused contraction in HL-1 atrial cardiomyocytes within 5 minutes of application. Photomicrographs (100 x magnification) were captured with a Nikon TMS microscope. A) Control HL-1 cells prior to addition of diabetic PD autoantibodies; B) corresponding images of the same HL-1 cells 5 minutes after the application of diabetic PD (Pt 4) autoantibodies. Higher magnification of cells (A) shown in inset by rectangle (C, D), diamond (E, F) or arrow (G, H) before (C, E, G) and five minutes after (D, F, H) the addition of PD autoantibodies. Similar results were obtained in experiments with two different diabetic PD patients autoantibodies.
Discussion
The present data are the first to suggest that subsets of older adult type 2 diabetes with co-morbid Parkinson’s disease harbor plasma agonist 5-HT2A receptor autoantibodies which cause accelerated neuroblastoma cell loss via activation of intracellular IP3R/Ca2+ signaling. These findings are consistent with a prior report that patients suffering with diabetic depression harbored similar kinds of agonist 5-HT2A receptor autoantibodies [4]. Depression, dementia and Parkinson’s disease are neurodegenerative disorders which all increase significantly in older adult type 2 diabetes. Our novel data suggest that agonist 5-HT2A receptor autoantibodies may be a shared feature in Parkinson’s disease, dementia, and depression affecting older adult men with diabetes.
Several lines of evidence have implicated altered intracellular calcium homeostasis in the unknown etiology of sporadic PD. First, alpha synuclein, a hallmark in PD neuropathology, can be cleaved by the calcium-dependent protease calpain I, which causes alpha synuclein to aggregate into high MW, β-rich conformational structures [12]. Calpain I expression was reported to increase in the substantia nigra (SN) in PD patients [18]. Second, SN neurons containing the calcium-buffering protein calbindin 1 (CALB1) were relatively more resistant to neurodegeneration compared to CALB1-negative neurons [19]. Third, inositol triphosphate kinase B (ITPKB), a critical regulator of the IP3/Ca2+ signaling pathway, was identified as a novel PD gene candidate(s) in a recent large genome wide association study [20]. Altered ITPKB gene expression was also reported in the postmortem brains of Alzheimer’s disease patients [21]. Taken together, these results suggest that deregulated calcium homeostasis perhaps induced (in part) by 5-HT2A receptor agonist autoantibodies might contribute to neuron loss in the SN and other brain regions affected in Parkinson’s disease, depression, and dementia.
Animal models and human studies have implicated neuro-inflammation and humoral immune mechanisms in the pathogenesis of idiopathic Parkinson’s disease [22–24]. For example, the post-mortem brains of PD patients demonstrated immunoglobulin G coating the cell surface (of perikarya and neurites) in SN neurons; nearby activated microglia contained Lewy bodies and IgG perhaps indicative of ingested IgG-coated neurons [23]. The Fcγ receptor on microglia is a candidate for mediating destruction of neurons coated by IgG [23]. Heat-aggregated IgG and circulating immune complexes which bound to the Fcγ receptor on lymphocytes were reported to modulate apoptosis and alter cytokine and HLA DR expression [14]. Our finding of reduced N2A cell apoptosis following heat treatment of IgG in plasma from a subset of PD patients with co-morbid bladder, prostate cancer, or CLL is consistent with previous reports of increased circulating immune complexes in these cancers [13, 14]. Non-rheumatoid factor, anti-IgG autoantibodies were reported in a substantially higher proportion (~50%) of chronically-ill patients (aged 50–70 years old) having Parkinson’s disease or diabetes compared to other chronic illnesses [25] suggesting a specific role for such autoantibodies in PD associated with diabetes.
Aging and vascular injury may contribute to circulating autoantibodies a subset of which could target antigens expressed on vascular surfaces and in the brain. Long-standing systemic inflammation may give rise to altered expression of certain receptors, including possibly 5-HT2AR, whose expression was reported to be substantially increased on CD3 positive T cells and in the vessels from involved skin in psoriasis patients [26]. Persistent (7 days) exposure to the pro-inflammatory cytokine IL-β in murine tracheal rings in vitro was reported to increase 5-HT2AR-mediated airway muscle contractility [27]. The 5-HT2AR was also reported to have played a role in sustained, increased nitric oxide production in animal models of LPS-induced inflammation [28, 29]. Taken together, these results suggest possible involvement of the 5-HT2A receptor in mediating several of the vascular and neurotoxic effects of chronic inflammation.
Hypercontractility in cerebral resistance arterioles was reported in the postmortem brains from Alzheimer’s-type dementia patients [30]. The RhoA/Rho kinase signaling pathway in N2A cells activated by 5-HT2A agonist autoantibodies [4] causes Ca2+ sensitization of contraction in vascular smooth muscle cells [31]. Our preliminary experiments in HL1 atrial cardiomyocytes suggest that vascular smooth muscle cells harboring 5-HT2A receptors may be an important peripheral target of circulating agonist, 5-HT2A receptor autoantibodies.
The origin of circulating 5-HT2A receptor autoantibodies in subsets of diabetes suffering with one or more co-morbid neurodegenerative disease(s) is unknown. Persistent inflammation is a shared risk factor in obese type 2 diabetes and in PD. In obese adult type 2 diabetes, the elaboration of visceral adipocytokines (IL-1, interferon gamma, IL-6) has been implicated in the loss of blood: brain barrier integrity, and may promote humoral and cell-mediated immune responses to sequestered antigens perhaps released as a result of diabetic vascular injury. The 5-HT2A receptor subtype expressed on platelets contributes to platelet activation resulting in 10-fold or higher concentrations of 5-HT and polyphosphates than those which normally circulate. It is possible that post-translationally modified circulating form(s) of 5-HT or 5-HT2A receptor may be target(s) for autoimmunity leading to the development of agonist 5-HT2A receptor autoantibodies.
The striking ability of Liposorber (dextran sulfate cellulose) to remove peak neurotoxic diabetic dementia and PDD autoantibodies in hyperlipidemic (TG-rich) plasma in vitro suggests 5-HT2A receptor autoantibodies may partition into triglyceride-rich lipoprotein particles, e.g. very low density (vLDL) or remnant lipoproteins [32], whose levels increase in viscerally-obese type 2 diabetes. The low- density lipoprotein receptor related protein-1 LRP is present on vascular cells comprising the blood brain barrier where it is thought to play a role in promoting the efflux of neurotoxic amyloid beta peptide from the brain into the peripheral circulation [33]. Heparan sulfate proteoglycan (HSPG) is not only a co-receptor for LRP, but it mediates an LRP-independent cellular lipoprotein uptake pathway. Since diabetic autoantibodies in patients suffering with depression displayed increased affinity for neuronal HSPG [10], the HSPG/LRP pathway(s) may be involved in how neurotoxic autoantibodies gain access to the CNS.
Neuroblastoma cells were exquisitely sensitive to low nanomolar concentrations of the Liposorber/protein-A- purified diabetic dementia autoantibodies whereas substantially higher (100 nM) concentrations of soluble PD autoantibodies [4] or oligomeric β-amyloid (1–42) were reportedly required to adversely affect dentate gyrus neuron survival [4] or long-term potentiation [34], respectively. These data suggest a possible role for the lipoprotein microenvironment in potentiation of agonist 5-HT2AR autoantibody toxicity. Fasting plasma triglyceride concentrations in diabetic dyslipidemia typically occur in the low millmolar concentration range. Thus TG-rich lipoproteins may provide an enormous reservoir of highly neurotoxic autoantibodies capable of mediating direct endothelial cell [4] and neuronal toxicity [4]. Since total IgG attains 100 μM concentrations in blood, specific neurotoxic diabetic IgG present at ~ 0.1% of total IgG concentrations might be able to compete with neurotoxic β-amyloid peptide for binding to shared components of the HSPG/LRP pathway mediating traffic of neurotoxic peptides and proteins into and out of the brain. These findings suggest a possible synergistic ‘toxic’ effect of the autoantibodies in mediating both direct toxicity and potentiation of β-amyloid toxicity (by slowing its efflux from brain) in older-onset dementia patients.
Our study was small and the results may be limited to men who had experienced long-standing type 2 diabetes. Thus in three of three patients having a diagnosis of pre-diabetes and co-morbid Parkinson’s disease plasma neurotoxic autoantibodies were present in each case, however in only one of the three patients tested was the autoantibody-induced neurotoxicity neutralized by co-incubating with (200 nM) concentrations of the selective 5-HT2AR antagonist M100907. Diabetes is associated with a two-fold increased risk for age-related cognitive decline or dementia [35]. In a prior study of cognitive decline in seventeen hundred ninety-one older adult type 2 diabetes patients from the Veterans Affairs Diabetes Trial, post-baseline TG-lowering was associated with significant protection against the prospective (5 year) decline in cognitive processing speed [36].
The present findings provide the first evidence that a subset of older men with type 2 diabetes and Parkinson’s disease harbored agonist 5-HT2A receptor autoantibodies which promoted accelerated neuroblastoma cell loss via long-lasting activation of IP3/Ca2+ signaling. The 5-HT2A receptor is a druggable G-protein coupled receptor (GPCR) target for which a number of highly specific FDA-approved antagonists already exist. More study is needed to determine if 5-HT2AR agonist autoantibodies precede the development of PD symptoms and may serve as a useful biomarker in drug development aimed at early prevention of PD- or diabetic dementia- neurodegeneration.
Conclusion
In conclusion, autoantibodies in twelve of twelve older adult type 2 diabetes patients tested having co-morbid Parkinson’s disease or dementia caused accelerated neuron loss via a mechanism involving the 5-hydroxytryptamine 2 receptor positively coupled to inositol triphosphate receptor-mediated sustained cytosolic Ca2+ release.
Acknowledgments
Supported by a grant from the Veterans Biomedical Research Institute (East Orange, New Jersey) to Mark B. Zimering, MD, PhD.
Footnotes
Conflict of Interest
The author reports no financial conflict of interest that would affect the objectivity of the presented findings.
References
- 1.Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157(11):1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- 2.Sauerbier A, Jenner P, Todorova A, Chaudhuri KR. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat Disord. 2016;22(Suppl 1):S41–S46. doi: 10.1016/j.parkreldis.2015.09.027. [DOI] [PubMed] [Google Scholar]
- 3.Hu G, Jousilahti Pekka, Bidel Siamak, Antikainen Riitta, Tuomilehto Jaakko. Type 2 Diabetes and the Risk of Parkinson’s Disease. Diabetes Care. 2007;30(4):842–847. doi: 10.2337/dc06-2011. [DOI] [PubMed] [Google Scholar]
- 4.Zimering MB. Diabetes Autoantibodies Mediate Neural- and Endothelial Cell- Inhibitory Effects Via 5-Hydroxytryptamine- 2 Receptor Coupled to Phospholipase C/Inositol Triphosphate/Ca2+ Pathway. J Endocrinol Diab. 2017;4(4):1–10. doi: 10.15226/2374-6890/4/4/00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu T, Pandey SC. Cellular localization of serotonin (2A) (5HT (2A)) receptors in the rat brain. Brain Res Bull. 2000;51(6):499–505. doi: 10.1016/s0361-9230(99)00278-6. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Cognitive impairment in Parkinson’s disease: amyloid plaques, neurofibrillary tangles, and neuropil threads in the cerebral cortex. J Neural Transm Park Dis Dement Sect. 1990;2(1):45–57. doi: 10.1007/BF02251245. [DOI] [PubMed] [Google Scholar]
- 7.Goetz CG, Poewe W, Rascol O, Sampaio C, Stebbins GT, Counsell C, et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord. 2004;19(9):1020–1028. doi: 10.1002/mds.20213. [DOI] [PubMed] [Google Scholar]
- 8.Zimering MB, Alder J, Pan Z, Donnelly RJ. Anti-endothelial and anti-neuronal effects from auto-antibodies in subsets of adult diabetes having a cluster of micro vascular complications. Diabetes Res Clin Pract. 2011;93(1):95–105. doi: 10.3389/fendo.2013.00058. [DOI] [PubMed] [Google Scholar]
- 9.Zimering MB. Recurrent macular edema and stroke syndrome in type 1 diabetes mellitus with potent endothelial cell inhibitory autoantibodies. Endocr Pract. 2010;16(5):842–850. doi: 10.4158/EP10148.CR. [DOI] [PubMed] [Google Scholar]
- 10.Zimering MB, Mirkovic N, Pandya M, Zimering JH, Behnke JA, et al. Toxic Immunoglobulin Light Chain Autoantibodies are Associated with a Cluster of Severe Complications in Older Adult Type 2 Diabetes. J Endocrinol Diab. 2016;3(1):1–14. doi: 10.15226/2374-6890/3/1/00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asahi T, Yamamoto T, Kutsuki H. Blood purification therapies using dextran sulfate cellulose columns Liposorber and Selesorb. Ther Apher Dial. 2003;7(1):73–77. doi: 10.1046/j.1526-0968.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- 12.Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, et al. Calpain-Cleavage of α-Synuclein: Connecting Proteolytic Processing to Disease-Linked Aggregation. Am J Pathol. 2007;170(5):1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi K, Umeda T, Akaza H, Niijima T, Aikawa T, Tanimoto K. Circulating immune complexes in patients with bladder cancer and other malignancies of the urogenital tract. Urol Int. 1984;39(4):232–235. doi: 10.1159/000280982. [DOI] [PubMed] [Google Scholar]
- 14.Gamberale R, Geffner JR, Giordano M. Immune complexes and apoptosis in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2002;43(2):251–255. doi: 10.1080/10428190290006008. [DOI] [PubMed] [Google Scholar]
- 15.de la Torre JC. Cardiovascular Risk Factors Promote Brain Hypoperfusion Leading to Cognitive Decline and Dementia. Cardiovascular Psychiatry and Neurology. 2012;2012:367516. doi: 10.1155/2012/367516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuemmerle JF, Murthy KS, Grider JR, Martin DC, Makhlouf GM. Coexpression of 5-HT2A and 5-HT4 receptors coupled to distinct signaling pathways in human intestinal muscle cells. Gastroenterology. 1995;109(6):1791–1800. doi: 10.1016/0016-5085(95)90745-9. [DOI] [PubMed] [Google Scholar]
- 17.Alsip NL, Harris PD, Durrani GE. Multiple Serotonin Receptors on Large Arterioles in Striated Muscle. J Vasc Res. 1991;28(6):537–541. doi: 10.1159/000158900. [DOI] [PubMed] [Google Scholar]
- 18.Mouatt-Prigent A1, Karlsson JO, Agid Y, Hirsch EC. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: a role in nerve cell death? Neuroscience. 1996;73(4):979–987. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- 19.Yamada T, McGeer PL, Baimbridge KG, McGeer EG. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990;526(2):303–307. doi: 10.1016/0006-8993(90)91236-a. [DOI] [PubMed] [Google Scholar]
- 20.Chang D, Nalls MA, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nature Genetics. 2017;49(10):1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emilsson L, Saetre P, Jazin E. Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis. 2006;21(3):618–625. doi: 10.1016/j.nbd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Kannarkat GT, Boss JM, Tansey MG. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):493–514. doi: 10.3233/JPD-130250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain. 2005;128(Pt 11):2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- 24.Kannarkat GT, Boss JM, Tansey MG. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):493–514. doi: 10.3233/JPD-130250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litwin SD, Singer JM. Studies of the incidence and significance of anti-gamma globulin factors in the aging. Arthritis Rheum. 1965;8:538–550. doi: 10.1002/art.1780080408. [DOI] [PubMed] [Google Scholar]
- 26.Nordlind K, Thorslund K, Lonne-Rahm S, Mohabbati S, Berki T, Morales M, et al. Expression of serotonergic receptors in psoriatic skin. Arch Dermatol Res. 2006;298(3):99–106. doi: 10.1007/s00403-006-0652-6. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Cardell LO, Adner M. IL-1beta induces murine airway 5-HT2A receptor hyper responsiveness via a non-transcriptional MAPK-dependent mechanism. Respir Res. 2007;8(1):29. doi: 10.1186/1465-9921-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voronova IP, Khramova GM, Kulikova EA, Petrovskii DV, Bazovkina DV, Kulikov AV. 5-HT2A receptors control body temperature in mice during LPS-induced inflammation via regulation of NO production. Pharmacol Res. 2016;103:123–131. doi: 10.1016/j.phrs.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 29.Liu C, Zhang X, Zhou JX, Wei W, Liu DH, Ke P, et al. The protective action of ketanserin against lipopolysaccharide-induced shock in mice is mediated by inhibiting inducible NO synthase expression via the MEK/ERK pathway. Free Radic Biol Med. 2013;65:658–666. doi: 10.1016/j.freeradbiomed.2013.07.045. [DOI] [PubMed] [Google Scholar]
- 30.Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, et al. Serum response factor and myocardin mediate arterial hyper contractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci U S A. 2007;104(3):823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 32.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40(1):1–16. [PubMed] [Google Scholar]
- 33.Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr Pharm Des. 2008;14(16):1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H-W, Pasternak JF, Kuo H, Ristic H, Lambert MP, et al. Soluble oligomers of β-amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924(2):133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 35.Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63(7):1181–1186. doi: 10.1212/01.wnl.0000140291.86406.d1. [DOI] [PubMed] [Google Scholar]
- 36.Zimering MB, Knight J, Ge L, Bahn G VADT Investigators. Predictors of cognitive decline in older adult type 2 diabetes from the veterans affairs diabetes trial. Front Endocrinol (Lausanne) 2016;7:123. doi: 10.3389/fendo.2016.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]