

Abstract
Multiple nonmalignant cell types in the tumor microenvironment (TME) impact breast cancer risk, metastasis, and response to therapy, yet most heritable mechanisms that influence TME cell function and breast cancer outcomes are largely unknown. Breast cancer risk is ~30% heritable and >170 genetic loci have been associated with breast cancer traits. However, the majority of candidate genes have poorly defined mechanistic roles in breast cancer biology. Research indicates that breast cancer risk modifiers directly impact cancer cells, yet it is equally plausible that some modifier alleles impact the nonmalignant TME. The objective of this review is to examine the list of current breast cancer candidate genes that may modify breast cancer risk and outcome through the TME.
Keywords: Breast Cancer, Tumor Microenvironment, Angiogenesis, Genetic, Consomic
Intersection of Breast Cancer Heritability and the Tumor Microenvironment
Breast cancer is the most common female malignancy and is the second most common cause of cancer death among females in the U.S., with more than 40,000 deaths each year. Approximately 30% of breast cancer risk is heritable [1], of which 5-10% of cases can be attributed to rare alleles (such as BRCA1, BRCA2, PTEN and TP53) that are highly penetrant and others that are moderately penetrant (such as ATM, BRIP1, CHEK2, and PALB2) [2, 3]. A larger group of 182 common alleles have been identified by genome-wide association studies (GWAS) [4–36], which confer lower relative risks (RR) of breast cancer (<1.5 fold RR) compared with risk modifiers that are highly penetrant (>5 fold RR) and moderately penetrant (1.5-5 fold RR) [2, 3]. Only a small fraction of breast cancer heritability can be explained by the current list of genetic candidates [37], indicating that additional rare and common modifier alleles likely exist. As might be expected, modifier alleles with high to moderate penetrance are typically linked directly with the malignant transformation of breast epithelial cells through disruption of pathways regulating DNA damage, cell cycle, and apoptosis, whereas GWAS candidates fall within a diverse range of molecular and cellular pathways [38]. Adding to the complexity is the preponderance of loci with multiple candidates in linkage disequilibrium (LD), which are “co-inherited” and therefore considered equally culpable candidates in the development of breast cancer [39].
Although breast cancer risk modifiers directly impact cancer cells, some modifier alleles can plausibly impact breast cancer risk through the nonmalignant tumor microenvironment (TME). To date, at least two host TME modifier loci of breast cancer have been experimentally validated, and evidence of several more TME modifier loci exist. In one example, the Mcs5a rat mammary tumor risk locus was shown to modify mammary carcinoma progression via the immune system, which was driven by FBXO10 and was dependent upon T lymphocytes [40]. A homologous mechanism has been replicated in human T lymphocytes [41] and associated with human breast cancer risk [42]. In another example, a newly developed genetic mapping strategy, Consomic/Congenic Xenograft Model (CXM), was used to identify a host TME modifier locus that is linked with DLL4 and impacts breast cancer growth and metastasis in the rat by inducing dysfunctional angiogenesis, which was independent of tumor cell changes [43–45]. Further evidence of host TME modifiers exist in mouse genetic mapping studies, including three modifier loci (Mmtg1-3) that were linked with mammary tumor angiogenesis [46] and PTPRJ, a mediator of angiogenesis [47] that was originally discovered for its role in susceptibility to colon cancer, and has since been linked with breast cancer risk [48]. In addition, the MHC-linked modifier loci in an MMTV-induced mammary tumor model were found to be largely dependent on systemic factors, such as infiltrating immune cells and inflammatory cytokines [49]. As in human breast cancer, many mammary tumor modifier loci in the mouse and rat remain uncharacterized and overlap with quantitative trait loci (QTL) for TME-related phenotypes, such as angiogenesis and immunity. Thus, there are likely many more uncharacterized host TME modifiers of breast tumor risk and progression.
Role of the TME in Breast Cancer Risk and Outcome
The breast TME is comprised of multiple nonmalignant cell types that interact with malignant tumor cells at all disease stages, including tumor initiation, metastatic progression, and response to therapy [50–54]. Expression studies of human breast tumor samples have identified stromal networks that predict breast cancer risk and outcome, demonstrating the importance of the breast TME [55–57]. Fibroblasts are a major component of the TME and are essential for maintaining normal mammary gland homeostasis [58]. In the malignant setting, cancer-associated fibroblasts (CAFs) modulate multiple aspects of tumor pathophysiology, including malignant progression of cancer cells (proliferation, survival, and invasion), fibrosis, angiogenesis, and tumor-associated immunity [59]. Tumor angiogenesis is necessary for growth and progression of breast tumors and is coordinated by cancer cells and multiple nonmalignant TME cell types, including endothelial cells, fibroblasts, and infiltrating leukocytes [60, 61]. A denser tumor vasculature is correlated with increased tumor growth and hematogenous metastasis, which is due to enhanced oxygen supply, nutrients, and routes for metastatic dissemination [62]. Likewise, tumor lymphatic vessels provide routes for tumor cell metastasis, and invasion of tumor-associated lymphatic vessels highly correlates with poor clinical outcomes [61, 63]. Tumor-associated blood and lymphatic vessels are also the primary routes for trafficking innate and adaptive immune cells, which play both pro- and anti-tumorigenic roles during tumor initiation, progression, and response to therapy [64]. It is now widely accepted that the TME affects most aspects of breast cancer biology, yet we do not fully understand the heritable genetic modifiers that influence human breast cancer through the host TME.
Challenges to Identifying Host TME Modifiers of Breast Cancer
Despite the evidence that genetic modifiers of the host TME impact breast cancer risk and outcome, the focused research on TME modifiers is very limited and there are many unresolved questions (see Outstanding Questions). One salient point from the existing literature is that host TME modifier candidates are likely to have complex interactions across multiple molecular pathways, cell types, and physiological functions (see Figure 1, Key Figure). It is also highly plausible that some genetic modifiers impact both cancer cells and multiple TME cell types. For example, multiple breast cancer candidates (e.g., FGFR2, TGFβR2, and MKL1) [13, 65–70] have physiological roles in mammary epithelial cell function and at least one TME cell type, whereas other candidates (e.g. eNOS and TLRs) [35, 71–75] are typically restricted to the TME and are aberrantly upregulated in cancer cells [76–78]. Thus, it is possible that a single genetic modifier might elicit complex physiological changes across multiple cell types and the combined effects of these cell type-specific alterations is ultimately manifested at the phenotypic level. One could also envision seemingly unrelated TME modifiers that are not connected at the molecular level, but might interact at the cellular or tissue levels by modifying the density or physiological poise of cellular mediators within the TME. For example, the phenotypic effects of a genetic modifier of cytotoxic T lymphocyte function might be dampened or amplified in a patient that has co-inherited a modifier of lymphocyte trafficking.
Figure 1. Key Figure. Schematic of host TME modifier candidates.
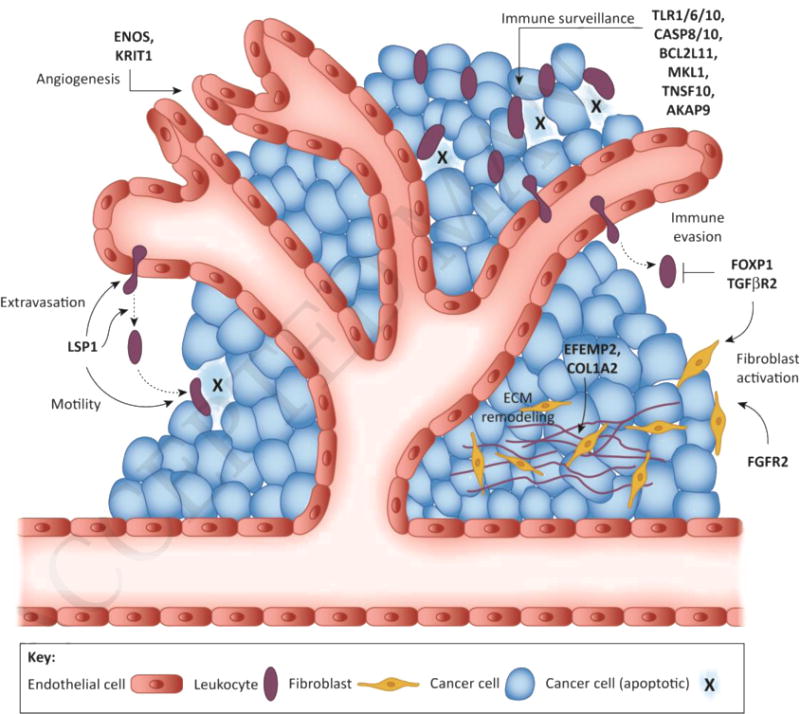
Multiple nonmalignant cell types in the TME impact breast cancer risk and progression, yet the underlying heritable mechanisms that alter TME cell function and influence breast cancer risk and outcome are frequently overlooked and largely unknown. Depicted here are polymorphic genes from established human breast cancer risk loci that likely function as host TME modifiers of breast cancer. ECM, extracellular matrix.
The challenges to disentangling the complexities of host TME modifiers are further compounded by limitations to the current tools for assessing the heritable genetic modifiers of breast cancer. Genetic association and mapping studies of breast cancer risk and outcome are suitable for nominating candidate regions, but are unable to establish the cell type specificity of a genetic modifier without functional testing. However, despite the preponderance of studies that have experimentally validated cancer cell-autonomous mechanisms [79–81], very few experimental models exist to identify and test the genetic modifiers that might impact the host TME. Another common method for identifying genetic modifiers of breast cancer is to scan for expression QTL (eQTL) [82]. However, a drawback of eQTL analyses is their basis upon mixed RNA extracted from tumor biopsies that contain variable amounts of cancer cells and TME cell types. Thus, similar to GWAS and other genetic mapping strategies, eQTL analyses are limited in their ability to distinguish host TME modifiers. Finally, because eQTL analyses of tumor biopsies are based on RNA that is derived from multiple cell types, it is also foreseeable that differences in cell type-specific expression of the same gene might mask the detection of eQTL that exist within only a specific TME cell type.
We propose that there are several existing strategies that could be adapted for discovering and characterizing host TME modifiers of breast cancer risk and outcome. One such strategy is to combine eQTL analyses with cell purification techniques or laser-capture microscopy. Both techniques have previously been used to quantify cell type-specific RNA expression in the breast TME [55–57, 83]; however, to our knowledge, none of the previous studies incorporated genotypic information and therefore TME-specific eQTL analyses have yet to be reported. Another promising strategy to identify host TME modifiers is to perform a modified eQTL analysis at the protein level, using multiplex immunofluorescent assays to correlate cell type-specific protein expression with patient genotypes. The capacity of this approach could be expanded using high density tissue microarrays and quantitative immunofluorescent imaging, which offers highly sensitive and spatially resolved detection of protein expression at the cellular and subcellular levels [84–87]. Finally, we recently developed CXM as the first experimental strategy for genetic mapping of host TME modifiers [43–45]. In CXM, human breast cancer cells or patient-derived xenografts (PDX) are orthotopically implanted into genetically-engineered consomic or congenic xenograft host strains (mice or rats), which are derived from two parental strains with different susceptibilities to breast cancer. Because the host strain backgrounds are different, whereas the inoculated tumor cells are the same, any phenotypic variation can be mapped to TME modifier(s) on the substituted chromosome (i.e., consomic) or subchromosomal region (i.e., congenic) of the host’s germline DNA. Once a host TME modifier has been localized by CXM, it can then be functionally tested by gene-editing and other experimental strategies.
Candidate Modifiers of the Breast TME
As the strategies for discovering host TME modifiers continue to develop, it is possible to begin leveraging the existing breast cancer association data [4–36] to interrogate which candidate genes are potentially modify breast cancer risk and outcome through the host TME. Here, we provide the biological context for 24 breast cancer risk modifiers that likely function, at least partially, through the host TME (Table 1). This list of TME modifier candidates is by no means exhaustive, and several TME modifier candidates may also impact malignant tumor cells directly. Whenever possible, we provide the genetic, molecular, and biological context for the variants in strong LD (r2>0.8) with the “tagged” polymorphism using bioinformatics resources such as HaploReg [88], RegulomeDB [89], Variant Effect Predictor (VEP) [90], TCGA/OncoLnc [91], and GTEx [92]. We also provide the haplotype-specific information and biological context for each candidate, with the hope of providing a roadmap for empirically testing host TME modifiers of breast cancer in the future.
Table 1.
Host TME modifier candidates of breast cancer risk and outcome
| Gene Symbol | Risk Allele | MAF (Cohort)* | Phenotype | Potential Cellular Mediator(s) | Functional Evidence** |
|---|---|---|---|---|---|
| TGFβR2 | rs12493607[C] | 0.33 (CEU) | Risk [68]*** | Cancer cells, fibroblasts, leukocytes | eQTL in leukocytes [104] |
| TGFβR2 | rs4522809 [G] | 0.47 (CEU) | Risk [69] | Cancer cells, fibroblasts, leukocytes | Functional consequences of variant are unknown |
| TGFβR2 | rs1078985[G] | 0.33 (CEU) | Risk [70] | Cancer cells, fibroblasts, leukocytes | eQTL in leukocytes [104] |
| FGFR2 | rs2981578 [T] | 0.47 (CEU) | Risk [13, 65–67] | Cancer cells, fibroblast | eQTL in fibroblasts and correlation with fibroblast activation [114] |
| eNOS/NOS3 | rs1799983 [T] | 0.32 (CEU) | Risk [71–75] | Cancer cells, endothelial cells | Missense variant (E298D); evidence altered proteolytic processing of eNOS [123], which has since been disputed [124, 125] |
| eNOS/NOS3 | rs2070744 [C] | 0.41 (CEU) | Risk [101–105] | Cancer cells, endothelial cells | Disrupted eNOS promoter activity in human endothelial cells [126, 127] |
| LSP1 | rs3817198 [C] | 0.32 (CEU) | Risk [66, 133], survival [134], mammographic density [135, 136] | Endothelial cells, leukocytes | Functional consequences of variant are unknown; LSP1 transcript expression is significantly associated with outcome in TCGA dataset [91] |
| MKL1 | rs6001930 [C] | 0.10(CEU) | Risk [68] | Cancer cells, myoepithelial cells, endothelial cells, leukocytes | eQTL in muscle cells [66]; functional consequences of variant in breast cancer-related cells unknown; MKL1 transcript expression is significantly associated with outcome in TCGA dataset [91] |
| MKL1 | rs17001868 [C] | 0.13(CEU) | Mammographic density [150] | Cancer cells, myoepithelial cells, endothelial cells, leukocytes | eQTL in muscle cell and thyroid cells [66]; functional consequences of variant in breast cancer-related cells unknown; MKL1 transcript expression is significantly associated with outcome in TCGA dataset [91] |
| TLR1/6/10 | rs6815814 [C] | 0.25 (CEU) | Risk [35] | Leukocytes, fibroblasts | eQTL for TLR1/6/10 in leukocytes [92, 104, 105]; eQTL for TLR6 in fibroblasts [92]; TLR10 transcript expression is significantly associated with outcome in TCGA dataset [91] |
| CASP8 | rs1830298[C] | 0.30 (CEU) | Risk [166, 167] | Cancer cells, leukocytes | eQTL in multiple tissues, including mammary epithelial cells and leukocytes [92] |
| CASP8 | rs1045485 [C] | 0.47 (CEU) | Risk [84, 149, 150, 152–154] | Cancer cells, leukocytes | missense variant (D302H) with unknown functional consequence |
| CASP8 | rs3834129 [T] | 0.19 (ASN) | Risk [162, 164] | Cancer cells, leukocytes | eQTL in mammary epithelial cells [92] and leukocytes [104]; six nucleotide deletion (AGTAAG/T) in the CASP8 promoter that disrupts SP1-mediated CASP8 expression and apoptosis in T- cells [162, 171] |
| CASP10 | rs13010627[A] | 0.06 (CEU) | Risk [162] | Cancer cells, leukocytes | Missense variant (V410I) that is predicted to be damaging [90]; potentially addititively modifies risk with rs1045485 [163] |
| BCL2L11 | rs71801447 [C] | 0.06 (CEU) | Risk [35] | Cancer cells, leukocytes | Alters 3′ UTR with unknown consequences on gene expression [88] |
| BCL2L11 | 2,903bp deletion | 0.12(ASN) | Risk of early onset metastatic disease and survival [176] | Cancer cells, leukocytes | Alters splicing of exons 3 and 4, resulting in preferential exclusion of the pro-apoptotic BH3 domain from the variant allele [177] |
| EFEMP2 | rs3903072 [T] | 0.46 (CEU) | Risk [68] | Fibroblasts, endothelial cells, smooth muscle cells | eQTL in fibroblasts [92] and leukocytes [81] |
| EFEMP2 | rs200340088[C] | 0.001 (ASN) | Risk [183] | Fibroblasts, endothelial cells, smooth muscle cells | Missense mutation that is predicted to be potentially damaging [90] |
| EFEMP2 | rs200995432[G] | 0.002 (ASN) | Risk [183] | Fibroblasts, endothelial cells, smooth muscle cells | Missense mutation that is predicted to be potentially damaging [90] |
| SDF-1 | rs1801157 [T] | 0.25 (CEU) | Risk [188, 189] and survival [186] | Cancer cells, fibroblasts, leukocytes | Alters 3′ UTR and is associated with lower circulating SDF-1 levels [172]. |
| IL6 | rs1800795[C] | 0.42 (CEU) | Risk [190] and survival [191, 192] | Cancer cells, fibroblasts, leukocytes | Alters putative regulatory region(s) of the IL6 promoter and affects IL6 expression |
| IL8 | rs4073 [A] | 0.41 (CEU) | Risk [193] | Cancer cells, fibroblasts, leukocytes | eQTL in leukocytes [104] |
| TNFa | rs1800629 [A] | 0.06 (ASN) | Risk [194] | Cancer cells, fibroblasts, leukocytes | eQTL in leukocytes [104] |
| COL1A2 | rs17268829[C] | 0.28 (CEU) | Risk [35] | Cancer cells, fibroblasts, | Functional consequences of variant are unknown |
| AKAP9 | rs6964587 [T] | 0.39 (CEU) | Risk [35] | Cancer cells, fibroblasts, endothelial cells, leukocytes | eQTL in leukocytes [92, 104] |
| KRIT-1 | rs6964587 [T] | 0.39 (CEU) | Risk [35] | Endothelial cells, leukocytes | eQTL in leukocytes [104] |
| TNSF10 | rs58058861 [A] | 0.19(CEU) | Risk [35] | Cancer cells, fibroblasts, endothelial cells, leukocytes | Functional consequences of variant are unknown |
| PDCD6 | rs116095464 [C] | 0.05 (CEU) | Risk [35] | Cancer cells, fibroblasts, endothelial cells, leukocytes | eQTL in fibroblasts |
| HIVEP3 | rs79724016[G] | 0.03 (CEU) | Risk [35] | Cancer cells, leukocytes | Functional consequences of variant are unknown. HIVEP3 transcript expression is significantly associated with outcome in TCGA dataset [91] |
| CEBPB | rs6122906[G] | 0.20 (CEU) | Risk [35] | Cancer cells, fibroblasts, endothelial cells, leukocytes | eQTL in leukocytes [104] |
| FOXP1 | rs6805189 [C] | 0.48 (CEU) | Risk [35] | Cancer cells, fibroblasts, endothelial cells, leukocytes | Functional consequences of variant are unknown |
MAF (mean allelic frequency) is based on the reported allelic frequences in the 1000 Genomes Project.
Functional evidence was obtained from HaploReg [88], RegulomeDB [89], Variant Effect Predictor [90], TCGA/OncoLnc [91], and GTEx [99]. eQTL, expression quantitative trait loci.
Cited references
Tumor growth factor beta receptor type 2 (TGFβR2)
TGFβR2 is expressed in mammary epithelial cells and multiple TME cell types (such as CAFs, vascular endothelium, and infiltrating leukocytes), and TGFβR2 signaling regulates stages of mammary tumorigenesis through epithelial-TME crosstalk, as demonstrated in several tissue-specific knockout models [93–99] (for a comprehensive review see [100]). TGFβR2 signaling in mammary epithelial cells suppresses early tumorigenesis [95–99, 101]. Paradoxically, TGFβR2 suppresses or promotes metastatic progression, depending on whether epithelial mammary TGFβR2 signaling is partially inhibited [95–99] or completely ablated [101], respectively. TGFβR2 signaling in mammary fibroblasts is also critical for maintaining homeostasis in the mammary gland, as evidenced by fibroblast-specific deletion of TGFβR2, which elevated normal mammary epithelium branching [102] and increased angiogenesis, growth, and invasiveness of breast tumors [93, 94]. TGFβR2 signaling in T lymphocytes also suppresses antitumor immunity, and upregulation of TGFβ (a ligand of TGFβR2) by malignant tumor cells is a mechanism for evading immune surveillance [103].
A recent GWAS [68] and two candidate gene studies [69, 70] correlated three independent TGFβR2 haplotypes with breast cancer risk (rs12493607 [68], rs4522809 [69], and rs1078985 [70]), yet the functional variants within these haplotypes remain unknown. All three haplotypes only included intronic SNPs in LD (r2 < 0.8), with variants in each haplotype predicted to overlap with putative transcriptional regulatory regions of TGFβR2 in mammary epithelial cells, fibroblasts, and peripheral blood leukocytes [88]. Both rs12493607 and rs1078985 were associated with altered TGFβR2 expression in peripheral blood leukocytes [104], suggesting that these risk alleles might modify breast cancer risk by altering TGFβR2-mediated antitumor immunity. The modifying effect(s) of these alleles on TGFβR2-dependent functions in other TME cells types has yet to be established.
In addition to the TGFβR2 haplotypes [68–70], a missense SNP in TGFβ (rs1800470; L10P) was linked with breast cancer risk [105–109] caused by higher levels of TGFβ secretion [109]. Collectively, the evidence indicates that breast cancer risk is associated with both receptor (TGFβR2) and ligand (TGFβ). In addition, there are equally strong implications of TGFβR2/TGFβ-dependent mammary epithelium-TME crosstalk, which suggests that complex genetic interactions between TGFβR2 and TGFβ risk alleles might also exist. Correctly interpreting the risk alleles affecting both malignant and nonmalignant cellular compartments within breast tumors will likely improve stratification of the risk associated breast cancer incidence and adverse outcomes.
A breast cancer GWAS candidate region containing the FOXP1 transcription factor [35] is yet another locus with ties to TGFβR2/TGFβ signaling [110], in addition to a cancer cell autonomous role in modulating ERα signaling and regulates breast cancer cell invasiveness [111, 112]. FOXP1 is also a critical downstream mediator of TGFβR2/TGFβ-dependent suppression of antitumor T lymphocytes [110], a key mechanism that is used by cancer cells to evade immune surveillance [103]. Since TGFβR2, TGFβ, and FOXP1 have all been linked with breast cancer risk, it is possible that interactions between these modifier alleles might have amplifying or dampening effects on TME-mediated breast cancer risk and outcome. In the CEU population, the genetically unlinked risk haplotypes for TGFβR2 (rs12493607; mean allelic frequency [MAF] = 0.33), TGFβ (rs1800470, MAF = 0.40), and FOXP1 (rs6805189; MAF = 0.48) are expected to be co-inherited in ~6% of breast cancer cases. If all three TME modifier alleles are hypermorphic (i.e., increasing protein expression or activity), one might predict that elevated TGFβ secretion by cancer cells and CAFs would elicit a more robust TGFβR2-mediated suppression of antitumor T lymphocytes, which in turn might increase breast cancer risk, limit the effectiveness of antitumor immune therapies, and potentially worsen outcome.
Fibroblast growth factor receptor 2 (FGFR2)
FGFR2 is a receptor tyrosine kinase that is expressed in malignant breast epithelial cells, CAFs, vascular endothelial cells, and possibly other TME cell types (for a comprehensive review see [113]). Multiple GWAS [13, 65–67] and candidate gene studies [114] linked the FGFR2 locus with breast cancer risk. Of the SNPs in LD (r2 < 0.8) with the tagged FGFR2 risk allele (rs2981582 [G > A]), a functional SNP (rs2981578 [C > T]) in the FGFR2 promoter was correlated with FGFR2 mRNA expression in normal and malignant breast tissues [115, 116]. Notably, these studies did not differentiate mRNA expression between cancer cells and the TME [115, 116]. However, another study demonstrated that rs2981578 correlated with FGFR2 expression in patient-derived fibroblasts, which also coincided with altered FRS2α and ERK1/2 phosphorylation in response to stimulation by the FGFR2 ligand, FGF10 [114]. Unexpectedly, another study found no effects of the same variant (rs2981578) on proliferation or cell cycle progression of MCF7 breast cancer cells [117]. Collectively, these data suggest that the breast cancer risk associated with rs2981578 might depend on altered FGFR2 signaling in CAFs [114] rather than malignant breast cancer cells [117], though this has yet to be fully explored. Thus, although FGFR2 regulates mammary epithelial cell physiology and pathophysiology, it is equally plausible that the FGFR2 polymorphisms modify breast cancer risk through the actions of FGFR2 signaling in CAFs [114] and vascular endothelial cells [118] within the host TME.
Endothelial Nitric Oxide Synthesis (eNOS/NOS3)
eNOS colocalizes primarily to the tumor blood and lymphatic vasculature [76, 119], but it is also aberrantly expressed in some breast cancer cells [76, 77]. Nitric oxide (NO) production by eNOS substantially mediates tumor angiogenesis [120] and lymphangiogenesis [119, 120], with both positive and negative roles that are dependent upon NO production duration and extent [120–122]. To date, at least two eNOS polymorphisms (rs1799983 [G > T] and rs2070744 [T > C]) have been correlated with breast cancer risk [71–75]. The rs1799983 risk allele causes an E298D substitution that was originally associated with proteolytic cleavage of eNOS and lower NO levels [123]; however, several subsequent studies failed to demonstrate that the E298D substitution has a major effect on eNOS activity [124, 125]. The rs2070744 risk allele is an intergenic SNP that colocalizes with a transcriptional regulatory region of the eNOS locus, which is correlated with decreased eNOS promoter activity and lower NO production [126, 127]. Although the majority of association studies have correlated minor eNOS alleles (rs1799983 [G > T] and rs2070744 [T > C]) with increased breast cancer risk [71, 73, 75], other studies showed weak or opposite effects [72, 74]. It has been noted that a potential limitation of previous eNOS association studies has been a failure to consider complex haplotypes with multiple variants that stratify eNOS function [128, 129]. Likewise, dual expression of eNOS in the cancers cells and TME has also potentially confounded the failed attempts to correlate overall eNOS expression in tumors with breast cancer outcomes [77], which should be considered in future studies.
Leukocyte Specific Protein 1 (LSP1)
LSP1 is an F-actin binding protein that is expressed by vascular endothelial cells and all hematopoietic cells [130]. In hematopoietic cells, LSP1 regulates chemotaxis, trans-endothelial migration, and motility via remodeling of F-actin (for a comprehensive review see [130]). In the vascular endothelium, LSP1 is activated by ICAM-1-mediated leukocyte adhesion [131] and maintains the endothelial barrier integrity and permeability during leukocyte extravasation [132]. LSP1 expression has not been reported in the malignant epithelial cells of breast tumors. However, LSP1 is widely associated with breast cancer risk [66, 133], outcome [134], and mammographic density [135, 136], which is a predictor of breast cancer incidence [137]. All studies of LSP1 to date have reported the strongest correlations with rs3817198 [T > C], an intronic SNP with three proximal intronic SNPs (rs72843959, rs112907808, and rs11041665) that are in LD (r2 > 0.8). Functional characterization of the rs3817198 haplotype has not been reported and variants within the haplotype have not been associated with LSP1 expression. All four SNPs colocalized within transcriptional regulatory regions of the LSP1 locus [88, 138], including putative binding sites for three key transcriptional regulators of leukocyte function: GFI1, GFI1B, and GATA1 [139–141]. Moreover, TCGA-BRCA expression data suggest that LSP1 expression is associated with breast cancer outcome [91], warranting further analysis of the functional impact of the rs3817198 haplotype in the relevant TME cell types (leukocytes and vascular endothelium). LSP1 expression is likely cell-type dependent and modulated in response to multiple stimuli under different physiological and pathophysiological conditions [130]. Thus, differentiating LSP1 expression from multiple LSP1+ leukocyte cell types and vascular endothelium in different physiological contexts, might be critical to unraveling the LSP1-dependent mechanisms that underlie the rs3817198 risk haplotype.
Megakaryoblastic Leukemia (Translocation) 1 (MKL1)
MKL1 is a ubiquitously expressed SRF-binding transcription factor that is critical for normal function of multiple cellular compartments including the mammary gland [142, 143], vascular endothelium [144, 145], and several leukocyte lineages (such as neutrophils and macrophages) [146–149]. Two SNPs in moderate LD (r2 = 0.41) [150] and in close proximity to the MKL1 locus are associated with breast cancer risk (rs6001930 [T > C]) [68] and mammographic density (rs17001868 [A > C]) [150].Both haplotypes contain only intronic SNPs (a combined 83 in LD; r2 > 0.8), which span MKL1 and neighboring genes, SGSM3 and TNRC6B. The currently unknown mechanisms underlying the haplotypes are most likely driven by altered expression of MKL1, rather than expression of SGSM3 and TNRC6B. Of the three genes, only expression of MKL1 is associated with breast cancer survival in the TCGA-BRCA dataset [91], and MKL1 has been directly linked with both normal [142, 143] and malignant breast epithelium biology [151]. Both haplotypes are significantly associated with MKL1 expression [92] and overlap with transcriptional regulatory regions in several cell types, including mammary epithelium, leukocytes, and vascular endothelium [88, 138]. Although MKL1 potentially modifies breast cancer risk and outcome through its direct actions on breast myoepithelial cells [142, 143, 151], it is equally plausible that MKL1 also mediates its effects through multiple TME cell types. For example, MKL1 regulates NF-κB-dependent inflammatory signaling in macrophages [147–149] and the vascular endothelium [144, 145], which mediates breast cancer development and progression [152]. Like LSP1, the modifier effects of MKL1 are likely dependent upon the cell type and physiological context, which should be accounted for in future studies of the MKL1 haplotypes to establish the underlying cellular and molecular mechanisms.
Toll Like Receptors (TLR1, TRL6, TRL10)
The TLR family consists of pattern recognition receptors that respond to exogenous pathogen-associated molecular patterns (PAMPs)or endogenous damage/danger-associated molecular patterns (DAMPs), which are released by injured or dying cells of tumors and other necrotic tissues (for a comprehensive review see [153]). In normal physiology, the TLRs are predominantly expressed by mucosal epithelium and leukocytes within the innate and adaptive immune systems [153]. TLRs are also aberrantly expressed in multiple malignancies, including breast cancer [78]. An intronic SNP (rs6815814 [A > C]) residing within the tightly grouped locus that contains TLR1, TLR6, and TLR10 is associated with breast cancer risk [35]. The rs6815814 haplotype block (r2 > 0.8) includes 23 SNPs and physically spans the TLR1 and TLR10 genes, yet it is significantly associated with expression of all three genes (TLR1/6/10) in leukocytes [92, 104, 154]. Moreover, multiple SNPs within the haplotype block overlap with putative promoter and enhancer regions of all three genes [88, 138]. TLR1, TLR6, and TLR10 are structurally similar and form heterodimers with TLR2 [155, 156], although TLR2/1, TLR2/6, and TLR2/10 heterodimers are in large part functionally distinct. For example, TLR2/6-dependent production of IL6 and TNFα by tumor-associated macrophages was recently linked with metastasis of multiple cancer types, whereas the TLR2/1 heterodimer had no effect and the TLR2/10 heterodimer was not tested [157]. Compared with TLR1 and TLR2, the role of TLR10 in DAMP-mediated signaling is less defined, as no definitive ligands of TLR10 have been identified and TLR10 has only recently been characterized as a predominantly anti-inflammatory mediator [158, 159]. Nonetheless, TLR10 is a particularly enticing TME modifier candidate, as expression of TLR10 is significantly associated with breast cancer survival (unlike TLR1 and TLR6) in the TCGA-BRCA dataset [91]. The next step to defining the underlying mechanisms of the risk locus containing TLR1, TLR6, and TLR10 (rs6815814) will be to identify the molecular and cellular mediators, which might include mammary epithelial cells or multiple leukocyte cell-types within the TME.
Caspases 8 and 10 (CASP8, CASP10)
CASP8 and CASP10 are cysteine-aspartic acid proteases involved in apoptotic programmed cell death (for comprehensive reviews see [160, 161]). Although CASP8- and CASP10-mediated apoptosis is a ubiquitous mechanism that can be induced in any cell type, it is particularly critical in maintaining immune homeostasis through activation-induced cell death (AICD) in lymphocytes, NK cells, and possibly other leukocytes [160]. To date, at least three distinct CASP8 haplotypes and one CASP10 haplotype have been associated with breast cancer risk [108, 162–170] and survival [164]. The first haplotype is tagged by two SNPs (rs1830298 [T > C] [166] and rs10931936 [T > C] [167]) and includes 17 additional SNPs in LD (r2 > 0.8) with multiple associations with CASP8 and CASP10 expression across various tissues [92, 104]. The second CASP8 haplotype is a missense SNP (rs10454485) that has no predicted functionally effects [90], yet it is associated with breast cancer risk through unknown mechanism(s) in at least six independent studies [108, 165, 166, 168–170]. Interestingly, the rs1045485 risk allele has a potential interaction with a CASP10 haplotype (rs13010627) [163], an unlinked missense variant (V410I) in CASP10 that is potentially damaging [90] and have additive effects in patients with the rare coincidence of both rs10454485 and rs13010627 risk alleles [163]. The third CASP8 haplotype that is associated with breast cancer risk includes the -652 6N InsDel variant (rs3834129) [162, 164], a common variant that disrupts SP1-mediated CASP8 expression in T-cells [162, 171] and modifies CASP8-dependent apoptosis of T lymphocytes [162]. CASP8 and CASP10 modulate both cancer cell apoptosis and maintain immune cell homeostasis. Therefore, it is critical to define the modifying effects of the CASP8 and CASP10 haplotypes within different cellular and pathophysiological contexts in order to disentangle the multiple CASP8- and CASP10-dependent genetic mechanisms that might alter breast cancer risk and outcome.
BCL2-like 11 (BCL2L11)
BCL2L11 (also known as Bim) is a ubiquitous pro-apoptotic mediator of programmed cell death that blocks survival signals (such as BCL2 and BCL2L21) and activates apoptosis signals (such as BAX and BAK). In addition to regulating cancer cell apoptosis [172], BCL2L11 is also critical for maintaining immune system homeostasis via apoptotic programmed cell death [173] (for comprehensive reviews see [174, 175]). To date, at least two distinct haplotypes within the BCL2L11 locus have been associated with breast cancer risk [176] and overall survival [176]: an 8-base pair (bp) deletion found in the 3′ UTR of BCL2L11 in ~6% of Caucasians (rs71801447) [35] and a 2,903-bp deletion found in intron 2 of BCL2L11 in ~13% of East Asians [176]. The mechanism(s) underlying the Caucasian risk variant are currently unknown, but are likely driven by altered expression of BCL2L11. In comparison, the East Asian variant allele of BCL2L11 (a 2,903-bp intron 2 deletion) was shown to drive alternative splicing of exons 3 and 4, resulting in preferential exclusion of the pro-apoptotic BH3 domain from the variant allele [177]. Similar to CASP8 and CASP10 risk alleles, it appears plausible that the BCL2L11 risk alleles might directly modify breast cancer cell apoptosis, while also potentially modifying breast cancer risk and outcome by altering immune cell homeostasis and subsequently antitumor immune surveillance of cancer cells.
EGF-Containing Fibulin-Like Matrix Protein 2 (EFEMP2)
EFEMP2 (also known as fibulin-4) is a secreted ECM glycoprotein that mediates assembly of both elastin and collagen fibrils [178], which is critical for ECM homeostasis and maintaining vascular structure and function (for a comprehensive review see [179]). In a nonmalignant setting, EFEMP2 is diffusely expressed by fibroblasts [180], smooth muscle cells [181], and endothelium [178]. EFEMP2 expression in cancer is less clear, with EFEMP2 likely expressed by both malignant and nonmalignant cells. For example, EFEMP2 is upregulated in the ECM of metastatic breast tumors, although the source of EFEMP2 is unknown [182]. Multiple EFEMP2 haplotypes (rs3903072 [G > T], rs200340088 [T > C], and rs200995432 [T > G]) have been associated with breast cancer risk [68, 183]. The common rs3903072 haplotype [68] includes 21 additional SNPs in LD (r2 > 0.8) and is associated with EFEMP2 expression in fibroblasts [92] and leukocytes [104]. The rare haplotypes, rs200340088 and rs200995432 [183], are missense mutations that are potentially damaging [90] and are possibly hypomorphic or functionally distinct from other rare deleterious mutations linked with vascular defects in cutis laxa patients [179]. Although the effects of EFEMP2 polymorphisms in breast cancer are unknown, EFEMP2 is correlated with vascular density and worse outcome in cervical cancer [184]. Combined with the vascular defects attributed to rare EFEMP2 mutations [179], these data suggest that hypomorphic EFEMP2 alleles might alter tumor angiogenesis. Another possible EFEMP2-dependent mechanism is the disruption of ECM homeostasis in the mammary gland, because EFEMP2 regulates the TGFβ and LOX/LOXL pathways [179] that are widely implicated in breast cancer risk and progression [185].
Miscellaneous TME modifier candidates
The abovementioned genetic polymorphisms represent plausible breast TME modifier candidates that likely impact one or more breast TME cell-types: fibroblasts (FGFR2, TGFβR2, and EFEMP2), leukocytes (LSP1, MKL1, TLR1, TLR6, TLR10, CASP8, CASP10, BCL2L11, and EFEMP2), and vascular endothelium (eNOS/NOS3, LSP1, and EFEMP2). Less-defined but equally plausible candidates exist and are worthy of future detailed discussions. For example, CAF-derived SDF-1 drives the selective enrichment of breast cancer cells with higher bone-metastatic potential [186] and a SDF-1 risk allele has been associated with circulating SDF-1 levels [187] and breast cancer risk [187–189]. There are many other polymorphic factors that are expressed by at least one TME cell type and reside in a candidate region that is associated with at least one parameter of breast cancer risk, including IL6 [190–192], IL8 [193], TNFα [194], COL1A2 [35], AKAP9 [35], KRIT-1 [35], TNSF10 [35], PDCD6 [35], HIVEP3 [35], CEBPB [35], and FOXP1 [35]. Future mechanistic studies will be necessary to establish whether these factors influence breast cancer risk and outcomes through the host TME.
Concluding Remarks
In this review, we have highlighted the broad evidence in the literature that suggests multiple genetic modifiers of the breast TME likely exist in the human genome. However, the majority of these host TME modifier candidates have poorly defined mechanistic roles in breast cancer biology and much of the current evidence implicating host TME modifier candidates is only circumstantial. Bridging this gap will require new experimental strategies to empirically identify and test host TME modifiers of breast cancer, as well as association studies that are designed to capture cell-type-specific functions of candidate genes and their interactions with germline genetic modifiers. Finally, we posit that identifying the host TME modifiers of cancer will improve the accuracy of patient prognosis and aid in developing novel precision therapies that are matched to a patient’s risk profile in both the germline and somatic genomes.
Trends Box.
Breast tumors are interactive systems that consist of malignant cancer cells and nonmalignant cell types within the TME (such as endothelial cells, fibroblasts, and leukocytes). Crosstalk between cellular compartments influences disease risk and outcome.
Although genetic modifiers of the TME have long been suspected, they remain largely uncharacterized. It is unclear what portion of heritable breast cancer risk is influenced by host TME modifiers.
In this review, a candidate host TME modifier is defined as having a significant association with breast cancer and a reported biological role in at least one TME cell type (such as endothelial cells, fibroblasts, and leukocytes). In a careful review of >170 genetic loci associated with human breast cancer, 24 candidates were identified to likely impact breast cancer risk through the TME.
Outstanding Questions Box.
How should we systematically identify and test the existing genetic modifiers of breast cancer that act through the host TME? The current breast cancer association studies rarely differentiate host TME modifiers from mechanisms that are cancer cell-autonomous. There is a preponderance of studies that experimentally validate cancer cell-autonomous mechanisms, whereas very few experimental models exist to identify and test host TME modifiers.
Do some genetic modifiers impact both cancer cells and the host TME? The answer is probably yes. Many breast cancer candidate genes (like FGFR2, TGFβR2 and MKL1) are linked with both mammary epithelial cell function and at least one TME cell-type. Other candidates (like eNOS/NOS3 and TLRs) are typically linked with functions of TME cell types in normal physiology, yet can be aberrantly expressed in malignant cancer cells.
What is the impact of host TME modifiers on the genetic evolution of breast cancer cells that adapt to distinct host TME characteristics? A recent analysis of TCGA data (Carter et al, Cancer Discovery. 2017 Apr;7(4):410-423) suggested that some germline polymorphisms can be correlated with somatic mutations. Although, the statistical power to do so remains limited and the specific role of host TME modifiers remains to be explored.
What is the impact of host TME modifiers on responses to therapy and patient outcome? With the emergence of new antitumor immune therapies, stratifying patients based upon host TME modifiers of infiltrating immune cells will likely impact patient response to therapy. Likewise, multiple TME-secreted factors amplify or dampen the effects of conventional chemotherapy and radiation treatments. However, the impact of TME modifiers on these therapies remains largely unexplored.
Can newly designed risk association tests discover host TME modifiers in human breast cancer patients? Addressing this question will require systems approaches that overcome the limitations of current association studies. We developed one such analytical approach called HistoQTL (histological quantitative trait loci), which is a multivariable regression model that integrates germline genetic variants with cell type-specific quantitative traits in both cancer cells and the TME to identify genetic modifiers that impact disease outcome through the host TME.
Acknowledgments
We apologize to the authors whose work was not cited due to space limitations. This work was supported by a seed grant from the Wisconsin Breast Cancer Showhouse, the MCW Cancer Center, the Advancing a Healthier Wisconsin Endowment, and the Dr. Nancy Sobczak Fund for Breast Cancer (M.J.F and C.B.). Support was also received from the NCI (R01CA193343 (M. J. F)); the Mary Kay Foundation (Grant No. 024-16 (M.J.F) and 017-29 (C.B.)); a Susan G. Komen Grant #CCR17483233 (C.B); an American Cancer Society Institutional Research Grant (#86-004-26 (C.B.)); and the National Center for Research Resources, the National Center for Advancing Translational Sciences, and the Office of the Director of the NIH via the Clinical & Translational Science Institute (#8KL2TR000056 (C.B.)).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
References
- 1.Moller S, et al. The Heritability of Breast Cancer among Women in the Nordic Twin Study of Cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2016;25:145–150. doi: 10.1158/1055-9965.EPI-15-0913. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nature genetics. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 3.Foulkes WD. Inherited susceptibility to common cancers. The New England journal of medicine. 2008;359:2143–2153. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed S, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009;41:585–590. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antoniou AC, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet. 2010;42:885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Q, et al. Genome-wide association study identifies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium. Hum Mol Genet. 2011;20:4991–4999. doi: 10.1093/hmg/ddr405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox A, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39:352–358. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- 8.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher O, et al. Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study. J Natl Cancer Inst. 2011;103:425–435. doi: 10.1093/jnci/djq563. [DOI] [PubMed] [Google Scholar]
- 10.Ghoussaini M, et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet. 2012;44:312–318. doi: 10.1038/ng.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haiman CA, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat Genet. 2011;43:1210–1214. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hein R, et al. Comparison of 6q25 breast cancer hits from Asian and European Genome Wide Association Studies in the Breast Cancer Association Consortium (BCAC) PLoS One. 2012;7:e42380. doi: 10.1371/journal.pone.0042380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter DJ, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nature genetics. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siddiq A, et al. A meta-analysis of genome-wide association studies of breast cancer identifies two novel susceptibility loci at 6q14 and 20q11. Hum Mol Genet. 2012;21:5373–5384. doi: 10.1093/hmg/dds381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stacey SN, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–869. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- 16.Stacey SN, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008;40:703–706. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- 17.Thomas G, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbull C, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng W, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Closas M, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45:392–398. doi: 10.1038/ng.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–361. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Q, et al. Genome-wide association analysis in East Asians identifies breast cancer susceptibility loci at 1q32.1, 5q14.3 and 15q26.1. Nat Genet. 2014;46:886–890. doi: 10.1038/ng.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long J, et al. Genome-wide association study in east Asians identifies novel susceptibility loci for breast cancer. PLoS Genet. 2012;8:e1002532. doi: 10.1371/journal.pgen.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michailidou K, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47:373–380. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milne RL, et al. Common non-synonymous SNPs associated with breast cancer susceptibility: findings from the Breast Cancer Association Consortium. Hum Mol Genet. 2014;23:6096–6111. doi: 10.1093/hmg/ddu311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudet MM, et al. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet. 2013;9:e1003173. doi: 10.1371/journal.pgen.1003173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer KB, et al. Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am J Hum Genet. 2013;93:1046–1060. doi: 10.1016/j.ajhg.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orr N, et al. Fine-mapping identifies two additional breast cancer susceptibility loci at 9q31.2. Hum Mol Genet. 2015;24:2966–2984. doi: 10.1093/hmg/ddv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.French JD, et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunning AM, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48:374–386. doi: 10.1038/ng.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couch FJ, et al. Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nature communications. 2016;7:11375. doi: 10.1038/ncomms11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrenson K, et al. Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nature communications. 2016;7:12675. doi: 10.1038/ncomms12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyszynski A, et al. An intergenic risk locus containing an enhancer deletion in 2q35 modulates breast cancer risk by deregulating IGFBP5 expression. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michailidou K, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milne RL, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nature genetics. 2017;49:1767–1778. doi: 10.1038/ng.3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature genetics. 2013;45:353–361. 361e351–352. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nielsen FC, et al. Hereditary breast and ovarian cancer: new genes in confined pathways. Nature reviews Cancer. 2016;16:599–612. doi: 10.1038/nrc.2016.72. [DOI] [PubMed] [Google Scholar]
- 39.Flister MJ, et al. Identifying multiple causative genes at a single GWAS locus. Genome research. 2013;23:1996–2002. doi: 10.1101/gr.160283.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smits BM, et al. The non-protein coding breast cancer susceptibility locus Mcs5a acts in a non-mammary cell-autonomous fashion through the immune system and modulates T-cell homeostasis and functions. Breast cancer research : BCR. 2011;13:R81. doi: 10.1186/bcr2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, et al. Human MCS5A1 candidate breast cancer susceptibility gene FBXO10 is induced by cellular stress and correlated with lens epithelium-derived growth factor (LEDGF) Molecular carcinogenesis. 2014;53:300–313. doi: 10.1002/mc.21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samuelson DJ, et al. Rat Mcs5a is a compound quantitative trait locus with orthologous human loci that associate with breast cancer risk. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6299–6304. doi: 10.1073/pnas.0701687104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flister MJ, et al. CXM - a new tool for mapping breast cancer risk in the tumor microenvironment. Cancer research. 2014 doi: 10.1158/0008-5472.CAN-13-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flister MJ, et al. Host genetic modifiers of nonproductive angiogenesis inhibit breast cancer. Breast cancer research and treatment. 2017 doi: 10.1007/s10549-017-4311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagtap J, et al. Methods for detecting host genetic modifiers of tumor vascular function using dynamic near-infrared fluorescence imaging. Biomed Opt Express. 2018;9:543–556. doi: 10.1364/BOE.9.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Voyer T, et al. Three loci modify growth of a transgene-induced mammary tumor: suppression of proliferation associated with decreased microvessel density. Genomics. 2001;74:253–261. doi: 10.1006/geno.2001.6562. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi T, et al. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Molecular and cellular biology. 2003;23:1817–1831. doi: 10.1128/MCB.23.5.1817-1831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lesueur F, et al. Allelic association of the human homologue of the mouse modifier Ptprj with breast cancer. Human molecular genetics. 2005;14:2349–2356. doi: 10.1093/hmg/ddi237. [DOI] [PubMed] [Google Scholar]
- 49.Dux A, Demant P. MHC-controlled susceptibility to C3H-MTV-induced mouse mammary tumors is predominantly systemic rather than local. International journal of cancer. Journal international du cancer. 1987;40:372–377. doi: 10.1002/ijc.2910400315. [DOI] [PubMed] [Google Scholar]
- 50.Liu W, et al. Microenvironmental Influences on Metastasis Suppressor Expression and Function during a Metastatic Cell’s Journey. Cancer microenvironment : official journal of the International Cancer Microenvironment Society. 2014 doi: 10.1007/s12307-014-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olson OC, Joyce JA. Microenvironment-mediated resistance to anticancer therapies. Cell research. 2013;23:179–181. doi: 10.1038/cr.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harbor perspectives in biology. 2010;2:a003244. doi: 10.1101/cshperspect.a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nature cell biology. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H, et al. Discovery of Stromal Regulatory Networks that Suppress Ras-Sensitized Epithelial Cell Proliferation. Developmental cell. 2017;41:392–407 e396. doi: 10.1016/j.devcel.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saleh SMI, et al. Identification of Interacting Stromal Axes in Triple-Negative Breast Cancer. Cancer research. 2017 doi: 10.1158/0008-5472.CAN-16-3427. [DOI] [PubMed] [Google Scholar]
- 57.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nature medicine. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 58.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalluri R. The biology and function of fibroblasts in cancer. Nature reviews Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 60.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 61.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 62.Nico B, et al. Evaluation of microvascular density in tumors: pro and contra. Histology and histopathology. 2008;23:601–607. doi: 10.14670/HH-23.601. [DOI] [PubMed] [Google Scholar]
- 63.Ran S, et al. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology : the official journal of the International Society for Pathophysiology/ISP. 2010;17:229–251. doi: 10.1016/j.pathophys.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fridman WH, et al. The immune contexture in human tumours: impact on clinical outcome. Nature reviews Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 65.Udler MS, et al. FGFR2 variants and breast cancer risk: fine-scale mapping using African American studies and analysis of chromatin conformation. Human molecular genetics. 2009;18:1692–1703. doi: 10.1093/hmg/ddp078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barnholtz-Sloan JS, et al. FGFR2 and other loci identified in genome-wide association studies are associated with breast cancer in African-American and younger women. Carcinogenesis. 2010;31:1417–1423. doi: 10.1093/carcin/bgq128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature genetics. 2013;45:353–361. 361e351–352. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scollen S, et al. TGF-beta signaling pathway and breast cancer susceptibility. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2011;20:1112–1119. doi: 10.1158/1055-9965.EPI-11-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma X, et al. Pathway analyses identify TGFBR2 as potential breast cancer susceptibility gene: results from a consortium study among Asians. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2012;21:1176–1184. doi: 10.1158/1055-9965.EPI-12-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghilardi G, et al. Vascular invasion in human breast cancer is correlated to T–>786C polymorphism of NOS3 gene. Nitric oxide : biology and chemistry. 2003;9:118–122. doi: 10.1016/j.niox.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 72.Schneider BP, et al. Association of polymorphisms of angiogenesis genes with breast cancer. Breast cancer research and treatment. 2008;111:157–163. doi: 10.1007/s10549-007-9755-9. [DOI] [PubMed] [Google Scholar]
- 73.Choi JY, et al. Genetic polymorphisms of eNOS, hormone receptor status, and survival of breast cancer. Breast cancer research and treatment. 2006;100:213–218. doi: 10.1007/s10549-006-9245-5. [DOI] [PubMed] [Google Scholar]
- 74.Lee KM, et al. Genetic polymorphisms of NOS3 are associated with the risk of invasive breast cancer with lymph node involvement. Breast cancer research and treatment. 2007;106:433–438. doi: 10.1007/s10549-007-9506-y. [DOI] [PubMed] [Google Scholar]
- 75.Lu J, et al. Promoter polymorphism (-786t>C) in the endothelial nitric oxide synthase gene is associated with risk of sporadic breast cancer in non-Hispanic white women age younger than 55 years. Cancer. 2006;107:2245–2253. doi: 10.1002/cncr.22269. [DOI] [PubMed] [Google Scholar]
- 76.Tschugguel W, et al. Presence of endothelial calcium-dependent nitric oxide synthase in breast apocrine metaplasia. British journal of cancer. 1996;74:1423–1426. doi: 10.1038/bjc.1996.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Loibl S, et al. Immunohistochemical evaluation of endothelial nitric oxide synthase expression in primary breast cancer. Breast. 2005;14:230–235. doi: 10.1016/j.breast.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 78.Yang H, et al. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. Journal of experimental & clinical cancer research : CR. 2010;29:92. doi: 10.1186/1756-9966-29-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Freedman ML, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nature genetics. 2011;43:513–518. doi: 10.1038/ng.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nature genetics. 2013;45:371–384. 384e371–372. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.French JD, et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. American journal of human genetics. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Q, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Allinen M, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 84.LeBaron MJ, et al. In vivo response-based identification of direct hormone target cell populations using high-density tissue arrays. Endocrinology. 2007;148:989–1008. doi: 10.1210/en.2006-1219. [DOI] [PubMed] [Google Scholar]
- 85.LeBaron MJ, et al. Ultrahigh density microarrays of solid samples. Nat Methods. 2005;2:511–513. doi: 10.1038/nmeth772. [DOI] [PubMed] [Google Scholar]
- 86.Rui H, LeBaron MJ. Creating tissue microarrays by cutting-edge matrix assembly. Expert review of medical devices. 2005;2:673–680. doi: 10.1586/17434440.2.6.673. [DOI] [PubMed] [Google Scholar]
- 87.Peck AR, et al. Validation of tumor protein marker quantification by two independent automated immunofluorescence image analysis platforms. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2016;29:1143–1154. doi: 10.1038/modpathol.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40:D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome research. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McLaren W, et al. The Ensembl Variant Effect Predictor. Genome biology. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science. 2016;2:e67. [Google Scholar]
- 92.Consortium G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Busch S, et al. TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene. 2015;34:27–38. doi: 10.1038/onc.2013.527. [DOI] [PubMed] [Google Scholar]
- 94.Cheng N, et al. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muraoka-Cook RS, et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer research. 2004;64:9002–9011. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- 96.Gorska AE, et al. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. The American journal of pathology. 2003;163:1539–1549. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tang B, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. The Journal of clinical investigation. 2003;112:1116–1124. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muraoka-Cook RS, et al. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–3423. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- 99.Siegel PM, et al. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nature reviews Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 101.Forrester E, et al. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer research. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 102.Jensen BC, McLeod HL. Pharmacogenomics as a risk mitigation strategy for chemotherapeutic cardiotoxicity. Pharmacogenomics. 2013;14:205–213. doi: 10.2217/pgs.12.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 104.Westra HJ, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma X, et al. Transforming growth factorbeta1 L10P variant plays an active role on the breast cancer susceptibility in Caucasian: evidence from 10,392 cases and 11,697 controls. Breast cancer research and treatment. 2010;124:453–457. doi: 10.1007/s10549-010-0843-x. [DOI] [PubMed] [Google Scholar]
- 106.Qi X, et al. Transforming growth factor-beta1 polymorphisms and breast cancer risk: a meta-analysis based on 27 case-control studies. Breast cancer research and treatment. 2010;122:273–279. doi: 10.1007/s10549-010-0847-6. [DOI] [PubMed] [Google Scholar]
- 107.Qiu LX, et al. TGFB1 L10P polymorphism is associated with breast cancer susceptibility: evidence from a meta-analysis involving 47,817 subjects. Breast cancer research and treatment. 2010;123:563–567. doi: 10.1007/s10549-010-0781-7. [DOI] [PubMed] [Google Scholar]
- 108.Cox A, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nature genetics. 2007;39:352–358. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- 109.Dunning AM, et al. A transforming growth factorbeta1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer research. 2003;63:2610–2615. [PubMed] [Google Scholar]
- 110.Stephen TL, et al. Transforming growth factor beta-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity. 2014;41:427–439. doi: 10.1016/j.immuni.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ijichi N, et al. FOXP1 and estrogen signaling in breast cancer. Vitamins and hormones. 2013;93:203–212. doi: 10.1016/B978-0-12-416673-8.00006-X. [DOI] [PubMed] [Google Scholar]
- 112.Oskay Halacli S. FOXP1 enhances tumor cell migration by repression of NFAT1 transcriptional activity in MDA-MB-231 cells. Cell biology international. 2017;41:102–110. doi: 10.1002/cbin.10702. [DOI] [PubMed] [Google Scholar]
- 113.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 114.Huijts PE, et al. Allele-specific regulation of FGFR2 expression is cell type-dependent and may increase breast cancer risk through a paracrine stimulus involving FGF10. Breast cancer research : BCR. 2011;13:R72. doi: 10.1186/bcr2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun C, et al. rs2981582 is associated with FGFR2 expression in normal breast. Cancer genetics and cytogenetics. 2010;197:193–194. doi: 10.1016/j.cancergencyto.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meyer KB, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS biology. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Robbez-Masson LJ, et al. Functional analysis of a breast cancer-associated FGFR2 single nucleotide polymorphism using zinc finger mediated genome editing. PloS one. 2013;8:e78839. doi: 10.1371/journal.pone.0078839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Presta M, et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine & growth factor reviews. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 119.Lahdenranta J, et al. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer research. 2009;69:2801–2808. doi: 10.1158/0008-5472.CAN-08-4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fukumura D, et al. The role of nitric oxide in tumour progression. Nature reviews Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 121.Jones MK, et al. Dual actions of nitric oxide on angiogenesis: possible roles of PKC, ERK, and AP-1. Biochemical and biophysical research communications. 2004;318:520–528. doi: 10.1016/j.bbrc.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 122.Burke AJ, et al. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34:503–512. doi: 10.1093/carcin/bgt034. [DOI] [PubMed] [Google Scholar]
- 123.Tesauro M, et al. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs. glutamate at position 298. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2832–2835. doi: 10.1073/pnas.97.6.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fairchild TA, et al. Acidic hydrolysis as a mechanism for the cleavage of the Glu(298)–>Asp variant of human endothelial nitric-oxide synthase. The Journal of biological chemistry. 2001;276:26674–26679. doi: 10.1074/jbc.M103647200. [DOI] [PubMed] [Google Scholar]
- 125.McDonald DM, et al. Functional comparison of the endothelial nitric oxide synthase Glu298Asp polymorphic variants in human endothelial cells. Pharmacogenetics. 2004;14:831–839. doi: 10.1097/00008571-200412000-00006. [DOI] [PubMed] [Google Scholar]
- 126.Nakayama M, et al. T-786–>C mutation in the 5′-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation. 1999;99:2864–2870. doi: 10.1161/01.cir.99.22.2864. [DOI] [PubMed] [Google Scholar]
- 127.Wang J, et al. Haplotype-specific effects on endothelial NO synthase promoter efficiency: modifiable by cigarette smoking. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:e1–4. doi: 10.1161/01.ATV.0000016248.51577.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moreno H., Jr Genetic polymorphisms and haplotypes of eNOS in breast cancer. Breast cancer research and treatment. 2008;109:181–182. doi: 10.1007/s10549-007-9630-8. [DOI] [PubMed] [Google Scholar]
- 129.Casas JP, et al. Endothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: a HuGE review. American journal of epidemiology. 2006;164:921–935. doi: 10.1093/aje/kwj302. [DOI] [PubMed] [Google Scholar]
- 130.Jongstra-Bilen J, Jongstra J. Leukocyte-specific protein 1 (LSP1): a regulator of leukocyte emigration in inflammation. Immunologic research. 2006;35:65–74. doi: 10.1385/IR:35:1:65. [DOI] [PubMed] [Google Scholar]
- 131.Hossain M, et al. ICAM-1-mediated leukocyte adhesion is critical for the activation of endothelial LSP1. American journal of physiology Cell physiology. 2013;304:C895–904. doi: 10.1152/ajpcell.00297.2012. [DOI] [PubMed] [Google Scholar]
- 132.Petri B, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. 2011;117:942–952. doi: 10.1182/blood-2010-02-270561. [DOI] [PubMed] [Google Scholar]
- 133.Antoniou AC, et al. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Human molecular genetics. 2009;18:4442–4456. doi: 10.1093/hmg/ddp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barrdahl M, et al. Association of breast cancer risk loci with breast cancer survival. International journal of cancer. Journal international du cancer. 2015;137:2837–2845. doi: 10.1002/ijc.29446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stone J, et al. Novel Associations between Common Breast Cancer Susceptibility Variants and Risk-Predicting Mammographic Density Measures. Cancer research. 2015;75:2457–2467. doi: 10.1158/0008-5472.CAN-14-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Keller BM, et al. Associations between breast density and a panel of single nucleotide polymorphisms linked to breast cancer risk: a cohort study with digital mammography. BMC cancer. 2015;15:143. doi: 10.1186/s12885-015-1159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2006;15:1159–1169. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- 138.Roadmap Epigenomics C, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic acids research. 2014;42:2976–2987. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang DY, et al. GATA-1 mediates auto-regulation of Gfi-1B transcription in K562 cells. Nucleic acids research. 2005;33:5331–5342. doi: 10.1093/nar/gki838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hu G, et al. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome research. 2011;21:1650–1658. doi: 10.1101/gr.121145.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li S, et al. Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Molecular and cellular biology. 2006;26:5797–5808. doi: 10.1128/MCB.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sun Y, et al. Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Molecular and cellular biology. 2006;26:5809–5826. doi: 10.1128/MCB.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hayashi K, et al. A novel inhibitory mechanism of MRTF-A/B on the ICAM-1 gene expression in vascular endothelial cells. Scientific reports. 2015;5:10627. doi: 10.1038/srep10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weinl C, et al. Endothelial SRF/MRTF ablation causes vascular disease phenotypes in murine retinae. The Journal of clinical investigation. 2013;123:2193–2206. doi: 10.1172/JCI64201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Scharenberg MA, et al. Megakaryoblastic leukemia protein-1 (MKL1): Increasing evidence for an involvement in cancer progression and metastasis. The international journal of biochemistry & cell biology. 2010;42:1911–1914. doi: 10.1016/j.biocel.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 147.Song M, et al. MKL1 is an epigenetic mediator of TNF-alpha-induced proinflammatory transcription in macrophages by interacting with ASH2. FEBS letters. 2017;591:934–945. doi: 10.1002/1873-3468.12601. [DOI] [PubMed] [Google Scholar]
- 148.Yu L, et al. MKL1 defines the H3K4Me3 landscape for NF-kappaB dependent inflammatory response. Scientific reports. 2017;7:191. doi: 10.1038/s41598-017-00301-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yu L, et al. MRTF-A mediates LPS-induced pro-inflammatory transcription by interacting with the COMPASS complex. Journal of cell science. 2014;127:4645–4657. doi: 10.1242/jcs.152314. [DOI] [PubMed] [Google Scholar]
- 150.Lindstrom S, et al. Genome-wide association study identifies multiple loci associated with both mammographic density and breast cancer risk. Nature communications. 2014;5:5303. doi: 10.1038/ncomms6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Medjkane S, et al. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nature cell biology. 2009;11:257–268. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nature reviews Immunology. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 153.Bhatelia K, et al. TLRs: linking inflammation and breast cancer. Cellular signalling. 2014;26:2350–2357. doi: 10.1016/j.cellsig.2014.07.035. [DOI] [PubMed] [Google Scholar]
- 154.Lappalainen T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ozinsky A, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Guan Y, et al. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. Journal of immunology. 2010;184:5094–5103. doi: 10.4049/jimmunol.0901888. [DOI] [PubMed] [Google Scholar]
- 157.Kim S, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jiang S, et al. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. Journal of immunology. 2016;196:3834–3841. doi: 10.4049/jimmunol.1502599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Oosting M, et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4478–4484. doi: 10.1073/pnas.1410293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Green DR, et al. Activation-induced cell death in T cells. Immunological reviews. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 161.Feltham R, et al. Caspase-8: not so silently deadly. Clinical & translational immunology. 2017;6:e124. doi: 10.1038/cti.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun T, et al. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nature genetics. 2007;39:605–613. doi: 10.1038/ng2030. [DOI] [PubMed] [Google Scholar]
- 163.Frank B, et al. Association of the CASP10 V410I variant with reduced familial breast cancer risk and interaction with the CASP8 D302H variant. Carcinogenesis. 2006;27:606–609. doi: 10.1093/carcin/bgi248. [DOI] [PubMed] [Google Scholar]
- 164.Kuhlmann JD, et al. Prognostic relevance of caspase 8 -652 6N InsDel and Asp302His polymorphisms for breast cancer. BMC cancer. 2016;16:618. doi: 10.1186/s12885-016-2662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Engel C, et al. Association of the variants CASP8 D302H and CASP10 V410I with breast and ovarian cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:2859–2868. doi: 10.1158/1055-9965.EPI-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lin WY, et al. Identification and characterization of novel associations in the CASP8/ALS2CR12 region on chromosome 2 with breast cancer risk. Human molecular genetics. 2015;24:285–298. doi: 10.1093/hmg/ddu431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Turnbull C, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nature genetics. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Frank B, et al. Re: Association of a common variant of the CASP8 gene with reduced risk of breast cancer. Journal of the National Cancer Institute. 2005;97:1012. doi: 10.1093/jnci/dji178. author reply 1012-1013. [DOI] [PubMed] [Google Scholar]
- 169.MacPherson G, et al. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. Journal of the National Cancer Institute. 2004;96:1866–1869. doi: 10.1093/jnci/dji001. [DOI] [PubMed] [Google Scholar]
- 170.Shephard ND, et al. A breast cancer risk haplotype in the caspase-8 gene. Cancer research. 2009;69:2724–2728. doi: 10.1158/0008-5472.CAN-08-4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liedtke C, et al. The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be up-regulated by a p53-dependent mechanism. The Journal of biological chemistry. 2003;278:27593–27604. doi: 10.1074/jbc.M304077200. [DOI] [PubMed] [Google Scholar]
- 172.Merino D, et al. Pro-apoptotic Bim suppresses breast tumor cell metastasis and is a target gene of SNAI2. Oncogene. 2015;34:3926–3934. doi: 10.1038/onc.2014.313. [DOI] [PubMed] [Google Scholar]
- 173.Bouillet P, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 174.Hata AN, et al. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer discovery. 2015;5:475–487. doi: 10.1158/2159-8290.CD-15-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sionov RV, et al. Regulation of Bim in Health and Disease. Oncotarget. 2015;6:23058–23134. doi: 10.18632/oncotarget.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin CH, et al. High Prevalence of the BIM Deletion Polymorphism in Young Female Breast Cancer in an East Asian Country. PloS one. 2015;10:e0124908. doi: 10.1371/journal.pone.0124908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ng KP, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nature medicine. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 178.Igoucheva O, et al. Fibulin-4 E57K Knock-in Mice Recapitulate Cutaneous, Vascular and Skeletal Defects of Recessive Cutis Laxa 1B with both Elastic Fiber and Collagen Fibril Abnormalities. The Journal of biological chemistry. 2015;290:21443–21459. doi: 10.1074/jbc.M115.640425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Papke CL, Yanagisawa H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix biology : journal of the International Society for Matrix Biology. 2014;37:142–149. doi: 10.1016/j.matbio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kobayashi N, et al. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. The Journal of biological chemistry. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- 181.Huang J, et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circulation research. 2010;106:583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Naba A, et al. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife. 2014;3:e01308. doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang Y, et al. Rare coding variants and breast cancer risk: evaluation of susceptibility Loci identified in genome-wide association studies. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2014;23:622–628. doi: 10.1158/1055-9965.EPI-13-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chen J, et al. Overexpression of fibulin-4 is associated with tumor progression and poor prognosis in patients with cervical carcinoma. Oncology reports. 2014;31:2601–2610. doi: 10.3892/or.2014.3139. [DOI] [PubMed] [Google Scholar]
- 185.Bonnans C, et al. Remodelling the extracellular matrix in development and disease. Nature reviews Molecular cell biology. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang XH, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–1073. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hassan S, et al. Plasma stromal cell-derived factor-1: host derived marker predictive of distant metastasis in breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:446–454. doi: 10.1158/1078-0432.CCR-07-1189. [DOI] [PubMed] [Google Scholar]
- 188.Zhu K, et al. The CXCL12 G801A polymorphism is associated with cancer risk: a meta-analysis. PloS one. 2014;9:e108953. doi: 10.1371/journal.pone.0108953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Razmkhah M, et al. Stromal cell-derived factor-1 (SDF-1) alleles and susceptibility to breast carcinoma. Cancer letters. 2005;225:261–266. doi: 10.1016/j.canlet.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 190.Hefler LA, et al. Interleukin-1 and interleukin-6 gene polymorphisms and the risk of breast cancer in caucasian women. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:5718–5721. doi: 10.1158/1078-0432.CCR-05-0001. [DOI] [PubMed] [Google Scholar]
- 191.Iacopetta B, et al. The -174 G/C gene polymorphism in interleukin-6 is associated with an aggressive breast cancer phenotype. British journal of cancer. 2004;90:419–422. doi: 10.1038/sj.bjc.6601545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.DeMichele A, et al. Interleukin-6 -174G–>C polymorphism is associated with improved outcome in high-risk breast cancer. Cancer research. 2003;63:8051–8056. [PubMed] [Google Scholar]
- 193.Snoussi K, et al. Combined effects of IL-8 and CXCR2 gene polymorphisms on breast cancer susceptibility and aggressiveness. BMC cancer. 2010;10:283. doi: 10.1186/1471-2407-10-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li HH, et al. Tumour Necrosis Factor-alpha Gene Polymorphism Is Associated with Metastasis in Patients with Triple Negative Breast Cancer. Scientific reports. 2015;5:10244. doi: 10.1038/srep10244. [DOI] [PMC free article] [PubMed] [Google Scholar]