

SUMMARY
Hfq is an RNA chaperone and an important posttranscriptional regulator in bacteria. Using chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq), we show that Hfq associates with hundreds of different regions of the Pseudomonas aeruginosa chromosome. These associations are abolished when transcription is inhibited, indicating that they reflect Hfq binding to transcripts during their synthesis. Analogous ChIP-seq analyses with the post-transcriptional regulator Crc reveal that it associates with many of the same nascent transcripts as Hfq, an activity we show is Hfq dependent. Our findings indicate that Hfq binds many transcripts co-transcriptionally in P. aeruginosa, often in concert with Crc, and uncover direct regulatory targets of these proteins. They also highlight a general approach for studying the interactions of RNA-binding proteins with nascent transcripts in bacteria. The binding of post-transcriptional regulators to nascent mRNAs may represent a prevalent means of controlling translation in bacteria where transcription and translation are coupled.
In Brief
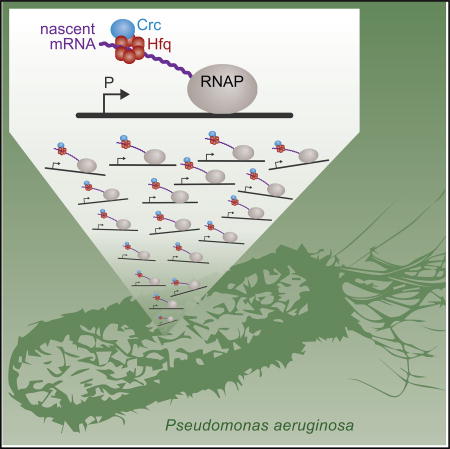
Using a modified ChIP-seq procedure, Kambara et al. find that the post-transcriptional regulator Hfq associates with hundreds of nascent transcripts in Pseudomonas aeruginosa. These findings suggest that Hfq exerts its regulatory effects on many target transcripts as soon as they emerge from RNA polymerase.
INTRODUCTION
In the face of changing environmental conditions, bacteria deploy post-transcriptional regulators as part of their rapid cellular response. Prominent among this class of regulator is Hfq, an RNA-binding protein that in many bacteria plays critical roles in modulating cellular processes such as growth, virulence, and metabolism (reviewed in Chao and Vogel, 2010; Hajnsdorf and Boni, 2012). Although Hfq is perhaps best known for its ability to facilitate base-pairing between small regulatory RNAs (sRNAs) and their target mRNAs (reviewed in Vogel and Luisi 2011; Updegrove et al., 2016), recent studies have shown that this small hexameric protein can act on mRNAs independently of sRNAs as a translational repressor (Večerek et al., 2005; Ellis et al., 2015; Chen and Gottesman 2017; reviewed in Kavita et al., 2018). In enteric bacteria, co-immunoprecipitation experiments have identified RNA-binding partners for Hfq (Zhang et al., 2003; Sittka et al., 2008; Chao et al., 2012; Tree et al., 2014; Holmqvist et al., 2016;Melamed et al., 2016), and an A-rich motif (referred to as the ARNmotif) has emerged as a specific binding site for Hfq that can be found on target mRNAs (Link et al., 2009; Tree et al., 2014). Still, questions remain about the mechanistic details of Hfq’s interaction with RNA-binding partners, particularly from a gene regulation standpoint. Indeed, transcription and translation are coupled in bacteria with RNA polymerase (RNAP) making direct contacts with the translation machinery (Miller et al., 1970; Kohler et al., 2017; Demo et al., 2017). Although the binding of Hfq to nascent transcripts could allow this regulator to exert its effects co-transcriptionally (Persson et al., 2013; Sedlyarova et al., 2016), the extent to which Hfq binds target transcripts as they are being made by RNAP is not known.
Here, in the opportunistic pathogen Pseudomonas aeruginosa, where Hfq is important for virulence (Sonnleitner et al., 2003), we use chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq) with cells grown in the presence and absence of an RNAP inhibitor to show that Hfq acts on hundreds of transcripts co-transcriptionally. Furthermore, we present evidence that in P. aeruginosa Hfq associates with many nascent transcripts together with a second post-transcriptional regulator called Crc. Our findings demonstrate the utility of using ChIP to study the association of RNA-binding proteins with nascent transcripts in bacteria and have important implications for how post-transcriptional regulators function in P. aeruginosa as well as in other bacteria.
RESULTS
Hfq Acts Co-transcriptionally on Hundreds of Transcripts in P. aeruginosa
We reasoned that if Hfq bound to transcripts emerging from RNAP, then it might be in sufficient proximity to the DNA to be crosslinked to it with formaldehyde in a ChIP assay (Figure 1A). Indeed, ChIP has been used to localize RNA-binding proteins to chromatin in both yeast and mammalian cells (Lei et al., 2001; Swinburne et al., 2006). We further reasoned that any crosslinking between Hfq and the DNA that occurred co-transcriptionally would be prevented by treatment of cells with rifampicin prior to crosslinking (Figure 1A). Therefore, to test whether we could detect the association of Hfq with nascent transcripts, we performed chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq) in the presence and absence of rifampicin. To do this, we first constructed a derivative of P. aeruginosa strain PAO1 that synthesized Hfq with a C-terminal vesicular stomatitis virus-glycoprotein (VSV-G) epitope tag (Hfq-V) from the endogenous hfq locus. ChIP-seq of Hfq-V in cells grown to mid-log in lysogeny broth (LB) identified that Hfq associates with 656 different regions of the chromosome (Figure 1B; Table S1). When we performed ChIP-seq with Hfq-V in the same cells grown to mid-log but treated with rifampicin for 30 min prior to crosslinking, we found that rifampicin either completely abolished or drastically reduced the association of Hfq-V with the vast majority of these regions (Figures 1B and 1C; Table S1). Similar findings were obtained when the association of Hfq with a particular genomic location was assessed by ChIP and qPCR (Figure 1D). Western blotting revealed that treatment of cells with rifampicin prior to crosslinking had no effect on the abundance of Hfq (Figure 1E). In addition, ChIP with a mutant version of Hfq-V (containing amino acid substitution Y25D) that is predicted to be specifically defective for binding ARN motifs in target mRNAs (Link et al., 2009; Zhang et al., 2013) resulted in little to no detectable enrichment of Hfq at three different chromosomal regions, even though the abundance of this mutant was similar to that of the wild-type protein (Figures S1A and S1B). Furthermore, the association of Hfq with genomic regions appears to occur in proximity to the translation start site of target genes (Figure 1F). Taken together, these findings indicate that Hfq associates with hundreds of different regions of the chromosome during mid-logarithmic growth in a manner that is sensitive to treatment with rifampicin. This association can be dependent upon the distal surface of Hfq that has been shown to be involved in binding ARN motifs on mRNA species (Link et al., 2009; Zhang et al., 2013). We infer from these findings that Hfq binds a plethora of transcripts co-transcriptionally in P. aeruginosa. We subsequently refer to performing ChIP with cells grown in the presence or absence of rifampicin as ChIPPAR.
Figure 1. Hfq Associates with Hundreds of Nascent Transcripts in P. aeruginosa.

(A) Schematic of ChIPPAR approach.
(B) Hfq-V enrichment peaks at the indicated genomic locations in mid-log grown cells prior to rifampicin treatment (−rif) or following rifampicin treatment (+rif). Genomic location in base pairs is provided at the top of each of the three panels. Genes are indicated by yellow bars at the bottom of each panel. Significantly enriched peaks are indicated by dark gray boxes below the read density plot (red lines within these dark gray boxes indicate site of maximum enrichment).
(C) Effect of rifampicin on enrichment of Hfq-V at 200 locations as determined by ChIP-seq. Maximum fold enrichment of Hfq-V at 200 most highly enriched regions in cells that were not treated with rifampicin (−rif, dark red bars) overlayed with fold enrichment at the corresponding genomic location in cells following rifampicin treatment (+rif, light red bars).
(D) Fold enrichment of Hfq-V at the indicated locations as determined by ChIP-qPCR in cells with (+) or without (−) rifampicin (rif). Throughout the figures, error bars denote SD.
(E) Western blot analysis of the effect of rifampicin treatment (indicated +rif) on the abundance of Hfq-V. RNAP α subunit was used as a loading control.
(F) Location of Hfq-V enrichment peaks in a 1 kb window relative to the predicted translation start (indicated ATG) of closest gene. Peaks are shown from top to bottom in decreasing order of maximum fold enrichment. Darker color represents greater fold enrichment values according to the key at the bottom of the figure.
See also Figures S1 and S4 and Tables S1 and S2.
Chromatin immunoprecipitation with cells grown in the presence or absence of rifampicin followed by high-throughput DNA sequencing (ChIPPAR-seq) of Hfq-V indicated that Hfq associated with 615 different nascent transcripts in cells grown to stationary phase (Table S2). Although many of the same nascent transcripts were associated with Hfq in both stationary phase and mid-log grown cells, many were not (Tables S1 and S2). Furthermore, for some transcripts, Hfq appeared to be associated preferentially at a particular phase of growth, likely reflecting, at least in part, growth phase-specific differences in gene expression. Our findings thus indicate that Hfq associates with hundreds of different transcripts co-transcriptionally in cells of P. aeruginosa grown to both the mid-log and stationary phases of growth.
Crc Associates with Nascent Transcripts Targeted by Hfq
Among the nascent transcripts that were most highly enriched for Hfq by ChIP-seq were some that were known to be controlled by the catabolite repression control protein Crc, a post-transcriptional regulator that plays an important role in carbon catabolite repression; these included bkdA1 encoding the α-subunit of 2-oxoisovalerate dehydrogenase, acsA encoding acetyl-coA synthetase, and estA encoding an esterase (Tables S1 and S2) (Sonnleitner et al., 2012; Sonnleitner and Bläsi, 2014). Crc is thought to function in concert with Hfq to repress the translation of target transcripts and has been shown in several instances to bind complexes formed between Hfq and RNA in vitro (Moreno et al., 2015; Wirebrand et al., 2018; Sonnleitner et al., 2018). We therefore next asked whether Crc, like Hfq, could associate with nascent transcripts in P. aeruginosa. To address this question, we constructed a strain of P. aeruginosa PAO1 in which the native copy of the crc gene had been modified such that it specified Crc with a C-terminal VSV-G epitope tag (Crc-V). ChIPPAR-seq with cells of the PAO1 Crc-V strain grown to mid-log revealed that Crc-V associated with 104 different regions of the chromosome, with the vast majority of these associations being sensitive to treatment with rifampicin (Figures 2A and S2) (Table S3). Similar findings were obtained when the association of Crc with specific genomic regions was assessed by ChIP and qPCR (Figure 2B). Western blotting revealed that treatment of cells with rifampicin did not alter the abundance of Crc (Figure 2C). Similar to our findings with Hfq, Crc appeared to be located close to the translation start site of target genes (Figure 2D). These findings indicate that like Hfq, Crc binds (either directly or indirectly) nascent transcripts in P. aeruginosa. Furthermore, although Crc does not detectably associate with many transcripts that are targeted by Hfq, essentially all of the Crc-associated nascent transcripts we identified (100 out of 104) were also found to be associated with Hfq (Tables S3 and S1).
Figure 2. Crc Associates with Nascent Transcripts Targeted by Hfq.

(A) Crc-V enrichment peaks at the indicated genomic locations in mid-log grown cells prior to rifampicin treatment (−rif) or following rifampicin treatment (+rif). Genomic location in base pairs is provided at the top of each of the three panels. Genes are indicated by yellow bars at the bottom of each panel. Significantly enriched peaks are indicated by dark gray boxes below the read density plot (red lines within dark gray boxes indicate site of maximum enrichment).
(B) Fold enrichment of Crc-V at the indicated locations as determined by ChIP-qPCR in cells with (+) or without (−) rifampicin (rif).
(C) Western blot analysis of the effect of rifampicin treatment (indicated +rif) on the abundance of Crc-V. RNAP α subunit was used as a loading control.
(D) Location of Crc-V enrichment peaks in a 1-kb window relative to the predicted translation start (indicated ATG) of closest gene. Peaks are shown from top to bottom in decreasing order of maximum fold enrichment. Darker color represents greater fold enrichment values according to the key at the bottom of the figure.
Association of Crc with Nascent Transcripts Is Hfq Dependent
In vitro studies indicate that the ability of Crc to associate with RNA is dependent upon Hfq (Milojevic et al., 2013; Moreno et al., 2015; Wirebrand et al., 2018; Sonnleitner et al., 2018). To determine whether or not this is the case for nascent transcripts, we performed ChIP with Crc-V in otherwise wild-type and Δhfq mutant cells. The results depicted in Figure 3A reveal that Crc did not appreciably associate with nascent transcripts in the absence of Hfq. Consistent with previous findings western blotting revealed that Hfq had no effect on the abundance of Crc in P. aeruginosa (Figure 3B) (Sonnleitner and Bläsi, 2014). These findings indicate that Crc associates with nascent transcripts in an Hfq-dependent fashion.
Figure 3. Hfq and Crc Influence One Another’s Association with Nascent Transcripts.

(A) Fold enrichment of Crc-V at the indicated locations determined by ChIP-qPCR in cells with or without hfq. Values obtained with PAO1 Crc-V cells are indicated WT (dark blue bars), whereas values obtained with PAO1 Δhfq Crc-V mutant cells are indicated Δhfq (light blue bars).
(B) Western blot comparing Crc-V abundance in cells used for qPCR in (A).
(C) Fold enrichment of Hfq-V at the indicated locations determined by ChIP-qPCR in cells with or without crc. Values obtained with PAO1 Hfq-V cells are indicated WT (dark red bars), whereas values obtained with PAO1 Δcrc Hfq-V mutant cells are indicated Δcrc (light red bars).
(D) Western blot comparing Hfq-V abundance in cells used for qPCR in (C). RNAP α subunit was used as a loading control (B and D).
Crc Increases the Occupancy of Hfq on Nascent Transcripts
Crc can enhance the negative effects of Hfq on the translation of target transcripts (Sonnleitner and Bläsi, 2014; Moreno et al., 2015; Wirebrand et al., 2018). To test whether Crc influences the ability of Hfq to associate with target nascent transcripts, we compared the ability of Hfq to associate with nascent transcripts by ChIP in the presence and absence of Crc. Comparison of the degree of Hfq-V enrichment as determined by ChIP and qPCR in otherwise wild-type (i.e., Crc+) and Δcrc mutant cells indicated that at multiple genomic locations, Hfq-V was more enriched in the presence of Crc than in its absence. Specifically, we found that the enrichment of Hfq at the Crc-associated bkdA1, PA1759, and estA regions was ~2 times higher in Crc+ cells than in cells of the Δcrc mutant (Figure 3C). These differences in Hfq enrichment could not be explained by differences in Hfq abundance in Crc+ and Δcrc mutant cells, as western blotting indicated that Crc had no appreciable effect on the abundance of Hfq-V (Figure 3D). These findings suggest that Crc increases the occupancy of Hfq on target nascent transcripts.
ChIP-Seq Identifies Regulatory Targets of Hfq and Crc
Translation of several of the transcripts we found to be associated with both Hfq and Crc using ChIPPAR-seq has been shown to be negatively controlled by these regulators, including that of amiE, estA, and acsA (Sonnleitner et al., 2012; Sonnleitner and Bläsi, 2014). Furthermore, many transcripts whose abundance was found previously to be altered in cells of a P. aeruginosa hfq or crc mutant (Sonnleitner et al., 2012; Sonnleitner and Bläsi, 2014; Sonnleitner et al., 2018) were identified as direct co-transcriptional targets of Hfq and Crc in our study (Tables S1 and S3). Nevertheless, many of the transcripts found to be associated with Hfq and Crc were not known previously to be subject to control by either of these proteins. To determine whether transcripts we found to be associated with both Hfq and Crc using ChIPPAR-seq might represent regulatory targets of these proteins, we asked whether Hfq and Crc influenced the translation of rbsB (encoding a putative ribose transporter), and phhA (encoding phenylalanine-4-hydroxylase), neither of which has been shown previously to be controlled at the level of translation by either Crc or Hfq.
We first made a reporter plasmid that contained a translational fusion of rbsB to lacZ. As a control, we made a reporter plasmid that contained a transcriptional fusion of the rbsB promoter to lacZ. We then introduced each of these plasmids into wild-type, Δcrc, or Δhfq mutant cells of P. aeruginosa strain PAO1 and assayed β-galactosidase activity in cells grown to mid-log. Although both Crc and Hfq repressed the expression of the rbsB translational-lacZ reporter fusion (Figure 4A), neither Crc nor Hfq had an appreciable effect on the expression of the rbsB promoter-lacZ fusion (Figure 4B). Furthermore, Crc was found to exert a smaller regulatory effect than Hfq on the expression of the rbsB translational-lacZ fusion, consistent with Crc acting to increase the occupancy of Hfq on target transcripts but itself not being absolutely required for Hfq to bind and control the translation of target transcripts (Figure 4A) (Sonnleitner and Bläsi, 2014; Moreno et al., 2015).
Figure 4. rbsB Is Subject to Control by Hfq and Crc.

(A) β-Galactosidase activity of PAO1 wild-type (WT), PAO1 Δcrc, and PAO1 Δhfq mutant cells containing a plasmid harboring an rbsB-lacZ translational fusion.
(B) β-Galactosidase activity of PAO1 wild-type (WT), PAO1 Δcrc, and PAO1 Δhfq mutant cells containing a plasmid harboring a rbsB-lacZ transcriptional fusion.
(C) Western blot of RbsB-V abundance in cells containing the native rbsB CA site. Cells used are PAO1 RbsB-V (WT), PAO1 Δcrc RbsB-V (Δcrc), PAO1 Δhfq RbsB-V (Δhfq), and PAO1 Δcrc hfq RbsB-V (Δcrc hfq). PAO1 wild-type cells (indicated cntrl) do not contain RbsB-V and were used as a negative control. Bar graph shows quantitation of RbsB-V abundance in indicated cells relative to levels in WT from a representative experiment performed with biological triplicate samples.
(D) Western blot of RbsB-V abundance in cells of the indicated strain background containing the mutant rbsB CA site (CA*). Cells used are PAO1 CA* RbsB-V (CA*), PAO1 Δcrc CA* RbsB-V (Δcrc CA*), PAO1 Δhfq CA* RbsB-V (Δhfq CA*), and PAO1 Δcrc hfq CA* RbsB-V (Δcrc hfq CA*). PAO1 wild-type cells (indicated cntrl) were used as a negative control. Bar graph shows quantitation of RbsB-V abundance in indicated cells relative to levels in CA* from a representative experiment performed with biological triplicate samples.
(E and F) Enrichment of Hfq-V (E) or Crc-V (F) as determined by ChIP-qPCR in cells containing either the WT or mutant rbsB CA motif sequence. Enrichment at bkdA1 was assessed as a negative control.
See also Figure S3.
As an additional test of whether rbsB is subject to control by Hfq and Crc, we constructed a derivative of P. aeruginosa strain PAO1 that synthesized RbsB with a C-terminal VSV-G epitope tag (RbsB-V) from the endogenous rbsB locus. We then made derivatives of this strain that lacked Crc, Hfq, or both Crc and Hfq. Quantitative western blotting revealed that RbsB-V was 7-fold more abundant in Δcrc mutant cells and 68-fold more abundant in Δhfq mutant cells than in wild-type cells (Figure 4C). Moreover, the abundance of RbsB-V was similar in cells of our Δhfq mutant and in cells of our Δcrc Δhfq double mutant, suggesting that Crc exerts its effects through Hfq (Figure 4C). These findings, taken together with those obtained with our rbsB-lacZ translational fusion and by ChIP-seq, indicate that repression of rbsB translation involves the binding of Crc and Hfq to the rbsB mRNA.
We next made a reporter plasmid that contained a translational fusion of phhA to lacZ and another that contained a transcriptional fusion of the phhA promoter to lacZ. Although Crc and Hfq repressed the expression of both these reporters, the phhA translational reporter was repressed to a much greater degree than the phhA promoter-lacZ fusion (Figures S3A and S3B). These findings, taken together with those obtained by ChIPPAR-seq, indicate that the repression of phhA mRNA translation by Crc and Hfq involves the direct binding of Hfq and Crc to the phhA transcript. They also suggest that the activity of the phhA promoter is repressed by both Crc and Hfq, possibly indirectly through effects on the abundance or activity of a transcription regulator. More generally, our findings with rbsB and phhA suggest that many of the associations we detect between nascent transcripts and Hfq/Crc could be regulatory in nature.
The Presence of a CA Site Is Important for the Association of Both Hfq and Crc with Nascent Transcripts
A common feature of known Crc-regulated genes is that they contain a so-called catabolite activity (CA) site in their 5′ UTRs, often in close proximity to the Shine-Dalgarno sequence (Sonnleitner et al., 2009; Moreno et al., 2015). This site has been reported to be AAnAAnAAnAA, where n is any nucleotide (Sonnleitner et al., 2009; Moreno et al., 2015); this sequence is essentially an ARN motif, which is recognized by the distal surface of Hfq (Link et al., 2009). Consistent with the CA site functioning as a binding site for Hfq in P. aeruginosa, post-transcriptional control of the amiE transcript, which contains a CA site, is abolished in cells synthesizing Hfq(Y25D) that cannot recognize ARN motifs (Sonnleitner and Bläsi, 2014). We noticed that many of the nascent transcripts we found to be associated with both Hfq and Crc, including rbsB, contained sequences that resembled a CA site. For rbsB, the putative CA site is AAcGAtAAgAAa positioned −28 to −17 relative to the predicted translation start site. To test whether this putative CA site was required for Crc and Hfq to control the translation of rbsB, we mutated it in cells that synthesized RbsB-V from its native chromosomal location. Consistent with this site functioning as a CA site, the synthesis of RbsB-V in cells in which this site was mutated (indicated CA*) was no longer subject to control by Crc or Hfq (Figure 4D).
To test whether the CA site we had identified for rbsB was required for the association of Hfq or Crc with the rbsB nascent transcript, we created a strain in which this site had been mutated on the PAO1 chromosome (referred to as PAO1 rbsB CA*). ChIP followed by qPCR revealed that mutation of the rbsB CA site abolished the associations of both Hfq and Crc with the rbsB region (Figures 4E and 4F). As expected, the occupancy of Hfq as well as Crc was unaltered at the bkdA1 region in cells containing a mutated rbsB CA site (Figures 4E and 4F). These findings indicate that the CA site we have defined for rbsB is required for the association of Hfq and Crc with the rbsB nascent transcript and are consistent with the idea that the association of Crc with the rbsB nascent transcript is dependent upon Hfq. We also note that the introduction of mutations into the predicted Shine-Dalgarno sequence for rbsB that are expected to abolish translation did not reduce the association of Hfq with the rbsB region, suggesting that the association of Hfq with the rbsB nascent transcript does not require ribosome binding (Figure S1C).
As additional tests of whether CA sites are important for the association of Hfq and Crc with nascent transcripts, we constructed a strain in which the putative CA site for phhA (AAcAAcAAa positioned −43 to −35 relative to the predicted translation start) had been mutated on the PAO1 chromosome. We also made a strain in which the previously defined CA site for estA (Sonnleitner et al., 2012) had been mutated on the PAO1 chromosome. ChIP followed by qPCR revealed that mutation of the putative phhA CA site dramatically reduced the associations of both Hfq and Crc with the phhA region (Figures S3C and S3D). We also found that mutation of the estA CA site abolished the association of Hfq with this region (Figure S3E). Taken together, our findings with rbsB, phhA, and estA lend further support to the notion that the associations of both Hfq and Crc with the DNA that we detect by ChIP are dependent upon the interaction of these proteins (possibly indirectly in the case of Crc) with nascent transcripts.
The sRNA PhrS Is Not Required for the Association of Hfq with a Target Nascent Transcript
Hfq can promote the base-pairing between sRNAs and their target mRNAs (Vogel and Luisi, 2011). In P. aeruginosa, the sRNA PhrS promotes translation of the mvfR transcript by targeting the transcript of a small open reading frame (referred to here as a sORF) that lies immediately upstream of mvfR (Sonnleitner et al., 2011). ChIPPAR-seq of Hfq suggested that Hfq associates directly with the nascent transcript of the sORF immediately adjacent to mvfR in cells grown to both mid-log phase and stationary phase (Figure S4A). We therefore asked whether this association is dependent upon PhrS. The results depicted in Figures S4B and S4C suggest that Hfq associates with the nascent transcript of the sORF in both wild-type cells and cells in which phrS is deleted, during both mid-log and stationary phases of growth. Note that PhrS is preferentially made during stationary phase (Sonnleitner et al., 2011) and that ectopic expression of phrS in cells grown to mid-log did not appreciably influence the association of Hfq with the sORF (Figure S4B). Thus, PhrS does not appear to be required for the association of Hfq with a target nascent transcript.
DISCUSSION
We found that Hfq and Crc act co-transcriptionally in P. aeruginosa. The binding of these two important post-transcriptional regulators to nascent transcripts may represent an efficient means of controlling the translation of specific mRNAs in this organism where transcription and translation are coupled. Our findings also demonstrate the utility of performing ChIP in the presence and absence of rifampicin for studying the interactions of RNA-binding proteins with nascent transcripts in bacteria.
Using ChIPPAR-seq, we found that Hfq associates with more than 600 different nascent transcripts in P. aeruginosa. Thus, targeting of nascent transcripts by Hfq appears to be widespread in this organism; this co-transcriptional activity of Hfq has not previously been shown to occur on such an extensive scale. Through binding nascent mRNAs, Hfq is able to exert its regulatory effects at the earliest available opportunity and possibly inhibit translation by preventing the association of the translation machinery with transcripts before they are fully mature. It is also possible that the association of Hfq with nascent transcripts could influence the activity of RNAP synthesizing a particular transcript, perhaps by preventing or facilitating the formation of an intrinsic transcription terminator or by modulating access by the transcription termination factor Rho (Figueroa-Bossi et al., 2014). Our findings suggest that in other bacteria, Hfq and other post-transcriptional regulators that function through contact with the RNA are likely to act on nascent transcripts.
Hfq associates with essentially all of the nascent transcripts that are associated with Crc, suggesting these proteins act together on these target transcripts (Tables S1 and S3). This finding is consistent with models of catabolite repression in both P. aeruginosa and Pseudomonas putida, where Hfq and Crc are thought to act together to exert their regulatory effects (Moreno et al., 2015; Wirebrand et al., 2018; Sonnleitner et al., 2018). Indeed, our ChIPPAR-seq findings with Hfq and Crc are consistent with the possibility that Crc may act together with Hfq on more than the ~100 nascent transcripts we find associated with both regulators (Tables S1 and S3). Our inability to detect Crc at many Hfq-associated nascent transcripts could simply reflect our limit of detection with ChIP of Crc; the fold enrichment of Crc at certain regions, as determined by ChIP, is in general much lower than what we find for Hfq (Tables S1 and S3).
Many of the nascent transcripts that we have identified as associated with Hfq and Crc (especially those most highly enriched by ChIP-seq) encode factors involved in the transport or utilization of carbon sources, consistent with these proteins playing key roles in the control of carbon catabolite repression (Tables S1–S3). However, the connection of other Hfq- and Crc-enriched nascent transcripts to carbon source utilization is less clear. It is striking that certain nascent transcripts we find associated with Hfq and Crc encode transcription regulators linked to the control of virulence gene expression in P. aeruginosa, including PtxS, AmrZ, and ExsA (Tables S1–S3) (Colmer and Hamood, 1998; Waligora et al., 2010; Frank and Iglewski, 1991). Thus, the regulatory effects of Hfq and Crc in P. aeruginosa may extend beyond the control of carbon catabolite repression and could reflect the need to coordinate the control of carbon source utilization with the synthesis of virulence factors.
We found that the association of Crc with nascent transcripts is Hfq dependent and that Crc can increase the occupancy of Hfq on nascent transcripts. These findings are consistent with the activities of Hfq and Crc observed in vitro with purified transcripts (Moreno et al., 2015; Wirebrand et al., 2018; Sonnleitner et al., 2018). Although we imagine that many of the regulatory effects of Crc could be explained through its ability to stabilize the binding of Hfq to specific sites on the RNA (Sonnleitner et al., 2018), it is conceivable that the regulatory role of Crc involves more than just influencing the degree with which Hfq occupies a particular transcript.
In Escherichia coli, Hfq plays a prominent role as a riboregulator by facilitating the base-pairing between sRNAs and their target mRNA species (reviewed in Vogel and Luisi, 2011). Furthermore, in E. coli, sRNAs have been shown to be able to prevent Rho-dependent transcription termination in a manner that requires the sRNA to be bound to a nascent transcript (Sedlyarova et al., 2016). It is therefore possible that the enrichment of Hfq we detect on certain nascent transcripts in P. aeruginosa might mark sites at which Hfq promotes the association of an sRNA. sRNAs are not necessarily required for Hfq to bind the mRNA species they target, and we have obtained evidence that the sRNA PhrS is not required for the association of Hfq with the corresponding nascent mRNA target in P. aeruginosa. However, in principle, the association of Hfq with a particular nascent transcript might be entirely dependent upon the action of an sRNA. Indeed, in E. coli at least one sRNA is thought to influence translation by recruiting Hfq to a specific site on the target mRNA (Desnoyers and Massé, 2012).
ChIPPAR involves enrichment of DNA that is crosslinked, either directly or indirectly, to an RNA-binding protein that is in turn bound to a nascent transcript. The rifampicin-sensitive enrichment of DNA we observed through ChIP with Hfq may reflect the crosslinking of RNA-bound Hfq directly to the DNA or reflect the crosslinking of RNAP to the DNA, which is enriched because the nascent transcript emerging from RNAP is bound to Hfq. In either case, the rifampicin-sensitive enrichment of DNA following ChIP of Hfq evidently reflects the association of Hfq with nascent transcripts.
We used ChIP-seq in combination with rifampicin treatment to identify nascent transcripts targeted by Hfq and Crc in P. aeruginosa. This approach uncovered known and previously unknown regulatory targets of Hfq and Crc in this organism and provided a compendium of potential direct regulatory targets of Hfq and Crc. We anticipate this same approach could be used to study the interactions of other RNA-binding proteins with nascent transcripts in P. aeruginosa as well as other bacteria and prove useful as a general approach in bacteria for identifying the regulatory targets of RNA-binding proteins.
EXPERIMENTAL PROCEDURES
Bacterial Strains
All experimental strains were derived from P. aeruginosa strain PAO1 and are listed in Supplemental Experimental Procedures. Cells were grown in LB at 37°C, with the following exception. For experiments that included strains lacking hfq, all strains were grown on No Carbon E (NCE) media supplemented with 20 mM succinate (Davis et al., 1980) when struck on plates or when setting up overnight cultures. These cells were then backdiluted in LB for growth prior to harvesting for experiments. Where appropriate, antibiotics were used at the following concentrations: gentamicin, 30 µg/mL (15 µg/mL in E. coli cloning and mating strains), tetracycline, 100 µg/mL (25 µg/mL in E. coli cloning and mating strains).
Plasmid and Strain Construction
All primers and plasmids used in this study, as well as details of plasmid and strain construction, are described in Supplemental Experimental Procedures.
ChIP
ChIP-seq was performed in triplicate in PAO1 Hfq-V FRT and PAO1 Crc-V. Cells were diluted from overnight cultures and grown to mid-log in 200 mL LB at 37°C with shaking, and 80 mL were collected. The remaining cells were treated with 150 µg/mL rifampicin and returned to the incubator for 30 min before another 80 mL was collected. The same experiment was performed in cells grown to stationary phase for PAO1 Hfq-V FRT and wildtype PAO1 (mock immunoprecipitation [IP] control). Upon collection, cells were immediately crosslinked with formaldehyde (final concentration 1%) for 30 min and then treated with 250 mM glycine to quench the reaction. Cells were lysed, and DNA was sheared with a Biorupter water bath sonicator (Diagenode). Lysates were combined with anti-VSV-G agarose beads for IP. After extensive washing and overnight crosslink reversal, DNA was isolated with a PCR purification kit (QIAGEN). DNA yields were measured by Nanodrop or Bioanalyzer (Agilent). For ChIP-qPCR, cells were grown in triplicate from single colonies in 3-mL volumes, and a tip sonicator was used instead of the Biorupter. For ChIP-qPCR experiments with cells ectopically expressing the sRNA PhrS, PAO1 Hfq-V FRT and PAO1 ΔphrS Hfq-V FRT cells were transformed with either pKH6 (empty vector) or pKH6-PhrS and recovered on LB plates containing gentamicin. Cells were diluted from overnight cultures grown from three individual colonies from each transformation and grown to midlog in liquid LB containing gentamicin. In the last 20 min of growth, 0.2% L-arabinose was added to cultures to induce expression of PhrS. Cells were then collected and processed for ChIP as described above.
ChIP-Seq Library Preparation and Sequencing
Sequencing libraries were constructed with the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) following the manufacturer’s instructions. Approximately 1–40 ng immunoprecipitated DNA was used, and adaptors were diluted 1:10 prior to ligation. For Hfq-V FRT and PAO1 (mock), size selection was performed using AMPure XP beads (Beckman Coulter) to yield inserts ~150 bp in length, followed by 7 rounds of PCR amplification. Crc-V and PAO1 (mock) samples underwent 10 rounds of amplification without size selection. Libraries were sequenced by Elim Biopharmaceuticals (Hayward, CA) on an Illumina HiSeq2500 producing 50-bp paired-end reads.
Data Analysis
Paired-end sequencing reads were mapped to the PAO1 genome (NCBI RefSeq NC_002516) using bowtie2 version 2.3.1 (Langmead and Salzberg, 2012). For Hfq-V FRT and corresponding PAO1 samples, only reads corresponding to fragments of 400 bp or less were used; fragments of 200 bp or less were used for Crc-V and corresponding PAO1 samples. A custom script was used to extract only read 1 from each pair and regions of enrichment were identified using QuEST, version 2.4 (Valouev et al., 2008). The appropriate PAO1 mock biological replicates were merged and used as a background control for each biological replicate. The following criteria were used to identify regions of enrichment (peaks) in the Hfq-V samples: they are 2-fold enriched in reads compared to background, have a positive peak shift and strand correlation, and have a q-value of less than 0.01. The same criteria were used to identify peaks in the Crc-V samples, only using a minimum of a 3-fold enrichment compared to background. Peaks for each immunoprecipitated protein were defined as the maximal region identified in at least two biological replicates. Data were visualized using the Integrative Genomics Viewer (IGV), version 2.3.9 (Thorvaldsdóttir et al., 2013). Peak analyses used custom scripts and BEDtools, version 2.26.0 (Quinlan and Hall, 2010). For heatmaps, the annotated gene start closest to each peak maximum (within 500 bp) was identified; if more than one peak was close to a single translation start site only the closest peak was used. For each immunoprecipitated protein, the fold enrichment value at each base pair 500 bp upstream to 500 bp downstream of the annotated gene start was identified. RStudio custom scripts and gplots were used to create graphics (R Core Team, 2016, Warnes et al., 2016).
qPCR
qPCR was performed on DNA isolated from ChIP experiments using FastStart Essential DNA Green Master (Roche) and a LightCycler 96 (Roche). Primer efficiencies were calculated by melting curve analyses. Data analyses were supported by LightCycler software version 1.1.0.1320 (Roche). Fold enrichment indicates the relative abundance of a DNA region of interest relative to a negative control region (here, we use a sequence in the gene PA2155) and the amount of DNA in the input. Specifically, we calculate fold enrichment = 1.9ΔΔCt, where ΔΔCt = (Ct_ChIPPA2155 − Ct_ChIPtarget) − (Ct_InputPA2155 − Ct_Inputtarget). Reported fold enrichments are the average of three biological replicates, and error bars denote SD. All data shown are representative of at least 2 independent experiments.
Western Blotting
Cell lysates from biological triplicates were separated by SDS-PAGE on either 4%–12% or 12% Bis-Tris NuPAGE gels in MES or MOPS running buffer (Thermo Fisher). Proteins were transferred to nitrocellulose or polyvinylidene fluoride (PVDF) membranes with the iBlot dry blotting system or the XCell II Blot Module (Thermo Fisher). Membranes were blocked overnight with SuperBlock Blocking Buffer supplemented with 0.25% Surfact-Amps (Thermo Fisher) or Odyssey Blocking Buffer diluted 1:5 in PBS (LI-COR). Membranes were probed with anti-VSV-G and/or anti-RNA polymerase α subunit and then re-blocked for up to 30 min. For qualitative western blots, membranes were then incubated with polyclonal goat anti-rabbit and/or polyclonal goat anti-mouse and proteins were detected by SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher). For quantitative western blots, membranes were incubated with near-infrared secondary antibodies, 680LT donkey anti-mouse, and 800CW donkey anti-rabbit (LI-COR). Imaging was performed on a LI-COR Odyssey CLx imager, and fluorescence intensity was quantified using Image Studio software (LI-COR).
β-Galactosidase Assays
PAO1, PAO1 Δcrc, and PAO1 Δhfq cells were transformed with pME6014-rbsB, pME6016-rbsB, pME6014-phhA, or pME6016-phhA. Three individual colonies from the resulting transformants were inoculated and grown to midlog phase. Cells were collected, mixed with Z-buffer, permeabilized with SDS and chloroform, and assayed for β-galactosidase activity using 2-nitrophenyl β-D-galactopyranoside (ONPG). Reactions were quenched with Na2CO3, and Miller units were calculated using optical density 420 (OD420) and OD550 readings as described previously (Dove and Hochschild, 2004). Reported values are the average of three biological replicates, and error bars denote SD. Representative data are shown, and experiments were performed at least twice on separate occasions.
Quantification and Statistical Analysis
For qPCR experiments, fold enrichment values shown are the mean of three biological replicates, and error bars represent SD. Experiments were performed on at least two independent occasions. For quantitative western blots, protein abundances were calculated as the mean fold change of three biological replicates. Error bars represent SD. All three replicates of all strains under comparison were run on the same blot. For β-galactosidase assays, values shown are the mean of three biological replicates, and error bars represent SD. Assays were performed at least twice on separate occasions.
Supplementary Material
Highlights.
ChIPPAR-seq identifies nascent transcripts that associate with RNA-binding proteins
RNA chaperone Hfq associates with hundreds of nascent transcripts in P. aeruginosa
Accessory factor Crc associates with many of the same nascent transcripts as Hfq
The post-transcriptional regulators Hfq and Crc frequently act co-transcriptionally
Acknowledgments
We thank Ian Hill and other members of the Dove lab for important discussions, Renate Hellmiss for artwork, and Ann Hochschild and Joseph Mougous for comments on the manuscript. We also thank Stephen Lory and Christoph Keel for plasmids and Josh Sharp for constructing the hfq deletion plasmid. Bioanalyzer analysis was performed in the Boston Children’s Hospital IDDRC Molecular Genetic Core that is supported by NIH award NIH-P30-HD 18655. This work was supported by NIH grants AI105013 and AI125876 (to S.L.D.). T.K.K. was supported by a Graduate Research Fellowship from the NSF (DGE1745303).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE111037.
Supplemental Information includes Supplemental Experimental Procedures, four figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.134.
AUTHOR CONTRIBUTIONS
T.K.K. and S.L.D. conceived and designed experiments. T.K.K. performed the experiments. T.K.K. and K.M.R. analyzed the data. T.K.K., K.M.R., and S.L.D. wrote the paper.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 2010;13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 2012;31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Gottesman S. Hfq links translation repression to stressinduced mutagenesis in E. coli. Genes Dev. 2017;31:1382–1395. doi: 10.1101/gad.302547.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmer JA, Hamood AN. Characterization of ptxS, a Pseudomonas aeruginosa gene which interferes with the effect of the exotoxin A positive regulatory gene, ptxR. Mol. Gen. Genet. 1998;258:250–259. doi: 10.1007/s004380050729. [DOI] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR. Advanced Bacterial Genetics: A Manual for Genetic Engineering. Cold Spring Harbor Laboratory; 1980. [Google Scholar]
- Demo G, Rasouly A, Vasilyev N, Svetlov V, Loveland AB, Diaz-Avalos R, Grigorieff N, Nudler E, Korostelev AA. Structure of RNA polymerase bound to ribosomal 30S subunit. eLife. 2017;6:e28560. doi: 10.7554/eLife.28560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers G, Massé E. Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev. 2012;26:726–739. doi: 10.1101/gad.182493.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SL, Hochschild A. A bacterial two-hybrid system based on transcription activation. Methods Mol. Biol. 2004;261:231–246. doi: 10.1385/1-59259-762-9:231. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Trussler RS, Haniford DB. Hfq binds directly to the ribosome-binding site of IS10 transposase mRNA to inhibit translation. Mol. Microbiol. 2015;96:633–650. doi: 10.1111/mmi.12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Bossi N, Schwartz A, Guillemardet B, D’Heygère F, Bossi L, Boudvillain M. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev. 2014;28:1239–1251. doi: 10.1101/gad.240192.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DW, Iglewski BH. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 1991;173:6460–6468. doi: 10.1128/jb.173.20.6460-6468.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnsdorf E, Boni IV. Multiple activities of RNA-binding proteins S1 and Hfq. Biochimie. 2012;94:1544–1553. doi: 10.1016/j.biochi.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Holmqvist E, Wright PR, Li L, Bischler T, Barquist L, Reinhardt R, Backofen R, Vogel J. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 2016;35:991–1011. doi: 10.15252/embj.201593360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavita K, de Mets F, Gottesman S. New aspects of RNA-based regulation by Hfq and its partner sRNAs. Curr. Opin. Microbiol. 2018;42:53–61. doi: 10.1016/j.mib.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Mooney RA, Mills DJ, Landick R, Cramer P. Architecture of a transcribing-translating expressome. Science. 2017;356:194–197. doi: 10.1126/science.aal3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei EP, Krebber H, Silver PA. Messenger RNAs are recruited for nuclear export during transcription. Genes Dev. 2001;15:1771–1782. doi: 10.1101/gad.892401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link TM, Valentin-Hansen P, Brennan RG. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl. Acad. Sci. USA. 2009;106:19292–19297. doi: 10.1073/pnas.0908744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S, Peer A, Faigenbaum-Romm R, Gatt YE, Reiss N, Bar A, Altuvia Y, Argaman L, Margalit H. Global mapping of small RNA-target interactions in bacteria. Mol. Cell. 2016;63:884–897. doi: 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller OL, Jr, Hamkalo BA, Thomas CA., Jr Visualization of bacterial genes in action. Science. 1970;169:392–395. doi: 10.1126/science.169.3943.392. [DOI] [PubMed] [Google Scholar]
- Milojevic T, Grishkovskaya I, Sonnleitner E, Djinovic-Carugo K, Blasi U. The Pseudomonas aeruginosa catabolite control protein Crc is devoid of RNA binding activity: false positive results caused by Hfq impurities. PLoS ONE. 2013;8:e64609. doi: 10.1371/journal.pone.0064609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R, Hernández-Arranz S, La Rosa R, Yuste L, Madhushani A, Shingler V, Rojo F. The Crc and Hfq proteins of Pseudomonas putida cooperate in catabolite repression and formation of ribonucleic acid complexes with specific target motifs. Environ. Microbiol. 2015;17:105–118. doi: 10.1111/1462-2920.12499. [DOI] [PubMed] [Google Scholar]
- Persson F, Lindén M, Unoson C, Elf J. Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat. Methods. 2013;10:265–269. doi: 10.1038/nmeth.2367. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2016. https://www.R-project.org/ [Google Scholar]
- Sedlyarova N, Shamovsky I, Bharati BK, Epshtein V, Chen J, Gottesman S, Schroeder R, Nudler E. sRNA-mediated control of transcription termination in E. coli. Cell. 2016;167:111–121. e13. doi: 10.1016/j.cell.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Bläsi U. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet. 2014;10:e1004440. doi: 10.1371/journal.pgen.1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Hagens S, Rosenau F, Wilhelm S, Habel A, Jäger KE, Bläsi U. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 2003;35:217–228. doi: 10.1016/s0882-4010(03)00149-9. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Abdou L, Haas D. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2009;106:21866–21871. doi: 10.1073/pnas.pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 2011;80:868–885. doi: 10.1111/j.1365-2958.2011.07620.x. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Valentini M, Wenner N, Haichar FZ, Haas D, Lapouge K. Novel targets of the CbrAB/Crc carbon catabolite control system revealed by transcript abundance in Pseudomonas aeruginosa. PLoS ONE. 2012;7:e44637. doi: 10.1371/journal.pone.0044637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Wulf A, Campagne S, Pei XY, Wolfinger MT, Forlani G, Prindl K, Abdou L, Resch A, Allain FH, et al. Interplay between the catabolite repression control protein Crc, Hfq and RNA in Hfq-dependent translational regulation in Pseudomonas aeruginosa. Nucleic Acids Res. 2018;46:1470–1485. doi: 10.1093/nar/gkx1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinburne IA, Meyer CA, Liu XS, Silver PA, Brodsky AS. Genomic localization of RNA binding proteins reveals links between pre-mRNA processing and transcription. Genome Res. 2006;16:912–921. doi: 10.1101/gr.5211806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree JJ, Granneman S, McAteer SP, Tollervey D, Gally DL. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014;55:199–213. doi: 10.1016/j.molcel.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove TB, Zhang A, Storz G. Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 2016;30:133–138. doi: 10.1016/j.mib.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Večerek B, Moll I, Bläsi U. Translational autocontrol of the Escherichia coli hfq RNA chaperone gene. RNA. 2005;11:976–984. doi: 10.1261/rna.2360205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waligora EA, Ramsey DM, Pryor EE, Jr, Lu H, Hollis T, Sloan GP, Deora R, Wozniak DJ. AmrZ beta-sheet residues are essential for DNA binding and transcriptional control of Pseudomonas aeruginosa virulence genes. J. Bacteriol. 2010;192:5390–5401. doi: 10.1128/JB.00711-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GW, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. gplots: Various R Programming Tools for Plotting Data. R package version 3.0.1. 2016 https://cran.r-project.org/web/packages/gplots/index.html.
- Wirebrand L, Madhushani AWK, Irie Y, Shingler V. Multiple Hfq-Crc target sites are required to impose catabolite repression on (methyl) phenol metabolism in Pseudomonas putida CF600. Environ. Microbiol. 2018;20:186–199. doi: 10.1111/1462-2920.13966. [DOI] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J. Mol. Biol. 2013;425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.