

Abstract
Objective
Small cell carcinoma of the ovary-hypercalcemic type (SCCOHT) is a rare disease with a poor prognosis. SCCOHT has recently been shown to be associated with SMARCA4 gene mutations as well as molecular and genetic similarities to malignant rhabdoid tumors (MRT). The objective of our study is to describe the clinical characteristics, treatment modalities and outcomes of 47 patients with SCCOHT.
Methods
We performed a retrospective analysis of 47 patients with SCCOHT evaluated at MD Anderson Cancer Center between 1990 and 2014. Medical records were reviewed for demographic information, pathologic findings, treatment regimens and outcomes.
Results
Median age at diagnosis was 30 years (range 5-46). All patients underwent surgery with unilateral salpingo-oophorectomy (USO) performed in 26 patients (55%), and hysterectomy with bilateral salpingooophorectomy (BSO) in 21 patients (45%). Sixteen patients (34.0%) had stage I disease, six (12.8%) stage II, 23 (48.9%) stage III, and two patients (4.3%) had stage IV disease. Information on adjuvant treatment was available for 43 patients: 83.3% received chemotherapy alone, 9.5% chemotherapy followed by radiotherapy, 2.4% chemoradiation, and 4.8% did not receive any adjuvant therapy. Median follow-up was 13.2 months (range, 0.1 to 210.7) with a median overall survival of 14.9 months. Multi-agent chemotherapy and radiotherapy were associated with a better prognosis.
Conclusion
Our findings suggest that aggressive therapy including multi-agent chemotherapy and possibly radiotherapy may extend survival. Further study is needed to improve outcomes in these patients including the adoption of systemic therapies used in MRT as well as the development of novel agents targeting specific mutations.
Keywords: small cell carcinoma of the ovary, hypercalcemic type, VPCBAE, radiotherapy, SMARCA4, malignant rhabdoid tumor
INTRODUCTION
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a rare disease with fewer than 300 cases reported in the literature. SCCOHT primarily affects young adult and pediatric patients with a mean age at diagnosis of 24 years.1–4 Most patients are diagnosed with advanced stage disease and do not respond to chemotherapy. Regardless of tumor stage, most patients relapse and die of disease within a short period of time, with an estimated one-year overall survival rate of 50% and a five-year overall survival rate of less than 10%.5
Recent studies have shown SCCOHT to be highly associated with germline or somatic mutations in the SMARCA4 gene; however, no therapies are yet available targeting these mutations.6–8 Furthermore, it is now known that SCCOHT has clinical, histopathological, molecular and genetic similarities to malignant rhabdoid tumors, suggesting that SCCOHT may represent a malignant rhabdoid tumor of the ovary (MRTO).8,10 Despite these recent findings, there is currently no consensus regarding the recommended treatment of SCCOHT, and management of this disease remains a challenge. In this report, we describe our experience of 47 patients with SCCOHT evaluated at a single institution. The clinical and pathologic characteristics, treatment modalities as well as outcomes are reviewed with the aim of providing hypothesis- generating data to help inform further studies and treatment protocols.
MATERIALS & METHODS
We performed a retrospective review of 47 patients with histologically confirmed SCCOHT evaluated at The University of Texas MD Anderson Cancer Center (MD Anderson) between 1990 and 2014. Institutional Review Board approval was obtained with a waiver of informed consent. Eligible patients were identified using databases from the Departments of Gynecologic Oncology, Pharmacy and Pathology. Pathologic diagnosis was confirmed by a pathologist at MD Anderson with expertise in gynecologic malignancies. Medical records were reviewed for demographic information, clinical data, pathologic findings, treatment modalities and outcomes. The follow-up period was defined as the time between initial SCCOHT diagnosis and the date of last contact.
Descriptive statistics were used to summarize patient demographic and clinical characteristics. Fisher’s exact test was used to compare categorical variables between patients with and without recurrence, while a t-test was used to compare means and the Wilcoxon test was used to compare medians of continuous variables. Overall survival (OS) was estimated using the product-limit estimator of Kaplan and Meier.11 Study data were collected and managed using the Research Electronic Data Capture (REDCap) electronic data capture tools hosted at MD Anderson.12
RESULTS
Forty-seven patients with SCCOHT were identified and included in the study. Patient demographics are shown in Table 1. The median age at diagnosis was 30 years (mean 30, range 5 to 46 years). The majority of patients were Caucasian (n=40, 85.1%). None of the patients reported a family history of SCCOHT. The majority of the patients were symptomatic and presented with abdominal pain (n=30, 63.8%) and/or distention (n=25, 53.2%). There were no reports of the tumor being found on routine pelvic examination. Preoperative serum calcium levels were available for 11 patients (23.4%) and were elevated in 10 of these 11 patients (90.1%). Preoperative CA-125 levels were available for 15 patients (31.9%) and were elevated in all 15 patients.
Table 1.
Patient demographics (N=47)
| N (%) | |
|---|---|
|
| |
| Age at diagnosis (years): | |
| Mean | 30 |
| Median | 30 |
| Range | 5–46 |
| Distribution by decade: | |
| 0–9 years | 1 (2.1%) |
| 10–19 years | 2 (4.3%) |
| 20–29 years | 19 (40.4%) |
| 30–39 years | 19 (40.4%) |
| 40–49 years | 6 (12.8%) |
|
| |
| Race: | |
| Caucasian | 40 (85.1%) |
| Hispanic | 5 (10.6%) |
| Black | 1 (2.1%) |
| Asian | 0 (0.0%) |
| Unknown | 1 (2.1%) |
|
| |
| Body Mass Index (kg/m2): | |
| Mean | 24.2 |
| Median | 22.5 |
| Range | 16.2 – 38.6 |
|
| |
| Parity: | |
| Nulliparous | 16 (34.0%) |
| Multiparous | 26 (55.3%) |
| Unknown | 5 (10.6%) |
Clinical and pathologic findings are shown in Table 2. The tumor was unilateral in the majority of cases (96.3%), with a higher proportion being on the right side (n=31, 66.0%). In 14 cases (29.8%), the tumors were initially misinterpreted as other ovarian neoplasms on final pathology read including neuroendocrine carcinoma (n=5), juvenile granulosa cell tumor (n=2), unclassified sex cord stromal tumor (n=1), embryonal carcinoma (n=2), yolk sac tumor (n=2), mixed mesodermal tumors (n=1), and desmoplastic small round cell tumor (n=1). The diagnosis of SCCOHT was subsequently confirmed at MD Anderson in all cases. A large cell component was noted in 12 patients (25.5%) and necrosis in eight (17.0%). Of note, two patients (4.3%) had a personal history of a malignant contralateral ovarian tumor: one patient had a SCCOHT diagnosed 10 years prior, and the other had a mixed germ cell tumor diagnosed 17 years prior. Unfortunately, the material from the previous diagnosis was not available for pathology review at MD Anderson to exclude SCCOHT in these original tumors.
Table 2.
Clinical and pathologic characteristics (N=47)
| N (%) | |
|---|---|
|
| |
| Tumor laterality (N=47): | |
| Right | 31 (66.0%) |
| Left | 16 (34.0%) |
|
| |
| Tumor size (cm): | |
| Mean | 15.7 |
| Median | 16.0 |
| Range | 3.6–30.0 |
|
| |
| Stage: | |
| I | 16 (34.0%) |
| II | 6 (12.8%) |
| III | 23 (48.9%) |
| IV | 2 (4.3%) |
All patients were initially treated with surgery. Twenty-six patients (55.3%) underwent unilateral salpingo-oophorectomy (USO) with or without hysterectomy, and 21 patients (44.7%) underwent total abdominal hysterectomy with bilateral salpingo-oophorectomy (TAH-BSO). Additional procedures including omentectomy, debulking of extra-ovarian tumor, lymph node dissection, and peritoneal biopsies were performed in 85% of cases. Thirty-four patients (72.3%) underwent lymph node sampling or full dissection, with positive lymph nodes were found in 19 (55.8%). Of note, 35.5% of the patients with positive nodes also had evidence of other extraovarian disease. The median tumor size was 16 cm (range 3.6 to 30.0 cm). Sixteen patients (34.0%) had stage I disease, six (12.8%) had stage II disease, 23 (48.9%) had stage III disease, and two patients (4.3%) had stage IV disease. Six patients (12.8%) had additional surgery post chemotherapy. This included three patients with stage I disease and two patients with stage II disease who had no evidence of disease in any of the specimens removed. One additional patient, initially found to have stage III disease, had a tumor debulking surgery following chemotherapy for persistent disease.
Adjuvant therapy information was available for 43 patients. One patient died three days following surgery of widespread disease and multi-organ failure prior to initiating any adjuvant therapy and is excluded from the following analysis. Thirty-five of the 42 remaining patients (83.3%) received adjuvant chemotherapy alone, 4 (9.5%) received chemotherapy followed by radiotherapy, one (2.4%) received chemoradiation, and two (4.8%) were observed (stage IA disease). The various chemotherapy regimens used as primary adjuvant therapy are shown in Table 3. The most common regimens included cisplatin/carboplatin and etoposide (CE) (n=15, 38.5%) and a multi-agent regimen including vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin and etoposide (VPCBAE) (n=10, 25.6%). The median time from surgery to initiation of chemotherapy was 30 days (mean 36, range: 12 to 79 days).
Table 3.
Primary adjuvant chemotherapy regimens (N=39)
| Chemotherapy Regimen | Number of Patients (%) |
|---|---|
| Cisplatin/carboplatin and etoposide (CE) | 15 (38.5%) |
| Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin and etoposide (VPCBAE) | 10 (25.6%) |
| Carboplatin and paclitaxel | 7 (17.9%) |
| Carboplatin, paclitaxel and bevacizumab | 1 (2.6%) |
| Bleomycin, etoposide and cisplatin (BEP) | 3 (7.7%) |
| Cisplatin, cyclophosphamide, doxorubicin and etoposide (CPAE) | 1 (2.6%) |
| Cisplatin and etoposide followed by paclitaxel and carboplatin | 1 (2.6%) |
| High dose chemotherapy (induction with carboplatin and paclitaxel × 3 cycles; mobilization with cyclophosphamide, etoposide and cisplatin × 1 cycle, followed by hyper-fractioned cyclophosphamide, doxorubicin and vincristine; followed by autologous stem cell transplant | 1 (2.6%) |
Thirty-five patients (74.5%) developed recurrent disease. The most common sites of recurrence included the pelvis, retroperitoneal lymph nodes, and the contralateral ovary. Treatment information for recurrence was available for 23 patients (65.7%) and included: chemotherapy (n=21, 91.3%), radiotherapy (n=5, 21.7%), and surgery (n=8, 34.8%). Median time to recurrence or progression was 6.5 months (range 2.3-125.4 months) for the 25 patients with date of recurrence available. There was no association between development of recurrent disease and age or stage at diagnosis (Table 4). However, a trend towards lower recurrence rates was noted in patients who received radiation as part of their primary adjuvant therapy (2/5, 40.0%) compared with those that did not (30/37, 81.1%) (p=0.0782). Median follow-up for all patients was 13.2 months (range 0.1-210.7). At last follow-up, 11 patients (23.4%) were alive without evidence of disease; six patients (12.8%) were alive with disease or receiving adjuvant treatment; and 30 (63.8%) had died of disease. Median PFS could not be calculated due to the lack of information regarding recurrence date for many patients. However, median OS was 14.9 months (95% CI 11.8 to 23.5) (Figure 1). Survival by stage is shown in Table 5.
Table 4.
Comparison of patients with and without recurrent disease
| Recurrence (N=35, 74.5%) | No Recurrence (N=12, 25.5%) | p-value | |
|---|---|---|---|
|
| |||
| Age at diagnosis (N=47): | |||
| Mean | 30.6 | 27.8 | 0.3191 |
| Median | 31.0 | 28.5 | 0.2930 |
|
| |||
| Stage (N=47): | 0.4384 | ||
| I | 10 (28.6%) | 6 (50.0%) | |
| II | 4 (11.4%) | 2 (16.7%) | |
| III | 19 (54.3%) | 4 (33.3%) | |
| IV | 2 (5.7%) | 0 (0.0%) | |
|
| |||
| Primary adjuvant therapy (N=42*): | 0.1006 | ||
| None | 2 (6.3%) | 0 (0.0%) | |
| Chemotherapy | 28 (87.5%) | 7 (70.0%) | |
| Chemotherapy followed by radiotherapy | 1 (3.1%) | 3 (30.0%) | |
| Primary chemoradiation | 1 (3.1%) | 0 (0.0%) | |
Five patients were excluded: four because the primary adjuvant treatment is unknown, and one because she died before receiving any adjuvant therapy.
Figure 1.
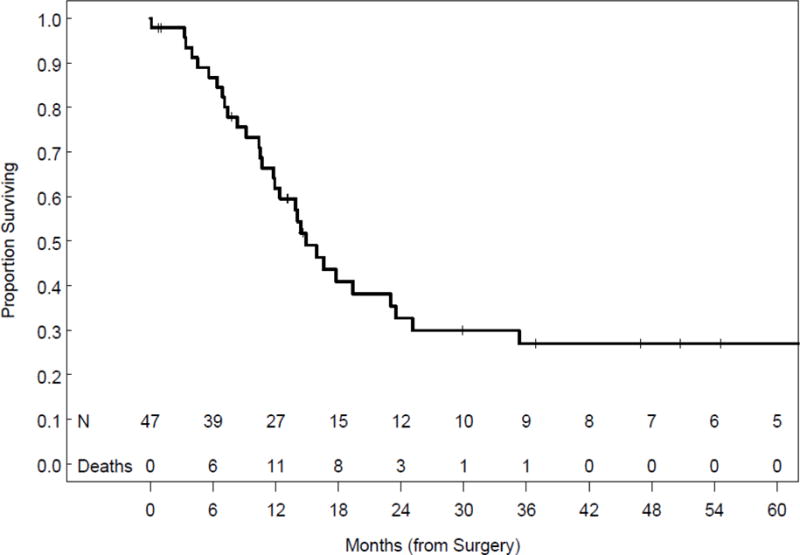
Overall survival
Table 5.
Survival by stage (N=47)
| Stage | Mean overall survival (months) |
|---|---|
| I (n=16) | 35.3 |
| II (n=6) | 12.4 |
| III (n=23) | 10.4 |
| IV (n=2) | 3.3 |
| All (n=47) | 14.9 |
A subset analysis was performed of the 16 patients with stage I disease. Median age at diagnosis was 31.0 years (mean 30.5, range 14-43). All patients underwent primary surgery, with fertility sparing USO performed in eight patients (50%). Adjuvant chemotherapy was given to 12 patients (75.0%) and consisted of VPCBAE (n=6, 50%); EP (n=5, 41.7%); and carboplatin and paclitaxel (n=1, 8.3%). Two patients did not receive adjuvant chemotherapy, and adjuvant treatment was unknown in two patients. Ten of the 16 patients (62.5%) developed recurrent disease: one of the six patients (16.7%) treated with VPCBAE, 5/5 (100.0%) treated with EP, 2/2 (100.0%) who did not receive adjuvant chemotherapy, and 2/2 (100%) where adjuvant treatment was unknown. VPCBAE was associated with a decreased rate of recurrence (p=0.008). With a median follow up of 24.3 months (mean 44.8, range 1 month – 17.5 years), seven patients (43.8%) died of disease, two patients (12.3%) were alive with disease and 7 (43.8%) were alive without evidence of disease. Of the eight patients with stage I disease who underwent USO only to retain fertility, only two patients (25%) are alive without evidence of disease. To date, there have been no reported pregnancies following treatment.
An additional analysis was performed of 12 patients with prolonged survival who were alive more than two years from time of diagnosis. Eight of these patients (67.0%) had stage I disease, two (16.5%) had stage II disease, and two (16.5%) had stage III disease. All patients underwent surgery, and seven (58.3%) received adjuvant treatment with chemotherapy alone, four (33.3%) received chemotherapy followed by radiotherapy and one (8.3%) patient received no adjuvant therapy. Five patients (41.7%) developed recurrent disease a median of 24 months (range, 8-125) from diagnosis. Treatment for recurrence included surgery (n=5, 100.0%), chemotherapy (n=3, 60.0%), and/or radiotherapy (n=3, 60.0%). Radiotherapy was used in six (50.0%) of the 12 patients with survival >2 years as adjuvant primary therapy or treatment for recurrent disease, compared with three (8.6%) of the 35 patients with survival <2 years (p=0.005).
DISCUSSION
The principle findings of our study are that women with SCCOHT are diagnosed at a young age (median = 30 years) and have a poor prognosis with a median overall survival of only 14.9 months. However, our findings suggest that aggressive therapy including multi-agent chemotherapy and possibly radiotherapy may extend survival. Similar to previous studies, favorable prognosis was shown to be associated with earlier stage disease. In our study, median overall survival was 35.3 months for patients with stage I disease compared with only 3.3 months for patients with stage IV disease. The largest study regarding SCCOHT is by Young et al.1, who performed a clinicopathological analysis of 150 patients with this disease. In their series, 50% of patients had stage I disease (33% Stage IA), 5% stage II, 43% stage III and 1% stage IV disease. Of note, 33% of the patients with stage IA disease were alive without evidence of disease with a mean follow-up of 5.7 years post surgery. In contrast, only 10% of patients with stage IC disease and 6.5% of patients with stages II, III and IV disease were also disease free (p=0.0009). Although stage IA disease was associated with a better prognosis, approximately 67% of patients with stage IA disease recurred regardless of treatment type.1
Many patients diagnosed with SCCOHT are of childbearing age and potentially interested in fertility preservation. In our study, the median age at diagnosis was 30 years (range: 5 to 46 years), which is higher than previous reports which have described a mean age of 24 years (range: 14 months to 58 years).1–4 Due to the rarity and poor prognosis of SCCOHT, fertility sparing surgery with USO has been controversial. Small case series have reported good outcomes with patients undergoing USO and adjuvant chemotherapy.5,13 However, an analysis of the various types of therapy in the previously mentioned series by Young et al.1 suggests a trend for better outcome after a procedure that includes BSO, inclusive of patients with stage IA tumors. Their report showed that 8 of 14 patients (57%) with stage IA disease who underwent BSO survived without recurrence in comparison with 5 of 21 patients (23%) who had a USO (p=0.075).1 In our cohort, 26 patients (55%) underwent USO. However, there are no reports of any of the patients having a subsequent successful pregnancy. This observation is not surprising given the small number of patients alive without disease following fertility-sparing surgery and that many of the patients received a combination of chemotherapy and/or radiotherapy, likely affecting their fertility. None of the patients had bilateral ovarian involvement, which is similar to the previously reported rate of less than 5%.1 Further research is needed to determine whether or not fertility-sparing surgery is indicated in patients with early stage SCCOHT.
The primary treatment for SCCOHT is usually surgical resection with adjuvant chemotherapy, but there is currently no standard regimen. In our study, two patients with stage IA disease did not undergo adjuvant treatment and died of recurrent disease. Many patients in our cohort and in other reports have been treated with cisplatin and etoposide based on studies of small-cell lung cancer. However, a better understanding of the biology of SCCOHT now suggests that there are no pathological or molecular similarities between these two tumor types. Several reports have also evaluated multi- agent chemotherapy for SCCOHT. Senekjian et al.15 reported the results of five patients with SCCOHT with stage IA-IIIA disease who received the combination chemotherapy regimen of VPCBAE. An objective response was observed in two patients who had measurable disease. One patient with stage IA disease was alive and disease-free at 29 months. The other four patients died of disease with a mean OS of 14 months (range, 11-18) from time of surgery.15 Long-term survival has also been reported with this regimen in a case report of a patient with stage IIIC SCCOHT diagnosed during pregnancy. She underwent optimal tumor debulking surgery followed by VPCBAE chemotherapy and was alive and without evidence of disease five and a half years from diagnosis.16 Our group also published a case series of three patients, included in this report, with stage IA disease treated with USO followed by six cycles of VPCBAE.17 The patients were alive and without evidence of disease at 37, 55 and 76 months after completing treatment. Major toxicities associated with VPCBAE include severe myelosuppression, neutropenic fever, nausea/vomiting and polyneuropathy. In the current study, VPCBAE was associated with a decreased rate of recurrence compared with cisplatin/etoposide or no adjuvant chemotherapy in a subset analysis of 16 patients with stage I disease.
High-dose chemotherapy with stem cell transplantation has also been studied in SCCOHT. Pautier et al.18 performed a prospective study of 27 patients with stage IA to IV SCCOHT who underwent surgical debulking followed by a dose-intensive regimen of four to six cycles of cisplatin, doxorubicin, etoposide and cyclophosphamide every 21 days. Patients with a complete response then underwent high-dose consolidation chemotherapy with carboplatin, etoposide and cyclophosphamide followed by autologous hematopoietic stem-cell transplantation. Eighteen patients (66.6%) achieved a complete response to the primary treatment and 37.0% of all patients (n=10) received high-dose chemotherapy and stem-cell transplantation. The median follow-up was 37 months (8–166) and the OS at one and three years was 58% and 49%, respectively. In our study, one patient underwent a similar treatment. She was diagnosed with stage IIIC disease and underwent primary surgical debulking followed by induction chemotherapy with three cycles of carboplatin and paclitaxel and then mobilization with cyclophosphamide, etoposide, and cisplatin. This was followed by hyper-fractionated cyclophosphamide, adriamycin, vincristine and autologous stem cell transplant. She developed recurrent peritoneal disease approximately one month after completing the treatment.
The role of radiotherapy in the treatment of SCCOHT is largely unknown, but there is limited information to suggest a potential benefit.1,19–21 In the series by Young et al., five of the 14 patients with stage IA disease received adjuvant radiotherapy, and four (80%) were long-term survivors.1 A study by Dickersin et al.19 described 11 patients with stage I/II SCOOHT. All patients underwent surgery, which was followed by chemotherapy in four patients (36.4%), by radiotherapy in four patients (36.4%), and by observation with no adjuvant treatment in three patients (27.3%). Interestingly, two of the patients experienced durable responses with radiotherapy alone after initial surgery.19 A subsequent study by Harrison et al.20 reported on 17 patients with SCCOHT who underwent surgery followed by adjuvant platinum-based chemotherapy. Seven of the patients also received adjuvant radiotherapy, and five (71.4%) are long-term survivors. In comparison, three of the four patients with stage I disease who did not receive radiotherapy developed recurrent disease.20 In our cohort, radiotherapy was used in six (50.0%) of the 12 patients as adjuvant primary therapy or treatment for recurrent disease with survival >2 years, compared with three (8.6%) of the 35 patients with survival <2 years (p=0.005). The results of these studies suggest that radiotherapy may play an important role in the treatment of SCCOHT. Interestingly, radiotherapy has been shown to be effective in the treatment of malignant rhabdoid tumors.22–24
There is no consensus regarding surveillance for patients with SCCOHT. In our cohort, 10 of 11 patients (90.1%) with serum calcium levels available at diagnosis were found to have hypercalcemia. Similarly, Young et al.1 reported that 62% of patients were noted to have hypercalcemia at diagnosis and that it is believed to be due to tumoral secretion of parathyroid hormone related peptide (PTHrp). Furthermore, in our cohort CA125 levels were elevated in all 15 patients who had results available. Unfortunately, we had limited and inconsistent information regarding both serum calcium and CA125 levels at the time of recurrence for the majority of patients. Despite these limited data, we would suggest using both serum calcium and CA125 levels as markers for both response and surveillance in patients with SCCOHT.
There has been significant recent progress in the understanding of the biology of SCCOHT. In 2014, three independent groups showed SCCOHT to be associated with germline or somatic deleterious mutations in the SMARCA4 gene.6–8 This gene produces a protein that is a central catalytic subunit, along with several others including SMARCB1, of the human ATP-dependent switching and sucrose non-fermenting (SWI/SNF) complex, a chromatin-remodeling complex critically involved in the control of the gene transcription within the cell.9 When the data from these three studies were combined, 64 of 69 patients (92.8%) with SCCOHT demonstrated immunohistochemical loss of tumoral SMARCA4 protein expression.6–8 In contrast, this mutation is rare in other solid tumors, having been identified in only 0.4% (2/485) of other primary ovarian cancer types.6,7 Somatic SMARCB1 alterations are also associated with the development of rhabdoid tumors including atypical teratoid/rhabdoid tumors as well as extracranial malignant rhabdoid tumors.9 In addition to sharing this genetic similarity, both SCCOHT and rhabdoid tumors have a similar histopathological appearance, are associated with hypercalcemia, as well as having a poor response to conventional therapy. Furthermore, new data on whole-exome sequencing propose that SCCOHT may actually represent a new entity known as malignant rhaboid tumour of the ovary (MRTO).8,10 These exciting recent findings are leading to a better understanding of this rare tumor and may eventually lead to improved treatment for SCCOHT.
In summary, SCCOHT is a rare and aggressive disease with a poor prognosis. Our study is limited by retrospective data collection, a small number of patients, a long study period, and possible referral bias. Despite these limitations, this report of 47 women provides information to help in the current management of SCCOHT as well as the design of future studies. Based on our findings, we currently recommend that adjuvant multi-agent chemotherapy be considered for all patients following surgery, including women with stage I disease. Furthermore, our results suggest that VPCBAE may be more effective than a platinum doublet or no treatment, even in women with stage I disease. Our study and others show that prolonged survival is rare but possible in patients with SCCOHT, even with advanced stage disease. Radiotherapy may play an important role, and consolidation radiotherapy should be considered in patients with stage I/II disease who have a good response to surgery and adjuvant chemotherapy. Finally, the newly discovered association between SCCOHT and SMARCA4 gene mutations may lead to novel agents targeting these specific mutations. In the interim, treatment regimens similar to those used to treat MRT should be considered. A prospective trial is currently under development that will use ifosfamide-carboplatin- etoposide (ICE) alternating with vincristine-doxorubicin-cyclophosphamide (VDC), a regimen used in patients with MRT, compared with our current regimen of VPBCAE in women with newly diagnosed SCCOHT.
Highlights.
SCCOHT is a rare disease that affects young women and has a poor prognosis.
Adjuvant treatment with multi-agent chemotherapy may improve survival.
Radiotherapy may improves outcomes for women with SCCOHT.
Acknowledgments
The research was supported in part by the National Institutes of Health through MD Anderson’s Cancer Center Support Grant CA016672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that there are no conflicts of interest.
References
- 1.Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol. 1994;18:1102–16. doi: 10.1097/00000478-199411000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Estel R, Hackethal A, Kalder M, et al. Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch Gynecol Obstet. 2011;284:1277–82. doi: 10.1007/s00404-011-1846-5. [DOI] [PubMed] [Google Scholar]
- 3.Florell SR, Bruggers CS, Matlak M, et al. Ovarian small cell carcinoma of the hypercalcemic type in a 14 month old: the youngest reported case. Med Pediatr Oncol. 1999;32:304–7. doi: 10.1002/(sici)1096-911x(199904)32:4<304::aid-mpo13>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 4.Shrimali RK, Correa PD, Reed NS. Dose-dense and dose-intense chemotherapy for small cell ovarian cancer: 2 cases and review of literature. Med Oncol. 2011;28:766– 70. doi: 10.1007/s12032-010-9509-0. [DOI] [PubMed] [Google Scholar]
- 5.Dykgraaf RH, de Jong D, van Veen M, et al. Clinical management of ovarian small- cell carcinoma of the hypercalcemic type: a proposal for conservative surgery in an advanced stage of disease. Int J Gynecol Cancer. 2009;19:348–53. doi: 10.1111/IGC.0b013e3181a1a116. [DOI] [PubMed] [Google Scholar]
- 6.Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427–9. doi: 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jelinic P, Mueller JJ, Olvera N, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424–6. doi: 10.1038/ng.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438–43. doi: 10.1038/ng.2931. [DOI] [PubMed] [Google Scholar]
- 9.Witkowski L, Lalonde E, Zhang J, et al. Familial rhabdoid tumour ‘avant la lettre’— From pathology review to exome sequencing and back again. J Pathol. 2013;231:35–43. doi: 10.1002/path.4225. [DOI] [PubMed] [Google Scholar]
- 10.Foulkes WD, Clarke BA, Hasselblatt M, et al. No small surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol. 2014;233:209–14. doi: 10.1002/path.4362. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958;53:457–481. [Google Scholar]
- 12.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG, Research electronic data capture (REDCap) A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woopen H, Sehouli J, Pietzner K, et al. Clinical experience of young patients with small cell ovarian carcinoma of the hypercalcemic type (OSCCHT) Eur J Obstet Gynecol Reprod Biol. 2012;165:313–7. doi: 10.1016/j.ejogrb.2012.07.034. [DOI] [PubMed] [Google Scholar]
- 14.Reed WC. Small cell carcinoma of the ovary with hypercalcemia: report of a case of survival without recurrence 5 years after surgery and chemotherapy. Gynecol Oncol. 1995;56:452–5. doi: 10.1006/gyno.1995.1081. [DOI] [PubMed] [Google Scholar]
- 15.Senekjian EK, Weiser PA, Talerman A, et al. Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin, and etoposide in the treatment of small cell carcinoma of the ovary. Cancer. 1989;64:1183–7. doi: 10.1002/1097-0142(19890915)64:6<1183::aid-cncr2820640603>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 16.Tewari K, Brewer C, Cappuccini F, et al. Advanced-stage small cell carcinoma of the ovary in pregnancy: long-term survival after surgical debulking and multiagent chemotherapy. Gynecol Oncol. 1997;66:531–4. doi: 10.1006/gyno.1997.4801. [DOI] [PubMed] [Google Scholar]
- 17.Wallbillich JJ, Nick AM, Ramirez PT, et al. Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin, and etoposide (VPCBAE) in the management of three patients with small-cell carcinoma of the ovary. Gynecol Oncol Case Rep. 2012;2:58–60. doi: 10.1016/j.gynor.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pautier P, Ribrag V, Duvillard P, et al. Results of a prospective dose-intensive regimen in 27 patients with small cell carcinoma of the ovary of the hypercalcemic type. Ann Oncol. 2007;18:1985–9. doi: 10.1093/annonc/mdm376. [DOI] [PubMed] [Google Scholar]
- 19.Dickersin GR, Kline IW, Scully RE. Small cell carcinoma of the ovary with hypercalcemia: a report of eleven cases. Cancer. 1982;49:188–97. doi: 10.1002/1097-0142(19820101)49:1<188::aid-cncr2820490137>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 20.Harrison ML, Hoskins P, du Bois A, et al. Small cell of the ovary, hypercalcemic type – analysis of combined experience and recommendation for management. A GCIG study. Gynecol Oncol. 2006;100:233–8. doi: 10.1016/j.ygyno.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 21.Callegaro-Filho D, Burke TW, Eifel PJ, et al. Radiotherapy for recurrent small cell carcinoma of the ovary: A case report and review of the literature. Gynecol Oncol Rep. 2014;11:23–5. doi: 10.1016/j.gore.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vujanić GM, Sandstedt B, Harms D, et al. Rhabdoid tumour of the kidney: a clinicophathological study of 22 patients from the International Society of Paediatric Oncology (SIOP) nephroblastoma file. Histopathology. 1996;28:333–340. doi: 10.1046/j.1365-2559.1996.d01-436.x. [DOI] [PubMed] [Google Scholar]
- 23.Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23:7641–7645. doi: 10.1200/JCO.2004.00.8110. [DOI] [PubMed] [Google Scholar]
- 24.Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer. 2010;54:35–40. doi: 10.1002/pbc.22285. [DOI] [PubMed] [Google Scholar]