

The maintenance of nucleolar integrity during interphase is essential for proper cell cycle progression to mitosis.
Abstract
The nucleolus is a dynamic nuclear body that has been demonstrated to disassemble at the onset of mitosis; the relationship between cell cycle progression and nucleolar integrity, however, remains poorly understood. We studied the role of nucleolar proteins in mitosis by performing a global analysis using small interfering RNAs specific to nucleolar proteins; we focused on nucleolar protein 11 (NOL11), with currently unknown mitotic functions. Depletion of NOL11 delayed entry into the mitotic phase owing to increased inhibitory phosphorylation of cyclin-dependent kinase 1 (Cdk1) and aberrant accumulation of Wee1, a kinase that phosphorylates and inhibits Cdk1. In addition to effects on overall mitotic phenotypes, NOL11 depletion reduced ribosomal RNA (rRNA) levels and caused nucleolar disruption during interphase. Notably, mitotic phenotypes found in NOL11-depleted cells were recapitulated when nucleolar disruption was induced by depletion of rRNA transcription factors or treatment with actinomycin D. Furthermore, delayed entry into the mitotic phase, caused by the depletion of pre-rRNA transcription factors, was attributable to nucleolar disruption rather than to G2/M checkpoint activation or reduced protein synthesis. Our findings therefore suggest that maintenance of nucleolar integrity during interphase is essential for proper cell cycle progression to mitosis via the regulation of Wee1 and Cdk1.
INTRODUCTION
The nucleolus is the largest nuclear body, and its structure changes dynamically in higher eukaryotes. The canonical function of the nucleolus is to serve as the site for ribosome biogenesis. The nucleolus forms around clusters of tandemly repeated ribosomal DNA (rDNA), where RNA polymerase I (Pol I) transcribes the rDNA repeats and generates 47S rRNAs (pre-rRNAs). The initially transcribed pre-rRNAs undergo processing to form mature 28S, 18S, and 5.8S rRNAs, which are assembled with ribosomal proteins to generate ribosomes (1). Nucleolar structure is governed by RNA levels within the nucleolus because pre-rRNAs and/or intermediate rRNAs form the scaffold for nucleolar proteins (2). Inhibition of rRNA transcription reduces the total rRNA levels within the nucleolus, thereby altering the architecture and composition of the nucleolus. In response to inhibition of rRNA transcription, some nucleolar proteins form a nucleolar cap at the nucleolar periphery, and some of the proteins translocate to the nucleoplasm—this is known as nucleolar disruption (3, 4). On the other hand, loss of rRNA processing increases nucleolar RNA levels owing to the accumulation of pre-rRNAs or intermediate rRNAs within the nucleolus, leading to enlargement of the nucleolus (5).
Recent studies have shown that cellular stress and nucleolar integrity are closely linked. Various types of intra- or extracellular stimuli decrease rRNA transcription, which in turn translocates nucleolar proteins to the nucleoplasm (6). The translocated nucleolar proteins induce stress-response events such as p53 activation (7), autophagy (8, 9), and cell differentiation (10, 11). Thus, the nucleolus may coordinate stress response by altering its architecture and composition.
The nucleolus is tightly connected to cell cycle progression. During interphase, the enzymes involved in posttranslational modifications such as ubiquitination, sumoylation, and phosphorylation are predominantly localized in the nucleolus (1). Therefore, it is possible that translocation of these enzymes in response to altered nucleolar structure would influence cell cycle progression. The most well-characterized examples of such enzymes are Cdc14B (12) and PP1γ (13), both of which respectively regulate G2/M checkpoint and chromosome segregation, in that they are sequestrated to the nucleolus and released to function at a proper time during the cell cycle. At the onset of mitosis, rRNA transcription is suppressed by cyclin-dependent kinase 1 (Cdk1) activation, leading to the disassembly of the nucleolus. When cells exit mitosis and Cdk1 is inactivated, rRNA transcription resumes and the nucleolus is reassembled. As the nucleolus is disassembled during mitosis, many nucleolar proteins translocate to the cytoplasm, a phenomenon that is similar to nucleolar disruption during interphase. A previous study used comprehensive proteome analysis to classify the molecular function of 700 nucleolar proteins and showed that some nucleolar proteins are involved in cell cycle control as well as DNA replication and repair (14). Therefore, it is reasonable to assume that some of the nucleolar proteins influence cell cycle progression by regulating cell cycle events and/or by changing their localization during mitosis.
Here, we performed a nucleolar RNA interference screen and found that nucleolar protein 11 (NOL11) controlled mitotic progression. Depletion of NOL11 also reduced rRNA transcription and nucleolar disruption during interphase. In particular, we found that the mitotic phenotypes were recapitulated when nucleolar disruption was induced by the depletion of other rRNA transcription factors. Our results showed that the temporal coordination between the cell cycle and nucleolar integrity is crucial for the proper progression of mitosis.
RESULTS
NOL11 depletion delays mitotic entry by inhibiting Cdk1 activation via abnormal Wee1 accumulation
Recent studies suggested that the nucleolus has multiple functions, including regulation of mitosis (15–17). We performed a large-scale screen with small interfering RNAs (siRNAs) against mRNAs encoding 591 nucleolar proteins (14) to identify nucleolar proteins whose knockdown increased mitotic cells in asynchronous cultures. Of the 59 candidate proteins (table S1), we focused on NOL11, whose mitotic function remained unknown.
First, we ascertained whether NOL11 was localized in the nucleolus, using HeLa cells stably expressing FLAG- and hemagglutinin-tagged NOL11. The nucleolus consists of three distinct compartments: the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC), each having a unique protein composition (fig. S1A). NOL11 largely colocalized with FBL and MYBBP1A, both of which are DFC markers (fig. S1B, middle). NOL11 also partially colocalized with UBF, an FC marker, but was excluded from the UBF foci within the nucleolus (fig. S1B, top) and slightly overlapped with nucleolin and NPM, which are GC markers (fig. S1B, bottom). These results showed that NOL11 predominantly localized in the DFC region within the nucleolus.
Next, we tested the effect of NOL11 depletion by using two different siRNAs specific to NOL11 (Fig. 1A). We found that NOL11 depletion increased the population of mitotic cells in asynchronous cultures (fig. S2A), which was in accordance with the screen results. To further analyze the effect of NOL11 depletion on cell cycle progression, we synchronized HeLa cells at the G1/S phase by thymidine blocking after NOL11 depletion. NOL11 depletion had negligible influence on the progression of the cells from 2N (G1 phase) to 4N (G2/M phase; Fig. 1B, 0 to 9 hours); however, it arrested the cell cycle with respect to the 4N cell population (Fig. 1B, 10.5 and 12 hours). Most of the control cells exited mitosis and entered the G1 phase by 12 hours after the release from G1/S synchronization, whereas most of the NOL11-depleted cells remained in the G2/M phase. To analyze cell cycle progression in further detail, we performed dual-color analysis with fluorescence-activated cell sorting (FACS), using propidium iodide (PI) staining in combination with anti–cyclin B1, anti-MPM2, and anti–H3-pS10 antibodies. This method revealed that NOL11 depletion delayed the accumulation of H3-pS10, a hallmark of mitosis (Fig. 1C, blue line). The fluctuation pattern of the MPM2 epitopes (Fig. 1C, gray line), which showed phosphorylated Cdk1 substrates and was indicative of mitosis, was similar to that of H3-pS10. In particular, NOL11 depletion did not affect the accumulation of cyclin B1 at the onset of mitosis (Fig. 1C, orange line). Nine hours after the release from G1/S synchronization, cyclin B1 accumulated normally; however, MPM2 epitopes were low in NOL11-depleted cells. We therefore assumed that NOL11 depletion delayed mitotic entry by suppressing Cdk1 activation without affecting cyclin B1 expression.
Fig. 1. NOL11 depletion delayed mitotic entry irrespective of proper accumulation of cyclin B1.
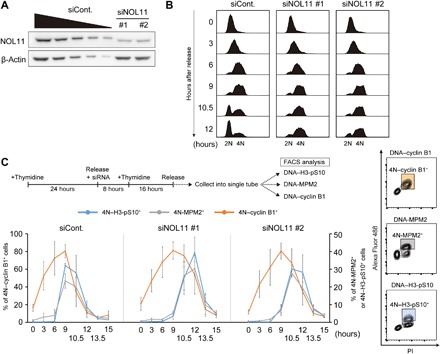
(A) The efficiency of NOL11 depletion using siRNA against NOL11. HeLa cells were transfected with luciferase siRNA (siCont.) or two distinct NOL11-specific siRNAs (siNOL11 #1 and #2). Forty-eight hours after transfection, whole-cell extracts were immunoblotted using the indicated antibodies. A dilution series of cell extracts from control cells (siCont.; lanes 1 to 5; 100, 50, 25, 12.5, and 6.3%, respectively) were performed simultaneously to estimate depletion efficiency. β-Actin was used as a loading control. (B) Arrest at the G2/M phase in NOL11-depleted cells. HeLa cells transfected with the indicated siRNAs were synchronized to the G1/S phase. After release from the thymidine block, the cells were collected at the indicated times, and the cell cycle distribution was measured by FACS using PI. (C) Delayed mitotic entry in NOL11-depleted cells. The top panel shows the scheme for siRNA treatment and cell cycle synchronization by double thymidine block. HeLa cells released from the G1/S phase were collected at the indicated times after release. The collected cells were divided into three aliquots, and each aliquot was analyzed by dual-color FACS using a combination of PI and anti–cyclin B1, anti-MPM2, or anti–H3-pS10 antibodies. The populations of 4N–cyclin B1+ cells, 4N-MPM2+ cells, and 4N–H3-pS10+ cells are indicated as colored squares in the representative plots (right panels). Line graphs indicate the percentage of these populations at the indicated time (lower left graphs). Values are shown as the means ± SD, n = 3.
We then synchronized the cells at the G2/M border using RO-3306, a potent Cdk1 inhibitor (18), and repeated the dual-color FACS experiments using anti-MPM2 antibodies to precisely examine the G2-M transition. At the G2/M border, the number of MPM2-positive cells was very low in control and NOL11-depleted cells. In control cells, removal of RO-3306 led to a rapid increase in the number of MPM2-positive cells, the proportion of which reached about 50% at 1 hour after release from the G2/M border. The number of MPM2-positive control cells gradually decreased 3 hours after release and returned to pre–RO-3306–removal levels (Fig. 2A). In contrast, in NOL11-depleted cells, MPM2-positive cells slowly increased in number over a period of 6 hours after release from the G2/M border (Fig. 2A), suggesting that Cdk1 activation is impaired by NOL11 depletion.
Fig. 2. NOL11 depletion impaired Cdk1 activation.
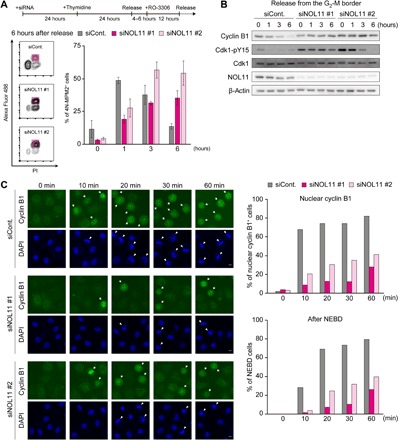
(A) Delayed Cdk1 activation after release from the G2/M border in NOL11-depleted cells. 4N-MPM2+ cells were identified by dual-color FACS. siRNA-treated HeLa cells synchronized at the G2/M border were released from the block and collected at the indicated times. Colored squares show 4N-MPM2+ cells in the representative plots at 6 hours after release (left panels). The right graph shows the percentage of 4N-MPM2+ cells at the indicated times after release from the G2/M border. Values are expressed as means ± SD, n = 3. (B) Increased Cdk1-pY15 in NOL11-depleted cells. Cells were synchronized and collected as shown in (A). The whole-cell extracts were immunoblotted with the indicated antibodies. (C) Delayed nuclear translocation of cyclin B1 and NEBD in NOL11-depleted cells. HeLa cells were released from RO-3306 synchronization. At the indicated times, cells were fixed and stained with anti–cyclin B1 antibody (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Arrows and arrowheads indicate cyclin B1 translocated into the nucleus and cells with NEBD, respectively. Scale bars, 10 μm. The percentage of cyclin B1 translocated into the nucleus (upper right graph) and NEBD (lower right graph) is shown. Over 200 cells were counted at each time point for each siRNA.
Cdk1 activity is regulated by removal of inhibitory phosphorylation of Cdk1 in addition to increased cyclin B expression. To examine the phosphorylation status of Cdk1 during the G2-M transition, we performed immunoblotting after synchronization at the G2/M border. When the cells were released from the G2/M border, cyclin B1 levels in control cells gradually decreased in a time-dependent manner, which is indicative of normal cell cycle progression (Fig. 2B). Cdk1 phosphorylation at Tyr15 (Cdk1-pY15) was very low or hardly detectable in control cells. NOL11-depleted cells, by contrast, showed substantially increased Cdk1-pY15 levels at the G2/M border, and there was no apparent difference in cyclin B1 levels before release. Furthermore, Cdk1-pY15 signals persisted even after removing RO-3306 in NOL11-depleted cells.
Nuclear translocation of cyclin B is required for the rapid activation of Cdk1 and subsequent key mitotic events such as nuclear envelop breakdown (NEBD) and chromosome condensation (19). Therefore, we examined the subcellular localization of cyclin B1 during the G2-M transition. In both control and NOL11-depleted cells synchronized at the G2/M border with RO-3306, cyclin B1 signals in the nucleus were barely detectable (Fig. 2C, 0 min). Removal of RO-3306 caused rapid cyclin B1 translocation from the cytoplasm to the nucleus in control cells, nearly 70% of which showed nuclear staining of cyclin B1 at 10 min and over 80% showed staining at 60 min. In contrast, NOL11 depletion severely delayed nuclear accumulation of cyclin B1; the number of cells showing cyclin B1 accumulation in the nucleus at 60 min was markedly lower than that of control cells at 10 min. Along with translocation of cyclin B1 into the nucleus, NEBD was also delayed in NOL11-depleted cells, indicating that cyclin B1 translocation and subsequent mitotic events were severely inhibited in NOL11-depleted cells. It is thus clear that suppression of Cdk1 activity and delayed mitotic entry were caused by both increased Cdk1-pY15 levels and blocking of cyclin B1 nuclear translocation in NOL11-depleted cells.
We next examined how NOL11 depletion delayed Cdk1 activation. Cdk1-pY15 is regulated by the balance between kinases, such as Wee1 and Myt1, and Cdc25 phosphatases (20). Although the functions of Myt1 remain unclear, Wee1 has been shown to play a critical role in controlling the G2-M transition based on experiments using Wee1-specific inhibitors, siRNA-mediated Wee1 depletion, and Wee1 knockout mice (21–23). During G2-M transition, Wee1 kinase undergoes ubiquitin-mediated degradation, while Cdc25 is activated, tilting the balance toward Tyr15 dephosphorylation and Cdk1 activation. Using RO-3306 synchronization, we checked the levels of Wee1 and Cdc25 at the G2/M border. The NOL11-depleted cells showed abnormally high levels of Wee1, along with increased Cdk1-pY15 (Fig. 3A). In contrast, levels of Cdc25 phosphatases changed only slightly following NOL11 depletion, if at all (Fig. 3A). These results suggest that accumulation of Wee1 is the cause of delayed Cdk1 activation and mitotic entry following NOL11 depletion. To examine fluctuation in the levels of Wee1 proteins during the cell cycle, we examined the levels of Wee1 after release from G1/S synchronization in the presence of RO-3306 to prevent mitotic entry. Wee1 levels gradually decreased in control cells but remained relatively stable in the NOL11-depleted cells (Fig. 3B), indicating that Wee1 was improperly stabilized during cell cycle progression in the NOL11-depleted cells.
Fig. 3. NOL11 depletion caused accumulation of Wee1.
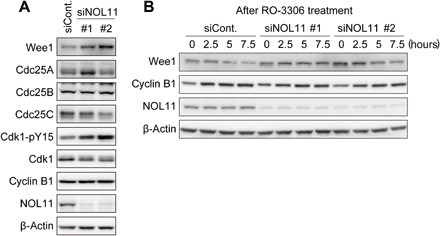
(A) Increased Wee1 levels caused by NOL11 depletion. HeLa cells were treated with the siRNAs for NOL11 and synchronized at the G2/M border. Whole-cell extracts were immunoblotted with the indicated antibodies. (B) Reduced Wee1 turnover by NOL11 depletion. NOL11-depleted cells were synchronized by single thymidine block. Six hours after release, cells were treated with 10 μM RO-3306 and collected at the indicated times for immunoblotting. The number in the upper immunoblot panels shows hours after the treatment of RO-3306.
NOL11 depletion causes nucleolar disruption
In addition to the occurrence of mitotic phenotypes, NOL11 depletion also down-regulated rRNA transcription (fig. S2, B and C), which is consistent with previously published findings (24, 25). Inhibition of rRNA transcription reportedly causes nucleolar disruption (3, 4), which is accompanied by two typical phenotypes: translocation of the nucleolar proteins to the nucleoplasm and formation of a nucleolar cap at the nucleolar periphery. To test whether NOL11 depletion causes nucleolar disruption, we examined the effect of NOL11 depletion on the localization of the nucleolar proteins by using siRNA for TIF-IA, an essential Pol I transcription factor, as a positive control for nucleolar disruption. NOL11 depletion induced the translocation of nucleolin and MYBBP1A into the nucleoplasm and the formation of nucleolar caps of UBF (fig. S2D), which was similar to the effects of TIF-IA depletion (fig. S2D), indicating that NOL11 depletion caused nucleolar disruption. An alternative possibility is that NOL11 depletion influenced mitotic progression, which in turn caused nucleolar phenotypes during interphase. However, this is unlikely because nucleolar disruption was induced despite NOL11 depletion without passing through the mitotic phase (fig. S3). We tested whether 59 siRNAs (table S1) that increased mitotic population in asynchronous cultures also caused nucleolar disruption during interphase (fig. S4). However, judging from the area of nucleolin and MYBBP1A in the nucleolus, nucleolar disruption was induced only by siNOL11 treatment (fig. S4).
Nucleolar disruption is accompanied by delayed mitotic entry
Previous reports have shown that nucleolar disruption induces a variety of cellular events such as p53-dependent cell cycle arrest and apoptosis, cell differentiation, and autophagy (7, 9, 11). Given that altered nucleolar morphology is involved in a variety of cellular events, it is possible that nucleolar disruption during interphase was the main cause of delay in the subsequent mitotic entry when NOL11 was depleted.
To test this hypothesis, we examined whether depletion of TIF-IA or UBF, both of which induced nucleolar disruption (Fig. 4A), also delayed mitotic entry. First, we tested cell cycle progression from G1/S synchronization, after the depletion of TIF-IA or UBF, using dual-color FACS analysis in combination with PI and anti–H3-pS10. Depletion of TIF-IA or UBF delayed mitotic entry, akin to NOL11 depletion (Fig. 4B). Furthermore, TIF-IA or UBF depletion strongly increased Cdk1-pY15 levels at all time points after the release from RO-3306 synchronization (Fig. 4C), in a manner similar to that following NOL11 depletion (Fig. 2B). In addition, nuclear accumulation of cyclin B1 was compromised by TIF-IA or UBF depletion (Fig. 4D). Then, we tested whether similar phenotypes were observed when nucleolar disruption was induced after treatment with low-dose actinomycin D (Act D) instead of siRNAs (fig. S5A). We found that Act D treatment also delayed mitotic entry, increased Cdk1-pY15 levels, and impaired nuclear accumulation of cyclin B1, in a similar way to the effects of NOL11, TIF-IA, or UBF depletion (fig. S5, B to D). These results support our hypothesis that nucleolar disruption during interphase causes delayed mitotic entry by inhibiting Cdk1 activation.
Fig. 4. Nucleolar disruption by TIF-IA or UBF depletion also delayed mitotic entry.
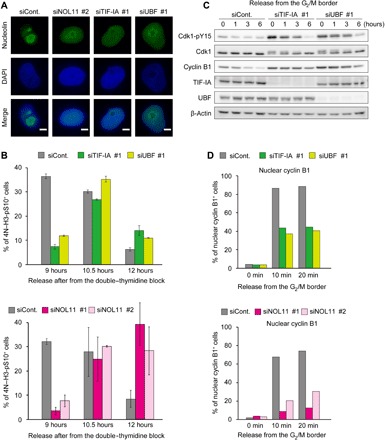
(A) Nucleolar disruption caused by TIF-IA or UBF depletion in addition to NOL11 depletion. HeLa cells were transfected with the indicated siRNAs and cultured for 48 hours. The cells were stained for immunofluorescence with anti-nucleolin antibodies (green). DNA was counterstained by DAPI (blue). Scale bars, 5 μm. (B) Delayed mitotic entry in cells with the disrupted nucleolus. HeLa cells were treated with the indicated siRNAs and synchronized at the G1/S boundary by double thymidine as shown in Fig. 1C. Nine, 10.5, and 12 hours after release from G1/S synchronization, the cells were collected for analysis of mitotic index by FACS. Upper histograms showed the results of TIF-IA or UBF depletion. Bottom histograms showed the results of NOL11 depletion, which is from the result in Fig. 1C. Values are shown as means ± SD, n = 3. (C) Increased Cdk1-pY15 in cells with the disrupted nucleolus. HeLa cells were treated with the indicated siRNAs and released from the G2/M border as the same protocol shown in Fig. 2A. The whole-cell extracts from the collected cells at the indicated times were immunoblotted with the indicated antibodies. (D) Delayed nuclear translocation of cyclin B in cells with the disrupted nucleolus. HeLa cells were transfected with the indicated siRNAs. The percentage of the cells in which cyclin B1 translocated into the nucleus is shown. Upper histograms showed the results of TIF-IA or UBF depletion. Bottom histograms showed the results of NOL11, which is from the result in Fig. 2C. Over 200 cells were counted at each time point for each siRNA.
We then tested how nucleolar disruption increased Cdk1-pY15 levels. We found that Wee1 accumulation was the cause of increased Cdk1-pY15 levels in the NOL11-depleted cells, which showed a disrupted nucleolus during interphase. Therefore, we conducted similar sets of the experiments as those shown in Fig. 3 (A and B) and checked the levels of Wee1 and Cdc25 following depletion of TIF-IA or UBF (Fig. 5, A and B). When cells were synchronized at the G2/M border using RO-3306, cells with nucleolar disruption caused by the depletion of TIF-IA or UBF contained high levels of Wee1, along with increased Cdk1-pY15 levels (Fig. 5A). In contrast, levels of Cdc25 phosphatases changed only slightly following TIF-IA or UBF depletion, and these changes were inconsistent for different siRNAs (Fig. 5A). When cells were released from G1/S synchronization in the presence of RO-3306, Wee1 levels gradually decreased in the control cells, but remained relatively stable in TIF-IA or UBF-depleted cells (Fig. 5B). Act D treatment also increased Wee1 levels at the G2/M border (fig. S5C). These results were in accordance with those of NOL11 depletion (Fig. 3, A and B), indicating that Wee1 was improperly stabilized during cell cycle progression in the cells whose nucleoli were disrupted.
Fig. 5. Nucleolar disruption caused aberrant Wee1 accumulation and delayed mitotic entry.
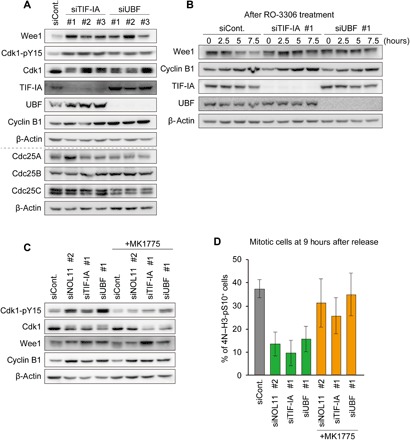
(A) Increased Wee1 levels caused by nucleolar disruption. HeLa cells were treated with the indicated siRNAs and synchronized at the G2/M border. Whole-cell extracts were immunoblotted with the indicated antibodies. (B) Reduced Wee1 turnover by nucleolar disruption. Cells were treated as Fig. 3B and examined by immunoblotting with the indicated antibodies. The number in the upper immunoblot panels shows hours after the treatment of RO-3306. (C) Reduction of enhanced Cdk1-pY15 by MK1775 treatment. Cells treated with indicated siRNAs were synchronized at the G2/M border. Subsequently, the cells were treated with 100 nM MK1775 for 3 hours and collected for immunoblotting using the indicated antibodies. (D) Restoration of mitotic entry by the Wee1 inhibitor. HeLa cells were treated with or without 100 nM MK1775 at 6 hours after release from the double thymidine block. The cells were collected at 9 hours after release from the double thymidine block, and mitotic index was examined using FACS. Values are shown as mean ± SD, n = 3.
To confirm that accumulation of Wee1 is the leading cause of abnormal Cdk1 phosphorylation and delayed mitotic entry, we treated cells with MK1775, an inhibitor of Wee1 kinase (26), at a concentration of 100 nM, at which cell cycle progression remains almost intact in control cells (fig. S6A). Treatment of MK1775 improved the abnormal phosphorylation of Cdk1 caused by depletion of NOL11, TIF-IA, or UBF without affecting Wee1 levels (Fig. 5C and fig. S6B). We also found that nuclear translocation of cyclin B1, which was delayed by NOL11, TIF-IA, or UBF depletion, was restored in the presence of MK1775 (fig. S6, C and D). Furthermore, treatment with MK1775 substantially suppressed the inhibited mitotic entry elicited by NOL11, TIF-IA, or UBF depletion (Fig. 5D). This finding suggests that the accumulation of Wee1 in the cells with nucleolar disruption is the leading cause of abnormal Cdk1 phosphorylation and delayed mitotic entry.
Nucleolar disruption is the main reason for delayed mitotic entry
Previous reports have shown that Wee1 degradation can be suppressed by activation of the G2/M checkpoint in the presence of DNA damage (12) that also causes nucleolar disruption (6). Therefore, it is possible that depletion of NOL11, TIF-IA, or UBF induces DNA damage, which in turn causes nucleolar disruption as well as Wee1 accumulation via G2/M checkpoint activation. To test this hypothesis, we examined the levels of Chk1 phosphorylation at Ser317 (Chk1-pS317), a marker of G2/M checkpoint activation, and γH2A.X, a marker of DNA double-strand breaks. The Chk1-pS317 signal was undetectable for cells with nucleolar disruption caused by depletion of NOL11, TIF-IA, or UBF, whereas the signal was clear for cells treated with etoposide, which induces DNA damage and G2/M checkpoint activation (fig. S7A). Furthermore, NOL11, TIF-IA, or UBF depletion caused nucleolar disruption but did not increase γH2A.X signal, even though etoposide treatment increased it (fig. S7B). In addition, neither the Chk1-pS317 signal nor the γH2A.X signal was detectable in the Act D–treated cells with disrupted nucleoli (fig. S7C). These results demonstrated that the mitotic phenotypes are induced by nucleolar disruption, independently of the activation of the G2/M checkpoint.
We cannot rule out the possibility that other nucleolar functions related to ribosome biogenesis cause the mitotic phenotypes found in the cells with nucleolar disruption. It might be possible that the inhibition of rRNA transcription, rather than nucleolar disruption, delays mitotic entry. To check this possibility, we attempted to eliminate NOL11 or TIF-IA without disrupting the nucleolus. To that end, we transfected siRNAs for NOL11 or TIF-IA in combination with siRNAs for RPL11, which is required for the export of pre-ribosomes with rRNA in yeast and humans (4, 27). Blocking the export of immature ribosomes from the nucleolus suppresses nucleolar disruption, even when rRNA transcription is down-regulated (4). Nucleolin, a marker of the nucleolus, was translocated from the nucleolus after NOL11 or TIF-IA depletion, whereas codepletion of RPL11 suppressed nucleolin translocation from the nucleolus (fig. S8, A to C), a finding consistent with the results of our previous work (4). Next, we examined the levels of Cdk1-pY15 and Wee1 when the nucleolar disruption occurred or was blocked. The lysates from the NOL11- or TIF-IA–depleted cells, in which the nucleolus was disrupted, showed high levels of Cdk1-pY15 and Wee1, which were restored to control levels by codepletion of RPL11 with NOL11 or TIF-IA (fig. S8, D and E).
However, we still cannot exclude the possibility that mitotic phenotypes of NOL11-, TIF-IA–, and UBF-depleted cells may be attributable to reduced protein synthesis, considering the fact that these proteins are involved in rRNA transcription and/or processing, inhibition of which possibly compromises ribosome biogenesis. To ascertain whether nucleolar disruption or reduced protein synthesis is the direct cause of mitotic phenotypes found in NOL11-, TIF-IA–, and UBF-depleted cells, we checked whether protein synthesis was inhibited without nucleolar disruption. We depleted rRNA processing factors such as PES1, DKC1, or RRP5 (Fig. 6A) and examined their effect on the nucleolar morphology (Fig. 6B), the levels of Cdk1-pY15 (Fig. 6C), mitotic entry (Fig. 6D), and protein synthesis (Fig. 6E). Immunofluorescence staining showed that depletion of these rRNA processing factors did not induce translocation of nucleolin and MYBBP1A (Fig. 6B), a finding consistent with the results of previous studies (5, 6). In addition, depletion of rRNA processing factors did not affect Wee1 levels, Cdk1-pY15 levels (Fig. 6C), or mitotic entry after release from double thymidine block (Fig. 6D), which contradicted the results of TIF-IA depletion that induced nucleolar disruption. We examined protein synthesis by measuring the incorporation of the amino acid analog l-homopropargylglycine (HPG). The depletion of PES1 or DKC1 reduced protein synthesis to an extent similar to that of NOL11, TIF-IA, or UBF depletion (Fig. 6E). It was not reduced protein synthesis, but nucleolar disruption that correlated with mitotic phenotypes such as delayed mitotic entry and elevated Cdk1-pY15 levels (Fig. 6, B to E), suggesting that nucleolar disruption during interphase is the cause of mitotic phenotypes. We therefore concluded that untimely nucleolar disruption during interphase induced improper stabilization of Wee1, which suppressed Cdk1 activation and delayed mitotic entry.
Fig. 6. Nucleolar disruption is a leading cause of delayed mitotic entry.
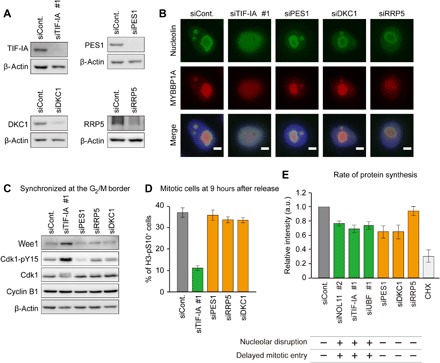
(A) Knockdown efficiency of siRNA for rRNA transcription factor or processing factors. HeLa cells transfected with the indicated siRNAs were cultured for 48 hours and immunoblotted by the indicated antibodies. (B) The alteration of nucleolar structure by depletion of rRNA transcription factor, not by processing factors. HeLa cells were transfected with the indicated siRNAs and cultured for 48 hours. The cells were stained with anti-nucleolin (green) and anti-MYBBP1A (red) antibodies. DNA was counterstained by DAPI (blue). Scale bars, 5 μm. (C) Increased Wee1 and Cdk1-pY15 in cells with the disrupted nucleolus. HeLa cells were synchronized at the G2/M border after siRNA transfection and immunoblotted using the indicated antibodies. (D) Delayed mitotic entry in cells with the disrupted nucleolus. HeLa cells were treated with the indicated siRNAs and synchronized by double thymidine as the same protocol shown in Fig. 1C. Nine hours after the release from G1/S synchronization, the cells were collected for analysis of mitotic index using flow cytometry. (E) Correlation between nuclear disruption and delayed mitotic entry. HeLa cells were transfected with the indicated siRNAs and cultured for 48 hours. Thereafter, cells were incubated in the presence of HPG for 2 hours and collected for the analysis by flow cytometry. Values are shown as means ± SD, n = 3. In parallel, the nucleolar disruption with reference to (B) and Fig. 4A and the delayed mitotic entry with reference to (D) and Figs. 1C and 4B are shown under the graph [+, nucleolar disruption induced (top); −, nucleolar disruption not induced (top); +, delayed mitotic entry induced (bottom); −, delayed mitotic entry not induced (bottom)]. a.u., arbitrary units; CHX, cycloheximide.
DISCUSSION
Nucleolar structure dynamically changes during the cell cycle in higher eukaryotes, especially during mitosis. Under normal conditions, cells contain several nucleoli, wherein continuous rRNA transcription occurs. Upon mitotic entry, the nucleolus disassembles in line with the activation of Cdk1, which phosphorylates the components of rRNA transcription machinery and suppresses RNA Pol I transcription (28, 29). At the end of mitosis, the nucleolus reassembles when rRNA transcription is reactivated by dephosphorylation of rRNA transcription factors (30). Thus, nucleolar disassembly during mitosis appears to be a programmed event because Cdk1 activity is tightly regulated during the cell cycle. In contrast to mitotic nucleolar disassembly, a variety of stressors such as DNA damage, hypoxia, and starvation suppress rRNA transcription and cause nucleolar disruption during interphase, suggesting that nucleolar disruption during interphase is an acute cellular response to stress and is induced by mechanisms distinct from those during mitosis. We speculated that interphase nucleolar disruption affects processes distinct from the programmed cell cycle in response to various stimuli. We therefore proposed a link between nucleolar integrity during interphase and cell cycle progression to mitosis.
Increased inhibitory phosphorylation of Cdk1 is the cause of delayed mitotic entry due to interphase nucleolar disruption
We showed that mitotic entry was delayed when nucleolar disruption was induced following the depletion of several rRNA transcription factors (Figs. 1C and 4B). Initiation of mitosis is achieved by Cdk1 activation, which is regulated by the accumulation of cyclin B1 and removal of inhibitory phosphorylation of Cdk1 by the dual-specificity phosphatase Cdc25 (20).
We showed that cyclin B1 accumulated normally during the cell cycle even in the presence of nucleolar disruption (Figs. 1C, 3B, and 5B), along with a reduction in protein synthesis (Fig. 6E). We believe that the reduction in protein synthesis was relatively mild owing to the long half-life of ribosomes, which is 5 to 10 days under normal physiological conditions (31). Therefore, severe reduction in protein synthesis did not occur in our study because our siRNA-based assays were performed between 48 and 72 hours after siRNA transfection. Additional synchronization at the G2/M border using RO-3306 showed that cells with or without disrupted nucleolus contained equal levels of cyclin B1, whereas nucleolus-disrupted cells showed increased levels of Cdk1-pY15 (Figs. 2B and 4C) and slow nuclear translocation of cyclin B1 (Figs. 2C and 4D). Thus, interphase nucleolar disruption appears to suppress Cdk1 activation by increasing the phosphorylation of Cdk1 without affecting cyclin B levels.
The activation of Cdk1–cyclin B is a switch-like process, as seen in Xenopus egg extracts (32) and in human cells (33). Once the activity of Cdk1–cyclin B exceeds the threshold, cells rapidly proceed from the G2 phase to mitosis. It was previously suggested that one of the earliest events to exceed the threshold for mitotic entry is dephosphorylation of Cdk1-pY15, which occurs before nuclear cyclin B translocation (34). If phospho-Cdk1 levels increase during the G2 phase, the threshold for mitotic entry likely shifts to the state that requires more Cdk1–cyclin B. We found that nucleolar disruption increased Cdk1-pY15 levels, which persisted even after RO-3306 removal (Figs. 2B and 4C). These results suggest that loss of nucleolar integrity delays the onset of mitosis by raising the threshold for mitotic entry.
Aberrant accumulation of Wee1 kinase is the cause of increased Cdk1-pY15 levels induced by nucleolar disruption
Inhibitory phosphorylation of Cdk1 is regulated by the balance between Wee1/Myt1 kinases and Cdc25 phosphatases (20, 35). Between Wee1 and Myt1, which are responsible for inhibitory phosphorylation, Wee1 is dominant in vertebrates (36). Our results indicate that aberrant accumulation of Wee1 is the cause of an increase in the inhibitory phosphorylation of Cdk1 when the nucleolus is disrupted during the interphase, based on the following observations. First, Wee1 levels increased along with those of Cdk1-pY15 during the G2 phase (Figs. 3A and 5A) when nucleolar disruption was induced by siRNAs against NOL11, TIF-IA, or UBF. However, the fluctuation profiles of the three isoforms of Cdc25 were not consistent with changes in the levels of Cdk1 phosphorylation (Fig. 3A and 5A). Second, up-regulation of both Wee1 and Cdk1-pY15, found in NOL11- and TIF-IA–depleted cells, was suppressed by RPL11 depletion, which inhibited nucleolar disruption (fig. S8). Finally, treatment with MK1775, a Wee1 inhibitor, abolished the Cdk1-pY15 up-regulation and delayed mitotic entry in cells with depleted NOL11, TIF-IA, or UBF (Fig. 5, C and D, and fig. S6, B to D).
The molecular mechanism underlying the increase in levels of Wee1 in response to nucleolar disruption during interphase remains to be elucidated. Our experimental results showed that Wee1 proteins were improperly stabilized and accumulated during cell cycle progression when the nucleolus was disrupted (Figs. 3B and 5B). One possible mechanism is that nucleolar proteins, redistributed from the nucleolus, mediate the stability of Wee1. This hypothesis is supported by previous studies stating that some of the nucleolar proteins are released from the nucleolus in response to several cellular stresses and, among those, some elicit cellular functions, for example, p53 activation. In general, Wee1 is down-regulated through proteasome-dependent degradation after ubiquitination by E3 ubiquitin ligase SCF-βTrCP during G2-M transition (37), and the ubiquitination of Wee1 is controlled by its phosphorylation. Therefore, ubiquitin ligases, deubiquitinating enzymes, kinases, and phosphatases in the nucleolus may regulate Cdk1 activity by controlling Wee1 levels in response to nucleolar disruption during interphase.
Nucleolar disruption is a leading cause of delayed mitotic entry
We found that mitotic entry was delayed when interphase nucleolar disruption was induced by the depletion of rRNA transcription factors or treatment of low-dose Act D. However, we cannot exclude the possibility that mechanisms other than nucleolar disruption, including the activation of the G2/M checkpoint, reduction in protein synthesis, and inhibition of rRNA transcription, induced mitotic phenotypes.
Cells that have experienced DNA damage are prevented by the G2/M checkpoint from initiating mitosis through suppression of Cdk1 (38). DNA damage also induces the down-regulation of rRNA transcription, which in turn induces nucleolar disruption (39). It is therefore possible that depletion of rRNA transcription induces DNA damage, which in turn suppresses Cdk1 activity and mitotic entry. However, neither NOL11, TIF-IA, or UBF depletion nor Act D treatment increased γH2A.X and Chk1 phosphorylation, the markers of DNA damage and G2/M checkpoint activation, respectively (fig. S7). Delayed mitotic entry and enhanced Cdk1-pY15 caused by nucleolar disruption are therefore independent of G2/M checkpoint signaling pathways.
siRNAs for NOL11 or TIF-IA were transfected in combination with siRNA for RPL11, which is required for the export of pre-ribosomes with rRNA. Blocking the export of immature ribosomes from the nucleolus suppresses nucleolar disruption, even when rRNA transcription is down-regulated. We found that codepletion of RPL11 suppressed nucleolar disruption and restored the levels of Cdk1-pY15 to control levels (fig. S8, D and E). These results show that it is not the inhibition of rRNA transcription but nucleolar disruption that elevates Cdk1-pY15 levels. Thus, nucleolar disruption is the main cause underlying delayed mitotic entry.
Nucleolar disruption caused by down-regulation of rRNA transcription potentially influences several biological processes such as ribosome biogenesis and protein synthesis. Therefore, a reduction in protein synthesis might cause delayed mitotic entry. However, as we have already discussed, reduction in protein synthesis was low when rRNA transcription factors were depleted (Fig. 6E). Considering that inhibition of rRNA processing increases the levels of rRNAs within the nucleolus (5, 40), we depleted several rRNA processing factors such as PES1, DKC1, and RRP5, which contribute to different steps of rRNA processing. Although these factors contribute differently to rRNA processing, nucleolar disruption did not occur, and mitosis was properly initiated (Fig. 6, B to D). Depletion of PES1 or DKC1 reduced protein synthesis to an extent similar to that following the depletion of NOL11, TIF-IA, or UBF1. Together, it is unlikely that reduction in protein synthesis is the cause of delayed mitotic entry.
In conclusion, this is the first study, to our knowledge, to provide evidence that the maintenance of nucleolar structure is essential for proper timing of mitotic entry. The loss of structural integrity induces stabilization of Wee1 proteins, which induces persistent Cdk1 phosphorylation without activating checkpoint signals.
MATERIALS AND METHODS
Cell culture
HeLa cells were grown at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 U/ml).
Cell cycle synchronization
HeLa cells were synchronized at the G1/S boundary by using the single thymidine block or double thymidine block methods. For the single thymidine block, the cells were treated with 2 mM thymidine (Sigma-Aldrich) for 24 hours. For the double thymidine block, single thymidine–arrested cells were replenished with fresh medium and cultured for 8 hours. Then, the cells were treated with 2 mM thymidine and cultured further for 16 hours. To synchronize the cells at the G2/M border, they were treated with 10 μM RO-3306 4 to 6 hours after releasing them from the single thymidine block, and then culturing for 12 hours.
siRNA screen and hit validation
A custom-made oligonucleotide siRNA library targeting 591 nucleolar proteins was purchased from Invitrogen. siRNAs were transfected into 3000 HeLa cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) in 96-well plates. After 72 hours, the cells were scored visually on an inverted microscope. The first screen showed that 207 siRNAs increased the number of spherical cells—the round shape is indicative of a mitotic cellular morphology. Following the primary screen, the cells were transfected with positive siRNA that increased spherical cells and lysed in RIPA buffer [20 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.8% NP-40, 0.1% SDS, 1% sodium deoxycholate, and 2 mM EDTA]. The lysates were analyzed by immunoblotting with anti–H3-pS10 antibodies. Among the 59 siRNAs that increased H3-pS10 levels, we chose NOL11 siRNA for further characterization, as NOL11’s mitotic function remains unexplored.
Antibodies
The following antibodies were used. Rabbit polyclonal antibodies against NOL11 (HPA022010, Sigma-Aldrich): Cdc25B (#9525, Cell Signaling Technologies), Cdc25C (#4688, Cell Signaling Technologies), Cdk1p-Y15 (#9111, Cell Signaling Technologies), Chk1-pS317 (#12302, Cell Signaling Technologies), FBL (#2639, Cell Signaling Technologies), and γH2A.X (ab2893, Abcam). Mouse monoclonal antibodies against α-β-actin (sc47778, Santa Cruz Biotechnology): cyclin B1 (sc245, Santa Cruz Biotechnology), UBF (sc13125, Santa Cruz Biotechnology), Cdk1 (sc45, Santa Cruz Biotechnology), Wee1 (sc5285, Santa Cruz Biotechnology), tubulin (DM1A, Millipore), nucleolin (M019-3, MBL), H3-pS10 (MABI0312, MBL), MPM2 (05-368, Millipore), NPM (FC61991, Thermo Fisher Scientific), and RPL11 (3A4A7, Thermo Fisher Scientific). In addition, a goat polyclonal antibody against TIF-IA (sc11805, Santa Cruz Biotechnology) was used. The rabbit polyclonal anti-MYBBP1A was prepared as previously described (4).
siRNA transfection
Cells were transfected with siRNAs using Lipofectamine RNAiMAX according to the manufacturer’s protocol. All siRNAs were purchased from Thermo Fisher Scientific. The sequences of the siRNA duplexes were as follows: NOL11 #1, 5′-CCAAACGCAUGUGCUUUCUUACAGU-3′; NOL11 #2, 5′-GUCUACUUCUGGAUGCGAAUU-3′; TIF-IA #1, 5′-CGACACCGUGGUUUCUCAUGCCAAU-3′; TIF-IA #2, 5′-GGGAUCACACCAAGCUCCUUUGACA-3′; TIF-IA #3, 5′-AGGAUGUCUGCUAUGUAGAUGGUAA-3′; UBF #1, 5′-UUCGGCUGCCUUCUUAAUCCACAUC-3′; UBF #2, 5′-AUCUCACUCAGCUCUCUCUCAUAUC-3′; UBF #3, 5′-UCUCCCUGGAAUUUCAUCUUCUUGG-3′; PES1, 5′-CCAUUGUCAACAAGUUCCGUGAAUA-3′; DKC1, 5′-GGCCAAGAUUAUGCUUCCAGGUGUU-3′; RRP5, 5′-GAACCAGGAGUGACUGGCCUUCUUU-3′; RPL11, 5′-GCUAGAUACACUGUCAGAUCCUUUG-3′. Stealth RNAi siRNA Luciferase Reporter Control (Thermo Fisher Scientific) was used as a negative control.
Immunoblotting and immunoprecipitation
The cells were lysed using the RIPA buffer supplemented with protease inhibitor (Nacalai Tesque) and phosphatase inhibitor cocktail (Nacalai Tesque) for 30 min on ice. The cleared lysates were separated on SDS-polyacrylamide gels and analyzed by immunoblotting with the previously mentioned antibodies.
Immunofluorescence
The cells were grown on a poly-l-lysine–coated coverslip. For immunostaining, the cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS; 140 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 8.1 mM Na2HPO4) for 15 min and permeabilized with 0.2% Triton X-100 in PBS for 5 min. The fixed cells were blocked with 3% bovine serum albumin (BSA)/PBS for 1 hour at room temperature. Then, the cells were incubated with the primary antibodies diluted with 3% BSA/PBS for 1 hour in a humidity box, stained with Alexa Fluor–conjugated secondary antibodies for 1 hour, counterstained with DAPI for 5 min, and mounted with VECTASHIELD (Vector Laboratories). Images were acquired using a BioRevo microscope (BZ-9000; Keyence) equipped with a 40× objective lens [Plan Apochromat, numerical aperture (NA) 0.95, Nikon] and a 100× oil objective lens (Plan Apochromat VC, NA 1.4). Cellular counting and image cropping were performed using ImageJ.
Classification of nucleolar morphology
To define nucleolar morphology as “without disruption” and with “nucleolar disruption” in figs. S3C and S8B, we performed a blinded test. One person prepared single-cell images and randomly numbered them. Then, these images were classified on the basis of nucleolin localization in a blinded manner by another person. We labeled the cells in which nucleolin was diffused into the nucleoplasm or localized at the nucleolar periphery as with “nucleolar disruption” and those in which nucleolin remained intact in the nucleolus as “without disruption.”
In addition, we analyzed the nucleolar area using ImageJ (figs. S3D, S4, and S8C). First, a raw image was split into nucleolin and DAPI-stained images, and the nuclear region was determined on the basis of DAPI signals. Second, we juxtaposed the nuclear region with the nucleolin image. Then, we measured the modal value of nucleolin signal intensity and subtracted this value from the whole image of nucleolin. Finally, we quantified the area in which nucleolin signals persisted and considered that as the nucleolar area.
RNA purification and reverse transcription quantitative polymerase chain reaction
RNA was isolated from cells by using the ReliaPrep RNA Cell Miniprep System (Promega) according to the manufacturer’s instructions. cDNA was synthesized from total RNA using RevatraAce reverse transcriptase (Toyobo) and random primers. The quantitative polymerase chain reaction (qPCR) analysis was performed to amplify fragments using the Thermal Cycler Dice TP800 (Takara) and SYBR Premix Ex Taq II (Takara). The specific primer sets for PCR amplification were 5′-GAACGGTGGTGTGTCGTTC-3′ and 5′-GCGTCTCGTCTCGTCTCACT-3′ for pre-rRNA.
Flow cytometry
The cells were fixed in 70% ethanol at −20°C. For staining intracellular proteins, the cells were washed with 0.1% Triton X-100/PBS. Primary antibodies were diluted with 3% BSA/PBS, with which the cells were incubated for 1 hour, followed by incubation with secondary antibodies for 1 hour. The cells were then stained with PI and RNase A solution (BD Biosciences) for 15 min. DNA content and the levels of intracellular proteins were measured using the FACSAria II (BD Bioscience) and analyzed using FlowJo.
Detection of newly synthesized RNA and proteins in cultured cells
For in situ detection of nascent rRNA, the cells were incubated for 30 min in the presence of 0.5 mM 5-ethynyluridine (EU) after 48 hours of siRNA transfection. The cells were fixed in 4% PFA/PBS for 15 min and permeabilized in 0.2% Triton X-100/PBS for 5 min at room temperature. The samples were then processed according to the manufacturer’s instructions. To measure the EU signals in the nucleolus, we counterstained the cells with an anti-UBF antibody to demarcate the nucleolar region. The fluorescence intensity of labeled RNAs in the nucleolus was measured using ImageJ. For analyzing protein synthesis, the cells were grown in methionine- and cysteine-free medium for 1 hour and were then treated with 25 μM HPG for 2 hours. After labeling the proteins with HPG, the cells were washed with PBS and fixed with 70% ethanol at −20°C. After fixation, Alexa Fluor 488 conjugation and subsequent procedures were carried out as described by the supplier (Thermo Fisher Scientific). Labeled cells were resuspended in PBS for flow cytometry analysis on the BD FACSAria II.
Quantification and statistical analysis
Each experiment was performed multiple times. The data from flow cytometry and reverse transcription qPCR were presented as means ± SD from a minimum of three independent repeated experiments. For analysis of immunofluorescence data, the presented values were from several experiments. The number of replication or the number of cells was annotated in the figure legends.
Supplementary Material
Acknowledgments
We would like to thank Enago (www.enago.jp) for the English language review. Funding: This work was supported by Grant-in-Aid for Scientific Research, KAKENHI [grants 25290064 (to K. Kimura), 15H02365 and 15H05977 (to T.H.), and 15J00599 (to Y. H.)] from the Japan Society for the Promotion of Sciences. Author contributions: Y.H., A.F., and K. Kimura designed the experiments. Y.H., A.F., K. Kato, R.U., and K. Kimura conducted the experiments. Y.H., T.H., and K. Kimura conducted the data interpretation. Y.H. and K. Kimura wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaap7777/DC1
fig. S1. NOL11 predominantly localized in the DFC region within the nucleolus.
fig. S2. NOL11 depletion induced down-regulation of rRNA transcription and nucleolar disruption.
fig. S3. NOL11 depletion induced nucleolar disruption without undergoing mitosis.
fig. S4. Nucleolar disruption is caused by NOL11 depletion alone.
fig. S5. Treatment with Act D delayed mitotic entry.
fig. S6. The treatment with MK1775 restored the levels of Cdk1-pY15 and the nuclear translocation of cyclin B1.
fig. S7. Depletion of NOL11, TIF-IA, or UBF and Act D treatment did not induce DNA damage nor activate G2/M checkpoint.
fig. S8. Codepletion of RPL11 suppressed nucleolar disruption and restored the levels of Cdk1-pY15 caused by NOL11 or TIF-IA depletion.
table S1. List of siRNAs that increased H3-pS10 levels in asynchronous cultures.
REFERENCES AND NOTES
- 1.Boisvert F.-M., van Koningsbruggen S., Navascués J., Lamond A. I., The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8, 574–585 (2007). [DOI] [PubMed] [Google Scholar]
- 2.McStay B., Nucleolar organizer regions: Genomic “dark matter” requiring illumination. Genes Dev. 30, 1598–1610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan X., Zhou Y., Casanova E., Chai M., Kiss E., Gröne H.-J., Schütz G., Grummt I., Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 19, 77–87 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Kuroda T., Murayama A., Katagiri N., Ohta Y.-m., Fujita E., Masumoto H., Ema M., Takahashi S., Kimura K., Yanagisawa J., RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J. 30, 1054–1066 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura K., Kumazawa T., Kuroda T., Katagiri N., Tsuchiya M., Goto N., Furumai R., Murayama A., Yanagisawa J., Kimura K., Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 10, 1310–1323 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Boulon S., Westman B. J., Hutten S., Boisvert F.-M., Lamond A. I., The nucleolus under stress. Mol. Cell 40, 216–227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James A., Wang Y., Raje H., Rosby R., DiMario P., Nucleolar stress with and without p53. Nucleus 5, 402–426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreiner G., Bierhoff H., Armentano M., Rodriguez-Parkitna J., Sowodniok K., Naranjo J. R., Bonfanti L., Liss B., Schütz G., Grummt I., Parlato R., A neuroprotective phase precedes striatal degeneration upon nucleolar stress. Cell Death Differ. 20, 1455–1464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katagiri N., Kuroda T., Kishimoto H., Hayashi Y., Kumazawa T., Kimura K., The nucleolar protein nucleophosmin is essential for autophagy induced by inhibiting Pol I transcription. Sci. Rep. 5, 8903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q., Shalaby N. A., Buszczak M., Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 343, 298–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi Y., Kuroda T., Kishimoto H., Wang C., Iwama A., Kimura K., Downregulation of rRNA transcription triggers cell differentiation. PLOS ONE 9, e98586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bassermann F., Frescas D., Guardavaccaro D., Busino L., Peschiaroli A., Pagano M., The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134, 256–267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andreassen P. R., Lacroix F. B., Villa-Moruzzi E., Margolis R. L., Differential subcellular localization of protein phosphatase-1 α, γ1, and δ isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 141, 1207–1215 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen J. S., Lam Y. W., Leung A. K., Ong S.-E., Lyon C. E., Lamond A. I., Mann M., Nucleolar proteome dynamics. Nature 433, 77–83 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Killian A., Le Meur N., Sesboüé R., Bourguignon J., Bougeard G., Gautherot J., Bastard C., Frébourg T., Flaman J.-M., Inactivation of the RRB1-Pescadillo pathway involved in ribosome biogenesis induces chromosomal instability. Oncogene 23, 8597–8602 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Ma N., Matsunaga S., Takata H., Ono-Maniwa R., Uchiyama S., Fukui K., Nucleolin functions in nucleolus formation and chromosome congression. J. Cell Sci. 120, 2091–2105 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Alawi F., Lin P., Dyskerin localizes to the mitotic apparatus and is required for orderly mitosis in human cells. PLOS ONE 8, e80805 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., Chen L., Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavet O., Pines J., Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hégarat N., Rata S., Hochegger H., Bistability of mitotic entry and exit switches during open mitosis in mammalian cells. Bioessays 38, 627–643 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Tominaga Y., Li C., Wang R.-H., Deng C.-X., Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int. J. Biol. Sci. 2, 161–170 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann B., Walter T., Hériché J.-K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U., Cetin C., Sieckmann F., Pau G., Kabbe R., Wünsche A., Satagopam V., Schmitz M. H. A., Chapuis C., Gerlich D. W., Schneider R., Eils R., Huber W., Peters J.-M., Hyman A. A., Durbin R., Pepperkok R., Ellenberg J., Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potapova T. A., Sivakumar S., Flynn J. N., Li R., Gorbsky G. J., Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol. Biol. Cell 22, 1191–1206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freed E. F., Prieto J.-L., McCann K. L., McStay B., Baserga S. J., NOL11, implicated in the pathogenesis of North American Indian childhood cirrhosis, is required for pre-rRNA transcription and processing. PLOS Genet. 8, e1002892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin J. N., Sondalle S. B., del Viso F., Baserga S. J., Khokha M. K., The ribosome biogenesis factor Nol11 is required for optimal rDNA transcription and craniofacial development in Xenopus. PLOS Genet. 11, e1005018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirai H., Iwasawa Y., Okada M., Arai T., Nishibata T., Kobayashi M., Kimura T., Kaneko N., Ohtani J., Yamanaka K., Itadani H., Takahashi-Suzuki I., Fukasawa K., Oki H., Nambu T., Jiang J., Sakai T., Arakawa H., Sakamoto T., Sagara T., Yoshizumi T., Mizuarai S., Kotani H., Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 8, 2992–3000 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Zhang J., Harnpicharnchai P., Jakovljevic J., Tang L., Guo Y., Oeffinger M., Rout M. P., Hiley S. L., Hughes T., Woolford J. L. Jr, Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes Dev. 21, 2580–2592 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heix J., Vente A., Voit R., Budde A., Michaelidis T. M., Grummt I., Mitotic silencing of human rRNA synthesis: Inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 17, 7373–7381 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sirri V., Roussel P., Hernandez-Verdun D., The mitotically phosphorylated form of the transcription termination factor TTF-1 is associated with the repressed rDNA transcription machinery. J. Cell Sci. 112, 3259–3268 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Klein J., Grummt I., Cell cycle-dependent regulation of RNA polymerase I transcription: The nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. U.S.A. 96, 6096–6101 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nikolov E. N., Dabeva M. D., Nikolov T. K., Turnover of ribosomes in regenerating rat liver. Int. J. Biochem. 15, 1255–1260 (1983). [DOI] [PubMed] [Google Scholar]
- 32.Pomerening J. R., Sontag E. D., Ferrell J. E. Jr, Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 5, 346–351 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Pomerening J. R., Ubersax J. A., Ferrell J. E. Jr, Rapid cycling and precocious termination of G1 phase in cells expressing CDK1AF. Mol. Biol. Cell 19, 3426–3441 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindqvist A., van Zon W., Karlsson Rosenthal C., Wolthuis R. M. F., Cyclin B1–Cdk1 activation continues after centrosome separation to control mitotic progression. PLOS Biol. 5, e123 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sur S., Agrawal D. K., Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 416, 33–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGowan C. H., Russell P., Cell cycle regulation of human WEE1. EMBO J. 14, 2166–2175 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe N., Arai H., Iwasaki J.-i., Shiina M., Ogata K., Hunter T., Osada H., Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 11663–11668 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Löbrich M., Jeggo P. A., The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 7, 861–869 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Rubbi C. P., Milner J., Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22, 6068–6077 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tafforeau L., Zorbas C., Langhendries J. L., Mullineux S. T., Stamatopoulou V., Mullier R., Wacheul L., Lafontaine D. L., The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of pre-rRNA processing factors. Mol. Cell 51, 539–551 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaap7777/DC1
fig. S1. NOL11 predominantly localized in the DFC region within the nucleolus.
fig. S2. NOL11 depletion induced down-regulation of rRNA transcription and nucleolar disruption.
fig. S3. NOL11 depletion induced nucleolar disruption without undergoing mitosis.
fig. S4. Nucleolar disruption is caused by NOL11 depletion alone.
fig. S5. Treatment with Act D delayed mitotic entry.
fig. S6. The treatment with MK1775 restored the levels of Cdk1-pY15 and the nuclear translocation of cyclin B1.
fig. S7. Depletion of NOL11, TIF-IA, or UBF and Act D treatment did not induce DNA damage nor activate G2/M checkpoint.
fig. S8. Codepletion of RPL11 suppressed nucleolar disruption and restored the levels of Cdk1-pY15 caused by NOL11 or TIF-IA depletion.
table S1. List of siRNAs that increased H3-pS10 levels in asynchronous cultures.