

Abstract
The Plexin family of transmembrane receptors are unique in that their intracellular region interacts directly with small GTPases of the Rho family. The Rho GTPase binding domain of plexin (RBD)—which is responsible for these interactions—can bind with Rac1 as well as Rnd1 GTPases. GTPase complexes have been crystallized with the RBDs of plexinA1, -A2, and -B1. The protein association is thought to elicit different functional responses in a GTPase and plexin isoform specific manner, but the origin of this is unknown. In this project, we investigated complexes between several RBD and Rac1/Rnd1 GTPases using multimicrosecond length all atom molecular dynamics simulations, also with reference to the free forms of the RBDs and GTPases. In accord with the crystallographic data, the RBDs experience more structural changes than Rho-GTPases upon complex formation. Changes in protein dynamics and networks of correlated motions are revealed by analyzing dihedral angle fluctuations in the proteins. The extent of these changes differs between the different RBDs and also between the Rac1 and Rnd1 GTPases. While the RBDs in the free and bound states have similar—if not decreased—correlations, correlations within the GTPases are increased upon binding. Mapping highly correlated residues to the structures, it is found that the plexinA1, -B1, and -A2 RBDs all have similar communication pathways within the ubiquitin fold, but that different residues are involved. Dynamic network analyses indicate that plexinA1 and -B1 RBDs interact with small GTPases in a similar manner, whereas complexes with the plexinA2 RBD display different features. Importantly complexes with Rnd1 have a considerable number of dynamic correlations and network connections between the proteins, whereas such features are missing in the RBD–Rac1 complexes. Overall, the simulations suggest mechanisms that are consistent with the experimental data on plexinB1 and indicate RBD and GTPase isoform specific changes in protein dynamics upon complex formation.
Graphical Abstract
INTRODUCTION
Plexins are a family of transmembrane receptors with 9 isoforms in humans (plexinA1–A4, B1–B3, C1, and D1) and are widely expressed in a variety of cells especially those belonging to the nervous, cardiovascular, skeletal, renal but also immune systems. Plexins regulate cell migration, adhesion, and targeting processes, for example in axon and blood vessel guidance, and misregulated or dysfunctional plexin also participates in cancer cell mobility (metastasis).1–5 To execute their functions, plexins receive guidance cues by binding semaphorin ligands to their extracellular domain, then transmitting this signal to the intracellular domains which participate in several cell signaling pathways that involve Ras and Rho GTPases. Highly unusual for transmembrane receptors is the direct binding of several different Rho family GTPases to an intracellular domain, named the Rho GTPase binding domain (RBD).
Several years ago we identified the Rho GTPase binding region of plexin-B1 as a domain with an ubiquitin fold. The 5 β-strands have an α-helix sitting on top and a short helix near the GTPase binding site, but by contrast to ubiquitin several long loops (L1, L2, and L4) are inserted as well as a short loop, L3, near the binding site (see Figure 1a).6,7 The secondary structure of RBD-B1 is shown on the top of Figure 1b. Rho GTPases have the Ras family superfold, consisting of 6–7 α-helices and 5–6 β-strands as the secondary structures shown on the top of Figure 1c, but special to this family, Rho GTPases have one helix inserted after α-helix4 (res. 110 in Rac1). The protein can be roughly divided into two subdomains: β, residues 1–86 and α, residues 87–end. The Rac1 GTPase Switch I (res. 27–44) and Switch II (res. 60–74) regions, shown in Figure 1a, are conformationally sensitive to the nature of the nucleotide that is bound.8 The GTP form is active and binds effector proteins, such as plexin.
Figure 1.

(a) Structures for Rac1 GTPase (left) and plexinB1 RBD (right) in the free form (red for Rac1, blue for loops and magenta for RBD, green for loops), and in the bound form (salmon for Rac1, light blue for loops and pink for RBD, limone/yellow for loops) after microsecond-length simulations. The Rac1 GTPase Switch I (res. 27–44), Switch II (res. 60–74), and the interswitch regions (res. 46–49) in Rac1 GTPase are shown in blue. The RBD-B1 loop regions, L1 (res. 16–26), L3 (res. 69–73), L4 (res. 77–95) are shown in green. RMSF for plexinB1 RBD (b) and Rac1 GTPase (c) in the bound form with HIS in HSD state (red), in the bound form with HIS in HSP state (blue); also shown is the free form (black). The larger fluctuations occur in the loop regions in the RBD and in the switch regions of Rac1 in both states with some considerable changes as discussed.
Cell biology studies have suggested that Rnd1 (a homologue of Rac1) and the Rac1 GTPase play different roles in plexinB1 signaling. While Rac1 and Rnd1 both bind to plexinB1, only clustering of the receptors in the presence of Rnd1 leads to the activation of its function in cell collapse.9,10 Rac1 may simply be bound and be removed (sequestered away) from other effectors, which signal to the cytoskeleton, such as PAK.11 By contrast to plexinB1, a sequential binding model was proposed for plexin-A1. Here Rac1 binds first and activates the plexinA1 for Rnd1 binding, which in turn stimulates function.12 These observations suggest an important conformational change takes place and together, imply an isoform/family specific function on the Rho GTPase-, as well as on the plexin side. However, the exact molecular mechanism for plexins to function in the different signaling pathways is still unknown. Especially the role of Rho GTPase binding is unclear, if not controversial.13,14
The “dimer-monomer/conformational change” model proposes binding events at the plexin intracellular and extracellular domains synergize to regulate the receptor in a manner that involves both outside-in as well as inside-out cellular signaling and that this bidirectional signaling is set up with the help of Rho GTPases that bind to the RBD.15 This complex formation may cause a substantial conformational change, such as a shift in a dimer to monomer equilibrium of the cytoplasmic region, as originally proposed by our laboratory for plexinB1.16 However, several crystal structures of plexinB1 and plexinA family members did not reveal large structural changes upon complex formation with Rac1 or Rnd1 GTPases at the domain level.17–20 Nevertheless, one crystal structure implied that the Rho GTPases may have a functional role in stabilizing a trimeric state of plexin-B120 via interdomain contacts.
It is not clear to what extent the crystal structures represent the available or relevant states for the cell signaling mechanism—even if the structural changes are subtle, it is anticipated that there are significant changes in protein dynamics upon GTPase binding to plexins. Some of these dynamical allosteric changes were revealed by an NMR relaxation study of the plexinB1 RBD:Rac1 complex.21 15N–1H NMR relaxation parameters for the plexinB1 RBD in solution for the monomer and dimer form as well as for the plexinB1 RBD:Rac1 complex have been reported.21–23 The data on the amplitude and correlation times of ps-ns time scale motions suggested the presence of coupling networks/correlated fluctuations that are altered upon complex formation.
Binding affinity measurements using isothermal titration calorimetry, comparing the formation of RBD-B1:Rac1 and RBD-B1:Rnd1 complexes, suggested a different thermodynamic origin for the interaction, with the latter being dominated by entropy changes upon binding. Furthermore, the measurements revealed a pH dependence of the interaction, with a lower pH of 6.0 leading to 2-fold greater binding than seen at a pH of 7.4.24 Since the pH can be lowered in cancer cells and at membranes, it has been suggested that low pH states of proteins may have a functional role in cell signaling [e.g., ref 25,26]. Preliminary NMR data on the RBD-B1:Rac1 complex at low pH suggest that this may well be the case as there are two histidines (His74 and 98) near the RBD-B1–GTPase contact interface (unpublished results).
The present report provides results from an ongoing project designed to reveal the allosteric nature of changes in plexin and GTPase dynamics by running microsecond long simulations on the Anton supercomputer.27 Previous work suggested that the plexinB1 RBD domain can transmit signals through allosteric mechanisms to faraway sites of the RBD domain.28,29 Also the bound Rac1 GTPase can have strong correlations in dynamics.21 Meanwhile, structures of several plexin RBD–GTPase complexes have been solved by crystallography, and we previously analyzed contacts in plexin:Rnd1 crystal structures.30 A similar analysis has not yet been carried out to the same level of detail for plexin:Rac1 complexes, forming an additional impetus for the work reported here. Following our published work and a recent report of allosteric networks in a GTPase–effector protein complex by Greg Buhrman and colleagues,22 we analyzed the structural and dynamics behavior of several plexin RBD and GTPase complexes. We have used the crystal structures of the complexes, but also of the free proteins structures as starting coordinates for long time-scale all atom MD simulations (either 1.0 or 2.4 μs). We examined the trajectories using dihedral angle cross-correlations as well as a network dynamics analysis. The simulations show that subtle isoform specific differences are propagated from the interfaces that differ in the details of contacts which are made between the proteins. As a consequence different patterns of correlated motions emerge across the complexes, showing that Rnd1 complexes are more tightly connected compared to Rac1 complexes. The possible biological implications of these findings are discussed.
MATERIALS AND METHODS
Simulation Setup
In total 14 simulations were performed in this project. For the initial plexinB1 and -A1 RBD bound with Rac1 GTPase, the structures were extracted from the X-ray coordinates by deleting the remainder of the plexin coordinates [of PDB ID of 3SU8, ref 20 and PDB ID 3RYT, ref 18, respectively]. In the case of the plexinA2 and -B1 RBD:Rnd1 complexes, the X-ray structures were used directly [PDB ID 3Q3J19 and 2REX,16 respectively]. Missing residues/side chains were filled using the Modeler program.31 For all complexes both the histidine in protonated state (HSP state) and in a deprotonated state (HSD state) were investigated. Besides those complexes, the free state of the RBDs and GTPases were simulated: plexinB1 RBD (res. 1548–1656; denoted here as res.6–114 to be consistent with previous studies) referred to as RBD-B1 [the higher electron density unit of dimer was used from PDB ID 2REX16]. As above, the RBD domains were taken from the full length intracellular domain structure for plexinA1. For Rac1 and Rnd1 (PDB IDs 1MH1 and 2CLS, respectively), GMPPNP, used as a nonhydrolyzable analogue in crystallography was replaced by GTP in this project, in the free as well as RBD bound GTPases. Table S1 gives an overview of all simulations and Figure S1 shows the amino acid sequences of all proteins used. For convenience we use a local numbering scheme for the RBD domains, relative to the numbering of the well characterized plexinB1 RBD, that is based on the alignment with the highly conserved turn between the second β-strand and the first α-helix.
The CHARMM program (version 36 or higher)32 was used to set up the systems by solvating the complex or the individual protein in a rectangular box of explicitly represented water (TIP3P), and initially placing the nearest protein atom at least 10 Å from a box wall. The box sizes only varied slightly, except for the RBD-B1:Rac1 complex (protonated form) which was rerun using a moderately increased cubic box. Counterions were added to neutralize each system and further Na+ and Cl–ions were added to bring up the NaCl concentration to 0.15 M. After brief minimization and gradual heating and equilibration, unrestrained all-atom molecular dynamics simulations were performed at a temperature of 300 K and a pressure of 1 atm using the NAMD program ver. 2.833 with a time step of 2 fs. The standard particle mesh Ewald method was used with periodic boundary conditions to calculate the long-range electrostatic interactions of the system. The CHARMM 22 all-atom potential function34 was used with CMAP correction.35 The GTP parameters were not available at the start of the project and were developed with the help of Dr. Alexander MacKerell at University of Maryland (these parameters are now part of CHARMM3636). For nonbonded calculations, a cutoff of 12 Å was used. All bonds involving hydrogen were kept rigid using the SHAKE algorithm. For each system listed above, at least 20 ns of NAMD simulations were performed to equilibrate the systems before the simulations were continued on Anton26 for 1.0 μs and in some cases to 2.4 μs.
Simulation Analysis
After aligning the molecule/complex on the starting X-ray structures or by aligning the individual proteins on their starting structure, root mean squared deviations (RMSD) were calculated for the entire mainchain of the complex and individual proteins. Similar to RMSD analysis, the root mean squared fluctuations (RMSF) were calculated for the mainchain by superposition of the individual proteins on the trajectory average structure using CHARMM.
Dihedral angle correlations20 were calculated as described in the Supporting Information.
Dynamical network analysis: To indicate which residues have the strongest interactions, a dynamical network analysis was carried out as done previously for the monomeric and dimeric RBD of plexinB1.29 The network model was built by the NetworkView plugin of VMD37 and the program Carma.38 In the network, nodes are single atoms or clusters of atoms, but here each Ca was treated as one node. The edge between nodes was defined if they are within at least 4.5 Å for at least 75% of the trajectory. An optimized communication pathway is derived from the shortest communication pathway, which is predicted based on the community network calculation. A community network is derived from dynamic cross-correlation calculations between nodes based on the microsecond long MD trajectories.
As a comparison, the Bio-3D program40,41 was also applied to predict the community nodes and communication pathways. Similar results were observed between Bio-3D and dynamic network analysis.
Analysis of residue contacts at the RBD–GTPase interfaces: The program ligplot/dimplot39 was used to examine both initial and final structures using a maximum D(donor)-A(acceptor) distance cutoff of 3.2 Å and an angle cutoff of 90 deg for hydrogen bonds, with no angular restrictions on other atom–atom contacts. Contact pairs in either structure were monitored over the course of the trajectory and the frequency of contact was tabulated based on the calculations from hydrogen bonds plugin from the VMD program using the same settings.
RESULTS
RBD-GTPase Complexes Are Stable in μs MD Simulations
In order to investigate the structural changes and dynamic fluctuations in RBD:GTPase complexes we carried out a total of 8 single microsecond or multi-microsecond all-atom simulations on four complexes and 6 reference simulations on the free proteins (see Table S1 in SI). Simulations of the protein complexes were carried out with neutral histidines (denoted HSD) and with positively charged groups (HSP), presenting a low pH state. As several of the histidines are near or at the RBD–GTPase interface (on both proteins), this allowed us to probe the likely effect of low pH interactions involving these histitines (see section Binding Interface Interactions below).
Root mean square deviations (RMSDs) provide a measure for how far the structures depart from the initial coordinate set. All the simulations appear to have reached equilibrated states and reasonably well maintained the X-ray determined structures of the complexes, as judged by a plateauing of the RMSD values. The time course of RMSDs for the unbound RBD of plexinB1, Rac1 GTPase in the free state, and their complexes are shown in Figure S2 in the Supporting Information.
The RMSDs are also given as averages over the last 500 ns of each simulation in Table S3. Free Rnd1 fluctuates more than free Rac1 (see also RMSF below), RBD-A2 and -B1 deviate more from their starting structures than free -A1. In the complexes this situation is reversed as far as the RBDs are concerned, but on average the GTPases deviate more from their starting structures than the free proteins. The complexes as a whole undergo larger fluctuations arising from changes in the orientations of the two proteins relatively to one another; still this is more pronounced in case of RBD-B1 complexes compared to the others. There are no consistent differences between protonated and deprotonated His simulations in terms of RMSD, except that the difference arises more from the RBDs at the different pH than from the GTPases.
Figure 1a shows a comparison of the final structures of RBD and Rac1 GTPase in complex with the final simulation coordinates of their free forms. It is apparent that the L1, L3, and L4 loops on the RBD show the largest structural differences from the free proteins. The same is true for a part of the Rac1 Switch I, Switch II and the β2–3 turn regions. It should be noted that all these regions are known to be flexible in the unbound states of both proteins.21 On binding it may be anticipated that the dynamics is reduced—an issue that was investigated by an RMSF analysis.
Larger Scale Fluctuations Are Localized in the Free Proteins and RBD-GTPase Complexes
In order to find out which regions in the plexinB1 RBD and in the Rac1 GTPase, for example, undergo larger structure fluctuations in the free and bound forms, root mean squared fluctuations (RMSFs) were calculated and are shown in Figure 1b,c. RMSF provides information about the local fluctuations relative to the simulation average structure. Here we superimposed by protein in the complex.
Relative to the free RBD, fluctuations of L3 and L4 are slightly attenuated in the complex as they are located in the RBD near the GTPase binding interface. By contrast, larger fluctuations occur in the switch regions of Rac1 when bound to the RBD. After binding, RMSF of L1 loop is increased. In RBD-A1 and -A2, the L1 loop is shorter and less flexible both in the free and bound states, showing no substantial changes. In order to quantitatively show the structural fluctuations in the RBDs and GTPases for the regions listed above, the average RMSF and the standard deviations for those regions are shown in the Table 1a,1b.
Table 1a.
RMSF Resultsa of the Complexes of RBD and Rac1/Rnd1 GTPases
| simulation | L1 (Å) | L3 (Å) | L4 (Å) | SW1 (Å) | β2–3 turn (Å) | SW2 (Å) | |
|---|---|---|---|---|---|---|---|
| RBD-B1:Rnd1 | sequence | 16–26 | 69–73 | 77–95 | 38–54 | 56–59 | 71–86 |
| HSD | 2.3 ± 0.6 | 1.0 ± 0.1 | 1.8 ± 1.0 | 1.2 ± 0.5 | 2.2 ± 0.5 | 1.0 ± 0.2 | |
| HSP | 4.8 ± 1.7 | 1.6 ± 0.1 | 2.9 ± 1.2 | 0.9 ± 0.3 | 2.2 ± 0.4 | 1.1 ± 0.1 | |
| RBD-B1:Rac1 | sequence | 16–26 | 69–73 | 77–95 | 28–44 | 46–49 | 61–76 |
| HSD | 2.6 ± 0.8 | 2.0 ± 0.3 | 2.5 ± 1.4 | 2.0 ± 0.8 | 2.3 ± 0.2 | 1.8 ± 0.6 | |
| HSP | 3.7 ± 1.3 | 2.0 ± 0.4 | 2.8 ± 1.3 | 1.2 ± 0.4 | 1.7 ± 0.17 | 1.1 ± 0.5 | |
| RBD-A1:Rac1 | sequence | 16–23 | 66–70 | 74–92 | 28–44 | 46–49 | 61–76 |
| HSD | 1.9 ± 0.5 | 1.7 ± 0.3 | 3.3 ± 1.6 | 1.4 ± 0.4 | 1.9 ± 0.2 | 0.7 ± 0.1 | |
| HSP | 2.3 ± 0.7 | 1.5 ± 0.3 | 1.7 ± 0.7 | 1.3 ± 0.4 | 2.1 ± 0.2 | 1.1 ± 0.3 | |
| RBD-A2:Rnd1 | sequence | 23–30 | 73–77 | 81–89 | 38–54 | 56–59 | 71–86 |
| HSD | 1.5 ± 0.4 | 1.2 ± 0.3 | 1.9 ± 0.6 | 0.9 ± 0.3 | 1.9 ± 0.1 | 0.9 ± 0.5 | |
| HSP | 1.5 ± 0.3 | 1.58 ± 0.4 | 1.1 ± 0.4 | 0.9 ± 0.3 | 1.7 ± 0.1 | 0.8 ± 0.2 |
Average of residue ranges from RMSF plots/data with standard deviation.
Table 1b.
RMSF Results for Free RBDs and GTPases
| simulation | L1 (Å) | L3 (Å) | L4 (Å) | SW1 (Å) | β2–3 turn (Å) | SW2 (Å) | |
|---|---|---|---|---|---|---|---|
| Rac1,free | sequence | 28–44 | 46–49 | 61–76 | |||
| 0.9 ± 0.3 | 1.5 ± 0.2 | 0.8 ± 0.1 | |||||
| Rnd1,free | sequence | 38–54 | 56–59 | 71–86 | |||
| 0.7 ± 0.1 | 2.3 ± 0.4 | 1.2 ± 0.4 | |||||
| RBD-A1,free | sequence | 16–23 | 66–70 | 74–92 | |||
| 1.7 ± 0.4 | 1.5 ± 0.3 | 1.6 ± 0.8 | |||||
| RBD-A2,free | sequence | 23–30 | 73–77 | 81–89 | |||
| 1.5 ± 0.2 | 2.3 ± 0.3 | 3.3 ± 1.1 | |||||
| RBD-B1,free | sequence | 16–26 | 69–73 | 77–95 | |||
| 2.9 ± 0.8 | 2.6 ± 0.5 | 3.7 ± 1.5 |
In summary, on average the bound form of the RBD in the protonated (HSP) state has a higher average RMSF compared to its free form, while its bound form in the deprotonated (HSD) state has a lower average RMSF than that of the free form. With exception of the RBD-B1:Rac1 (HSD) simulations, the Rac1/Rnd1 GTPases have on average slightly less structural fluctuations in the β2-β3 and SW2 in their free form, compared to their bound forms, but the trend in SW1 is the opposite. In several of the simulations the higher RMSF arises because the SW1 region opens up modestly/slightly and moves away from the nucleotide compared to the structure seen in the free protein.
Covariance Analysis in Dihedral Angle Changes Suggests Longer Range Correlations
Recently we analyzed the dynamics of the plexinB1 RBD monomer and dimer using a dihedral analysis,20 also described in the Supporting Information. This analysis avoids the long-range correlations in inter-residue distance analyses that are spuriously introduced due to changes in the overall frame of the protein, and especially in case of protein complexes. Figure 2 shows a comparison of the circular covariance analysis for the PlexinB1 RBD bound with Rac1 GTPase and the free plexinB1 RBD, Rac1 GTPase. As can be seen, the RBD in the free state has overall similar correlations to its bound state; the Rac1 GTPase in the free state has much less correlations than in its bound state. The results for plexinB1 RBD bound with Rac1 GTPase in HSD and HSP states are very similar to each other (shown in Figure S2). PlexinB1 RBD bound with Rnd1 GTPase in HSD and HSP states are very similar to each other too (as shown in Figure S3).
Figure 2.
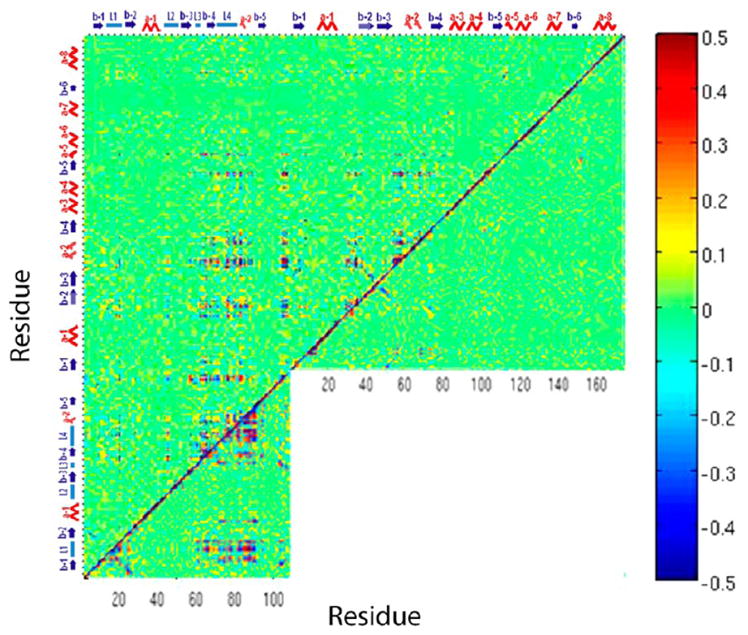
Dihedral angle covariance matrix for plexinB1 RBD bound with Rac1 in HSD state (above diagonal) compared to unbound plexin-B1 RBD (below diagonal, left) and the Rac1 GTPase in the free state (below diagonal, right). The color bar at the side indicates the level of maximal cross-correlation.
Figure 2 shows that L1, L3, L4 loop as well as other regions are highly correlated in the free RBD whereas no region shows significant long-range correlation in the free Rac1 GTPase. Upon complex formation correlations of L1 in the RBD are diminished but L3 and L4 and the SW I and SW II regions are highly correlated in both the HSD and HSP states (Figure S2). Remarkably those four regions are also highly correlated across the protein–protein interface in the complex.
At the residue level, however, changes are more diverse and we mapped some of the most correlated residues onto the structure: We observe correlations that are consistent with previous findings published in ref 29; for example, residue 21 in L1 of the RBD-B1 in the free state, has strong correlations with other regions. Once bound with Rac1, the correlations between residue 21 and other residues decreased significantly in HSP state, and even more so in the HSD state. By contrast, residue 81, another residue with strong correlations, located at the beginning of L4, shows strongly correlated motions with other regions in both free and bound states as shown in Figure 3. Do these individual correlations go across the protein–protein interface? In case of residue 21, this residue is far away from the interface and only very mild correlations are observed with residues in Rac1. By contrast, residue 81 is near the binding interface and correlations seen into the GTPase are much stronger, penetrating the protein up to 2–3 layers deep (e.g., residue 14 at the end of β-strand 1 of Rac1 GTPase).
Figure 3.

Covariance coefficient between residue 21 (left)/residue 81 (right) and other residues mapped on the structure of RBD-B1 in the free state (row (a)), in the Rac1 bound HSD state (row (b)), and in the Rac1 bound HSP state (row (c)). The complex with Rac1-GTPase is also shown in the HSD state in order to illustrate the cross-interface correlations (row (d)); the structure is slightly rotated to show the interface and the RBD residues clearly. Residues with the magnitude of their covariance coefficients higher than 0.1 are labeled and colored using the same scale as in Figure 2.
On the side of the Rac1-GTPase, we saw that the bound state has much stronger correlations than the free state. Results are shown in Figure 4, using residue 38 (in the SW1 region) and residue 63 as examples. Especially residue 63 shows good local cross correlations, which are substantially strengthened in the RBD-B1 bound HSP but not HSD state. Correlations involving residue 38 are strengthened and become longer range in the complex, especially in the HSD state (Figure 4d, left).
Figure 4.

Covariance coefficient between residue 38 (left)/residue 63 (right) and other residues mapped on the structure of Rac1-GTPase in the free state (row (a)), in the bound HSD state (row (b)), and in the bound HSP state (row (c)). The complex with Rac1-GTPase is also shown in the HSD state in order to illustrate cross-interface correlations (row (d)). Correlated residues with their magnitude of the covariance coefficients higher than 0.1 are labeled.
Analysis of covariance matrices and structure mapped results for plexinA1 RBD bound with Rac1 (Figures S4 and Figure S6) revealed results broadly similar to those above. Again, the RBD of plexinA1 has overall similar correlations in the bound and free forms, while Rac1 has stronger correlation in the bound form.
Results for the dihedral cross-correlation analysis of the PlexinA2 RBD:Rnd1 complex, in comparison with the free proteins are shown in the Figures S5 and Figure S7. Highly correlated regions were found in common between the HSP and HSD states, but they are stronger within the Rnd1 HSD state protein. Intriguingly the pattern of correlations is already strong in the free form of Rnd1 (compared to free Rac1 which had only very few correlations) and this pattern is slightly shifted upon RBD-A2 binding. However, by contrast to plexinB1 and -A1 RBDs, the correlations within the unbound -A2 RBD are weaker and become strengthened upon GTPase binding.
Compared to the free Rac1 protein (Figure 2), free Rnd1 has more regions with considerable cross-correlations as shown in Figure S7. However, in both the correlations mainly exist in the β-subdomain; correlations between β and α-subdomains are strengthened upon RBD binding. Similar to the RBD-A1 Rac1 complex as shown in Figures S4, S6, correlations in Rnd1 are strengthened by protein association in the GTPase but not in the RBD, where nearly all dihedral angle cross-correlations are diminished (again contrasting to Rac1 bound RBD-B1 where only L1 was diminished). While the cross-correlated motions are diminished in the RBD, the binding interface β3 region is cross-correlated with several regions of Rnd1 across the interface. Cross interface correlations are also observed for a region at the end of the C-terminal strand β5. An addition, for the RBD-B1:Rnd1 complex, there are correlations between the end of the RBD strand β1 and regions of the GTPase.
Focusing on the RBD domains only and extracting the highly correlated regions from the covariance matrixes, then mapping the correlation coefficients on the structures, we observed highly correlated residues in the structures of RBD-B1, RBD-A1, and RBD-A2, such as residue 81, 87, and 77, respectively, shown in Figure 5. It should be noted that the identity of the correlated residues in the RBDs is altered due to differences in the amino acid sequence and detailed residue intraprotein interactions. However, a similar pattern emerges, also consistent with our previous studies:28,29 The most flexible loop L4, whose motion is dampened by GTPase binding, is distantly correlated with motions of the L1 loop, with the β-strand 4 and the α-helices likely serving as conduits.
Figure 5.
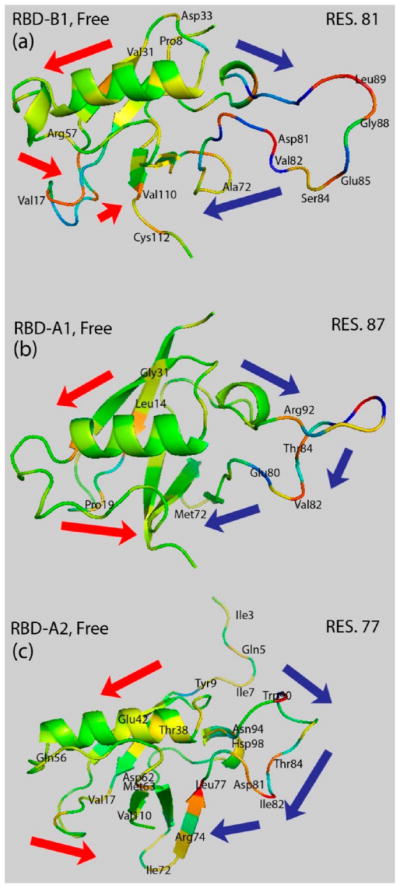
Possible communication pathways predicted based on highly correlated residues: residue 81 (top) in RBD-B1, residue 87 (middle) on RBD-A1, and residue 77 on RBD-A2 (bottom) and other residues mapped on structure of RBD-B1, RBD-A1, and RBD-A2 in their free state. Correlated residues with the covariance coefficient magnitude higher than 0.1 are labeled. Blue and red arrows show the possible communication pathway 1 and pathway 2 which were originally found for the unbound plexin-B1 RBD in the previous work.28,29
Based on Figures 2, S6, S7, and S8, but with some exceptions (e.g., Rnd1 bound RBD-B1), the RBDs in the bound state show highly correlated key regions similar to their free state although the correlations are stronger in the free state. Some neighboring residues are also modestly correlated in the bound state. Thus, the possible communication pathways in the bound state of the RBDs are overall expected to be similar to the free state.
Dynamic Network Analysis Reveals Significant Cross-Communication between RBD-Rnd1 Compared to RBD-Rac1 Complexes
The dihedral angle covariance matrixes revealed a set of highly correlated residues as shown in Figure 5 for the free state of RBD-B1, RBD-A1, and RBD-A2. A dynamic network analysis was performed on the three RBDs in both the free and bound forms to find out possible communication pathways which connect these residues. We followed the same dynamic network analysis method and settings as in ref 29 (see also Materials and Methods Section). The results are shown in Figure S9. Comparing to Figure 5, Figure S9 revealed similar possible communication pathways inside RBDs.
We also compared the community networks, which represents the dynamic correlation between nodes, for RBDs in the free and bound form, with results shown in the Figure 6.
Figure 6.

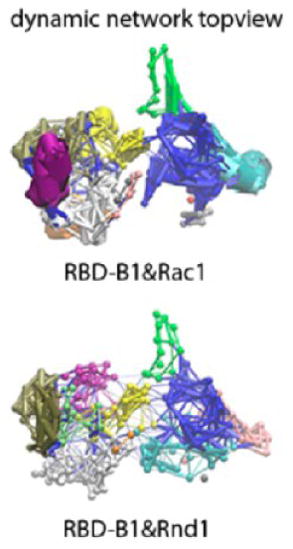
Community networks formed in the plexinB1 RBD (row (a)), plexinA1 RBD (row (b)), plexinA2 RBD (row (c)), and plexinB1 RBD (row (d)) in free form (left column), and in bound form to Rac1/Rnd1 (HSD) in face-view (middle column), and in topview (right column) based on MD simulation and dynamical network analysis. The free form and bound form network structures are oriented in the same direction as shown in the top of the figure using RBD-B1 (in green) bound with Rac1(in cyan) in the HSD form as example.
In the RBD-B1 free form, there are in total 10 communities (each shown in different colors), but only 12 communities are formed in the complex (Figure 6a). Consistent with the dihedral angle cross correlation analysis above, there is more extensive intracommunication within each protein after binding. Comparison of the thickness of edges shows that the RBD L2 loop becomes the interaction-intensive community in the bound form (orange), but not in the free form. It should be noted that the L2 loop is not adjacent to the GTPase binding site of the RBD. However, the binding site forms an adjacent community and the network couples it to other regions of the protein (for example the end of the α-helix and start of L2). Rac1 GTPase also forms around 5 intensive interaction communities. Cross-communication between RBD-B1 and Rac1 GTPase is not significant, possibly because the interactions fluctuate and are not as densely packed compared to the protein’s interior.
Comparison of the thickness of edges of the RBD-A1 communities (Figure 6b) also reveals that the RBD L2 loop and part of the β-sheet become an interaction-intensive community in the bound form, but not in the free form. The cross-communication between RBD-A1 and Rac1 GTPase increases slightly compared to RBD-B1:Rac1 complex. By contrast to these results, there are a considerable number of weak communication connections across the interface between RBD-A2 and Rnd1 GTPase after binding as shown in Figure 6c (right). Some of the intracommunication pathways inside each protein are also increased in extent, but not necessarily in intensity/strength, for example, for part of the L2 loop region in RBD-A2. Comparison of the thickness of edges reveals that α-helix 2 has an interaction-intensive community in the free RBD, but upon complex formation, this community was weakened. Since the RBD-A2:Rnd1 complexes also involve the N-terminal region (RBD β-strand 1) plus residues from residues 96 to 102 near the C-terminal RBD region, it is not surprising to see that there are more community networks for this complex. By contrast, the RBD-A1:Rac1 complex and the RBD-B1:Rnd1 complexes have fewer community networks involving the binding interface, as shown in Figure 6 (row b and d).
Comparing the nodes thickness, no intensive intraprotein communities were formed for RBD-B1 bound with Rnd1 GTPase (Figure 6d) compared to RBD-B1 bound with Rac1 GTPase (Figure 6a). Instead, there are many connections across the protein–protein interface in similar regions to those seen to be connected in the RBD-A2 Rnd1 complex (results shown in Figure S10d). Community networks are shown for the HSP protonated state of the complexes in Figure S10a–d in the SI, suggesting that histidine protonation state does not significantly influence the dynamic networks in this analysis.
For a comparison with the results above, we also used the Bio-3D program of Gray and colleagues,40,41 to carry out dynamic correlation and network analyses. Using each Ca atom as one node and based on μs long simulation trajectories, the community nodes were predicted using Bio-3D, which gives similar results to dynamic network analysis results shown above (data not shown).
Detailed Analysis of Protein–Protein Contacts Shows that the Interactions Are Considerably Dynamic and Slightly Different from the X-ray Structures
The number of contacts formed across the protein–protein interface during the microsecond simulations are shown in Figure S11a–d in the Supporting Information. The protonation state (HSD vs HSP) only slightly influenced the number of contacts across the binding interface (typically 7.8–9.1 vs 8.0–10.7) except for the RBD-B1:Rac1 complex. Here, there are more contacts in the HSD state (6.9 contacts on average) than in the HSP state (3.5).
Contacts at the binding interfaces of the four RBD-GTPase complexes were analyzed in detail (for both protonated and deprotonated histidine states) using both ligplot/dimplot39 and the hydrogen bond plugin of the VMD program.42 The residue pairs forming contacts with the occupancy of greater than 5% are listed in Tables S4–S11 in the Supporting Information.
Over the course of the simulations, residues on the binding interfaces in the HSD and HSP states are in similar locations. Generally the binding interface regions for RBD and small Rho GTPases include the L3 loop, β-strand 4, parts of the L4 loop, α-helix 2 and occasionally the C-terminus of β-strand 5 on the side of the RBDs, and mostly β2, β3, α2/α3 on the side of Rac1 and Rnd1. However, the exact residues forming contacts are not the same in the proteins. Key results that emerged are discussed below, while additional results, especially involving histidine residues at the interface are given for the 4 complexes in the Supporting Information.
DISCUSSION
Several features of RBD-GTPase complexes are of interest: The stability of the complexes (significant shifts at interfaces or even dissociation), effects of pH (as mimicked by de/protonated histidines), correlation of motions within protein structures and across the protein complexes, and finally, the most persistent interactions at the protein–protein interface. We have analyzed these features in all trajectories and discuss them in order below.
Global Features Are Similar Comparing Plexin RBD:Rnd1 and Rac1 Complexes
Using extensive all-atom molecular dynamics simulations in the study reported here, we reveal considerable similarities, but also some differences in the details of the structural and dynamic interaction of Rac1 and Rnd1 GTPases with the Rho GTPase binding domain (RBD) for several members of the human plexin family of trans-membrane receptors. From the MD simulations it is clear that the RBDs experience more changes to their structures (RMS deviations from starting structures) while GTPases experience more changes in their internal dynamics (see Table 1a,1b).
Overall the RBD-GTPases complexes are stable in the simulations, even at a lower pH (simulated by protonated histidine residues, HSP). The exact residues that are in contact can subtly change between the HSP and HSD states comparing the persistency of the contacts, but the binding interfaces are localized to highly similar regions on the protein surface. We had anticipated a considerable conformational transition upon histidine protonation, possibly akin to the dynamic protein complex of the EphA2-SHIP2 SAM-SAM interaction we recently characterized by simulations.43,44 However, in case of that system the changes in the configurational dynamics are on the sub-100 ns time scale, supported by NMR shifts that titrate upon protein binding.45 In the case of the plexinB1–RBD interaction with Rac1 there is considerable NMR signal broadening, suggesting an exchange process on the micro to millisecond time scale.16,21 Thus, even longer term simulations and further experimental studies are needed to reveal the configurational dynamics of RBD:GTPase complexes.
Similarities and Differences in Cross-Correlated Motions Across the Proteins and Protein–Protein Interfaces Point To Isoform Specificity
In a recent publication, we applied a dihedral angle cross-correlation analysis in preference to the more traditional distance based analysis, as the latter can be influenced by spurious longer range motions around a distant hinge and also ignore rotational movements.29 An issue with any correlation analysis is that there has to be considerable dynamics and also changes in dynamics to make them visible/stand out above the noise. This, therefore, confines the analyses to the most dynamic regions of the proteins, such as the RBD loops (L1–L4) and the GTPase Switch regions (SW I and II). Our previous study includes 400 ns trajectories for RBD-B1, while the current project has 1.0–2.4 μs long simulations trajectories performed on RBD-B1, RBD-A1, RBD-A2, and their complexes with small GTPases. Comparing RBD-B1 results between the short and the microsecond long trajectories, closely similar regions showed correlated dynamics and consistent communication pathways are suggested (Figures 2–5).
Calculating the backbone dihedral angle cross-correlation matrix, overall the 3 RBDs in the free states have similar correlations to their bound states. Mapping the highly correlated residues to the structure, we find that RBD-A1, RBD-B1, and RBD-A2 all have similar but diminished communication pathways, but with different key residues involved (Figure 5). Comparing the critical nodes in the dynamic networks, RBD-B1 and RBD-A1 have a greater number of nodes than RBD-A2. However, the pattern previously identified—a long-range coupling between L3, L4 with L2 and then the C-terminal region of the RBD α-helix (ref 28)—is evident in these simulations as well. A different analysis based on the critical nodes and optimized communication pathways for RBD domains was also carried out. By contrast to the dynamic network analysis, the community analysis is concerned about the persistency of interactions (and their packing). Nevertheless, the network communities overall agree with the communication pathways that are suggested by the correlation analysis. While this may be expected because our input data are the same trajectories, the correspondence, nevertheless, strengthens the general conclusion that all three RBDs transmit signals in a broadly similar pattern. The question arises how such networks are affected by GTPase binding to the RBD (and by RBD binding to the GTPase). For example, Fetics et al.22 found that mutation on residue 61 in Ras could switch the communication pathways from one pattern to another. The wild type Ras and bound Ras have an allosteric site and an active site in the same network community, while mutation of Q61L brings the allosteric site and active site into separate network communities. Because of the structural similarity of the GTPases, and since RBD-B1 binding with Rac1 GTPase also implicates an allosteric mechanism, the findings in the current project suggests that RBD-B1 and -A1:Rac1 GTPase complexes may have the active and allosteric sites in separate network communities, while RBD-B1 and -A2:Rnd1 complexes have the active binding sites and the allosteric sites in the same community. This observation is in accord with the suggestion that only Rnd1 activates the function of plexin-B1 while Rac1 binding could results in a different signaling mechanism, called GTPase sequestration (not directly involving the downstream regulation of plexin itself).
Detailed Analysis of Contacts Reveals the Dynamic Nature of the RBD:GTPase Interface and Shows Prevalent Interactions Involving Residues that Differ between the RBD as well as GTPase Isoforms
Tables S4–S11 and the results section above outlined the finding that the RBD–GTPase interactions experience considerable fluctuations over the course of the simulations. For example, at around 750 ns of the simulation, significant loop structure change was observed for plexin-B1 RBD bound with Rac1 in protonated state. The movie based on the 2.4 μs simulation trajectory is shown in the Supporting Information, and the complex structure at around 750 ns is shown in Figure S12. Depite this temporary change in the conformation of loop 4, the same regions of protein–protein contacts were broadly maintained. Based on Figure S11a–d, although the total number of contacts fluctuate during the simulations, the RBDs are bound stably with the small Rho GTPases. We noticed in the analysis of protein interface contacts, that residues with long side chains (Arg/Lys/Glu/Gln) often formed the more persistent contacts and we speculate that this may be due to length of these side chains. When the fluctuations allow a range of orientations of the side chains, such side chains are better suited to satisfy a 3.2 Å cutoff upon slight lateral movements of the interface, whereas small side chains, such as Ala, quickly fall outside the distance cutoff. Thus, overall there are considerable differences between the contacts identified for a static structure (such as the initial crystal structures) and those seen in the simulations. Our early solution NMR analysis of contacts between Rac1 and Rnd1 and the RBD of plexinB1, as monitored by a saturation transfer experiment to 15N labeled mainchain amides, also suggested that the C-terminal region of L4 was not in direct contact with the GTPases. This contrasted with data on the primary interface regions (partly involving the mainchain or being close to it), that showed them to be in direct contact.16 Remarkably none of the contacts in the dynamics analysis are 100% persistent; very few single contacts persist to approximately 90% with a handful at 40–60% while a similar number are in contact 15–40% of the time.
Isoform Specificity Is Confirmed by the Variation of Residues Involved in the Interactions Across the Plexin-Family RBDs
The differences (and similarities) of the structures and residues in contact can be largely explained by the sequence similarities/differences between Rnd1 vs Rac1 and RBD-B1 vs -A1 and -A2 (Figure 7a,b). Comparing the two complexes, that is RBD-B1 and –-A1 with Rac1, similar residues in switch I (SW I) and more extensively in switch II (SW II) are contacted.
Figure 7.

Sequence alignment of RBD and GTPase interface regions with the 9 members of the human plexin family in (a) and human Rac1 and Rnd1 in (b). * indicates contact with >20% occupancy in the neutral pH simulations (uncharged His), whereas * (blue) indicates additional contacts at low pH (charged His) from SI Tables S4–S11. Figure adapted from ref 18 “Structural Basis of Rho GTPase Rnd1 Binding to Plexin RBDs” by Wang et al, used under CC BY from original.
In the RBD-A1:Rac1 complex there is a greater tendency to interact with loop L4 (comprising β5 and β6) especially at lower pH. The latter may be explained by the change of Lys100 in RBD-B1 to a Gln in RBD-A1, making the interaction site that is adjacent to a region of L4 less positively charged (also RBD-B1 Arg91 is changed to Lys). Similarly, at the N-terminal side of L4, there are differences in the amino acid sequence of RBD-B1 and -A1: Asp85 is mutated to Lys and Gln87 to Asp, as well as Leu89 to Asp in going from RBD-B1 to -A1; these charged residues are forming some of the most persistent contacts in the simulations of the respective complexes (Table S3–S6), a feature that was not appreciated from the crystal structures.
The polar (hydrogen bonding and charge–charge) contacts identified by ligplot/dimplot in the crystal structure of the RBD-A2 Rnd1 complex11 are mostly maintained in the simulations (Gln100/HSD98 on side of RBD and Tyr74/Asn76 on side of Rnd1). Again, the shift in contacts toward those involving L4, compared to the RBD-B1:Rac1 complex, above, can be explained by their difference in sequence. The mutations are nearly the same in L3 and L4 (Gln87 is changed to Glu in going from RBD-B1 to -A2, instead of Asp in -A1) while the RBD-A1 and -A2 sequences are highly similar for the interfaces. For the complex of RBD-B1 with Rnd1 again, some of these contacts are seen in the original crystal structure, but due to the fluctuations at the interface, the overall persistency or contact time is always less than 100%, as noted above, allowing additional contacts compared to the static structure.
Noting that the most persistent and different contacts involve residues that differ between the RBD-B1, -A1, and -A2 isoforms, it is important to examine the sequence difference between Rac1 and Rnd1 (Figure 7b). There are many differences away from the interface (including especially in the inset helix24), but very few are located in the regions the GTPase uses to make contact with the RBDs. Specifically Ser41 to Thr51 and Ser81 to Cys91 do not feature in contacts, but alongside Rac1 residues Gly54/Ala59/Glu62 which are changed to Ser64/Ser69/Pro72, they could provide decreased flexibility, in the latter instance to the SW II region. Other changes from Rac1 to Rnd1 that stand out because they are used for GTPase isoform specific interactions are Asp38 to Glu48, a longer side chain at the end of switch region 1. At SW I, the Glu62/Asp62 change to Pro72/Tyr73 is significant, as is the change of Rac1 Arg66 to Asn76 in Rnd1. Arg66 is a key residue for interactions with both RBD-A1 and -B1 and forms many flexible contacts. Its lengthy side chain is the primary reason why early docking attempts using the initial RBD-B1:Rnd1 complex structures to model the RBD-B1: Rac1 complex flailed due to steric clashes (L.Z. and M.B. unpublished data). Finally, the swap of two histines (103/104) on the Rac1 α2 helix—which is adjacent to the SW II region and close to the RBD L4 and to Asp113/Tyr114—play a role; in the Rac1 structures, this region forms strong interactions with RBD-A1 in the low pH state (with the residues Lys85 and Asp87 that differ at in L4, as noted). In the HSD RBD-B1: Rac1 complex the interactions are shifted to Asp81/Glu85, whereas in the HSP state there is no contact involving these regions. The Rnd1 complexes have weak, pH independent contacts and experimentally it was noted that Tyr114, a residue that differs between Rnd1 and Rnd2 does not account for the difference between these two isoforms.18
pH Influence
The pH can influence the structure, dynamics and even function of proteins as mentioned in the Introduction Section. As constant pH simulations are not yet available on Anton (and are computationally more expensive), we explored the two extreme cases in this project (all His deprotonated and all His protonated). Despite these extreme conditions, the overall structures of RBD with/without bound Rho GTPases are stable in the simulation. The structure and dynamics of RBDs are also similar in both the protonated and deprotonated states. Specifically, there are only subtle differences between the HSD and HSP states concerning the types of contacts. Interactions in the HSP state of RBD-A1:Rac1 are strengthened (have higher persistency) than in the HSD state, whereas the situation in the RBD-B1:Rac1 complex is reversed. Interestingly, overall there is less increase in protonated histidine (HSP)–Asp/Glu interactions as might be anticipated, except for Asp87–Arg102/HSP103 in RBD-A1:Rac1 (65%) and HSP98–Arg66 in RBD-B1:Rac1 (down to 9% in HSP compared to 29% in HSD). For the plexinA2 RBD:Rnd1 complex, there is a slight shift in the contacts upon the protonation of His98, with fewer residues forming persistent contacts in the low pH state.
Biological Implications
The Rho GTPase binding domain (RBD) is unique to the plexin family of transmembrane receptors and, as we reported previously, provides a new GTPase recognition surface for signal transduction in biology (ref 16,7). Since its first structural characterization in 2005–2008, it has meanwhile become clear that this surface is quite versatile and binds a number of disparate Rho GTPase family members,16,46 and does so potentially in a GTPase family member specific manner (ref 19, S-J.K. and M.B, unpublished). With this understanding also came the appreciation, expanded here, that the original interface regions and motifs were too limited and that regions outside them—but still within the RBD—also play a role in the wider repertoire of RBD-GTPase interactions (e.g., ref 19). In cell and cell lysate based biochemical experiments by the Negishi9,10 and by the Kikutani11 laboratories indicate that the functional role of Rac1 and Rnd1 is different, both for plexin-B1 and again for plexin-A1, leading to the suggestion of isoform specificity both on the side of plexin and of the side of these Rho GTPases. The publication of four RBD:Rac1/Rnd1 complex crystal structures invited us to examine the possible molecular origin of these differences. We did not endeavor to estimate the overall thermodynamic parameters here for the different protein complexes, as this remains a complicated issue (e.g., see ref 43).
Interactions that were not appreciated from the initial NMR and crystal structure analysis concern the dimerization loop, L4. The overall shift in the simulations seen here is toward greater interactions with this loop, compared to those seen in the X-ray structures.16,18 This could be explained if the L4 region were fixed by additional contacts; either crystal lattice contacts (in case of the RBD-A2:Rnd1 structure) or due to dimerization involving the loop (RBD-B1:Rnd1) and/or binding of a coupling loop to this region (in case of RBD-A1:Rac1 and -B1:Rac1 complexes).16 The RBD-A2:Rnd1 complex structure, for example, has no electron density for four residues (EGDW) in L4, suggesting that these residues are disordered, if not flexible, in the crystal structure. Similarly, NMR relaxation data of L4 in the RBD-B1:Rac1 complex showed this region was still mobile in the complex on the ps-ns time scale, with slightly decreased dynamics at the termini but slightly increased flexibility in the middle when placed in a complex with Rac1.21 Here we find that several of the residues at the N- and C-termini of the L4 loop are involved in interactions with the GTPases and, importantly that these residues are different in the different RBD family members (see Figure 7, above). The interactions, newly discovered in this simulation project, are transient but involve these distinctive residues, and, are thus likely provide a molecular basis and origin for the isoform specific behavior of the different RBDs in the RBD-GTPase complexes. There are further differences also in how Rac1 and Rnd1 engage the RBDs but the details will need to await further analysis of GTPase internal dynamics.
RBD-Rnd1 complexes show a significant amount of interprotein correlations of the dynamic fluctuations. This observation may be related to the role of Rnd1 in activating the receptor for function. Furthermore, with exception of plexinA2, the intraprotein correlations in the plexin-B1 and -A1 RBDs remain strong upon complex formation. We could speculate that these pathways for dynamic allostery27,28,30 might be involved in the activation mechanism; they communicate to the other side of the RBD, to regions which are attached to the catalytic GAP domain of plexin while others are available for binding additional partner proteins. A similar argument can be made for the GTPases which undergo changes in their internal dynamics upon binding; overall the level of dynamic cross-correlations is increased, apparently in a manner which brings together the two β- and α-subdomains of Rac1/Rnd1. While there is some evidence for these changes from experimental NMR relaxation measurements,47,21 this MD simulation study focused more on the RBDs and we will further analyze and discuss GTPase dynamics in another study (in preparation), especially dealing with allostery within the GTPase fold.48 We and others suspect that interactions of the GTPases are not confined to the RBD alone, but that other regions of plexin may interact with them. Furthermore, interactions between GTPases and the lipid bilayer of the plasma membrane, to which they are anchored, also could play a major role.49 Thus, future investigations will be needed to shed light on such possible interactions.
CONCLUSION
In this project, microsecond simulations were performed on RBDs of three plexin family members, plexinB1, -A1, and -A2; small Rho GTPases Rac1 and Rnd1; and of their RBD:GTPase complexes. It was found that during the long time-scale simulations, both the free RBD and GTPases and their complexes are stable without any dramatic transitions observed, even for simulations in different protonation states. While the RBDs in the free states have overall similar dihedral angle correlations, these are slightly, and in case of plexinB1, considerably decreased when Rac1 or Rnd1 are bound. Correlations within the GTPases are increased upon binding. Mapping highly correlated residues to the structure, it was found that plexinA1-RBD, plexinB1-RBD, and plexinA2-RBD all have similar communication pathways within the plexin domains, but that different residues are involved. Dynamic network analysis on the microsecond long trajectories revealed that RBD-B1 and RBD-A1 could have similar binding mechanisms with small GTPases, but that binding of RBD-A2 likely has a different mechanism and possible functional role. Similarly, Rnd1 is seen to form many networks of dynamic correlations across the protein–protein interface with plexin RBDs, whereas the homologous GTPase Rac1 does not. Overall, the simulations suggest mechanisms that are consistent with the experimental data on plexinB1 and -A1. Our analysis indicates RBD and GTPase isoform specific changes in protein dynamics upon complex formation.
Supplementary Material
Acknowledgments
The project was started by Dr. Zhang when she was a postdoctoral fellow at Case Western Reserve University and all the simulations were carried out in Dr. Buck’s laboratory, while majority of the analysis was completed by Dr. Zhang in her laboratory in Tennessee Technological University. A summer undergraduate research student, Thomas Centa (then at Univ. of Cincinnati) helped with some of the analysis in the initial stages of the project. The work was funded by grants to M.B. (NIH R01GM112491, preceded by R01GM092851) and by a postdoctoral fellowship to L.Z. (NIH T32DK007470, PI: J. Sedor), and the new faculty startup package to L.Z. from Tennessee Technological University. The use of the Anton supercomputer at PSC is supported by an NIH award (RC2GM093307) to Carnegie Mellon University and part of the work was carried out on Lonestar under XEDES grant (MCB120047), as well as the High Performance Computing Cluster at Case Western Reserve University.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.6b11022.
Amino acid sequences for all the proteins used in simulations; overview of simulations performed in the project; main chain RMSD relative to the starting structure; residues forming contact on the binding interface of RBDs and Rho GTPases; RMSD for plexin-B1 RBD and Rac1 GTPase in free and bound forms; extra dihedral angle covariance matrix for RBD, Rho GTPase, and their complexes in HSD and HSP forms; extra analysis of RBD and Rho GTPase in free and bound form structures with covariance coefficients mapped on those structure; optimized communication pathways for RBDs; number of contacts for RBD and small Rho GTPases in HSD and HSP forms; dynamic network analysis result and figure for different RBDs bound with different GTPases; and additional explanation on the dihedral angle correlation calculation (PDF) Movie of the binding interface between proteins during the 2.4 μs all atom molecular dynamics simulation (MPG)
References
- 1.Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, et al. Plexins Are a Large Family of Receptors for Transmembrane, Secreted, and GPI-anchored Semaphorins in Vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- 2.Negishi M, Oinuma I, Katoh H. Plexins: Axon Guidance and Signal Transduction. Cell Mol Life Sci. 2005;62:1363–1371. doi: 10.1007/s00018-005-5018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel BN, Van Vactor DL. Axon Guidance: the Cytoplasmic Tail. Curr Opin Cell Biol. 2002;14:221–229. doi: 10.1016/s0955-0674(02)00308-3. [DOI] [PubMed] [Google Scholar]
- 4.Gitler AD, Lu MM, Epstein JA. PlexinD1 and Semaphorin Signaling Are Required in Endothelial Cells for Cardiovascular Development. Dev Cell. 2004;7:107–116. doi: 10.1016/j.devcel.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, et al. Class 3 Semaphorins Control Vascular Morphogenesis by Inhibiting Integrin Function. Nature. 2003;424:391–397. doi: 10.1038/nature01784. [DOI] [PubMed] [Google Scholar]
- 6.Tong Y, Buck M. 1H, 15N and 13C Resonance Assignments and Secondary Structure Determination Reveal that the Minimal Rac1 GTPase Binding Domain of Plexin-B1 Has a Ubiquitin Fold. J Biomol NMR. 2005;31(4):369–370. doi: 10.1007/s10858-005-0943-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong Y, Hota PK, Hamaneh MB, Buck M. Insights Into Oncogenic Mutations of Plexin-B1 Based on the Solution Structure of the Rho GTPase Binding Domain. Structure. 2008;16(2):246–258. doi: 10.1016/j.str.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vetter IR, Wittinghofer A. The Guanine Nucleotide-binding Switch in Three Dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 9.Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D Receptor Plexin-B1 is a GTPase Activating Protein for R-Ras. Science. 2004;305(5685):862–865. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- 10.Oinuma I, Katoh H, Negishi M. Molecular Dissection of the Semaphorin 4D Receptor Plexin-B1-stimulated R-Ras GTPase-activating Protein Activity and Neurite Remodeling in Hippocampal Neurons. J Neurosci. 2004;24(50):11473–11480. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vikis HG, Li W, Guan KL. The plexin-B1/Rac Interaction Inhibits PAK Activation and Enhances Sema4D Ligand Binding. Genes Dev. 2002;16(7):836–845. doi: 10.1101/gad.966402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toyofuku T, Yoshida J, Sugimoto T, Zhang H, Kumanogoh A, Hori M, Kikutani H. FARP2 Triggers Signals for Sema3A-mediated Axonal Repulsion. Nat Neurosci. 2005;8(12):1712–1719. doi: 10.1038/nn1596. [DOI] [PubMed] [Google Scholar]
- 13.Hota PK, Buck M. Peer Reviewed Review for Cell & Mol Life Sciences. Vol. 69. Birkhauser Press; 2012. Plexin Structures Are Coming! Multilevel Investigations of the Function of the Guidance Receptors and Their Mechanisms of Cell Signaling; pp. 3765–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Negishi M, Buck M. Ras/Rap GAP Function and GTPase Sequestration in Plexin Mediated Cell Signaling Mechanisms. Commentary in Science Signaling. 2012 http://stke.sciencemag.org/content/5/207/ra6.e-letters. Commentary on ref 19.
- 15.Vikis HG, Li W, He Z, Guan KL. The Semaphorin Receptor Plexin-B1 Specifically Interacts with Active Rac in a Ligand-dependent Manner. Proc Natl Acad Sci U S A. 2000;97(23):12457–12462. doi: 10.1073/pnas.220421797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong Y, Chugha P, Hota PK, Alviani RS, Li M, Tempel W, Shen L, Park H, Buck M. Binding of Rac1, Rnd1, and RhoD to a Novel Rho GTPase Interaction Motif Destabilizes Dimerization of the Plexin-B1 Effector Domain. J Biol Chem. 2007;282(51):37215–37224. doi: 10.1074/jbc.M703800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong Y, Hamaneh MB, Penachioni JY, Hota PK, Kim SJ, Alviani RS, Shen L, Tempel W, Tamagnone L, Park HW, et al. Structure and Function of the Intracellular Region of the Plexin-B1 Transmembrane Receptor. J Biol Chem. 2009;284:35962–35972. doi: 10.1074/jbc.M109.056275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Hota PK, Tong Y, Li B, Shen L, Nedyalkova L, Kim SJ, Tempel W, Buck M, Park HW, Borthakur S. Structural Basis of Rho GTPase Rnd1 Binding to Plexin RBDs. J Biol Chem. 2011;286:26093–26106. doi: 10.1074/jbc.M110.197053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, He H, Srivastava N, Vikarunnessa S, Chen YB, Jiang J, Cowan CW, Zhang X. Plexins are GTPase-activating Proteins for Rap and Are Activated by Induced Dimerization. Sci Signaling. 2012;5:ra6–ra6. doi: 10.1126/scisignal.2002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell CH, Aricescu AR, Jones EY, Siebold C. A Dual Binding Mode for RhoGTPases in Plexin Signalling. PLoS Biol. 2011;9(8):e1001134. doi: 10.1371/journal.pbio.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouguet-Bonnet S, Buck M. Compensatory and Long-range Changes in Picosecond-nanosecond Main-chain Dynamics upon Complex Formation: 15N Relaxation Analysis of the Free and Bound States of the Ubiquitin-like Domain of Human Plexin-B1 and the Small GTPase Rac1. J Mol Biol. 2008;377:1474–1487. doi: 10.1016/j.jmb.2008.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, Mattos C. Allosteric Effects of the Oncogenic RasQ61L Mutant on Raf-RBD. Structure. 2015;23:505–516. doi: 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hota P, Buck M. Thermodynamic Characterization of Two Homologous Protein Complexes: Association of the Semaphorin Receptor Plexin-B1 Rho GTPase Binding Domain with Rnd1 and Active Rac1. Protein Sci. 2009;18:1060–1071. doi: 10.1002/pro.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiche J, Brahimi-Horn MC, Pouyssègur J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J Cell Mol Med. 2010;14(4):771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Damaghi M, Wojtkowiak JW, Gillies RJ. pH Sensing and Regulation in Cancer. Front Physiol. 2013;4:370. doi: 10.3389/fphys.2013.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, et al. Anton, A Special-Purpose Machine for Molecular Dynamics Simulation. Commun ACM. 2008;51(7):91–97. [Google Scholar]
- 28.Zerbetto M, Anderson R, Bouguet-Bonnet S, Rech M, Zhang L, Meirovitch E, Polimeno A, Buck M. Analysis of 15N-1H NMR Relaxation in Proteins by a Combined Experimental and Molecular Dynamics Simulation Approach: Picosecond-nanosecond Dynamics of the Rho GTPase Binding Domain of Plexin-B1 in the Dimeric State Indicates Allosteric Pathways. J Phys Chem B. 2013;117:174–184. doi: 10.1021/jp310142f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Centa T, Buck M. Structure and Dynamics Analysis on Plexin-B1 Rho-GTPase Binding Domain Monomer and Dimer. J Phys Chem B. 2014;118(26):7302–7311. doi: 10.1021/jp503668k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerbetto M, Buck M, Meirovitch E, Polimeno A. Integrated Computational Approach to the Analysis of NMR Relaxation in Proteins: Application to ps-ns Main Chain 15N-1H and Global Dynamics of the Rho GTPase Binding Domain of Plexin-B1. J Phys Chem B. 2011;115(2):376–388. doi: 10.1021/jp108633v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Šali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 32.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the Biomolecular Simulation Program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Buck M, Bouguet-Bonnet S, Pastor RW, MacKerell AD., Jr Importance of the CMAP Correction to the CHARMM22 Protein Force Field: Dynamics of Hen Lysozyme. Biophys J. 2006;90:L36–L38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, MacKerell AD., Jr CHARMM36 All-atom Additive Protein Force Field: Validation based on Comparison to NMR Data. J Comput Chem. 2013;34(25):2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eargle J, Luthey-Schulten Z. NetworkView: 3D Display and Analysis of Protein·RNA Interaction Networks. Bioinformatics. 2012;28(22):3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glykos NM. Carma: a Molecular Dynamics Analysis Program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 39.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a Program to Generate Schematic Diagrams of Protein-ligand Interactions. Protein Eng, Des Sel. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 40.Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3D: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 41.Skjærven L, Yao X, Scarabelli G, Grant BJ. Integrating Protein Structural Dynamics and Evolutionary Analysis with Bio3D. BMC Bioinf. 2014;15:399. doi: 10.1186/s12859-014-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L, Buck M. Molecular Simulations of a Dynamic Protein Complex: Role of Salt-Bridges and Polar Interactions in Configurational Transitions. Biophys J. 2013;105(10):2412–2417. doi: 10.1016/j.bpj.2013.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Borthakur S, Buck M. Dissociation of a Dynamic Protein Complex Studied by All-Atom Molecular Simulations. Biophys J. 2016;110:877–886. doi: 10.1016/j.bpj.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee H, Hota PK, Chugha P, Miao H, Zhang L, Kim S, Guo H, Stetzik L, Wang B, Buck M. Refined NMR Structure of a Heterodimeric SAM:SAM Complex. Characterization and Manipulation of the EphA2 Interface Leads to New Cellular Functions of SHIP2. Structure. 2012;20:41–55. doi: 10.1016/j.str.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fansa EK, Dvorsky R, Zhang SC, Fiegen D, Ahmadian MR. Interaction Characteristics of Plexin-B1 with Rho Family Proteins. Biochem Biophys Res Commun. 2013;434(4):785–790. doi: 10.1016/j.bbrc.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Bouguet-Bonnet S, Buck M. Combining NMR and Molecular Dynamics Studies for Insights into the Allostery of Small GTPase-protein Interactions. Methods Mol Biol. 2012;796:235–259. doi: 10.1007/978-1-61779-334-9_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao XQ, Malik RU, Griggs NW, Skjærven L, Traynor JR, Sivaramakrishnan S, Grant BJ. Dynamic Coupling and Allosteric Networks in the α Subunit of Heterotrimeric G Proteins. J Biol Chem. 2016;291(9):4742–4753. doi: 10.1074/jbc.M115.702605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li ZL, Cao S. Buck MK-Ras at Anionic Membranes: Orientation Orientation···Orientation. Recent Simulations and Experiments. Biophys J. 2016;110(5):1033–1035. doi: 10.1016/j.bpj.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.