

Abstract
Background
Nephrotic syndrome can be caused by a subgroup of mitochondrial diseases classified as primary coenzyme Q10 (CoQ10) deficiency. Pathogenic COQ2 variants are a cause of primary CoQ10 deficiency and present with phenotypes ranging from isolated nephrotic syndrome to fatal multisystem disease.
Case Report
We report three pediatric patients with COQ2 variants presenting with nephrotic syndrome. Two of these patients had normal leukocyte CoQ10 levels prior to treatment. Pathologic findings varied from mesangial sclerosis to focal segmental glomerulosclerosis, with all patients having abnormal appearing mitochondria on kidney biopsy. In two of three patients treated with CoQ10 supplementation, the nephrotic syndrome resolved and at follow-up both have normal renal function and stable proteinuria.
Conclusions
COQ2 nephropathy should be suspected in patients presenting with nephrotic syndrome, although less common than disease due to mutations in NPHS1, NPHS2, and WT1. The index of suspicion should remain high and we suggest that providers consider genetic evaluation even in patients with normal leukocyte CoQ10 levels, as levels may be within normal range even with significant clinical disease. Early molecular diagnosis and specific treatment are essential in the management of this severe yet treatable condition.
Keywords: Nephrotic syndrome, coenzyme Q10 deficiency, COQ2 nephropathy, Mitochondrial proliferation in podocytes, kidney pathology, ubiquinone
Introduction
Nephrotic syndrome is caused by alterations of the glomerular capillary barrier leading to proteinuria, hypoalbuminemia and edema, and is a frequent reason for referral to pediatric nephrology. Monogenic causes are responsible for a minority of cases, with most frequent mutations found in the NPHS1, NPHS2, and WT1 genes, although a growing number of genes have been implicated in the pathogenesis of nephrotic syndrome [1, 2]. Recent registry-based studies suggest that mitochondrial disorders are responsible for 2% of genetically defined nephrotic syndromes due to mutations resulting in deficient coenzyme Q10 (CoQ10, ubiquinone) [3].
Primary CoQ10 deficiency is an autosomal recessive mitochondrial disorder with heterogeneous neurologic, renal, and muscular manifestations [4] caused by deficiency of coenzyme Q10, a lipid-like molecule involved in multiple cellular processes including mitochondrial respiratory chain activity [5]. In the mitochondrial respiratory chain, CoQ10 acts as an electron shuttle by donating reducing electrons from complexes I and II to complex III [6]. Clinical disease may result from pathogenic variants in one of at least 10 genes termed COQ1 through COQ10. In contrast to most mitochondrial disorders, some patients with primary CoQ10 deficiency show substantial clinical improvement with oral CoQ10 supplementation, making early diagnosis and treatment essential in the management of these patients [7].
COQ2 encodes the para-hydroxybenzoate-polyprenyl-transferase enzyme (EC 2.5.1.39) of the CoQ10 synthetic pathway. To date, 20 patients with COQ2 mutations have been reported, with phenotypes ranging from isolated nephrotic syndrome to multisystem disease (Table S1). We report the genotypes and phenotypes of 3 new patients with biallelic mutations in COQ2, including 3 previously unreported variants.
CASE PRESENTATIONS
Patient 1, a 2-year-old Asian-American male, presented at 9 months of age with severe nephrotic syndrome with hypoalbuminemia (0.6 g/dL) and proteinuria with a urine protein/creatinine ratio of 197 mg/mg (normal <0.5). Screening for infectious and metabolic disease was negative, and brain MRI and electroencephalogram were normal. His kidney biopsy demonstrated focal mesangial sclerosis and collapsing glomerulopathy, for which he was treated with angiotensin-converting enzyme inhibitor (ACEi) and indomethacin. Hemofiltration and human albumin infusions were required to treat his severe volume overload. A 27-gene nephrotic syndrome panel identified heterozygous variants in COQ2. He was started on high-dose ubiquinone 5 weeks following diagnosis of nephrotic syndrome at 30 mg/kg/day with subsequent increase to 50 mg/kg/day after 3 weeks. Baseline leukocyte CoQ10 level was 121 pmol/mg protein (normal range 66-183) and increased to 233 pmol/mg protein after 3 weeks of supplementation. Within 4 weeks of initiating treatment, he had decreased proteinuria and improved serum albumin levels, eliminating the need for albumin infusions (Fig S1). After 20 months of follow-up, he has stable renal function and is no longer nephrotic with an albumin of 3.4g/dL; he has stable proteinuria treated with an ACEi in addition to ongoing ubiquinone supplementation.
Patient 2 is a 12-year-old Caucasian male with normal birth and development. At 10 years of age, the patient developed severe steroid-resistant nephrotic syndrome, which rapidly progressed to end-stage renal disease within a year requiring hemodialysis. Renal biopsy demonstrated focal segmental glomerular sclerosis (FSGS). A 21-gene nephrotic syndrome panel identified compound heterozygous variants in COQ2. Leukocyte CoQ10 level was 1.64 mg/L (normal range 0.44 – 1.64 mg/L) at baseline. He was started on ubiquinone supplementation (30 mg/kg/day) 8 months after the diagnosis of nephrotic syndrome. This increased his leukocyte CoQ10 level to 259 pmol/mg protein after 3 months of supplementation but he did not show clinical improvement. Approximately 2 years following diagnosis, he underwent a successful renal transplantation and has not had recurrence of proteinuria.
Patient 3 is a 4-year-old male of mixed European ancestry who presented at 2 years of age with edema, hypoalbuminemia and nephrotic-range proteinuria. He was initially treated with steroids for 3 months without response. Cyclosporine and hydrochlorothiazide were subsequently used for steroid-resistant nephrotic syndrome. Urine protein to creatinine ratio was 22.5 mg/mg at initiation of cyclosporine. Following molecular diagnosis of COQ2-related disease, he was started on ubiquinol 9 months after his diagnosis of nephrotic syndrome at 30 mg/kg/day with subsequent increase to 50 mg/kg/day with incremental improvement of protein to creatinine ratio to less than 1.0 mg/mg. Cyclosporine and hydrochlorothiazide were discontinued without recurrence of significant proteinuria. After 1 year of follow-up, serum albumin is normal and urine protein to creatinine ratio remains around 1.0 mg/mg. Leukocyte CoQ10 levels were not measured prior to supplementation, but after supplementation were 305 and 498 pmol/mg protein (normal range 66-183 pmol/mg) at 3 and 6 months after initiation of supplementation, respectively.
Molecular Findings
All three patients were confirmed to have compound heterozygous biallelic variants in COQ2 by molecular testing (Table S2, Item S1). No mutations in NPHS1, NPHS2, or WT1 were detected in any of the patients. Patient 1 harbored a nonsense mutation, p.Arg387*, which has previously been reported as pathogenic [8]. Patients 2 and 3 each harbored the heterozygous missense variant p.Asn228Ser, which has previously been reported as pathogenic [9]. The other 2 missense variants in patients 1 and 2, p.Thr325Ala and p.Thr294Ile, have not been reported in the literature but both affect highly conserved amino acid positions and are predicted by in silico tools to be deleterious (Item S1). The p.Thr325Ala variant is not present in population databases, and p.Thr294Ile is a rare variant with an allelic frequency of 0.0015% in European populations (rs376333414, ExAC 0.0008%). The insertion mutation c.176dupT present in the third patient causes a frameshift and premature stop codon p.Ala60Argfs*33, which, as a functional null variant, is classified as pathogenic.
Pathologic findings
The renal biopsy from patient 1 showed global and segmental glomerulosclerosis involving approximately 20% of the glomeruli. Many glomeruli had mild mesangial hypercellularity associated with increased matrix resulting in sclerosis and solidification (Figure 1-a&b). Several glomeruli had global collapse associated with markedly hypertrophied visceral podocytes, occasionally forming ‘pseudocrescents’ (Figure 1-c). Electron microscopic (EM) examination demonstrated collapsed glomeruli with prominent podocytes and extensive foot process effacement. Few podocytes contained increased numbers of mitochondria, some dysmorphic (Figure 1-d).
Figure 1.
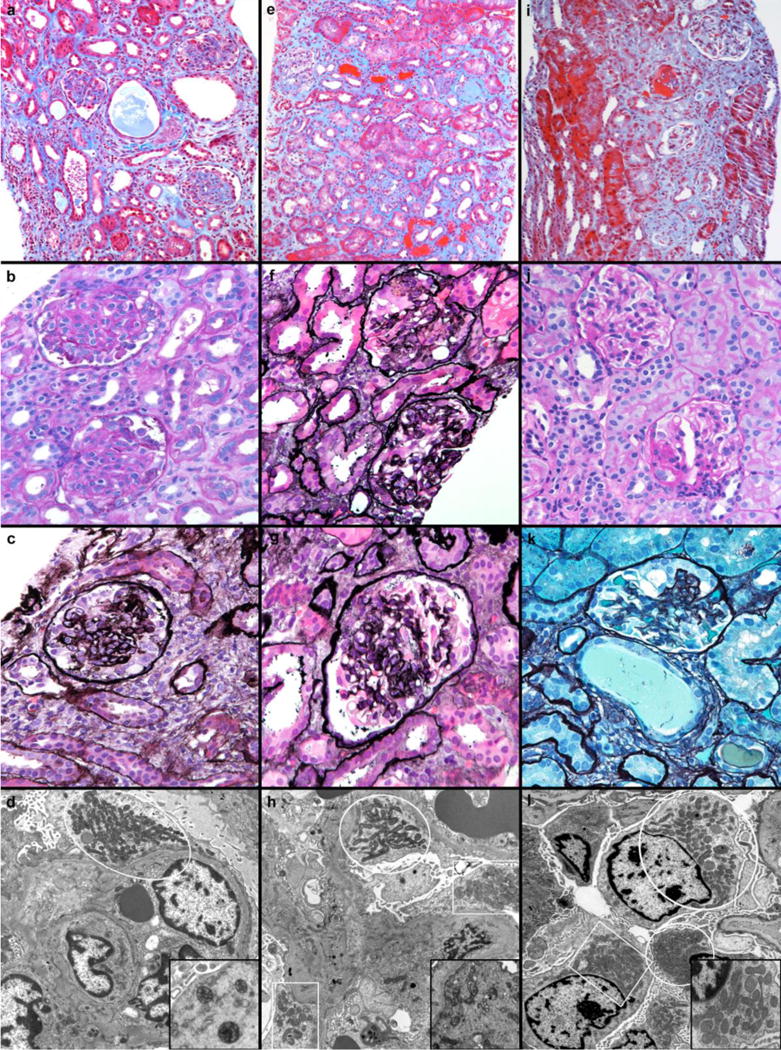
Patient 1, a-d. a) There was marked interstitial fibrosis associated with tubular atrophy and occasional tubular microcysts that separated sclerotic glomeruli (Trichrome, 100×). b) Tuft consolidation and mesangial sclerosis were widespread (Periodic acid Schiff, 200×). c) Collapsed glomerular segments were associated with hypertrophied podocytes (Jones methamine silver, 400×). d) Electron microscopic examination revealed extensive podocyte foot process effacement and collections of mitochondria (white outline), some containing aberrant cristae, inset (Uranyl acetate and lead citrate, 4800×). Patient 2, e-h. e) The fibrotic interstitium contained numerous glomeruli with global and segmental sclerosis (Trichrome, 100×). f) Tufts with segmental sclerosis (bottom) and collapse (top) bore voluminous podocytes (Jones methamine silver, 200×). g) Segments with patent capillaries (3 o’clock and 7 o’clock) were adjacent to collapsed segments where hypertrophied podocytes formed pseudocrescents (top) (Jones methamine silver, 400×). h) Several enlarged podocytes had fused foot processes and regional increases in mitochondria (white outlines) (Uranyl acetate and lead citrate, 4800×); dysmorphic mitochondria were irregular in shape and had oddly-oriented cristae (inset). Patient 3, i-l. i) Tubular atrophy and patchy fibrosis was associated with glomerular damage ranging from segmental (top) to global (bottom) sclerosis (Trichrome, 100×). j) Glomeruli within regions of intact parenchyma had variably increased mesangial matrix or segmental sclerosis (bottom) associated with synechia to Bowman’s capsule (Periodic acid Schiff, 200×). k) Occasional sclerotic segments bore hypertrophied podocytes (11 to 1 o’clock) (Jones methamine silver, 400×). l) Villous hyperplasia of enlarged podocytes was accompanied by extensive foot process effacement and increased numbers (white outlines) of variably misshapen mitochondria (inset) (Uranyl acetate and lead citrate, 4800×).
Forty percent of the glomeruli in the kidney biopsy from patient 2 were globally sclerotic (Figure 1-e). In addition to segmental sclerosis, scattered glomeruli had collapsed tufts which were surrounded by hypertrophic podocytes with voluminous vacuolated cytoplasm (Figure 1-f&g). In preserved regions, foot process effacement and occasional podocyte vacuolation were noted on EM. Few podocytes contained areas with increased numbers of mitochondria, some with dysmorphic features characterized by irregular shape and aberrant cristae, including “pinwheel” configuration (Figure 1-h). The pathology was compatible with the collapsing glomerulopathy variant of focal segmental glomerulosclerosis (FSGS).
The renal biopsy for patient 3 showed approximately 10% of glomeruli with global sclerosis; focal and segmental glomerulosclerosis was evident in 10%. The interstitium had focal fibrosis associated with tubular atrophy (Figure 1-i-k). Electron microscopy demonstrated extensive effacement of the visceral epithelial cell foot processes. In several fields, podocyte mitochondria were plentiful and occasionally dysmorphic (Figure 1-l). Immunoflorescence and EM examination exclude the presence of immune complexes in all of the cases.
DISCUSSION
Mitochondrial cytopathies resulting in renal dysfunction are rare. Primary CoQ10 deficiency due to COQ2 mutations has been described in patients presenting with isolated nephrotic syndrome, renal disease with neurologic symptoms, or multisystem disease (Table S1). Here we report 3 children with COQ2 deficiency presenting with nephrotic syndrome. We add 3 novel mutations, 2 missense (p.Thr325Ala and p.Thr294Ile), and one frameshift (c.176dupT), to those recognized as presenting with isolated nephrotic syndrome.
Consistent with previously described COQ2 nephropathy, all 3 of our patients presented with heterogeneous glomerular lesions characteristic of podocytopathy [7]. Two had changes of so-called “collapsing glomerulopathy” that is associated with „dedifferentiation’ of mature podocytes and has been observed secondary to various infections, medications, autoimmune disorders, or severe vascular disease. Focally increased and dysmorphic mitochondria were evident in podocytes which showed extensive foot process effacement. Normal mitochondrial function is necessary to supply the high energy demands of these specialized cells, including maintenance of structure and function. Similar changes are described in the CoQ deficient PDSSkd/kd murine model of collapsing glomerulopathy, which shows dramatic rescue of proteinuria and interstitial nephritis with oral CoQ10 supplementation [10]. COQ2 mutations likely interfere with normal podocyte metabolism via altered oxidative phosphorylation, and impairment of mitochondrial biogenesis [11]. Recent translational research has suggested that COQ2 genotype may predict the severity of CoQ10 deficiency [12].
Two patients had normal leukocyte CoQ10 levels at baseline prior to treatment (the third was not measured), and had increased CoQ10 levels with supplementation. This differs from low CoQ10 levels previously reported in patients with COQ2 variants and multisystem disease [13]. Normal leukocyte CoQ10 levels have never been reported, to our knowledge, in individuals with COQ2 nephropathy [7]. However, studies have described low CoQ10 levels and decreased activity in renal tissue, as the concentration of CoQ10 likely varies by tissue, and in patients with nephrotic syndrome, the concentration may be lower in podocytes than in leukocytes or fibroblasts [9]. Based on our experience, leukocyte CoQ10 levels alone cannot be relied upon to screen for primary CoQ10 deficiency in the setting of nephrotic syndrome, and providers should consider genetic evaluation even in patients with normal leukocyte CoQ10 levels.
Remarkably, two of our patients had resolution of nephrotic syndrome with CoQ10 treatment. Patient 2 had rapid progression of kidney disease, which suggests that early diagnosis and initiation of treatment with CoQ10 is important in treatment of patients with COQ2 nephropathy. Patients 1 and 3 showed normalization of serum albumin levels and decreased proteinuria within weeks of treatment with CoQ10, similar to one previously reported patient [14]. Both ubiquinone and ubiquinol may be used for treatment depending on availability and clinician preference, however these compounds differ in terms of bioavailability. While ubiquinol appears to have greater bioavailability, it is more challenging to obtain for clinical use [9]. The appropriate treatment dosage, measurable clinical response, and ranges of leukocyte CoQ10 levels indicative of treatment response remain to be established, but it appears that 30 to 50 mg/kg/day may be an appropriate starting dose.
In conclusion, we report 3 novel variants in COQ2 resulting in nephrotic syndrome in 3 unrelated individuals. COQ2 mutations cause renal disease with variable renal pathology resulting from podocyte injury. Our experience demonstrates resolution of nephrotic syndrome and attenuation of renal disease progression with high-dose CoQ10 supplementation, highlighting the importance of early genetic evaluation in cases of nephrotic syndrome [15]. In particular, leukocyte CoQ10 levels may be normal even with significant disease and cannot be relied upon to screen for this treatable cause of nephropathy. Given the potential efficacy of treatment, timely diagnosis with strong consideration for appropriate rapid comprehensive genetic testing and expedient initiation of CoQ10 therapy may confer clinical benefit.
Supplementary Material
Table S1: Genotype-phenotype correlation in inherited COQ2 mutations.
Table S2: Clinical course of kidney disease and genotype in 3 patients with biallelic COQ2 mutations manifesting as nephrotic syndrome
Figure S1: Urine protein/creatinine ratio (mg/g) at baseline and after ubiquinone supplementation (30 mg/kg/d initially, then increased to 50 mg/kg/d) for Patient 1.
Item S1: Detailed methods.
Acknowledgments
We would like to thank Dr. Carlos R. Ferreira at the National Institutes of Health for his contribution of expertise in CoQ10 deficiency-associated diseases, and Dr. Mark Lovell at Colorado Children’s Hospital for his assistance with obtaining renal pathology. We would also like to thank the patients and families for their agreement to participate in our manuscript. Supported by the National Institutes of Health (5T32GM007454 to IC) and (T32DK007662 to MS).
Footnotes
The authors declare that they have no other relevant financial interests.
Contributions: MS, IC, and CL generated ideal for case report. MS, IC, AS, AL, JG, CH, JVH, SH, and CL all participated in the clinical care of these patients. LF reviewed the renal pathology for all cases, assisted with interpretation of findings and provided images for publication. MS wrote the first draft of this paper. All authors discussed the results and implications, commented on the manuscript at all stages, and approved the final manuscript.
References
- 1.Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629–639. doi: 10.1016/S0140-6736(03)14184-0. [DOI] [PubMed] [Google Scholar]
- 2.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119:e907–919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 3.Dogra S, Kaskel F. Steroid-resistant nephrotic syndrome: a persistent challenge for pediatric nephrology. Pediatr Nephrol. 2017;32:965–974. doi: 10.1007/s00467-016-3459-5. [DOI] [PubMed] [Google Scholar]
- 4.Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J Inherit Metab Dis. 2015;38:145–156. doi: 10.1007/s10545-014-9749-9. [DOI] [PubMed] [Google Scholar]
- 5.Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014;5:156–162. doi: 10.1159/000362826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Rourke B, Cortassa S, Aon MA. Mitochondrial Ion Channels: Gatekeepers of Life and Death. Physiology (Bethesda) 2005;20:303–315. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emmanuele V, López LC, Berardo A, Naini A, Tadesse S, Wen B, D’Agostino E, Solomon M, DiMauro S, Quinzii C, Hirano M. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69:978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desbats MA, Vetro A, Limongelli I, Lunardi G, Casarin A, Doimo M, Spinazzi M, Angelini C, Cenacchi G, Burlina A, Rodriguez Hernandez MA, Chiandetti L, Clementi M, Trevisson E, Navas P, Zuffardi O, Salviati L. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet. 2015;23:1254–1258. doi: 10.1038/ejhg.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 10.Barisoni L, Madaio MP, Eraso M, Gasser DL, Nelson PJ. The kd/kd mouse is a model of collapsing glomerulopathy. J Am Soc Nephrol. 2005;16:2847–2851. doi: 10.1681/ASN.2005050494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imasawa T, Rossignol R. Podocyte energy metabolism and glomerular diseases. Int J Biochem Cell Biol. 2013;45:2109–2118. doi: 10.1016/j.biocel.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Desbats MA, Morbidoni V, Silic-Benussi M, Doimo M, Ciminale V, Cassina M, Sacconi S, Hirano M, Basso G, Pierrel F, Navas P, Salviati L, Trevisson E. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet. 2016;25:4256–4265. doi: 10.1093/hmg/ddw257. [DOI] [PubMed] [Google Scholar]
- 13.Mitsui J, Matsukawa T, Yasuda T, Ishiura H, Tsuji S. Plasma Coenzyme Q10 Levels in Patients With Multiple System Atrophy. JAMA Neurol. 2016;73:977–980. doi: 10.1001/jamaneurol.2016.1325. [DOI] [PubMed] [Google Scholar]
- 14.Montini G, Malaventura C, Salviati L. Early Coenzyme Q10 Supplementation in Primary Coenzyme Q10 Deficiency. N Engl J Med. 2008;358:2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 15.Gbadegesin RA, Winn MP, Smoyer WE. Genetic testing in nephrotic syndrome–challenges and opportunities. Nat Rev Nephrol. 2013;9:179–184. doi: 10.1038/nrneph.2012.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Genotype-phenotype correlation in inherited COQ2 mutations.
Table S2: Clinical course of kidney disease and genotype in 3 patients with biallelic COQ2 mutations manifesting as nephrotic syndrome
Figure S1: Urine protein/creatinine ratio (mg/g) at baseline and after ubiquinone supplementation (30 mg/kg/d initially, then increased to 50 mg/kg/d) for Patient 1.
Item S1: Detailed methods.