

Abstract
Repairing DNA double-strand breaks (DSBs) by non-homologous end-joining (NHEJ) requires multiple proteins to recognize and bind DNA ends, process them for compatibility, and ligate them together. We constructed novel DNA substrates for single-molecule nano-manipulation allowing us to mechanically detect, probe, and rupture in real-time DSB synapsis by specific human NHEJ components. DNA-PKcs and Ku allow DNA end synapsis on the 100 ms timescale, and addition of PAXX extends this lifetime to ~2s. Further addition of XRCC4, XLF and Ligase IV resulted in minute-scale synapsis and led to robust repair of both strands of the nanomanipulated DNA. The energetic contribution of the different components to synaptic stability is typically on the scale of a few kCal/mol. Our results define assembly rules for NHEJ machinery and unveil the importance of weak interactions, rapidly ruptured even at sub-picoNewton forces, in regulating this multicomponent chemomechanical system for genome integrity.
Introduction
NHEJ is a complex multicomponent process, which is conceptually divided into three steps: DNA end synapsis, processing, and ligation. Ku70–Ku80 heterodimer (Ku) recognizes DNA ends [1] and recruits DNA-PKcs to form the DNA-PK holoenzyme, which interacts with another equivalent holoenzyme to form a synaptic complex [2]. Through autophosphorylation, the holoenzyme then provides a platform for assembly and regulation of downstream components [3, 4, 5, 6, 7, 8]. For instance, DNA-PKcs recruits Artemis to open the DNA hairpin structure during V(D)J recombination [9, 10]. Although PAXX has recently been identified as important for NHEJ, its role remains unclear [11, 12, 13, 14]. XRCC4 and XLF interact with each other and form filament structures [15, 16, 17, 18, 19]. The XLF-XRCC4 complexes interact with DNA-PK assembled at DNA ends and are believed to bridge spatially two DNA ends so as to facilitate ligation of the break by Ligase IV [20, 21]. Although the overall process is understood with the key components identified and characterized through various approaches, critical information concerning the forces that hold together DNA ends, the kinetics of assembly and disassembly of NHEJ complexes, as well as the robustness of the repair pathway to fluctuations in complex composition remain unknown [22, 23]. Here we used single-molecule experimentation along with a novel DNA substrate to enable mechanical and energetic analysis of molecular interactions across a DNA break and address these questions.
Results
The DNA molecular forceps sustains a functional NHEJ reaction
To measure objectively and quantitatively the properties of complex molecular synapsis and interactions, we developed a DNA-scaffold based single-molecule assay that allows one to observe repeated cycles of synapsis and rupture. Microscopic properties such as kinetics of formation and lifetime of synapsis can be derived from such assays (Figure 1A). Briefly, the DNA system consists of two linear double-strand DNA segments ~1510 bp in length, connected to each other by a third double-strand segment, termed a “bridge,” of ~690 bp (see Online Methods and Supplementary Figure 1). The bridge is anchored 58 bp away from the ends of each DNA segment it tethers, allowing the ends to fluctuate freely as they face each other and providing ample space for loading of NHEJ components [6, 24, 25]. We attach this construct at one end to a digoxigenin-modified glass surface and at the other end to a streptavidin-coated magnetic bead via 1-kb digoxigenin- or biotin-labelled DNA fragments attached to either end of the ~3.6 kbp construct. With a magnetic trap and computer-aided video-microscopy we applied a vertical extending force, F, to the bead and hence the construct, all the while imaging the position of the magnetic bead above the surface [26]. This system allows real-time monitoring of DNA end synapsis by monitoring the molecule’s overall extension, l. If there is no synapsis between the DNA ends, then the extension of the ~3.6 kbp construct under a 1.4 picoNewton (pN) force (1 pN = 10−12 N) is predicted to be 1085 nm based on the worm-like chain (WLC) model which describes the mechanical properties of DNA [27]. If there is synapsis between the DNA ends, then the bridge no longer contributes to the extension of the system, which is now predicted to be only 913 nm, or 172 nm shorter than in the absence of synapsis.
Figure 1. Double-strand break repair by NHEJ proteins at single-molecule resolution.

(A) A 600 bp dsDNA segment (magenta) joins two 1.5 kbp dsDNA segments (blue, red), forming a construct in which two blunt ends face each other. Stars represent phosphate groups. The construct is tethered to a treated glass surface and a 1-micron magnetic bead. Magnets located above the sample generate a controlled extending force on the DNA (green arrow), and the DNA end-to-end extension is determined in real-time. Ligation is observed via a series of four steps: (1) at high force a high-extension state is initially observed, (2) the force is lowered allowing the DNA ends to interact, (3) the force is returned to its initial value but if end ligation has occurred the construct cannot recover its initial extension, (4) the initial extension is recovered upon specific cleavage of repaired DNA. (B) Time-trace obtained upon force-modulation (red) in the presence of Ku, DNA-PKcs, PAXX, XLF, XRCC4 and Ligase IV. Initially, DNA extension (blue points) is shown to alternate between a low and a high value upon force modulation. After addition of NHEJ components (black up arrow) the maximum extension displayed by the construct is reduced. After washing the sample with 0.2% SDS (break in time-trace), addition of SmaI (red up arrow) results in an increase, ∆l in DNA extension. (C) Histogram of ∆l values. Red line is a fit to a Gaussian distribution, with a maximum at 161 ± 8 nm (SD, n=28 cleavage events). (D) DNA ligation probability per traction cycle. We monitored 50 DNA molecules; of 36 molecules repaired 28 were monitored throughout the cleavage reaction. Red line: single-exponential fitting yields a time constant of ~0.8 ± 0.2 cycles (SEM, n=36) or ~175s.
We first show that this system can successfully recapitulate complete NHEJ of blunt-ended DSBs (Fig. 1B). We digested the DNA with SmaI restriction enzyme, assembled constructs in the magnetic trap (~25-50 constructs/field-of-view), and introduced into the reaction chamber the full complement of human NHEJ components (10 nM Ku, 100 pM DNA-PKcs, 20 nM PAXX, 20 nM XLF and 20 nM XRCC4–Ligase IV, see Supp. Fig. 1C,D). We repeatedly cycled the force applied to the DNA between a low and high value with 450 second period (Flow = 0.04 pN for ~225 seconds, Fhigh = 1.4 pN for ~225 seconds); in the low-force state the DNA ends are free to encounter each other whereas in the high-force state we can reliably interrogate the DNA as to the status of its ends. After one or two traction cycles we frequently observed a DNA state with reduced extension (Fig. 1B) and which never recovered initial extension, indicative of successful DNA ligation. Successful ligation of the blunt DNA ends of the construct generates a SmaI site. Thus, after disrupting residual NHEJ components with 0.2% SDS and then washing out the detergent (see Materials and Methods), we introduced SmaI into the experiment and the initial DNA state was restored via an abrupt increase in DNA extension <Δl> = 161 +/- 2 nm (SEM, n = 28, Fig. 1C). This repair-specific cleavage reaction provides us with a length-change calibration that we can then use as a robust signature of bonafide, specific interactions between the ends of the DNA construct. Most DNA molecules (36 out of 50, or 72%) were repaired within the first few traction cycles (Fig. 1D), underscoring the robust nature of DSB repair in this system when all components are present. Ligation was still observed, although at lower efficiency, if any one of PAXX, XLF or XRCC4 C-terminal domain were omitted from the reaction (Table 1). No ligation was observed if DNA-PKcs or Ligase IV was omitted (Table 1) or if DNA ends were dephosphorylated (see below).
Table 1. Efficiency of observed NHEJ ligation reactions as a function of NHEJ components present.
Ligation events were identified as an irreversible shortening of the DNA scaffold’s extension by Δl ~ 165 nm. For the complete reaction, 28 of the 36 molecules scored for repair were further confirmed by DNA cleavage via SmaI digest (Fig. 1C); the remaining 8 molecules were lost from tracking during SDS washes which can destabilize streptavidin and antidigoxigenin links between DNA and surfaces)
| Assay | # molecules | Number of molecules ligated at # pulling cycle | Total | Efficiency % | Normalized Efficiency % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 4th | 5th | 6th | 7th | 8th | 9th | |||||
| Complete reaction | 50 | 26 | 7 | 2 | 1 | N/A | N/A | N/A | N/A | N/A | 36 | 72 ± 12 | 100 |
| Complete but for DNA-PKcs | 40 | 0 | 0 | 0 | N/A | N/A | N/A | N/A | N/A | N/A | 0 | 0 | 0 |
| Complete but for Ku | 69 | 0 | 1 | 0 | 1 | N/A | N/A | N/A | N/A | N/A | 2 | 3 ± 2 | 4 |
| Complete but for PAXX | 57 | 6 | 5 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 16 | 28 ± 7 | 39 |
| Complete but for XLF | 184 | 0 | 1 | 0 | 1 | 1 | 3 | 1 | 2 | 0 | 9 | 5 ± 2 | 7 |
| Complete but for XRCC4 NTD | 193 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 2 ± 1 | 3 |
| Complete but for Ligase IV | 102 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
The ability of the ligated DNA to supercoil demonstrated that there was a double ligation with both strands of the DNA ligated to their respective counterpart in the other DNA end (Supp. Fig. 2A). We also show (Supp. Fig. 2B-D) that T4 DNA ligase, although capable of efficiently ligating the construct prepared with overhanging ends (via XmaI digestion), was unable to efficiently ligate the construct with blunt ends, underscoring the requirement in vivo for robust molecular pathways to repair non-complementary DSBs. We conclude that this DNA system functionally recapitulates human NHEJ at single-molecule resolution, providing us with a basis by which to evaluate the contribution of each reaction component to synaptic stability.
Stepwise Assembly of the human NHEJ machinery
We thus proceeded by progressively assembling the NHEJ machinery and testing for DNA end synapsis by the force-modulation method described above. We find that the minimal combination of the DNA-PK holoenzyme (DNA-PKcs plus Ku) and PAXX is necessary to observe frequent, second-scale interactions between the two DNA ends (Fig. 2A,B and Supp. Table 1). In this assay an interaction was observed as a transient plateau in the DNA extension signal obtained upon increasing the force from Flow to Fhigh; duration of the plateau reflects the lifetime of the end interaction. The histogram depicting the distribution of observed Δl values (Fig. 2C) again displays a Gaussian peak located close to the expected amplitude (166 ± 1 nm SEM, n =129). Comparing the change in DNA extension detected at the end of the plateau to our calibrated length-change obtained via specific recleavage of the repaired DNA ends confirms we are detecting bonafide interactions between the ends of the DNA construct; we specifically designate as “synaptic” those interaction events within three standard deviations of the likeliest value. In Fig. 2D we show that the lifetime distribution for synaptic events follows single-exponential behavior with an average, <tsynapsis>, of 2.2 ± 0.3 s (SEM, n = 98). Synapsis frequency, defined as the fraction of force-modulation cycles in which we observe a synaptic event, is roughly 6% (102 synaptic events out of 1611 force-modulation cycles collected on ~90 DNA substrates, see Supp. Table 1).
Figure 2. Ku, DNA-PKcs and PAXX are necessary to stabilize DNA-end synapsis.

(A) Experimental design. DNA is prepared with blunt ends using SmaI digest, stars represent phosphate groups. (B) Representative time-trace obtained upon application of the force-modulation pattern (red). DNA is prepared with blunt ends using SmaI digest. Inset shows an expanded view of an end-interaction rupture event, which can be characterized by both the change in DNA extension upon rupture, Δl, and the duration of the synaptic event prior to rupture, tsynapsis. (C) Histogram of DNA extension change, ∆l, upon rupture event. Red line is a fit to a Gaussian distribution, with a maximum (red arrow) at 166 ± 10 nm (SD). The entire histogram contains n=129 events, of which 98 are within three standard deviations from the peak. (D) Lifetime distribution of the synaptic state is fit to a single-exponential distribution (red line), giving a lifetime of 2.2 ± 0.3 s (SEM, n=98).
These synaptic events did not depend significantly on the phosphorylation state of the DNA ends (Fig. 3A). Notably, synaptic events were abolished when we replaced wild-type PAXX with a mutant (V199A and F201A) deficient in its ability to interact with the Ku subunit of the DNA-PK holoenzyme (Fig. 3B). XLF was unable to substitute for PAXX (Fig. 3C) despite its ability to interact with Ku [28, 29]. Neither DNA-PK holoenzyme nor PAXX on their own generates synapsis on the 2-second scale (Fig. 3D, E). We conclude that the “upstream” components Ku, DNA-PKcs and PAXX sustain formation of DNA end synapsis which can resist up to ~1 pN traction forces (0.15 kCal/mol·nm) for second time-scales, and that PAXX plays a central role in stable bridging of the gap between the two DNA ends.
Figure 3. DNA synapsis on the 2-second timescale in the presence of Ku, DNA-PKcs, and PAXX: control experiments.

(A) Synapsis by Ku, DNA-PKcs and PAXX does not require phosphorylated DNA ends. (Left) Time-trace showing rupture events. (Middle) Amplitude distribution of rupture events (n=69). Distribution peak is fit to a Gaussian (red line) with a maximum at 163 ± 2 nm (SEM; σ=12 nm; n=40). The distribution displays additional density for Δl values of 75 nm and 400 nm. Values of 400 nm are consistent with the extension of a 1500 bp DNA segment at the force employed, and thus likely correspond to “loop-back” interactions between the tip of a 1500 bp DNA segment and the surface to which that segment is anchored. The smaller peak is consistent with local bending or wrapping deformations of DNA with ~50 nm persistence length [37]. (Right) Lifetime distribution of synaptic events follows a single-exponential distribution (red line) with a mean of 1.9±0.5s (SEM, n=40). (B-E) Representative time-traces show that the combinations of (B) Ku, DNA-PKcs, and the PAXX mutant; (C) Ku, DNA-PKcs, and XLF; (D) Ku and DNA-PKcs; and (E) PAXX alone do not lead to 2-second synapsis. The well-known contamination of DNA-PKcs preparations by Ku [25, 38] means it is not formally possible to test DNA-PKcs alone.
To assess the role of the “downstream” components XRCC4, XLF and Ligase IV we added them in combination with the “upstream” components used earlier, to obtain a “complete” reaction (Fig. 4). Unlike in the prior assays with all components however, here we dephosphorylated the DNA ends so as to prevent ligation, allowing us to repeatedly interrogate the construct. We now observed long-lived synaptic interactions: <tsynapsis> = 66.4 ± 7.6s (SEM, n=175). Omitting any one of Ligase IV, XLF and the N-terminal portion of XRCC4 from the above reaction (Supp. Fig. 3A-C) resulted in the same short synaptic lifetime as was observed in the presence of Ku, DNA-PKcs and PAXX using dephosphorylated DNA substrate (Fig. 3A), i.e. of the order of 2-3 seconds. This supports and extends observations that XRCC4, XLF and Ligase IV are involved in a series of joint interactions that could lead them to act as a modular, functional unit of NHEJ; Ligase IV interacts with the coiled-coil (cc) region of XRCC4 (ccXRCC4, corresponding to residues 138-213), the N-terminal domain of which associates with XLF [21, 30]. Removing any one of these three components thus abolishes the stabilizing effects of this functional unit. The same results were obtained when these experiments were repeated using a phosphorylated DNA substrate (compare Supp. Fig. 3D-F and Fig. 2).
Figure 4. Maximum stabilization of DNA end synapsis by Ku, DNA-PKcs, PAXX, XLF, XRCC4 and Ligase IV.

(A) Experimental design. DNA is prepared with blunt ends using SmaI digest and then dephosphorylated (see Materials & Methods). (B) Representative time-trace obtained upon application of the force-modulation pattern (red) in the presence of Ku, DNA-PKcs, PAXX, XLF, XRCC4 and Ligase IV. Transient synapsis is observed as an intermediate DNA extension state, which spontaneously reverts to the maximum extension, allowing us to characterize the duration of the intermediate state and the change in extension upon reversion to the maximum extension. (C) Histogram of DNA extension change (∆l) upon rupture event. Red line is a fit to a Gaussian distribution, with a maximum (red arrow) at 165 ± 9 nm (SD, n=324 events in the histogram, of which 183 within three standard deviations from the peak). (D) Lifetime distribution of the synaptic state is fit to a single-exponential distribution (red line), giving a lifetime of 66.4 ± 7.6 s (SEM, n=183).
Replacing Ligase IV by a catalytically-inactive mutant (K273A, see Supp. Figs. 1D and 3G) resulted in a synaptic lifetime of 34.5±6.9 s (SEM, n = 82) reflecting a mild impact on synapsis lifetime and indicating this component could still sustain the web of interactions necessary for downstream components to stabilize synapsis by DNA-PK and PAXX. Surprisingly, stable interactions were observed when wild-type XLF was replaced by a mutant (XLF1-233) deficient in its interactions with Ku (44±12.6s SEM, n=84, see Supp. Fig. 3H). However, removing DNA-PKcs from the complete reaction essentially abolished synapsis (Supp. Fig. 3I). Synapsis was also abolished when both XLF and PAXX were omitted from the complete reaction (Supp. Fig. 3J), supporting and extending recently-described overlapping function between XLF and PAXX and resulting synthetic embryonic lethality in double-knockout mouse models [31, 32]. We note that the ternary combination of XLF, XRCC4, and Ligase IV does not support synapsis formation under the physiological salt conditions employed in this work (Supp. Fig. 4).
To investigate further the role of PAXX, we next removed it from the complete reaction, resulting in a seven-fold reduction in synaptic lifetime compared to when PAXX was present (9 ± 1.8 s SEM, n = 77, see Supp. Fig. 5A-C). We used these conditions to probe the effect of the traction force on synaptic lifetime. We thus observed that varying Fhigh from 0.1 pN to 4.7 pN had no significant effect on the lifetime of synapsis (Supp. Fig 5D). This indicates that the second-to-minute lifetimes we measure are relevant to the kinetics of NHEJ in vivo where the forces acting on broken DNA ends are essentially diffusive. We also note that substituting the PAXX mutant for wild-type PAXX resulted in roughly the same reduction in synaptic lifetime as removing PAXX entirely from the reaction (here to 5.4 ± 1.3s, see Supp. Fig. 5E-G).
Discussion
We thus find that synaptic junctions of varied composition can resist picoNewton-scale forces for times ranging from seconds to minutes. Presumably this is a reflection of actual commitment to repair. Based on these results we propose a simple model for NHEJ synapsis (Fig. 5). Taking the lifetime of the complete synaptic junction as a baseline (66s), we find that removing PAXX is responsible for a roughly seven-fold destabilization of the complete synaptic junction (9s synapsis without PAXX). We likewise determine that disabling the XRCC4–XLF–Ligase IV system is responsible for a roughly 30-fold destabilization of the complete synaptic junction (2s synapsis for just DNA-PK and PAXX, without XRCC4–XLF–Ligase IV). Because the lifetime of a complex is related to its free energy of activation by Boltzmann’s law, we estimate that PAXX contributes kBT ln (66s/9s) ~ 2 kBT or 1.2 kcal/mol to the stability of the complete synaptic junction, whereas XRCC4–XLF–Ligase IV contributes kBT ln (66s/2s) ~ 3.5 kBT or 2.1 kcal/mol to the stability of the complete synaptic junction. Together these two sets of components would stabilize the synaptic state by roughly 5.5 kBT, increasing its lifetime roughly two-hundred fold. These results suggest that the primary synapsis formed by the DNA-PK holoenzyme alone is very short-lived, in the range of hundreds of miliseconds. Finally therefore, to attempt to detect this short-lived interaction, we increased by a factor of ten the rate at which the force is switched between low and high values. The large number of traction cycles thus generated allowed us to detect rare and very short-lived synaptic events of expected synaptic amplitude (Supp. Fig. 6). The lifetime of this synapsis is, as predicted, in the 100 milisecond range. Control experiments show this short-lived and specific synapsis is only observed in the presence of DNA-PKcs and Ku (Supp. Fig. 6 and Supp. Table 2).
Figure 5. Model for multivalent stabilization of DNA-end synapsis by NHEJ machinery.
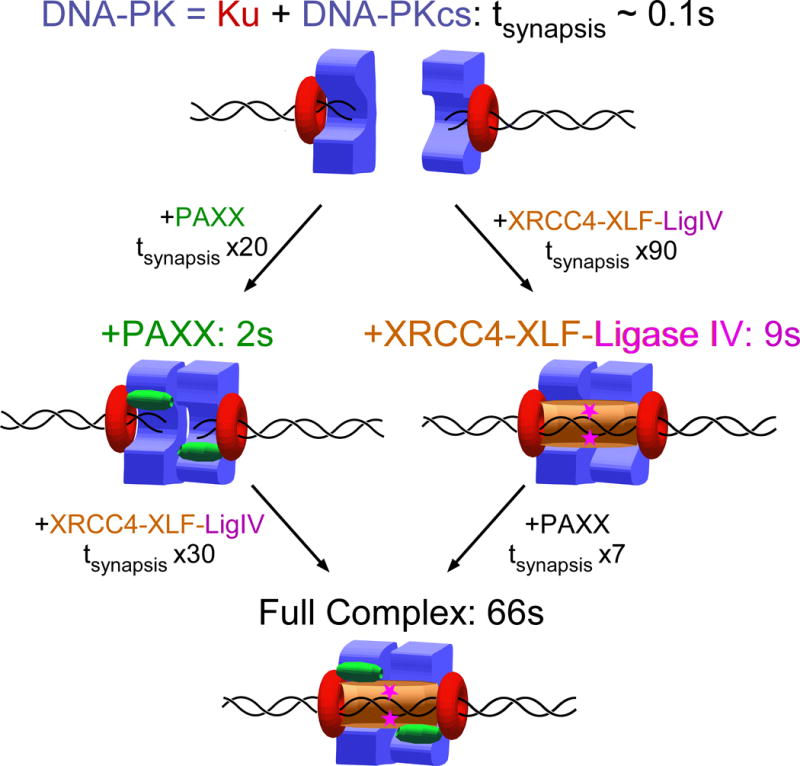
Schematic model for synapsis and repair involves (top) an initial synaptic complex formed by two DNA-PK holoenzymes but with a lifetime in the range of hundreds of milliseconds and which can be stabilized by incorporation of PAXX (left path) or XRCC4–XLF–Ligase IV (right path). A complete complex stabilized by both PAXX and XRCC4–XLF–Ligase IV has the longest lifetime (bottom) and leads to efficient ligation of the DSB.
The additivity of these short-lived and weak interactions, revealed by these single-molecule experiments, indicates a multivalent system where multiple protein interfaces stabilize DNA end-synapsis. Use of multiple weak interactions to obtain high affinity balanced by regulation is a critical emerging common theme in DNA repair systems, such as RPA whose sub-nanomolar affinity is built on multiple weak interactions [33]. Longer synapsis effectively increase ligation probability.
Our work supports and broadens the observation that PAXX stabilizes the core NHEJ proteins at damaged chromatin [11, 31, 32], and suggests PAXX as a target for cancer drug development. Indeed removing PAXX from the complete reaction reduces the probability of repair from 72% to 28% but does not completely abolish repair – as is the case when XLF, or XRCC4 are absent from the reaction (Table 1). Whether or not PAXX can incorporate into NHEJ complexes only at the initial stage of assembly, or also at later stages of assembly for instance once XRCC4–XLF–Ligase IV have assembled, remains to be determined. With this validated single-molecule system, it will be feasible and intriguing to examine the impacts of other components that act in NHEJ, such as Artemis and APLF [34] proteins as well as noncoding RNA [35], and relate their energetic and mechanistic roles to cancer predispositions resulting from mutational defects and aberrant regulation.
Currently, our results show that novel and functional DNA scaffolds provide unique and unexpected kinetic insights into the human protein machinery responsible for carrying out NHEJ. Our studies thus provide a solid foundation upon which to further explore the order of assembly and stoichiometry of functional repair complexes using fluorescently-labeled protein variants and correlative single-molecule nanomanipulation and fluorescence (NanoCOSM [36]). Importantly, the measurements of synapsis lifetime and kBT-scale energetics uncover the nature of multi-component DNA break repair complexes. This knowledge has broad scientific and biomedical implications including strategies for targeting the DNA-damage response by small molecules. Specifically, we propose a strategy of chemical inhibitors to compete with the functionally-important yet individually-weak interactions in this major system for break repair in humans.
Online Methods
Materials and Methods
DNA Construct
The construct is assembled from two precursor dsDNA molecules. For the first, we assemble two ssDNA oligos, a short and a long, into a covalently-linked, asymmetric branched structure. In a PCR reaction using a pair of such branched oligos, the longer oligos prime PCR while the shorter one remains unused. The branchpoints end up 58 bp from the end of dsDNA. This “Break” PCR product is 3 kb in length, and by hybridization one can convert the remaining ssDNA branches into dsDNA branches with 4-bp overhangs. This fragment can then be “circularized” by hybridization and ligation to a ~600 bp “Bridge” DNA fragment, which is simply a standard dsDNA fragment digested so as to have overhangs compatible with those of the short dsDNA branches. The circularized DNA can then be reopened with a pair of restriction enzymes and gel purified so as to obtain the desired product, namely two 1.5 kbp DNA segments connected to each other via an internal DNA “bridge” anchored 58 bp from the closest DNA ends. Finally, one of the 1.5 kbp fragments is ligated to a biotin-labeled dsDNA fragment (at the extremity distal to the anchor) while for the other 1.5 kbp fragment is ligated to a digoixgenin-labeled dsDNA fragment (at the extremity distal to the anchor). The construct, assembled to a micron-size magnetic bead at the biotin-labeled end and a treated glass coverslip at the dig-labeled end can then be handled and observed in a magnetic trap (~30-50 constructs per field-of-view). The ends can be processed into blunt or overhang ends by restriction digest either after ligation to the biotin- and dig-labeled fragments, or directly under the magnetic trap.
Oligonucleotides DBCO-Hyb1 5′ YCCATGGGCATACTGATCGGTAGGG and DBCO-Hyb2 5′ YGAGCCAAGACGCCTCCATCCATGCA, where Y indicates DBCO-dT (courtesy of John Randolph, Glen Research, USA), and oligos Az-Charomid-1456-SmaI (5′ GAGAGACCCGGGCACCGTCTCCTTCGAACTTATTCG CAATGGAGTGTCATTCATCAAGGACGCCGCZATCGCAAATGGTGCTATCC) and Az-Charomid-3778-SmaI (5′ GAGAGACCCGGGCACGACTTATCGCCACTGGCAGCAGCCACTGGTAACAGGATTAGCAGAGCGAGGZATGTAGGCGGTGCTACAGAG), where Z represents Azido-dT (Trilink, USA) and the underlined sequence is the SmaI/XmaI restriction site, were resuspended to 10 μg/μl in formamide at 37°C. 50 μg each of oligos DBCO-Hyb1 and Az-Charomid-1456 were combined. Similarly, 50 μg each of DBCO-Hyb2 and Az-Charomid-3778 were combined. Combined oligos were incubated 8h at 37°C, and products corresponding to covalently-coupled oligos were separated on a 8M Urea, 8% acrylamide:bisacrylamide (29:1) gel running at ~10V/cm in 1×Tris-borate-EDTA buffer. Gel slices corresponding to product were excised from the gel, using a sacrificial imaging lane to carry out UV shadowing to identify the relevant bands. DNA was purified by first eluting DNA from the crushed gel fragment overnight in 10 mM phosphate buffer pH 8.0 with agitation at 4°C. A SepPak C18 cartridge (Waters) was conditioned with first acetonitrile and then water; the eluate was loaded onto the cartridge which was then washed with water, and the oligo released from the cartridge by eluting with acetonitrile. Acetonitrile was evaporated under vacuum centrifugation, and oligo resuspended to ~10 μM. The coupled oligos were then used as primers in a PCR reaction using Charomid 9-5 ΔSbfI as template (i.e. a Charomid 9-5 plasmid derivative from which the native SbfI site has been removed by SbfI digestion, fill-in and religation), and ~3 kbp product DNA was purified using agarose gel electrophoresis and extraction (Macherey-Nagel).
Next, the covalently-coupled oligos which do not participate in the PCR reaction were annealed, respectively, to ssDNA oligos O1-Comp (5′ Pho-CGCGCCCTACCGATCAGTATGCCCATGGA, complementary to DBCO-Hyb1 but allowing formation of an AscI overhang compatible with ligation to an MluI overhang as well as an AscI overhang) and O2-Comp (5′ Pho-TGGATGGAGGCGTCTTGGCTCA, complementary to DBCO-Hyb2 but allowing formation of an NsiI overhang compatible with ligation to an SbfI overhang as well as an NsiI site) in equimolar ratio at a final concentration of ~300 nM each for one hour at room temperature in 1× SureCut buffer (New England Biolabs).
Precursor to the “bridge” DNA was obtained by PCR amplifcation of Charomid C9-5 ΔSbfI template with oligos Charo-3600-MluI (5′ GAGAGAACGCGTTACCTGTCCGCCTTTCTCCCTTCGGG) and Charo-4230-SbfI (5′ GAGAGACCTGCAGGC CTCACTGATTAAGCATTGGTAACTGTCAGACC), digesting with MluI and SbfI, and agarose gel separation and extraction (Macherey-Nagel).
“Break” DNA and precursor to “bridge” DNA prepared as above were combined at ~130 nM final concentration each in 1× SureCut buffer (New England Biolabs), along with 0.5 μl each of SbfI, MluI-HF, NsiI and AscI, HC-T4 DNA ligase (New England Biolabs), and 1 mM ATP and 1 mM DTT, and left overnight at room temperature. After 20′ heat inactivation at 65°C, the DNA was digested with XbaI and SacI. This resulted in a linear ~3.6 kbp DNA product consisting of the ~690 bp bridge segment in the center and two linear 1.5 kbp DNA segments at each end anchored to the bridge internally ~58 bp from a SmaI/XmaI restriction site. This 3.6 kbp fragment was purified by agarose gel separation and extraction (Macherey-Nagel). The DNA was finally ligated to biotin-labelled ~1 kbp DNA bearing an XbaI site and digoxigenin-labelled ~1 kbp DNA bearing a SacI site for attachment in the magnetic trap to, respectively, streptavidin-coated magnetic bead and antidigoxigenin-modified glass surface. In this 10 μl ligation reaction conducted in the manufacturer’s recommended conditions (New England Biolabs T4 DNA Ligase buffer 1×), the DNA was at a concentration of 3 nM, the biotin- and dig-labeled fragments were each at 10 nM concentration, and 200 units (0.5 μl) of T4 DNA ligase were used to ligate the DNA for 3h at room temperature before thermally inactivating the ligase.
To complete preparation of the DNA ends, we then carried out overnight digestion of 1 μl of this DNA preparation in a final reaction volume of 10 μl using either 10 units of XmaI (New England Biolabs) or 20 units of SmaI (New England Biolabs) restriction enzyme. Restriction enzyme was heat inactivated as per manufacturer recommendation, and construct was diluted 30-fold to a nominal DNA concentration of 100 pM and stored at −20°C. For experiments conducted using dephosphorylated DNA as a substrate for NHEJ, we dephosphorylated the construct by combining into a 10 μl reaction volume 2 units of antarctic phosphatase (New England Biolabs) and 0.6 fmoles of the ligation product obtained in the previous paragraph. The reaction was allowed to proceed for 1h at 37°C before being thermally inactivated as per manufacturer’s recommendation.
For assembly onto the microscope, the ligation reaction was first diluted sixty-fold to 50 pM nominal concentration of DNA in Tris buffer (10 mM TrisCl pH 8). Then, 0.5 μl of this dilution was mixed with 10 μl of Dynal MyOne C1 magnetic beads. The beads were prepared by taking 10 μl of stock solution, washing them with 100 μl of RB and concentrating the beads, discarding the supernatant, and resuspending the beads in 10 μl of RB. The bead+DNA mixture was diluted after 5-10 seconds with an additional 15 μl of RB and injected onto functionalized surfaces.
Functionalized surfaces consist in two glass coverslips (#1, ~180 μm thick) separated by two thicknesses of parafilm into which a channel (1mm × 50 mm) has been cut. Both coverslips are functionalized with antidigoxigenin and passivated as described recently [39], but one of the coverslips has 2 mm diameter holes located above each end of the channel and which are used for filling and draining the reagent-filled channel. The surface is mounted on a custom-designed holder and placed atop the oil-immersion objective of a magnetic trap, in which a pair of high-grade permanent magnets located above the sample can be translated (to vary the force) or rotated (to rotate the bead and supercoil the DNA) to modify the mechanical constraints applied to the DNA. Real-time particle tracking software (PicoJai SARL) allows for videomicroscopy based tracking of the 1-micron magnetic beads with nanometer resolution and in real-time (~30 Hz).
A typical sample consists in a microscope field-of-view containing ~30-50 individual DNA tethers which can be monitored simultaneously. These tethers are systematically verified to display an appropriate change in DNA extension when the applied force is varied from Flow=0.04 pN to Fhigh=1.4 pN, and when necessary are further verified to not change extension when supercoiling is changed.
Proteins
Constructs
Ku70 and Ku80 were both cloned into pFastBac Dual vector (Thermo Fisher) and co-expressed in insect Sf9 cells. Ku70 contains an N-terminal hexahistidine tag followed by a TEV cleavage site. XLF and XRCC4 were individually cloned into pHAT5 vector [40] and transformed into Rosetta™2 (DE3) cells (Invitrogen). Both XLF and XRCC4 contain a C-terminal hexahistidine tag. Cell cultures were grown in LB medium until OD600 was approximately 0.6. IPTG was added to a final concentration of 1 mM. Proteins were expressed at 16°C overnight for XLF and at 37°C for 4 hours for XRCC4. The PAXX and full-length LigaseIV-XRCC4 co-expression constructs are as described previously [11, 41]. XLF1-233 (C-terminal truncation mutant, which can not bind to Ku70/80) was cloned into pETG-41A (Gateway™ Destination vector, EMBL) and expressed as descripted before [42]. The catalytically-dead LigaseIV(K273A)-XRCC4 protein complex was generated from the LigaseIV-XRCC4 co-expression plasmid (A gift from Prof. Ming-Daw Tsai) by the QuikChange method (Agilent Technologies). Full-length Ligase IV fused with a hexahistidine tag at the C-terminus and ccXRCC4 (residue 138-213) were amplified from the Ligase IV-XRCC4 co-expression plasmid and an XRCC4 plasmid, of which cysteines were mutated to alanines, respectively and cloned into pRSFDuet1 vector (Novagen).
Protein purifications
Ku70/80
Insect Sf9 cells containing Ku70/80 were lysed by sonication in lysis buffer (50 mM Tris pH 8.0, 5% glycerol, 150 mM NaCl, 2 mM β-mercaptoethanol, 20 mM imidazole, protease inhibitor (Roche) and 40 μg/ml Deoxyribonuclease I (Sigma)). After 30 minutes incubation in 4°C after sonication, the salt concentration of the lysate was adjusted to 500 mM NaCl. After centrifugation, Ku70/80 supernatant was mixed with Ni-NTA affinity resin (Qiagen) pre-equilibrated with binding buffer (50 mM Tris pH 8.0, 5% glycerol, 500 mM NaCl, 2 mM β-mercaptoethanol, and 20 mM imidazole). After washing beads with binding buffer for 10× column volume, protein was eluted using elution buffer (50 mM Tris pH8.0, 5% glycerol, 500 mM NaCl, 2 mMβ-mercaptoethanol, and 100 mM imidazole). The N-terminal hexahistidine tag of Ku70 was cleaved by TEV protease. Uncleaved Ku70/80 and TEV (also hexahistidine-tagged) were removed by running through a gravity column containing Ni-NTA resin. The flow-through containing Ku70/80 was dialysed with Q column (GE healthcare) buffer A (20 mM Tris pH8, 50 mM NaCl, 5% glycerol, and 5 mM DTT) and loaded onto the column. Protein was eluted in a gradient against buffer B (20 mM Tris pH8, 1M NaCl, 5% glycerol, and 5 mM DTT). Eluted sample was further purified by running through Superdex 200 10/300 (GE Healthcare) equilibrated in Buffer GF (20 mM Tris pH8, 150 mM NaCl, 5% glycerol, and 5 mM DTT). Protein samples were analysed on 4-12% NuPAGE® Bis-Tris gels, concentrated and stored in −80°C.
XLF and XRCC4
XLF cells were purified by a similar process as for Ku70/80. The lysis buffer for XLF is 20mM HEPES pH8.0, 2M NaCl, 10mM imidazole, 2mM β-Mercaptoethanol and protease inhibitor). No cleavage was carried out for the elution sample from after Ni affinity purification. The Q column for XLF is buffer A (20 mM HEPES pH 8.0, 10 mM NaCl, and 5 mM DTT) and buffer B (20mM HEPES pH8.0, 1M NaCl, and 5 mM DTT). The final gel filtration buffer is 20 mM HEPES pH 8.0, 150 mM NaCl, and 5 mM DTT. Purification of XRCC4 was performed as described previously [43]. Purification of XLF1-233 was identical to that of wild-type XLF, except that the mutant contains an N-terminal 6-His-MPB tag followed by aTEV cleavage site. After Ni affinity purification, the tag was cleaved before proceeding to the Q column.
Ligase IV-XRCC4, PAXX and DNA-PKcs
Purifications of all Ligase IV-XRCC4 constructs, PAXX and DNA-PKcs were performed as described previously [5, 11, 41].
Bulk Ligation of DNA with overhang ends by XRCC4-LigIV System
pHAT4 was double-digested with XhoI and NcoI to generate four base-pair overhangs. 200 ng of the digested plasmid was incubated with 25 nM of the indicated proteins in 20 ml of reaction buffer containing 25 mM Tris-HCl pH 7.5, 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 10 μM ATP, 10% (w/v) PEG10,000, and 10 μg/ml BSA. Mixtures were incubated at 37 °C for 5 min before initiating ligation by XRCC4/LIG4 at 37 °C for 30 min. The mixtures were incubated at 50 °C for another 30 min after adding 2 μl of a reaction-stop solution (100 mM EDTA, 0.1% (w/v) SDS) and 0.2 μl of 20 mg/ml Proteinase K. Reaction mixtures were separated by electrophoresis on a 0.8% agarose gel in TBE buffer. The gel was stained with SYBR Gold, visualized using a UV imager and quantified using GeneTools (Synegene). We note that the presence of four base-pair overhangs makes this assay more permissive than the blunt ends used in the scaffold assay.
Experimental Conditions
All assays were conducted at 34°C in reaction buffer RB (20 mM K·Hepes pH 7.8, 100 mM KCl, 5 mM MgCl2, 1 mM ATP, 1 mM DTT, 0.05% Tween-20, and 0.5 mg/ml BSA), unless noted otherwise. When present, NHEJ components were used at a concentration of 10 nM Ku70/80 (dimer), 100 pM DNA-PKcs, 20 nM PAXX, 20 nM XRCC4, 20 nM XLF, and 20 nM Ligase IV. When mutated versions of proteins were used, they were at the same concentration as wild-type proteins.
To digest DNA molecules which have undergone NHEJ and repair under the magnetic trap, we first disrupt any remaining components of the repair machinery by rincing the capillary with Wash Buffer supplemented with 0.2% SDS (Wash Buffer: 20 mM Tris·Cl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.05% Tween-20, and 0.5 mg/ml BSA), then rinsing the capillary with Wash Buffer, then infusing the capillary with 100 μl of RB containing either 20 units of XmaI or 10 units of SmaI (New England Biolabs). Data presented in histograms of changes in DNA extension upon NHEJ repair or T4-based ligation are obtained by comparing DNA extension before and after cleavage.
Data Collection and Analysis
Single-molecule magnetic trapping data giving DNA extension as a function of time and under force-modulation cycles was obtained using the Picotwist software suite (Picotwist S.A.R.L). Magnetic trapping is carried out as has been described in the literature. In the magnetic trap used the magnetic bead z-position was tracked at 31 Hz to ~5 nm resolution for F = 1.4 pN.
Unless noted otherwise, force was repeatedly alternated between low (Flow = 0.04 pN) and high (Fhigh=1.4 pN) with a total cycle time of typically 450 seconds (~225 seconds at Flow followed by ~225 seconds at high force). For experiments presented in Supp. Fig. 6 and Supp.
2 the total cycle time was about 50 seconds (25 seconds at low force followed by 25 seconds at high force).
Time-traces of DNA extension vs. time were analyzed to determine the lifetime (tsynapsis) and extension change (Δl) upon rupture of individual interaction events observed on the scaffold after the extending force had been increased from low to high force.
Finally, to determine the frequency of synaptic events, we included all events with expected amplitude-change signature for specific end-end interactions (i.e. within three standard deviations of the mean, or Δl between 130 and 200 nm). In this calculation we also accounted for apparently irreversible events characteristic of ligation, as well as very long-lived events which end up reversing after apparently multiple traction cycles.
Events for which rupture did not take place within the same traction cycle in which the event was first observed, but instead ruptured in later traction cycles, could not be reliably assessed for continuity or scored for tsynapsis and were therefore excluded from (tsynapsis, Δl) analysis. Such long-lived events lasting thousands of seconds were observed in the presence of downstream components (XLF, XRCC4 and Ligase IV), and in particular in the “complete” reaction for which they represent approximately half of all events. When any one of the downstream components was removed, these long-lived events spanning at least two traction cycles were no more than 10% of all observed events. No such long-lived events were observed when only upstream components (Ku, DNA-PKcs, and PAXX) were present. We note that the extended lifetime of such events precludes their detection by fluorescence-based methods which are essentially unable to access such timescales. We further note that the amplitude of such long-lived events was systematically within the expected range for bonafide synaptic events, but the very long-lived nature of these events suggests an alternative conformation or state of one of the NHEJ components employed.
Statistics and Reproducibility
Data were collected from at least 50-100 monitored DNA molecules observed over the course of at least two independent experimental replicates, and events were distributed according to a Poisson law among DNA molecules, with ~1-3 events per DNA molecule generating interactions (with typically at least ~30% of the 50-100 DNA molecules monitored reporting interactions).
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
J.L.W is supported by a PhD scholarship from the China Scholarship Council; C.D. is supported by a PhD scholarship from the University of Paris V and the Frontieres du Vivant doctoral program. For funding and support we thank the Ligue Nationale Contre le Cancer “Equipe Labellisée” program and PSL University (NanoRep grant) as well as the University of Paris VII, the CNRS, and the Ecole normale supérieure (to T.R.S); the National Institute of Health (PO1CA92584 to S.P.L.-M. and J.A.T and R01GM110387 to S.E.T); and the Wellcome Trust for support for T.O. and Q.W. (093167MA and 200814/Z/16/Z to T.L.B). J.A.T. is partly supported by a Robert A. Welch Chemistry Chair, the Cancer Prevention and Research Institute of Texas, and the University of Texas System Science and Technology Acquisition and Retention.
Footnotes
Author Contributions
J.L.W, C.D., Q.W., T.O., S.L., S.E.T., S.P.L.-M., M.N., J.A.T., T.L.B., and T.R.S designed experiments; J.L.W, C.D., Q.W., T.O., S.L., and T.R.S. prepared reagents; J.L.W, C.D., Q.W., and T.R.S carried out experiments; J.L.W, C.D. and T.R.S analyzed data; J.L.W., C.D., Q.W., T.O., S.L., S.E.T., S.P.L-M., M.N., J.A.T., T.L.B. and T.R.S. wrote the paper.
Competing Financial Interests The authors declare no competing financial interests.
References
- 1.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 2.Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev. 1999;13:916–934. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- 3.Dvir A, Stein LY, Calore BL, Dynan WS. Purification and characterization of a template associated protein kinase that phosphorylates RNA polymerase II. J Biol Chem. 1993;268:10440–10447. [PubMed] [Google Scholar]
- 4.Carter T, Vancurova I, Sun I, Lou W, DeLeon SA. DNA-activated protein kinase from HeLa cell nuclei. Mol Cell Biol. 1990;10:6460–6471. doi: 10.1128/mcb.10.12.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, Lee BJ, Dubois RL, Liu C, Yu X, Lan L, Zha S. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Molecular Cell. 2015;58:172–185. doi: 10.1016/j.molcel.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science (New York, NY) 2017;355:520–524. doi: 10.1126/science.aak9654. [DOI] [PubMed] [Google Scholar]
- 9.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 11.Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, Blundell TL, Jackson SP. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015;347:185–188. doi: 10.1126/science.1261971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, Hou M, Dong C, Guo R, Gao G, Ji J, Zha S, Lan L, Liang H, Xu D. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun. 2015;6:6233. doi: 10.1038/ncomms7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craxton A, Somers J, Munnur D, Jukes-Jones R, Cain K, Malewicz M. XLS (c9orf142) is a new component of mammalian DNA double-stranded break repair. Cell Death Differ. 2015;22:890–897. doi: 10.1038/cdd.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tadi SK, Tellier-Lebegue C, Nemoz C, Drevet P, Audebert S, Roy S, Meek K, Charbonnier JB, Modesti M. PAXX is an accessory c-NHEJ factor that associates with Ku70 and has overlapping functions with XLF. Cell Rep. 2016;17:541–555. doi: 10.1016/j.celrep.2016.09.026. [DOI] [PubMed] [Google Scholar]
- 15.Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Marquez JA, Pietrement O, Guerois R, Callebaut I, Le Cam E, Revy P, de Villartay JP, Charbonnier JB. Structural characterization of filaments formed by human XRCC4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc Natl Acad Sci USA. 2011;108:12663–12668. doi: 10.1073/pnas.1100758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer J. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–32650. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Q, Ochi T, Matak-Vinkovic D, Robinson CV, Chirgadze DY, Blundell TL. Non-homologous end-joining partners in a helical dance: structural studies of XLF-XRCC4 interactions. Biochem Soc Trans. 2011;39:1387–92. doi: 10.1042/BST0391387. [DOI] [PubMed] [Google Scholar]
- 18.Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40:1868–1878. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, de Melo AJ, Zhang H, Normanno D, Modesti M, Peterman EJG, Wuite GJL. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature. 2016;535:566–569. doi: 10.1038/nature18643. [DOI] [PubMed] [Google Scholar]
- 20.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 21.Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair 1. Biochem Cell Biol. 2013;91:31–41. doi: 10.1139/bcb-2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang S, Esswein SR, Ochi T, Wu Q, Ascher DB, Chirgadze D, Sibanda BL, Blundell TL. Achieving selectivity in space and time with DNA double-strand-break response and repair: molecular stages and scaffolds come with strings attached. Struct Chem. 2017;28:161–171. [Google Scholar]
- 24.Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. The EMBO journal. 1997;16:5098–5112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West RB, Yaneva M, Lieber MR. Productive and nonproductive complexes of Ku and DNA-dependent protein kinase at DNA termini. Mol Cell Biol. 1998;18:5908–5920. doi: 10.1128/mcb.18.10.5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strick T, Allemand JF, Bensimon D, Bensimon A, Croquette V. The elasticity of a single supercoiled DNA molecule. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 27.Bouchiat C, Wang MD, Block SM, Allemand J-F, Strick TR, Croquette V. Estimating the persistence length of a Worm-Like Chain Molecule from Force-extension Measurements. Biophys J. 1999;76:409–413. doi: 10.1016/s0006-3495(99)77207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yano K-I, Morotomi-Yano K, Wang S-Y, Uematsu N, Lee K-J, Asaithamby A, Weterings E, Chen DJ. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grundy GJ, Rulten SL, Arribas-Bosacoma R, Davidson K, Kozik Z, Oliver AW, Pearl LH, Caldecott KW. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat Commun. 2016;7:11242. doi: 10.1038/ncomms11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4-Ligase IV complex. Structure (London, England 1993) 2010;18:1431–1442. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balmus G, Barrons AC, Wijnhoven PWG, Lescale C, Hasse HL, Boroviak K, le Sage C, Doe B, Speak AO, Galli A, Jacobsen M, Deriano L, Adams DJ, Blackford AN, Jackson SP. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016;30:2152–2157. doi: 10.1101/gad.290510.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Shao Z, Jiang W, Lee BJ, Zha S. PAXX promotes ku accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat Commun. 2017;8:13816. doi: 10.1038/ncomms13816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walther AP, Gomes XV, Lao Y, Lee CG, Wold MS. Replication protein A interactions with DNA. 1. functions of the DNA-binding and zinc-finger domains of the 70-kDa subunit. Biochemistry. 1999;38:3963–3973. doi: 10.1021/bi982370u. [DOI] [PubMed] [Google Scholar]
- 34.Hammel M, Yu Y, Radhakrishnan SK, Chokshi C, Tsai MS, Matsumoto Y, Kuzdovich M, Remesh SG, Fang S, Tomkinson AE, Lees-Miller SP, Tainer JA. An intrinsically disordered APLF links Ku, DNA-PKcs and XRCC4-DNA ligase iv in an extended flexible non-homologous end joining complex. J Biol Chem. 2016 doi: 10.1074/jbc.M116.751867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lees-Miller SP, Beattie TL, Tainer JA. Noncoding RNA joins Ku and DNA-PKcs for DNA-break resistance in breast cancer. Nat Struct Mol Biol. 2016;23:509–510. doi: 10.1038/nsmb.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graves ET, Duboc C, Fan J, Stransky F, Leroux-Coyau M, Strick TR. A dynamic DNA-repair complex observed by correlative single-molecule nanomanipulation and fluorescence. Nat Struct Mol Biol. 2015;22:452–457. doi: 10.1038/nsmb.3019. [DOI] [PubMed] [Google Scholar]
- 37.Bouchiat C, Mezard M. Elasticity model of a supercoiled DNA molecule. Phys Rev Lett. 1998;80:1556–1559. [Google Scholar]
- 38.Hammarsten O, Chu G. DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci USA. 1998;95:525–530. doi: 10.1073/pnas.95.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duboc C, Fan J, Graves ET, Strick TR. Preparation of DNA substrates and functionalized glass surfaces for correlative nanomanipulation and colocalization (NanoCOSM) of single molecules. Methods Enzymol. 2017;582:275–296. doi: 10.1016/bs.mie.2016.09.048. [DOI] [PubMed] [Google Scholar]
- 40.Peranen J, Rikkonen M, Hyvonen M, Kaariainen L. T7 vectors with modified T7lac promoter for expression of proteins in Escherichia coli. Anal Biochem. 1996;236:371–373. doi: 10.1006/abio.1996.0187. [DOI] [PubMed] [Google Scholar]
- 41.Ochi T, Wu Q, Chirgadze DY, Grossmann JG, Bolanos-Garcia VM, Blundell TL. Structural insights into the role of domain flexibility in human DNA ligase IV. Structure (London, England : 1993) 2012 Jul;20:1212–1222. doi: 10.1016/j.str.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Yi, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, Ahnesorg P, Jackson SP, Blundell TL. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. EMBO J. 2008 Jan;27:290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murray JE, van der Burg M, Ijspeert H, Carroll P, Wu Q, Ochi T, Leitch A, Miller ES, Kysela B, Jawad A, Bottani A, Brancati F, Cappa M, Cormier-Daire V, Deshpande C, Faqeih EA, Graham GE, Ranza E, Blundell TL, Jackson AP, Stewart GS, Bicknell LS. Mutations in the NHEJ component XRCC4 cause primordial dwarfism. Am J Hum Genet. 2015;96:412–424. doi: 10.1016/j.ajhg.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.