

Abstract
Introduction
Antigen-specific immunotherapy could provide a targeted approach for the treatment of multiple sclerosis that removes the need for broad-acting immunomodulatory drugs. ATX-MS-1467 is a mixture of four peptides identified as the main immune-dominant disease-associated T-cell epitopes in myelin basic protein (MBP), an autoimmune target for activated autoreactive T cells in multiple sclerosis. Previous animal studies have shown that ATX-MS-1467 treatment prevented the worsening of signs of disease in experimental autoimmune encephalitis (EAE) in the humanized (DR2 × Ob1)F1 mouse in a dose-dependent fashion.
Methods and Results
Our study extends these observations to show that subcutaneous treatment with 100 µg of ATX-MS-1467 after induction of EAE in the same mouse model reversed established clinical disability (p < 0.0001) and histological markers of inflammation and demyelination (p < 0.001) compared with vehicle-treated animals; furthermore, in longitudinal magnetic resonance imaging analyses, disruption of blood–brain barrier integrity was reversed, compared with vehicle-treated animals (p < 0.05). Chronic treatment with ATX-MS-1467 was associated with an enduring shift from a pro-inflammatory to a tolerogenic state in the periphery, as shown by an increase in interleukin 10 secretion, relative to interleukin 2, interleukin 17 and interferon γ, a decrease in splenocyte proliferation and an increase in interleukin 10+ Foxp3− T cells in the spleen.
Conclusion
Our results suggest that ATX-MS-1467 can induce splenic iTregs and long-term tolerance to MBP with the potential to partially reverse the pathology of multiple sclerosis, particularly during the early stages of the disease.
Funding
EMD Serono, Inc., a business of Merck KGaA.
Keywords: ATX-MS-1467, (DR2 × Ob1)F1 mouse, Experimental autoimmune encephalitis, Interleukin 10, iTregs, Multiple sclerosis
Introduction
Most currently prescribed treatments for multiple sclerosis (MS) have a broad mechanism of action, often leading to nonspecific immunomodulation and associated complications, such as infections, malignancy and disruption of natural regulatory mechanisms [1]. Therefore, there is an unmet need for antigen-specific treatments targeting key regulators in the breakdown of tolerance to self-antigens, such as antigen-presenting cells. The aim of such treatments is to silence or reprogram autoreactive T cells in the periphery to a regulatory phenotype and thus create a tolerogenic state to the targeted protein. By directly targeting the pathogenic mechanisms of MS, immunological tolerance to environmental and self-antigens, such as myelin basic protein (MBP), can be reinstated without disrupting normal immune function, and the hypersensitivity associated with repeated antigen-specific immunotherapy can also be avoided [1, 2].
Several studies have shown antigen-specific peptide immunotherapy suppresses central nervous system (CNS) inflammation in rodent models of MS [3–5]. Specifically, peptide-specific down-regulation of T-cell responses with a soluble acetylated form of the N-terminal peptide of MBP (Ac1-9) was protective against experimental autoimmune encephalitis (EAE), a T cell-mediated inflammatory disease that has similarities to MS and is the most commonly used experimental model for this disease [6, 7].
Nearly two decades of research on tolerance to T cell-restricted epitopes, more specifically in the context of the study of EAE in a murine model of MS, has led to the generation of the concept of antigen-processing-independent T-cell epitopes (apitopes), which have the potential to be used in the treatment of several autoimmune diseases [7–18]. ATX-MS-1467 is a mixture of four peptides of human MBP (MBP epitope 30–44: PRHRDTGILDSIGRF; MBP epitope 83–99: ENPVVHFFKNIVTPRTP; MBP epitope 130–144: RASDYKSAHKGFKGV; MBP epitope 140–154: GFKGVDAQGTLSKIF). These have been identified as disease-associated epitopes in patients with MS who have the human leukocyte antigen (HLA) DR2 haplotypes DRB1*1501 and DQB1*0602 [14], of which the DRB1*1501 haplotype may explain around 50% of cases of MS [15]. It is assumed that ATX-MS-1467 binds to naked MHC-II of immature dendritic cells [19] and engages the T-cell receptor of MBP-specific primed memory T cells, leading to the development of a state of peripheral tolerance to MBP [14].
Treatment with ATX-MS-1467 has previously been shown to prevent worsening of EAE scores in a dose-dependent manner in the humanized transgenic (DR2 × Ob1)F1 mouse model [14], despite the fact that the study did not investigate the effect of ATX-MS-1467 on established EAE. In addition, a phase 1 study in six adults with secondary progressive MS showed that treatment with ATX-MS-1467 was well tolerated. The immune response to MBP was neither dramatically enhanced nor inhibited, due to study group sizes being too small to allow conclusions to be drawn concerning the efficacy of this treatment in humans. IL-10 mRNA transcripts were, nevertheless, transiently induced by the treatment in one patient phenotyped as expressing DRB1*15, suggesting that the drug might be capable of inducing desirable cytokine changes [14].
The aim of this study is to extend the results from the EAE by assessing the efficacy of late (at the observed peak of disease) as well as early (in the prodromal phase) treatment with ATX-MS-1467 for reversing the neurological damage and deficits accompanying EAE in the (DR2 × Ob1)F1 mouse. These findings will be corroborated with an examination of treatment effect on blood–brain barrier (BBB) permeability, on known markers of the shift from a pro-inflammatory to a tolerogenic state following acute and chronic treatment with ATX-MS-1467, more specifically by looking at the effect of in vivo pharmacological treatment on: the ex vivo antigen-specific cytokine release from splenocytes of EAE mice; the in vivo cytokine production following antigenic stimulation; and the accompanying changes on IL-10-secreting FoxP3+/FoxP3− regulatory cell subpopulations.
Methods
Animals
(DR2 × Ob1)F1 transgenic mice homozygous for DRA*0101/DRB1*1501 (HLA-DR2) and MBP84–102-specific human T-cell receptor (TCR) clone (Ob1) were generated in C57Bl/6 mice [20] and maintained and bred at Charles River Laboratories (Wilmington, MA, USA); the F1 progeny of these animals were used in all experiments. Before each experiment, expression of human MHC and TCR were confirmed in peripheral blood by phenotyping with fluorescein isothiocyanate (FITC)-labelled mouse anti-human HLA-DR, HLA-DP, HLA-DQ (BD Pharmingen, San Jose, CA, USA) and IOTest anti-TCR VB2-PE clone MPB2D5 (Beckman Coulter, Indianapolis, IN, USA). Only double-positive mice with a frequency of human MHC and TCR expression > 15% in peripheral leukocytes were used in the studies.
On arrival, mice were group-housed in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited vivarium with a 12:12 light–dark cycle and food and water provided ad libitum. The mice were allowed to acclimatize for at least 48 h before any experiments were started. All procedures were approved by the EMD Serono Institutional Animal Care and Use Committees (IACUC) or by the Massachusetts General Hospital IACUC in the case of magnetic resonance imaging (MRI) studies.
Mice reaching a predefined clinical endpoint for humane withdrawal from the study (Table 1) and mice that were not required for tissue sampling were euthanized by slow replacement of the air in their cage by an air mixture containing CO2. Death by CO2 asphyxiation was confirmed by cervical displacement. All animal experiments complied with the National Institutes of Health guide for the care and use of laboratory animals. All efforts were made to minimize the number of animals used and to optimize their well-being.
Table 1.
Clinical score scale to assess extent of EAE
| Clinical score | Outcome |
|---|---|
| 0 | Normal |
| 0.5 | Partially limp tail |
| 1.0 | Completely limp tail |
| 1.5 | Slightly impaired righting reflex |
| 2.0 | Delayed righting reflex |
| 2.5 | Virtually absent righting reflex |
| 3.0 | Partial hind-limb paralysis |
| 3.5 | Complete unilateral hind-limb paralysis |
| 4.0* | Complete bilateral hind-limb paralysis |
| 4.5 | Moribund |
| 5.0 | Death due to EAE |
EAE experimental autoimmune encephalitis
*Humane endpoints and special care for mice with EAE were also observed. Once animals reached a score of 4.0 they were observed at least twice a day to ensure they did not progress further and did not get worse: animals that improved to a score of 3.5 or lower were allowed to continue in the study; if animals did not improve below a score of 4.0 within 72 h, they were euthanized. In addition, mice were also euthanized if they experienced a body weight loss of 20%. From the onset of paralysis, mice also received wet dietary supplement (Dietgel 76A, ClearH2O) on the cage bottom and subcutaneous sterile Ringer's glucose injections, as needed, for fluid replacement
Induction of EAE
Ten to 14-week-old male or female (DR2 × Ob1)F1 mice under isoflurane anesthesia were shaved and received a subcutaneous injection at the tail base of 100 µL of an emulsion containing 200 µg (wet weight) of mouse spinal cord homogenate (SCH, harvested in-house from syngeneic spinal cords) and heat-inactivated Mycobacterium tuberculosis (400 µg/mouse; Difco, Oxford, Oxon, UK) prepared using equal volumes of phosphate-buffered saline (PBS) and incomplete Freund’s adjuvant (Difco, Oxford, Oxon, UK). On the day of immunization [0 days post-induction (dpi)], and also 2 days later (2 dpi), mice received an intraperitoneal injection with 300 ng of pertussis toxin (ENZO, Farmingdale, NY, USA).
ATX-MS-1467 Treatment
All treatments with ATX-MS-1467 were performed from a reconstituted sterile, freeze-dried formulation of a mixture (1:1:1:1) of four synthetic peptides (MBP epitope 30–44: PRHRDTGILDSIGRF; MBP epitope 83–99: ENPVVHFFKNIVTPRTP; MBP epitope 130–144: RASDYKSAHKGFKGV; MBP epitope 140–154: GFKGVDAQGTLSKIF). After reconstitution of the lyophilized formulation to the original volume with sterile water, the solution was further diluted with PBS to the required doses. The concentration of the ATX-MS-1467 mixture was adjusted for a 100-µL injection volume, administered subcutaneously in all cases. The doses corresponded to the sum of each peptide in the mixture; therefore, a dose of 100 µg contained 25 µg of each peptide. The treatment regimen with ATX-MS-1467 varied; please refer to specific sections for the dose regimen used in each experiment.
HLA-Binding Peptide Treatment
In some experiments, HLA-binding peptide (HLAbp) was used as a control for ATX-MS-1467. Although HLAbp can bind directly to MHC on the surface of antigen-presenting cells without requiring antigen processing, the peptide sequence of HLAbp has no similarity to the epitopes involved in the adaptive immune responses to MS or EAE. The peptide sequence used (NPILLWQPIPVHTVPLSEDQ) was provided by Professor David Wraith (University of Bristol, UK).
Clinical Scoring, Animal Care and Humane Endpoints
Clinical score and body weight were monitored and recorded daily from 7 dpi by observers blind to treatment. Neurological disability and humane endpoints were monitored daily using a subjective 0–5-point clinical score scale (Table 1) [21]. Whenever possible, a single investigator scored the animals in each study, and in any single day the same investigator scored all groups of animals. However, because clinical scoring sometimes progressed for several weeks, including weekends, up to four different investigators potentially provided input into a single study. Each investigator’s scoring was routinely calibrated, to ensure the results were comparable across different investigators.
Magnetic Resonance Imaging
EAE was induced on 0 dpi (n = 35). On 9 or 10 dpi, MRI was obtained, and those animals with radiological evidence of leakage in the cerebellum/brainstem seen on T1-weighted gadolinium-enhanced (Gd+) MRI with a 9.4-Tesla magnet scanner (Bruker Biospin, Billerica, MA, USA) were randomized into two groups for treatment with either ATX-MS-1467 (100 µg/mouse, n = 7) or vehicle (n = 7). Treatment was given every other day, starting at 10 dpi (mice scanned on 9 dpi) or 11 dpi (mice scanned at 10 dpi). T1-weighted Gd+ MRI was repeated after three doses (15 or 16 dpi). The T1-weighted gradient echo sequence with a short echo time (TE) and a large flip angle [repetition time (TR)/TE/flip angle = 100 ms/2.7 ms/50 degrees] was used to assess Gd leakage.
Histology
Pathological analysis was done on samples of spinal cords from animals treated at 7 or 14 dpi with either ATX-MS-1467 or vehicle. Samples were obtained at the termination date of these experiments (30 post-immunization). A 1.6 g/mL 2,2,2-tribromoethanol (T48402, Sigma, St. Louis, MO, USA) stock solution was prepared in 2-methyl-2-butanol (A1685, Sigma, St. Louis, MO, USA). A working solution at 20 mg/mL was obtained by subsequent dilution with PBS. At predefined time points, mice were overdosed with anesthetic (400 mg/kg intraperitoneal 2,2,2-tribromoethanol) and perfused through the left cardiac ventricle with saline followed by 10% neutral buffered formalin. The vertebrae were partially removed to obtain the spinal cord, and the tissues were post-fixed with 10% neutral buffered formalin for 48 h and then stored in PBS before dissecting out the spinal cords. Spinal cords were cut into five segments, spanning the cervical (one piece), thoracic (two pieces) and lumbar (two pieces) regions, which were processed for paraffin embedding. Histological analyses were done on 10 cross-sections of the spinal cord for each animal. These cross-sections were 7 µm thick and at least 280 µm apart.
Hematoxylin and eosin (H&E) and luxol fast blue (LFB) staining were performed, as well as immunohistochemistry staining against CD3 (Abcam, Cambridge, MA, USA) and CD45R (BD Pharmingen, San Jose, CA, USA) surface antigens using Leica BOND-III autostainer (Leica, Wetzlar, Germany). High-quality images obtained with a Nanozoomer 2.0 HT digital slide scanner (Hamamatsu, Hamamatsu City, Shizuoka, Japan) were semi-quantitatively scored for each stain by an experimenter who was blinded to the treatment groups.
The H&E scoring was performed as set out in Table 2. CD3 and CD45R scoring were similar to the H&E scoring, except that only immunostained cells were scored. For the LFB scoring, each cross section of the spinal cord was analyzed as four independent regions (ventral, dorsal, right lateral and left lateral). In all cases, the scores for each of the ten sections was averaged and entered as a single data point for each mouse in the experiment.
Table 2.
Histology scoring of spinal cord sections
| Score | H&E/CD3 and CD45R | LFB |
|---|---|---|
| 0 | No visible signs of inflammation | No region presenting with demyelination |
| 0.5 | Immune cells found only in close association with meninges | Only one region exhibiting demyelination spanning < 50% of its area |
| 1.0 | Plus at least one clearly defined example of a perivascular cuff [at this stage, perivascular cuffs have limited cellularity (< 10 cells) and usually up to maximum of 3–4 perivascular cuffs can be found] | Two regions exhibiting demyelination spanning < 50% |
| 2.0 | Plus discrete numbers of immune cells beyond the perivascular cuffs (i.e., infiltrating into the parenchyma of the white matter) | Three regions exhibiting demyelination spanning < 50% |
| 3.0 | Plus extensive cell clusters (≥ 20 cells each) throughout the parenchyma of the white matter (i.e., not restricted to perivascular cuffs) | Four regions exhibiting demyelination spanning < 50% |
| 4.0 | Plus major infiltrations (typically clusters of ≥ 50 cells) all over the white matter | One region exhibiting demyelination spanning > 50% |
| 5.0 | Significant numbers of immune cells (> 30) dispersed throughout the grey matter, usually accompanied by massive hypercellular events in the white matter | Two regions exhibiting demyelination spanning > 50% |
H&E hematoxylin and eosin, LFB luxol fast blue
Ex-Vivo Antigen-Recall Assay
Mice were treated with either ATX-MS-1467 (100 µg/mouse, three times per week) or vehicle for 7 days. At 0 dpi, mice were also immunized as if for induction of EAE, but these mice were euthanized before they displayed signs of disease. Mice received the last dose of ATX-MS-1467 or vehicle at 7 dpi, 1 h before sacrifice for spleen removal and cell culture. On the day of sacrifice, mice were anaesthetized with 400 mg/kg intraperitoneal 2,2,2-tribromoethanol, and their spleens were harvested and processed in a gentle MACS Octo dissociator (Myltenyi Biotec, Birgisch Gladback, Germany). The splenocytes underwent several cycles of washing and centrifugation after lysis of red blood cells with red blood cell lysis buffer (Roche, Basel, Switzerland) and were counted manually in a Neubauer chamber using trypan blue solution to exclude dead cells. The concentration of the cell suspension was adjusted to 3 × 106 cells/mL in RPMI medium (Gibco, Waltham, MA, USA) supplemented with fetal calf serum (10%), penicillin–streptomycin (1:100; Gibco, Waltham, MA, USA), HEPES (final concentration 10 mM; Gibco, Waltham, MA, USA), MEM non-essential amino acid solution (1:100; Sigma, St. Louis, MO, USA), l-glutamine (final concentration 2 mM; Gibco, Waltham, MA, USA), sodium pyruvate (final concentration 1 mM; A&E Scientific PAA, San Jose, CA, USA) and β-mercaptoethanol (final concentration 2.5 µM; Sigma, St, Louis, MO, USA). For cell culture, 100 µL of the cell suspension was prepared as described, together with 100 µL of each appropriate stimulant (ATX-MS-1467 0.03–30 µg/mL final concentrations) diluted in medium and incubated at 37 °C for 48 or 72 h, corresponding to the optimum time points for evaluation of cytokine or proliferative responses, respectively. Cytokines were quantified in supernatants from 48-h splenocyte cultures with commercial enzyme-linked immunosorbent assay (ELISA) kits (mouse IL-2 Duoset ELISA, mouse IL-10 Quantikine ELISA, mouse IL-17 Duoset ELISA and mouse IFNγ Duoset ELISA, all purchased from R&D Systems, Minneapolis, MN, USA). Splenocyte proliferation was assessed by replacing 100 μL of the medium from wells that had been stimulated for 72 h with 100 μL of 3H thymidine solution and further incubating for 12 h. Incorporation of 3H thymidine was quantified in a scintillator. The results were expressed as counts per minute (cpm).
Antigen-Specific Cytokine Release In Vivo
The experiments that were conducted to assess antigen-specific cytokine release in vivo were performed in mice that had not been immunized with adjuvant in order to avoid background inflammation caused by this agent. Serum samples were collected from untreated mice or mice that had received repeated treatment with ATX-MS-1467 (100 µg/mouse, three times per week) or HLAbp (100 µg/mouse three times per week) receiving a total of 10 injections, followed or not by a wash-out period lasting from 2–42 days for assessment of the concentrations of IL-2, IL-10, IL-17 and IFNγ. Samples were collected 2 h after the last injection, which was experimentally predetermined as the time point at which the concentrations of most cytokines had reached a maximum. To ascertain the in vivo cytokine secretion of (DR2 × Ob1)F1 mice to an acute challenge with MBP, serum was collected 2 h after acute treatment with MBP at doses ranging 30–1000 µg/mouse. To ascertain the effect of chronic dosing with ATX-MS-1467 on in vivo pro-inflammatory cytokine secretion, mice received repetitive treatment (from one up to ten doses) with ATX-MS-1467 (100 µg/mouse, three times per week) with serum collected 2 h after the last dose. To ascertain the effect of chronic treatment with ATX-MS-1467 on the ratio of IL-10-to-pro-inflammatory cytokines after an acute challenge with MBP, mice received ten doses of ATX-MS-1467 (100 µg/mouse, three times per week) or ten doses of HLAbp (100 µg/mouse, three times per week). At 2, 7, 21 or 42 days after the last injection with ATX-MS-1467 or HLAbp, mice received a single injection of MBP (300 µg/mouse) and serum was collected 2 h later. Acute cytokine responses in serum were quantified using a customized MCYTOMAG-70 K | MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel—Immunology Multiplex Assay (Millipore, Billerica, MA, USA).
Effect of ATX-MS-1467 Treatment on Leukocyte Populations
Flow cytometric analysis was used to assess the effect of treatment with ATX-MS-1467 (100 µg/mouse, three times per week) or vehicle for 2 weeks on cell surface and intracellular markers associated with regulatory cell function. Two different staining panels were used (Table 3).
Table 3.
Antibody panels used for splenocyte staining
| Panel 1 (cell surface markers on freshly isolated splenocytes) | Panel 2 (cell surface and intracellular markers) |
|---|---|
| CD49b (BV421, 1:100, cat. # 63063, BD Pharmingen, San Jose, CA, USA) | CD49b (BV421, 1:100, cat. # 63063] BD Pharmingen, San Jose, CA, USA) |
| CD3 (V500, 1:100, cat. # 560771, BD Pharmingen, San Jose, CA, USA) | CD4 (V500, 1:100, cat. # 560782] BD Pharmingen San Jose, CA, USA) |
| CD4 (FITC, 1:100, cat. # 553729, BD Pharmingen, San Jose, CA, USA) | LAG3 (PE, 1:100, cat. # 125207, Biolegend, San Diego, CA, USA) |
| LAG3 (PE, 1:100, cat. # 125207, Biolegend, San Diego, CA, USA) | IL-10 (APC, 1:100, cat. # 554468, BD Pharmingen, San Jose, CA, USA) |
| B220 (PE-Cy7, 1:200, cat. # 552772, BD Pharmingen, San Jose, CA, USA) | CD8 (APC-Cy-7, 1:100, cat. # NC0079947, Biolegend, San Diego, CA, USA) |
| CD8 (BV605, 1:100, 563152, cat. # 563152, BD Pharmingen, San Jose, CA, USA) | CD45R/B220 (BV605 1:100, cat. # 563708, BD Pharmingen, San Jose, CA, USA) |
| FC block (CD16/CD32, unstained, 1:100, cat. # 553141, BD Pharmingen, San Jose, CA, USA) | FC block (CD16/CD32, unstained, 1:100, cat. # 553141, BD Pharmingen, San Jose, CA, USA) |
| FoxP3 (PerCP-Cy5.5 1:100, cat. # 35-5773-82, eBioscience, San Diego, CA, USA) |
CD45R/B220 and CD8 staining were used as a negative selection for the T lymphocyte subpopulations of interest. Listed are the specificities, fluorochromes, catalogue numbers and manufacturers, and dilutions for each of the antibodies
The first panel focused on cell surface markers and was applied in freshly isolated splenocytes. The second panel included intracellular markers, IL-10 and the transcription factor forkhead box P3 (Foxp3). To enable better identification of the IL-10-secreting cells, the second panel was applied in splenocytes incubated for 3 h with phorbol-myristate acetate (PMA, 5 ng/mL; Sigma, St. Louis, MO, USA), ionomycin (500 ng/mL; Sigma, St. Louis, MO, USA) and the protein transport-inhibitor monensin (4 µL/6 mL of cell culture; GolgiStop, BD Biosciences, San Jose, CA, USA) for 3 h. The splenocytes were isolated as previously described. B220 and CD8 staining were used as a negative control for the T lymphocyte subpopulations of interest. Samples were measured on a LSRFortessa X20 flow cytometer (BD Bioscience, San Jose, CA, USA) and analyzed in FlowJo version 10 (FlowJo, LLC, Ashland, OR, USA).
Statistical Analyses
In experiments with multiple groups, clinical score data were analyzed via daily comparison of group averages using the Kruskal–Wallis test followed by Dunn’s multiple comparison test. Clinical score data were also integrated as area under the curve, and the results analyzed with the Kruskal–Wallis test followed by Dunn’s multiple comparison test. MRI, EAE imaging and clinical score data were analyzed daily by Mann–Whitney test. The mean semi-quantitative histological scores of spinal cord sections were input as a single data point that was used to calculate group averages, which were compared by one-way analysis of variance (ANOVA) followed by the Bonferroni test. Data on concentration-dependent cytokines and proliferation response ex vivo were analyzed with two-way ANOVA followed by the Bonferroni test. In vivo cytokine-release data were analyzed with one-way ANOVA followed by Dunnett’s multiple comparison test.
Results
Effect of ATX-MS-1467 on EAE Clinical Disability
We treated (DR2 × Ob1)F1 mice with different doses of ATX-MS-1467, starting either 7 or 12 dpi and observed the clinical signs of disease. Compared with vehicle, ATX-MS-1467 dose-dependently reduced clinical disability from the empirically determined disease onset (7 dpi, Fig. 1a, c) and peak disease (12 dpi, Fig. 1b, d) time points, as assessed by the mean daily score and cumulative scores on the subjective 0–5-point clinical score scale during the treatment period. As can be observed in Fig. 1a, d, if treatment starts early in the course of disease, it is possible to completely prevent the appearance of clinical signs. Even if the treatment is initiated when mice exhibit full-blown disease (tail–hand–hind limb paralysis, matching an average score of 3), partial, albeit significant, recovery from the natural course of EAE was seen in this mouse strain (Fig. 1b, d). The optimum dose (100 µg) significantly improved the clinical score in mice showing advanced signs of neurological disability, and this dose was selected for subsequent experiments.
Fig. 1.
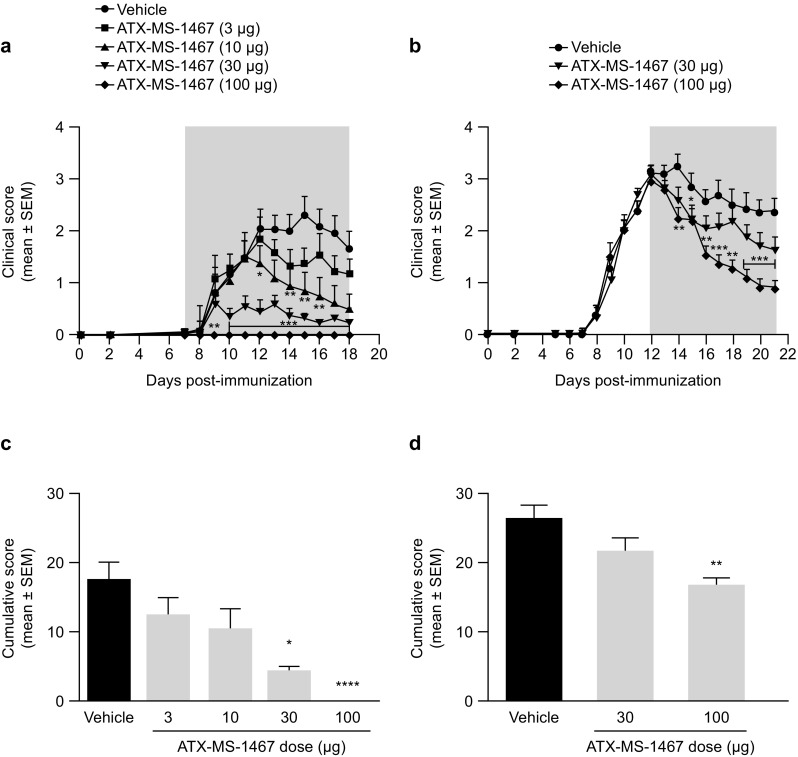
Effect of dose response of ATX-MS-1467 from disease onset or peak of clinical disability in a mouse model of EAE. EAE was induced at 0 dpi with spinal cord homogenate in (DR2 × Ob1)F1 mice, and treatment was initiated with ATX-MS-1467 at 7 dpi (a, c) or at 12 dpi (b, d) at doses ranging 3–100 µg, as indicated, subcutaneously three times per week. The grey area in graphs a, b indicates the treatment period. For the experiment shown in a, c, the group sizes were n = 10–14 mice, and for the experiment shown B and D, the data represent a pool from two independent experiments with a total of n = 21–22 mice. *, **, *** and **** indicate p < 0.05, < 0.01, < 0.001 and < 0.0001, respectively, by Kruskal–Wallis test followed by Dunn’s test versus vehicle (PBS) group. dpi days post-injection, EAE experimental autoimmune encephalomyelitis, PBS phosphate-buffered saline, SEM standard error of the mean
Effect of ATX-MS-1467 on CNS Infiltration and Inflammation
We performed two independent experiments, with treatment started at either 7 or 14 dpi. This allowed us to generate a large tissue bank to assess the effects of treatment with ATX-MS-1467: spinal cord samples were obtained from 20–28 mice from each treatment group, with each spinal cord being stained and analyzed at ten different levels for each staining. In this experiment, we used histological evaluation with subjective scoring (Table 2) to confirm the observed efficacy. The average score for the vehicle-treated groups, which varied from 2 to 3, depending on the marker being analyzed, could wrongly suggest that the level of tissue damage is not compatible with overt disease pathology. However, due to the unpredictable location where the lesions occur in this model, the average score is the result of both highly affected spinal cord sections and other sections that might have remained intact. Indeed, it was a common feature of all vehicle-treated mice that one or multiple areas with intense cellular infiltration and demyelination were observed at various, albeit not all, sections examined. In agreement with the attenuation in clinical score, treatment with ATX-MS-1467 from the empirically determined disease onset (7 dpi) or peak of disease (14 dpi) time points reduced spinal cord inflammation, demyelination and cellular infiltration, unsurprisingly with the greatest effect for all outcomes reported when treatment was initiated at disease onset (p < 0.001 for all measures; Fig. 2). The reduction in inflammation and demyelination seen in the CNS was also accompanied by an improvement in the clinical score, for which the greatest improvement was observed following early (7 dpi) treatment (Fig. 2a). Treatment with ATX-MS-1467 from the peak of disease (14 dpi) partially reduced the histological markers indicative of inflammation (H&E score; Fig. 2b), demyelination (LFB score; Fig. 2c) and T-cell infiltration (CD3 score; Fig. 2d; p < 0.05) and histology scores indicative of B-cell infiltration (p < 0.01; Fig. 2e).
Fig. 2.
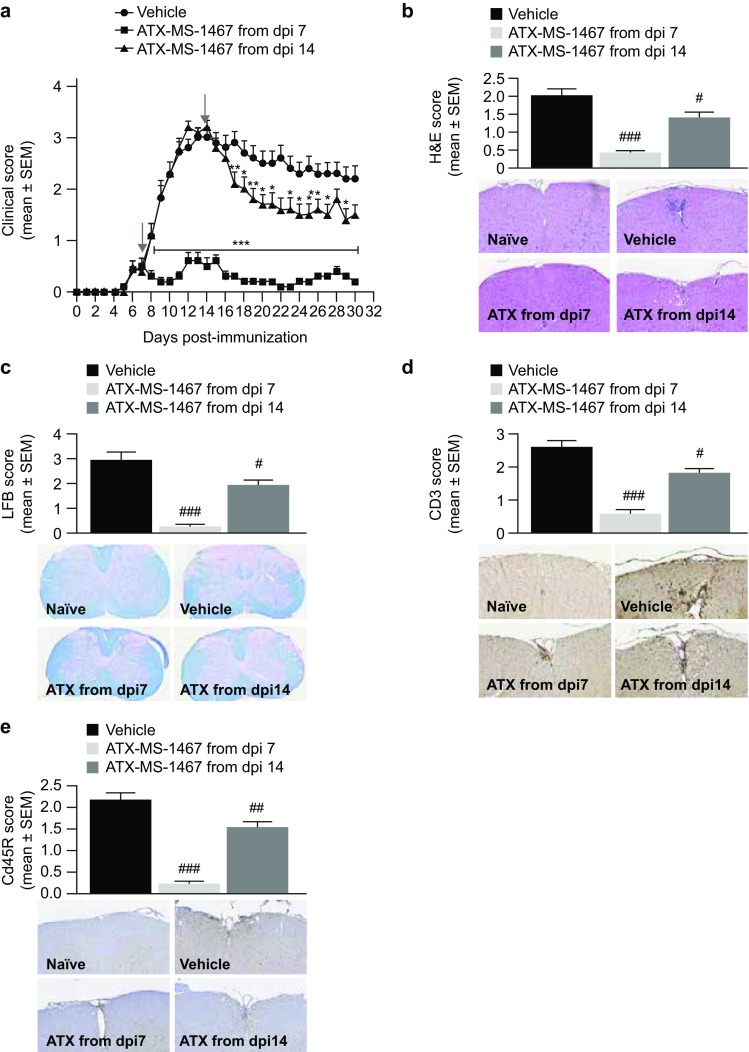
Effect of therapeutic treatment with ATX-MS-1467 on spinal cord tissue damage in spinal cord homogenate-induced EAE in (DR2 × Ob1)F1 mice. a The effect of 100-µg ATX-MS-1467 treatment starting at 7 or 14 dpi on daily clinical scores and on terminal (day 30) histological scores. The arrows indicate the beginning of the treatment period in each group. *, ** and *** indicate p < 0.05, < 0.01 and < 0.001, respectively, by Kruskal–Wallis test followed by Dunn’s test versus vehicle. H&E staining was applied to assess inflammation (b), luxol fast blue staining was applied to assess demyelination (c), and immunohistochemical staining for CD3 or CD45R was applied to assess T-cell (d) and B-cell infiltration (e). The inserts show representative images for each group with the respective staining for which the semi-quantitative evaluation is plotted. #, ## and ### indicate p < 0.05, < 0.01 and < 0.001, respectively, by ANOVA followed by Bonferroni versus mice treated with vehicle (PBS). The data represent a pool from 2 independent experiments with a total of 20–28 mice. Spinal cords from naïve mice were also processed, stained and analyzed along with the samples from the immunized/treated mice. The histological (as well as clinical) scores for naïve mice was zero for all cases; therefore, for the sake of simplicity, data from naïve mice was not included in the graphs. dpi days post-induction, EAE experimental autoimmune encephalitis, H&E hematoxylin & eosin, LFB luxol fast blue, PBS phosphate-buffered saline, SEM standard error of the mean
MRI was applied as a translational approach to investigate the efficacy of ATX-MS-1467 in mice. Gd administered intravenously during the acute phase of disease allows the detection of leakage, which can be seen as bright areas of the spinal cord in T1-weighted scans, indicating disruption of the BBB. Because BBB disruption tends to be more active during the beginning of the acute phase of disease, and tends to naturally subside at later phases, we opted for a shorter window of treatment, in order to better appreciate the drug effect. In a longitudinal MRI experiment (Fig. 3a), therapeutic dosing with ATX-MS-1467 (100 µg subcutaneous) every other day (three doses) from 10 dpi significantly (p < 0.05 on 13 and 15 dpi and p < 0.01 on 14 and 16 dpi) reversed disruption to the BBB, as measured by Gd leakage, compared with vehicle-treated animals (Fig. 3c). In group analyses at follow-up, Gd-leakage volume was significantly reduced in ATX-MS-1467-treated mice (Fig. 3e) compared with baseline but not in vehicle-treated mice [Fig. 3e; ATX-MS-1467-treated mice: 77.4% reduction (Gd-leakage volume 1.1 ± 0.3 mm3) from baseline, p < 0.01; vehicle-treated mice: 39.7% reduction (Gd-leakage volume 4.45 ± 0.3 mm3) from baseline, p = 0.23]. Figure 3b shows representative T1-weighed MRI scans of the spinal cords of the mice in this study.
Fig. 3.
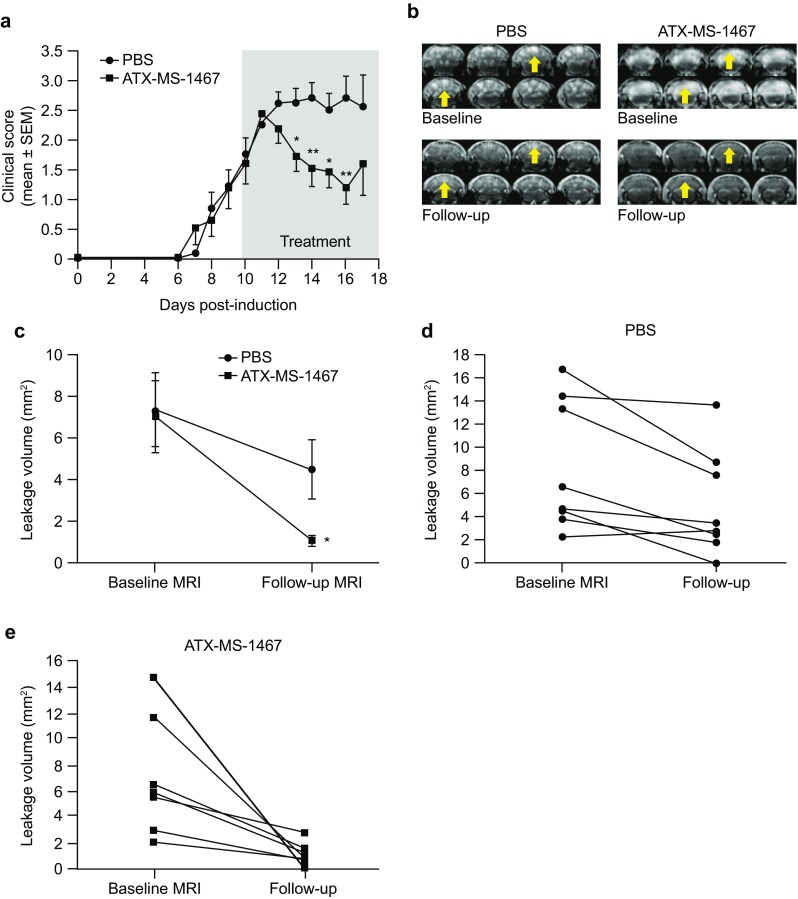
Effect of ATX-MS-1467 on EAE-induced blood–brain barrier leakage. The effect on clinical score of ATX-MS-1467 (n = 7) or vehicle (PBS; n = 7) given at 10 or 11 dpi, depending on the timing of MRI; the grey area shows the treatment period (a). Four serial images at baseline and follow-up for two animals (left: vehicle-treated mice; right: ATX-MS-1467-treated mice; b). The yellow arrows highlight the areas with changes in Gd leakage. Levels of Gd leakage volume at dosing onset and follow-up (c). Leakage volume for the individual mice with vehicle (d) or ATX-MS-1467 treatment (e) were compared using a paired t test. *p < 0.05, **p < 0.01. dpi days post-induction, EAE experimental autoimmune encephalomyelitis, Gd gadolinium, MRI magnetic resonance imaging, PBS phosphate-buffered saline, SEM standard error of the mean
Effect of ATX-MS-1467 Treatment on the Anti-Inflammatory Milieu in Ex Vivo Splenocytes
ATX-MS-1467 binds to surface-expressed MHC-II on antigen-presenting cells. Following binding, ATX-MS-1467 is hypothesized to drive either the differentiation of MBP-specific regulatory cells from naïve cells or to induce the conversion of effector into type 1 regulatory cells (Tr1). In both cases, inhibition of the release of the pro-inflammatory cytokines IL-17, and IFNγ from Th1 and Th17 T-cell subsets, which are known to promote the inflammatory processes in MS, along with an increase in the release of IL-10, which is indicative of a tolerogenic state, would be expected. In vivo and in vitro strategies were employed to demonstrate whether these changes occurred in mice treated with ATX-MS-1467. For the in vitro analysis, splenocytes were recovered from animals treated four times between 0 to 7 dpi with ATX-MS-1467 (on days 0, 2, 4 and 7); the last administration took place 1 h prior to take down. The spleens were processed, as described in the methods section, and the cytokine concentrations in the supernatant were assayed (Fig. 4a–d). The cultures were stimulated in the presence of a range of concentrations of each antigen, to allow the assessment of the response over a broad range of concentrations.
Fig. 4.
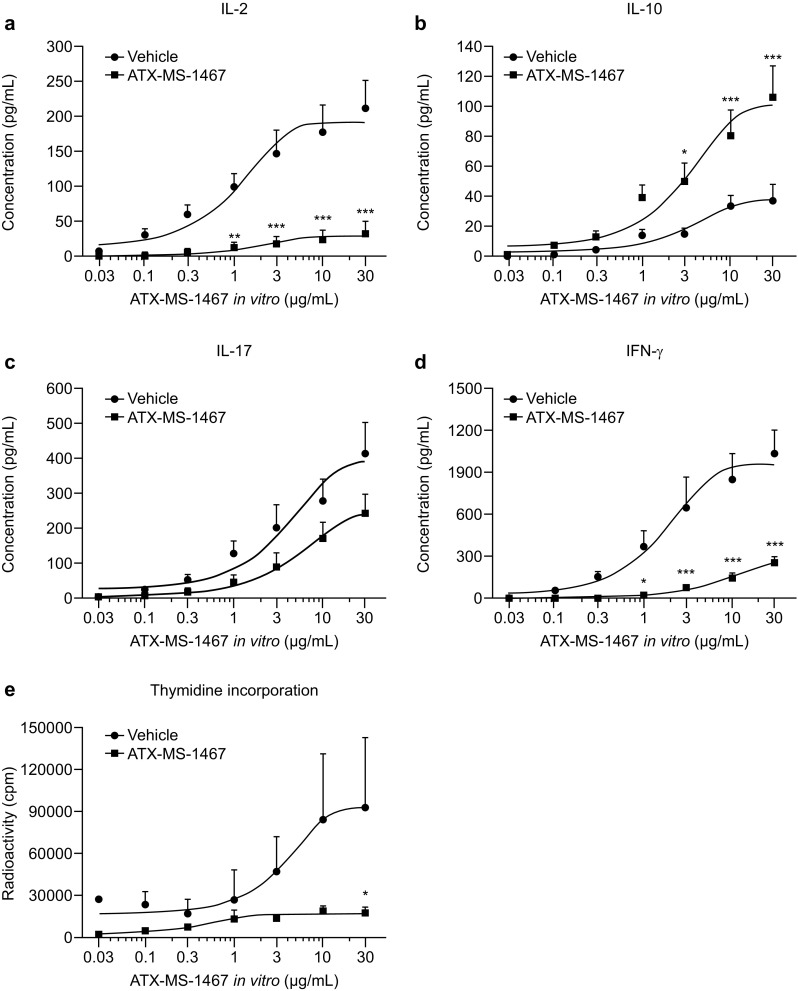
Effect of preventive treatment with ATX-MS-1467 on ex vivo splenocyte proinflammatory cytokine release and proliferation. (DR2 × Ob1)F1 mice were immunized with spinal cord homogenate and treated with 100 µg ATX-MS-1467 or vehicle (PBS) on days 0, 2, 5 and 7. Splenocytes were harvested at 7 dpi and stimulated in vitro with ATX-MS-1467 at 0.03, 0.1, 0.3, 1, 3, 10 and 30 µg/mL; cytokine concentrations were measured in the supernatants (a–d). Splenocyte proliferation was also measured by 3H thymidine uptake (cpm) following in vitro stimulation with ATX-MS-1467 (e). *, ** and *** indicate p < 0.05, < 0.01 and < 0.001, respectively, by two-way ANOVA followed by Bonferroni’s post-tests (n = 5–6). ANOVA analysis of variance, cpm counts per minute, dpi days post-induction, IFN interferon, IL interleukin, PBS phosphate-buffered saline
It is important to differentiate the purpose of ATX-MS-1467 as a treatment (Fig. 4a–e filled squares), when it was administered in vivo with a vehicle-administered group as a control (Fig. 4a–e filled circles), from its use here as an antigen-specific stimulus for cellular proliferation and cytokine release (x axis, various concentrations). The in vitro antigen recall assay showed that the in vivo treatment with ATX-MS-1467 caused a dose-dependent inhibition in pro-inflammatory cytokine (IL-17 and IFNγ) and IL-2 secretion and splenocyte proliferation. IL-10 secretion was significantly increased only after stimulation with concentrations of ATX-MS-1467 of 3 µg/mL (p < 0.05) and above (p < 0.001) in the ex vivo antigen recall assay (Fig. 4).
Effect of ATX-MS-1467 Treatment on the In Vivo Cytokine Profile
The effect of ATX-MS-1467 persists when the drug is cleared from the circulation. To extend the results for ex vivo cytokine secretion, we wanted to assess whether a favorable shift in cytokine balance also occurred in vivo. To achieve this, we performed three types of experiments.
The first type (Fig. 5) aimed to define the optimal dose of MBP needed to elicit robust serum cytokine changes. In the naïve wild-type mouse strain, MBP injection would not normally induce cytokine release at levels that can be detected in serum. However (DR2 × Ob1)F1 mice were genetically engineered to constitutionally express both MHC and TCR recognizing one MBP epitope; therefore, circulating effector and regulatory cells that respond to MBP are already present in (DR2 × Ob1)F1 mice, even when they have not been immunized against this protein. Untreated (DR2 × Ob1)F1 mice responded to an acute challenge with MBP with a dose-dependent increase in pro-inflammatory cytokines IL-17 (Fig. 5c) and IFNγ (Fig. 5d) and also IL-2 (Fig. 5a) compared with vehicle-treated mice. Although the concentration of IL-10 was also increased in response to low doses of MBP, this effect plateaued at MBP concentrations of 100–300 µg and then declined as the concentration of MBP increased (Fig. 5b).
Fig. 5.
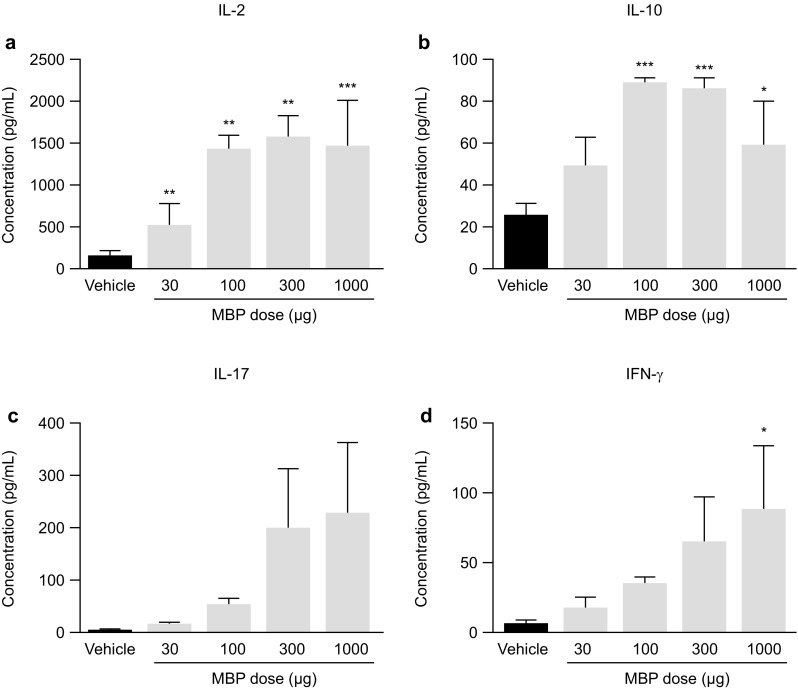
Response of (DR2 × Ob1)F1 mice to an acute challenge with MBP. Dose-dependent secretion of cytokines in the serum of (DR2 × Ob1)F1 mice 2 h after subcutaneous injection of MBP (a–d). Data were analyzed by ANOVA followed by Dunnett’s multi-comparison test. *, ** and *** indicate p < 0.05, < 0.01 and < 0.001, respectively, versus the vehicle (PBS)-treated group (n = 4–5). ANOVA analysis of variance, IFN interferon, IL interleukin, MBP myelin basic protein, PBS phosphate-buffered saline
The second type of experiment on in vivo cytokine release (Fig. 6) assessed how cytokine release shifted after repeat treatments. The insert summarizes how the injections were performed in each group (Fig. 6a). A single dose of ATX-MS-1467 elicited an increase in the concentrations of all cytokines in serum (Fig. 6b–e), and the concentrations returned to baseline values within a few hours after reaching their peak at 2 h after the injection (results not shown). Similar to explanation given above for MBP-induced cytokine release, naïve mice with the transgenic mouse construct also respond to ATX-MS-1467 with quantifiable cytokine levels in serum without the need of concomitant exposure with adjuvant. Further doses of ATX-MS-1467 did change the serum IL-2 (Fig. 6b), IL-17 (Fig. 6d) or IFNγ (Fig. 6e) cytokine concentrations in comparison with vehicle for up to ten consecutive doses of ATX-MS-1467. However, up to five consecutive doses of ATX-MS-1467 were still capable of inducing statistically significant drug-induced IL-10 release in serum (Fig. 6d). The use of ratios of signature cytokines for the predicted response is an accurate index of how an organism responds to antigens or disease: how the ratios change over time or how they compare between different groups under controlled conditions shows how a system is responding to the challenge [22]. If treatment with ATX-MS-1467 does indeed induce tolerance, the persistence of the tolerogenic effect, as deduced from changes in the ratios of IL-10-to-pro-inflammatory cytokines, could be assessed by the changes in these ratios. From the second dose to the fifth dose, the concentration of IL-10 relative to IL-17 and IFNγ, respectively, was higher than in vehicle-treated animals. However, the concentration of IL-10 relative to IL-2 was lower for vehicle-treated mice after one dose, and was not different from vehicle-treated animals from the second dose onwards (Fig. 6).
Fig. 6.
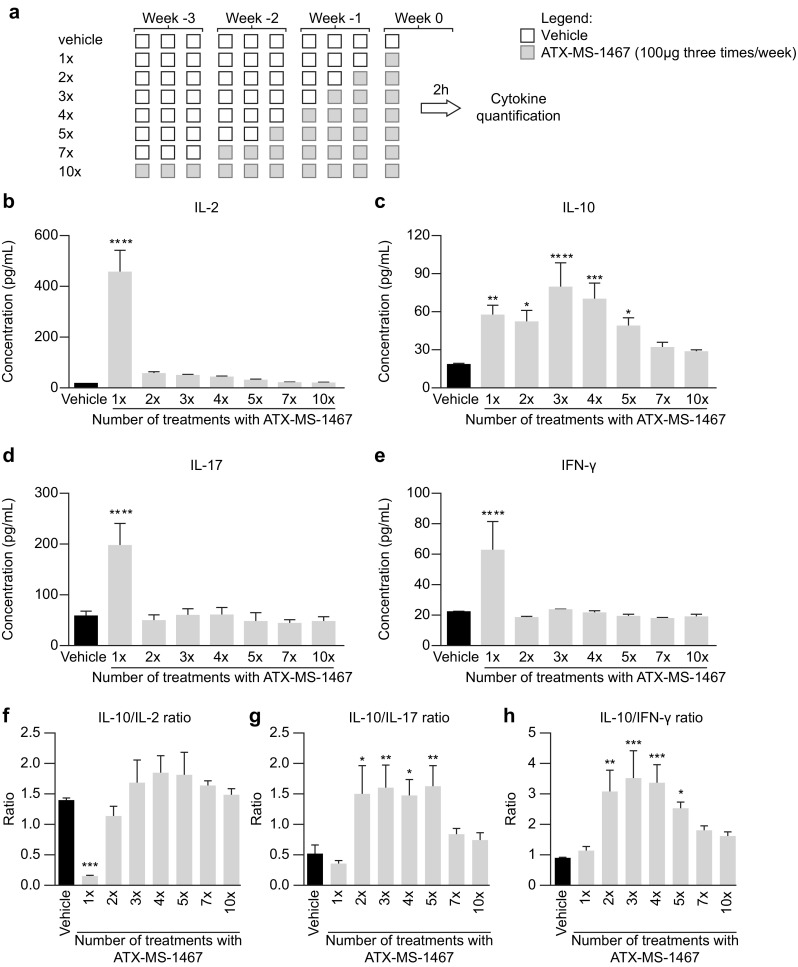
Effect of chronic dosing with ATX-MS-1467 on in vivo proinflammatory cytokine secretion. (DR2 × Ob1)F1 mice received from 1 to 10 treatments with 100 µg of ATX-MS-1467 or vehicle (PBS) on days 0, 2, 5, 7, 9, 12, 14, 16, 18 and 20. The top panel shows the overall dosing schedule (a). Serum cytokine concentrations were assayed 2 h after the last injection. The bottom panel shows the absolute concentrations of each cytokine in serum (b–e) used to generate ratios of interleukin 10 versus the other cytokines, shown in the insert (f–h). Data were analyzed by ANOVA followed by Dunnett’s multi-comparison test. *, **, *** and **** indicate p < 0.05, < 0.01, < 0.001 and < 0.0001, respectively, versus the vehicle (PBS)-treated group. ANOVA analysis of variance, IFN interferon, IL interleukin, PBS phosphate-buffered saline
Finally, a third type of experiment was done to assess how mice reacted to the native antigen (MBP) after they received a course of 10 injections with 300 µg of ATX-MS-1467, which was previously determined as the highest dose to elicit the maximum response (Fig. 7). Because all previous evidence indicates that ATX-MS-1467 treatment is associated with induction of Tregs, we hypothesized that the effect of the treatment is long-lasting; therefore, the effects should persist for at least as long as this pool of regulatory cells remained expanded, counteracting the natural emergence of Th1 or Th17 effector cells. To test this hypothesis, we challenged a group of mice with MBP following a wash-out period of 2 days (equivalent to the 3×/weekly regimen treatment with ATX-MS-1467), a group with wash-out periods of either 7, 21 or 42 days from the last ATX-MS-1467 treatment, and also mice in which HLAbp was used as a control for ATX-MS-1467. To help interpret this study (as depicted in Fig. 7), we compared the cytokine levels (or ratios) between the third bar, i.e., mice treated with control peptide (HLA-bp) then challenged with MBP, which could also be regarded as “untolerized”, against those levels in the four last columns representing mice treated chronically with ATX-MS-1467 prior to MBP challenge (performed from 2, 7, 21 or 43 days after the last treatment). When naïve mice were challenged with MBP (Fig. 7a–d; second column in each plot) there was always a large induction of serum cytokines. The same was true for the mice that received ten doses of HLA-bp (Fig. 7a–d; third column in each plot). However, for all cytokines tested, the serum cytokine concentrations were always lower for the groups of mice that received a course of ATX-MS-1467 than those mice that received HLA-bp treatment prior to the challenge, regardless of the duration of the wash-out period. Furthermore, the effector cytokines tended towards a slight increase after longer wash-out periods, whereas the opposite occurred for IL-10. This suggests a down-regulation of the pool of IL-10-secreting cells and a re-emergence of the pool of effector cytokine-secreting cells with time. From this observation, one question arises: at which point does the balance of regulatory/effector cytokines return to that seen in the absence of tolerization (equivalent to the HLB-bp group, third column in each plot)? This can be assessed by replotting the data as cytokine ratios (Fig. 7f–h). The results vary depending on the analyzed cytokine ratio. For the IL-10-to-IL-2 ratio, tolerization with ATX-MS-1467 can still induce higher ratios at wash-out periods of up to 7 days, but the ratios return to untolerized levels (HLA-bp treated) after a wash-out of 21 days (Fig. 7f). For the IL-10-to-IL-17 ratio, the return to baseline values occurs very quickly (Fig. 7g); the ratio has normalized after a wash-out of 7 days. However, the effects on the IL-10-to-IFNγ ratio are more persistent; this ratio did not return to baseline values until a wash-out period of 42 days. This result indicates that, at least with regard to this cytokine pair ratio, the effect of ATX-MS-1467 is long-lasting. On the other hand, the effect on the IL-10-to-IL-17 ratio was less persistent.
Fig. 7.
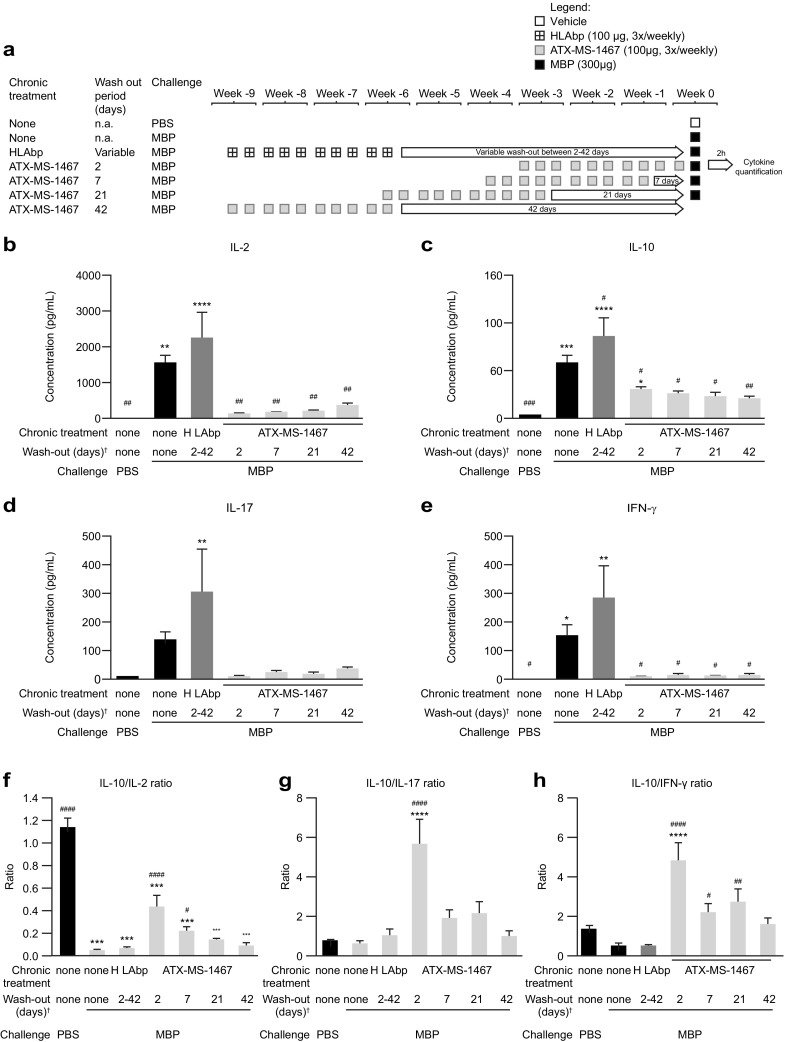
Effect of chronic treatment with ATX-MS-1467 on the ratio of interleukin 10-to-proinflammatory cytokines after an acute challenge with myelin basic protein. The length of the wash-out period is indicated by the arrows in the schematic representation of the study design (a). The absolute serum cytokine concentrations (b–e) were used to generate ratios of interleukin 10 versus the other cytokines shown in the insert (f–h). Data were analyzed by ANOVA followed by Dunnett’s multicomparison test. *, **, *** and **** indicates p < 0.05, < 0.01, < 0.001 and < 0.0001, respectively, versus naïve mice (first column). #, ##, ### and #### indicate p < 0.05, < 0.01, < 0.001 and < 0.0001, respectively, versus mice that were challenged with MBP without any prior tolerization (HLAbp column only), n = 6–8. †After 10 doses of ATX-MS-1467. ANOVA analysis of variance, HLAbp human leukocyte antigen-binding peptide, IFN interferon, IL interleukin, MBP myelin basic protein, na not applicable, PBS phosphate-buffered saline
Effect of ATX-MS-1467 Treatment on Regulatory T Cell Populations
The mechanism of tolerance induction can be investigated by quantification of the intracellular and/or extracellular markers unique to each of these Treg subsets (i.e., state-specific markers and subsets). We relied on two different approaches to define the induced regulatory T cells (iTreg) subset: one was the co-expression of LAG3 and CD49 in CD4+ lymphocytes; the other was the presence or absence of FOXP3 in IL-10 secreting CD4 lymphocytes. In the second approach, we assumed that the IL-10+FOXP3+ CD4 lymphocytes are natural T regulatory cells (nTregs), and the remaining IL-10+FOXP3− are, by exclusion, iTregs. Staining was performed in splenocytes of mice that had been treated in vivo with ATX-MS-1467 or HLA-bp for 2 weeks. Figure 8a, b shows the representative gating leading to assessment of the frequency of LAG3+CD49b+CD4+ lymphocytes from an HLA-bp-treated mouse and an ATX-MS-1467-treated mouse, respectively. Figure 8c shows mean data for both groups. The results indicate that chronic treatment with ATX-MS-1467 expanded the iTreg compartment in the spleen compared with mice treated with HLAbp. The spleens of mice treated with ATX-MS-1467 had a higher proportion of LAG3+C49b+ cells compared with HLAbp-treated mice (p < 0.05). Figure 8d–g shows the gating and the means for the upper-left and upper-right quadrants, denoting IL-10+FOXP3− and IL-10+FOXP3+ lymphocytes, respectively, for each group. In conclusion, in vivo ATX-MS-1467 treatment increased the pool of IL-10+Foxp3− (iTregs), but it had no effect on IL-10+Foxp3+ (nTregs).
Fig. 8.
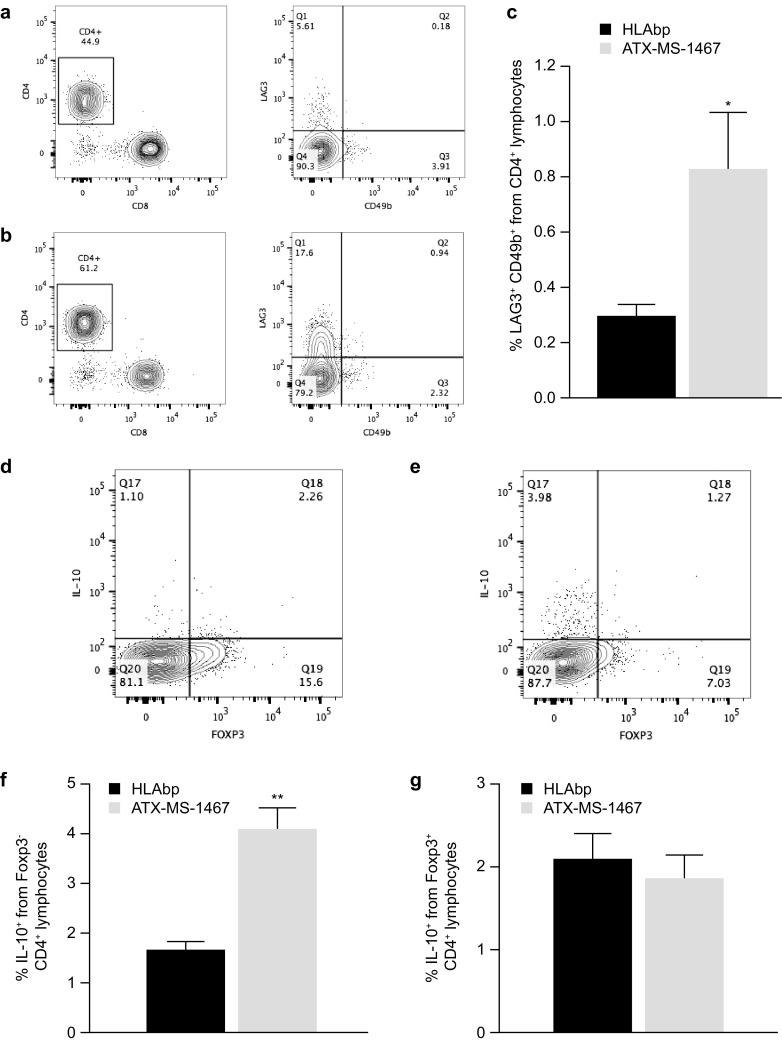
Effect of chronic treatment with ATX-MS-1467 on the splenic iTreg population of (DR2 × Ob1)F1 mice. For a–c, the spleens were harvested and fresh cells stained as described in the methods section. a, b are representative plots showing the populations of LAG3+CD49b+ cells (CD3+CD8−B220−) from total CD4+ lymphocytes, for which the averages are shown in c. For d–g, the harvested splenocytes were stimulated in vitro for 3 h in the presence of PMA, ionomycin and monensin, to allow intracellular accumulation and subsequent staining of IL-10. The contributions of Foxp3− and Foxp3+ cells to the total IL 10-producing CD4+ lymphocytes is shown in d and e, which are representative dot plots of the groups of mice treated with HLAbp (n = 8) or ATX-MS-1467 (n = 8), respectively. The averaged data are shown in f and g. * and ** indicate p < 0.05 and < 0.01, respectively, by unpaired t test with Welch’s correction versus the HLAbp group (n = 8). Foxp3 forkhead box p3, HLAbp human leukocyte antigen-binding peptide, IL interleukin, iTreg induced regulatory T cells, PMA phorbol myristate acetate
Discussion
Progressive axonal loss secondary to immune cell infiltration into the CNS, inflammation-mediated damage and demyelination, and diminishing repair are widely considered to be the pathological correlates of clinical disability in MS [23]. In the preclinical study published by Streeter et al., treatment with ATX-MS-1467 prevented the increase of neurological disability when the drug was administered prior to (dpi 4) or immediately after (dpi 8) the appearance of the first signs of disease [14]. To study this effect in more detail, mice were treated with four doses of ATX-MS-1467 at the onset and the peak of disease. As shown here, therapeutic treatment with ATX-MS-1467, both early and late after EAE induction in (DR2 × Ob1)F1 mice, reduced inflammation and T-cell and B-cell infiltration in the spinal cord, reduced demyelination and reduced BBB disruption. These effects were more profound in the animals treated early in the course of EAE than in those treated later, thus demonstrating the neuroprotective benefit of early intervention with ATX-MS-1467 in this animal model of MS. Although the exact association between the severity of disability and the extent of damage in the CNS is not fully understood [24], these results show good agreement between the abatement of inflammatory markers and demyelination and improvement in clinical score, particularly when treatment is started soon after the onset of EAE.
Autoantigen T-cell responses to MBP are involved in the pathogenesis of MS [25–28]. T-cell responses to MBP were initially identified in eight regions of MBP, of which five (MBP 30–44, MBP 83–99, MBP 130–144, MBP 140–154 and MBP 156–170) are the regions most frequently recognized by T cells. ATX-MS-1467 is a mixture of four of these peptides (MBP 30–44 [ATX-MS1]; MBP 131–145 [ATX-MS4]; 140–154 [ATX-MS6]; MBP 83–99 [ATX-MS7]), which act as apitopes and suppress EAE in a humanized mouse model [14].
Direct targeting of key regulators in the development and maintenance of autoreactivity aims to silence or reprogram autoreactive T cells in the periphery back to a regulatory phenotype, and thus promote a tolerogenic state to the targeted protein. Such targeted treatment may help to avoid many of the bystander effects seen with broad suppression of the immune system. An ex vivo antigen recall assay and in vivo quantification of cytokine concentrations in response to an acute challenge with MBP and also in response to chronic dosing with ATX-MS-1467 were used to investigate the effect of ATX-MS-1467 on the immune environment. ATX-MS-1467 elicited a dose-dependent switch to an IL-10-dominant environment after chronic treatment. Following repeated exposure to ATX-MS-1467, there was a positive and robust shift in the ratio of anti-inflammatory to pro-inflammatory cytokines, which persisted for as long as the mice were exposed to ATX-MS-1467. Some aspects of the effect induced by ATX-MS-1467 remained, at least partially, beyond 21 days, such as the shift in IL-10/IFNγ ratio; however, this effect was not as persistent as the effect on the ratio of other cytokine pairs (e.g., IL-10-to-IL-17). The shift to an anti-inflammatory cytokine profile was seen in treated mice in response to repeated doses of ATX-MS-1467 but not in mice treated with vehicle or in response to HLAbp, which binds to the MHC but contains none of the epitopes involved in the autoimmune response to MBP.
Consideration of ATX-MS-1467 as a tolerogenic approach presupposes that a rebalance of antigen-specific immunity occurs sometime after the initial treatment. It also presupposes that because the immune system is, at least temporally, reprogramed against a particular antigen, the effect of ATX-MS-1467 persists when the drug is cleared from the circulation. The induction of tolerance in type-1 helper (Th1) cells after repeated peptide immunization of MBP-specific-TCR transgenic mice led to a reduction in the capacity of these cells to proliferate and a shift to an IL-10-secreting phenotype [7]. IL-10 suppresses dendritic cell maturation and prevents Th1-cell differentiation, which is proposed as the negative arm of a feedback loop to limit Th1-driven immunopathology [11]. As shown here, with repeated ATX-MS-1467 treatment, the concentrations of the pro-inflammatory cytokines IL-17 and IFNγ diminished relative to the concentration of IL-10, but the relation between IL-2 and IL-10 was less clear. IL-2 is secreted by effector cells but not by regulatory cells, whereas it is removed, via receptor-mediated internalization, by both subtypes [29]. IL-2 is a broad regulator of Th-cell differentiation, notably in the differentiation of Th cells into both effector and cytolytic T cells, and is vital for the normal development of iTregs [29]. Therefore, IL-2 may have a gateway role in the early development of tolerance that diminishes with the development of a tolerogenic state following repeated exposure. Such a role would be consistent with the results reported here, as well as with other reports in which single peptide exposure led to an initial variable peripheral T-cell death that was transient and incomplete. Taken in isolation, the reduction in ex vivo secretion of IL-2 from splenocytes of mice treated in vivo with ATX-MS-1467 does not allow us to conclude whether the treatment led to an increase in MBP-specific Tregs, a reduction in MBP-specific effector Tregs or both, because, in each case, the observed result (i.e., reduced IL-2 concentration in culture supernatant) was to be expected. Nevertheless, any of these treatment effects would have a beneficial therapeutic impact. By comparison, multiple doses of Ac 1–9 of MBP completely protected TCR-transgenic mice inoculated with myelin, leading to down-regulation of the capacity of CD4+ T cells to proliferate or to produce IL-2, IFNγ and IL-4, but increased the production of IL-10. This effect was completely reversed by neutralization of IL-10 [7].
Treg cells are a crucial subpopulation of T cells that suppress the activity of several immune-cell lineages and are thought to have diverse and overlapping roles in the suppression of foreign-stimulated and autoantigen-stimulated immune reactions. Pharmacological induction of tolerance may, theoretically, occur by expansion of the nTreg compartment, by way of differentiation, expansion of induced Tregs (iTregs, also called type 1 regulatory T cells), or by both mechanisms. Chronic treatment with ATX-MS-1467 preferentially induced differentiation of naïve T cells into iTregs and there is also evidence of re-education of the immune system, with sparing of normal cellular immune surveillance. These findings are consistent with the development of a devolved tolerogenic state and the recruitment of resting Th cells from more than one compartment. This is also in keeping with the more consistent response seen following repeated exposure to peptides, as compared with the response to single exposure, which was transient, incomplete and resulted in only variable peripheral T-cell apoptosis [11].
Analysis of the phenotype of the IL-10-secreting cells showed that the splenic cell compartment contained iTregs that were derived from peripheral compartments. iTregs that co-express the LAG3+CD49+ phenotype represent a population of cells that migrate to the bone marrow following the effector stage of an immune response and provide a pool of long-lived resting cells that form the basis of the memory response to an antigen. ATX-MS-1467 treatment also led to greater secretion of IL-10 from iTregs that lacked the transcription factor Foxp3 (IL-10+Foxp3–) but not from IL-10+Foxp3+ nTregs. This indicated that the main source of IL-10 is from iTregs that originated in the periphery during the effector phase of the initial immune response to ATX-MS-1467, as opposed to nTregs of thymic origin [30]. The presence of iTregs with LAG3+CD49b+ and IL-10+Foxp3– phenotypes in the spleens of ATX-MS-1467-treated mice might indicate an overlapping cell population that express IL-10 transiently and at low concentrations during differentiation [31].
The results presented here raise some interesting questions for further investigation in preclinical studies and clinical trials in humans that were outside the scope of this manuscript. Future investigators should look at performing the ex vivo assay in co-cultures of purified dendritic cells and T cells, instead of whole-spleen cultures. Furthermore, ex vivo data could also be obtained only from leukocytes that infiltrated the CNS, which is more relevant for diseases such as EAE, and a wider complement of cytokines (such as IL-6 and transforming growth factor beta) could be measured. The regulatory effect of IL-10 could be further confirmed in the EAE mouse model by the use of IL-10-neutralizing antibodies or by adoptive-transfer experiments using purified iTregs, to gain further validation of the regulatory potential of the iTregs.
Although these current results show that ATX-MS-1467 can induce tolerance in (DR2 × Ob1)F1 mice in which EAE has been induced, this effect diminished with a 7-day wash-out period in the case of IL-10/IL-17. However, the effect on the IL-10-to-IFNγ ratio was more persistent. This observation is consistent with the inhibition of antigen-specific IFNγ release, but not of IL-17 release, in vitro from the splenocytes of mice treated with ATX-MS-1467. These data suggest that, in this model, ATX-MS-1467 is a more robust treatment to curb Th1-type responses than Th17, although this falling off may also be indicative of improved efficacy and a reduction in the need for high levels of cytokines over time. This finding could have a substantial effect on the regularity of dosing required in patients to achieve and maintain a state of tolerance to MBP. Studies in patients with MS will also determine whether the improvements in disability can be reproduced and whether the magnitude of the treatment effect will be sufficient to make this viable for the treatment of patients beyond the early stages of the disease. Experimental testing of ATX-MS-1467 has so far been restricted to animals designed to model the MS-associated HLA-DR2b allele; therefore, the generalizability of the treatment for patients who do not have genetic susceptibility and the efficacy in patients with different forms of MS will need to be established.
Furthermore, in the absence of a model that responds to all the peptides in the ATX-MS-1467 sequence, the model used in this study was constructed around only a small region of MBP: MS7 overlapping the MBP amino acids 81–102 [14]. This means that the efficacy of the drug in this model is restricted to this region, rather than the whole mix of peptides in ATX-MS-1467. It would be interesting to fully assess the efficacy of ATX-MS-1467 in a non-transgenic model, in which the MBP sequences in the drug are part of the encephalitic T-cell repertoire of the animal. We have already reported preliminary findings that ATX-MS-1467, given before the onset of EAE, delayed the onset (p < 0.04) and severity (p < 0.002) in non-transgenic Lewis rats [32]. A phase 1 first-in-human study in six patients with secondary progressive MS showed that ATX-MS-1467 was safe and well tolerated [14]. This study also showed a trend towards a transient induction of immunotolerance 1 month after chronic dosing with ATX-MS-1467. A phase 2 trial of ATX-MS-1467 in patients with relapsing/remitting MS to evaluate the clinical, biological and radiological effects and the safety profile of ATX-MS-1467 for up to 36 weeks has also been completed [33].
Conclusions
ATX-MS-1467 improved clinical disability and histological markers of inflammation and demyelination. It also restored the integrity of the BBB following induction of EAE in the humanized (DR2 × Ob1)F1 mouse. The greatest effectiveness was seen when treatment was given at the onset of EAE, but the effect is still significant and reproducible with dosing at the peak of disease. Chronic treatment with ATX-MS-1467 led to the development of tolerance to MBP in this model, accompanied by a shift to an immunotolerant state. The results of this study indicate that ATX-MS-1467 is a potential candidate in the management of MS.
Acknowledgements
ATX-MS-1467 is owned and being developed by Apitope Technology (Bristol) Limited. All of the data presented here was generated under a license from Apitope Technology (Bristol) Limited. The authors are grateful to David Wraith (University of Bristol) and Keith F. Martin (Apitope, Chepstow, Monmouthshire, UK) for scientific exchange. The authors would like to thank Professor Joseph Mandeville (Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital, Charlestown, MA, USA) who helped to design the study and provided guidance on the data analysis, and Fréderic Bernard (EMD Serono, Billerica, MA, USA), Anneli Savinainen (EMD Serono, Billerica, MA, USA) and Dongzi Yu (EMD Serono, Billerica, MA USA) for supporting the in vivo studies. The authors would also like to thank Paul Smith (Merck, Geneva, Switzerland) and Alla Zozoulya (EMD Serono, Billerica, MA, USA) for being involved in previous project management activities and model validation relevant to this work.
Funding
This study was funded by EMD Serono, Inc., a business of Merck KGaA, Darmstadt, Germany. At the time of the study, most of the authors were employees of EMD Serono, Inc.; therefore, the sponsor was involved in the study design, the collection, analysis and interpretation of the data and the decision to submit the study for publication. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by Steven Goodrick, inScience Communications, UK, and funded by Merck KGaA, Darmstadt, Germany.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Adriano Luís Soares de Souza wrote the manuscript, designed and performed the research and analyzed the data. Shuning Huang performed the research, MRI scans and analysis. Ji-Kyung Choi performed the MRI scans and analysis. Timothy Crandall performed the experiments. Stefan Rudin designed and performed the research and analyzed the data. Blake Tomkinson devised the strategy for the experiments and contributed to the research design. Rui Chang contributed to the histological analysis and analyzed the data. Keith Mitchell contributed to antigen-specific proliferation assays and analyzed the data. Shinji Okitsu contributed to the flow-cytometry experiments and analyzed the data. Danielle Graham designed the research and analyzed the data. Tammy Dellovade wrote the manuscript, designed the research and analyzed the data. Images were acquired by Shuning Huang and Ji-Kyung Choi. Gadolinium leakage was evaluated independently by Shuning Huang, Ji-Kyung Choi and Joe Mandeville. All authors critically reviewed the content of the manuscript and approved the final version for publication.
Disclosures
Adriano Luís Soares de Souza is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Stefan Rudin is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Rui Chang is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Keith Mitchell is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Timothy Crandall is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Shuning Huang is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Ji-Kyung Choi is an employee of Harvard Medical School at the Massachusetts General Hospital. The Massachusetts General Hospital received sponsorship from EMD Serono, Inc to allow access to magnetic resonance scanners and expertise in acquiring/analysing data, particularly for the MRI studies shown in this study. Shinji L. Okitsu is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Danielle L. Graham is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Blake Tomkinson is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Tammy Dellovade is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany.
Compliance with Ethics Guidelines
All procedures conducted were approved by the EMD Serono Institutional Animal Care and Use Committees (IACUC) or by the Massachusetts General Hospital IACUC in the case of MRI studies. All animal experiments complied with the National Institutes of Health guide for the care and use of laboratory animals. All efforts were made to minimize the number of animals used and to optimize their well-being.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Adriano Luís Soares de Souza
is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Stefan Rudin is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Rui Chang is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Keith Mitchell is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Timothy Crandall is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Shuning Huang is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Ji-Kyung Choi—is an employee of Harvard Medical School at the Massachusetts General Hospital. The Massachusetts General Hospital received sponsorship from EMD Serono, Inc to allow access to magnetic resonance scanners and expertise in acquiring/analysing data, particularly for the MRI studies shown in this study. Shinji L. Okitsu is an employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Danielle L. Graham is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Blake Tomkinson is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany. Tammy Dellovade is a former employee of EMD Serono, Inc, a business of Merck KGaA, Darmstadt, Germany.
Footnotes
Enhanced content
To view enhanced content for this article, go to 10.6084/m9.figshare.5919007.
References
- 1.Badawi AH, Siahaan TJ. Immune modulating peptides for the treatment and suppression of multiple sclerosis. Clin Immunol. 2012;144:127–138. doi: 10.1016/j.clim.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Immunology Garber K. A tolerant approach. Nature. 2014;507:418–420. doi: 10.1038/507418a. [DOI] [PubMed] [Google Scholar]
- 3.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159–1165. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 4.Anderton SM, Wraith DC. Hierarchy in the ability of T cell epitopes to induce peripheral tolerance to antigens from myelin. Eur J Immunol. 1998;28:1251–61. https://doi.org/10.1002/(SICI)1521-4141(199804)28:04<1251:AID-IMMU1251>3.0.CO;2-O. [DOI] [PubMed]
- 5.Liu JQ, Bai XF, Shi FD, Xiao BG, Li HL, Levi M, et al. Inhibition of experimental autoimmune encephalomyelitis in Lewis rats by nasal administration of encephalitogenic MBP peptides: synergistic effects of MBP 68-86 and 87-99. Int Immunol. 1998;10:1139–1148. doi: 10.1093/intimm/10.8.1139. [DOI] [PubMed] [Google Scholar]
- 6.Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) Br J Pharmacol. 2011;164:1079–1106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int Immunol. 1999;11(10):1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 8.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3(4):407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 9.Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4 + T cells. J Exp Med. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003;170(3):1240–1248. doi: 10.4049/jimmunol.170.3.1240. [DOI] [PubMed] [Google Scholar]
- 11.Gabrysova L, Nicolson KS, Streeter HB, Verhagen J, Sabatos-Peyton CA, Morgan DJ, et al. Negative feedback control of the autoimmune response through antigen-induced differentiation of IL-10-secreting Th1 cells. J Exp Med. 2009;206:1755–1767. doi: 10.1084/jem.20082118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhagen J, Burton BR, Britton GJ, Shepard ER, Anderton SM, Wraith DC. Modification of the FoxP3 transcription factor principally affects inducible T regulatory cells in a model of experimental autoimmune encephalomyelitis. PLoS One. 2013;8(4):e61334. doi: 10.1371/journal.pone.0061334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burton BR, Britton GJ, Fang H, Verhagen J, Smithers B, Sabatos-Peyton CA, et al. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat Commun. 2014;5:4741. doi: 10.1038/ncomms5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm. 2015;2:e93. doi: 10.1212/NXI.0000000000000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svejgaard A. The immunogenetics of multiple sclerosis. Immunogenetics. 2008;60(6):275–286. doi: 10.1007/s00251-008-0295-1. [DOI] [PubMed] [Google Scholar]
- 16.Anderton SM, Viner NJ, Matharu P, Lowrey PA, Wraith DC. Influence of a dominant cryptic epitope on autoimmune T cell tolerance. Nat Immunol. 2002;3(2):175–181. doi: 10.1038/ni756. [DOI] [PubMed] [Google Scholar]
- 17.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11(4 Suppl):S69–S76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 18.Wraith D, Streeter HB, inventors; Composition of myelin basic peptide peptides patent WO 2009056833 A3. 2009.
- 19.Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc Natl Acad Sci USA. 1999;96(26):15050–15055. doi: 10.1073/pnas.96.26.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet. 1999;23:343–347. doi: 10.1038/15525. [DOI] [PubMed] [Google Scholar]
- 21.Lewis ND, Patnaude LA, Pelletier J, Souza DJ, Lukas SM, King FJ, et al. A GPBAR1 (TGR5) small molecule agonist shows specific inhibitory effects on myeloid cell activation in vitro and reduces experimental autoimmune encephalitis (EAE) in vivo. PLoS One. 2014;9(6):e100883. doi: 10.1371/journal.pone.0100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oreja-Guevara C, Ramos-Cejudo J, Aroeira LS, Chamorro B, Diez-Tejedor E. TH1/TH2 Cytokine profile in relapsing-remitting multiple sclerosis patients treated with Glatiramer acetate or Natalizumab. BMC Neurol. 2012;12:95. doi: 10.1186/1471-2377-12-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol. 2005;252(Suppl 5):v3–v9. doi: 10.1007/s00415-005-5002-7. [DOI] [PubMed] [Google Scholar]
- 24.Gajofatto A, Calabrese M, Benedetti MD, Monaco S. Clinical, MRI, and CSF markers of disability progression in multiple sclerosis. Dis Markers. 2013;35:687–699. doi: 10.1155/2013/484959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponsford M, Mazza G, Coad J, Campbell MJ, Zajicek J, Wraith DC. Differential responses of CD45 + ve T-cell subsets to MBP in multiple sclerosis. Clin Exp Immunol. 2001;124:315–322. doi: 10.1046/j.1365-2249.2001.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazza G, Ponsford M, Lowrey P, Campbell MJ, Zajicek J, Wraith DC. Diversity and dynamics of the T-cell response to MBP in DR2 + ve individuals. Clin Exp Immunol. 2002;128:538–547. doi: 10.1046/j.1365-2249.2002.01831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etzensperger R, McMahon RM, Jones EY, Fugger L. Dissection of the multiple sclerosis associated DR2 haplotype. J Autoimmun. 2008;31:201–207. doi: 10.1016/j.jaut.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Ramagopalan SV, Ebers GC. Multiple sclerosis: major histocompatibility complexity and antigen presentation. Genome Med. 2009;1:105. doi: 10.1186/gm105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 2011;23:598–604. doi: 10.1016/j.coi.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Pillars article: immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995. J Immunol. 2011;186:3808–3821. [PubMed] [Google Scholar]
- 31.Gagliani C, Magnani CF, Huber SGM, Pala M, Licona-Limon P, Guo B, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2014;19:739–746. doi: 10.1038/nm.3179. [DOI] [PubMed] [Google Scholar]
- 32.Dellovade T, Rudin S, Zozulya A, Tomkinson B. ATX-MS-1467, and immunotolerizing agent, halts disease progression and reduces CNS inflammation in rodent models of multiple sclerosis. Neurology. 2015;82(10):216. [Google Scholar]
- 33.ClinicalTrials.gov. ATX-MS-1467 in multiple sclerosis. https://clinicaltrials.gov/ct2/show/NCT01973491?term=ATX-MS-1467&rank=1. Accessed February 29, 2016.