

Abstract
The ability of a cell to undergo mitochondrial apoptosis is governed by pro- and anti-apoptotic members of the BCL-2 protein family. The equilibrium of pro- versus anti-apoptotic BCL-2 proteins ensures appropriate regulation of programmed cell death during development and maintains organismal health. When unbalanced, the BCL-2 family can act as a barrier to apoptosis and facilitate tumour development and resistance to cancer therapy. Here we discuss the BCL-2 family, their deregulation in cancer and recent pharmaceutical developments to target specific members of this family as cancer therapy.
Keywords: BCL-2 family, apoptosis, BAX/BAK
1. Introduction
Apoptosis is a form of regulated cell death that is triggered in response to developmental cues or cellular stress. This selective cell suicide plays an essential role in numerous physiological and pathological processes including development, immunity and disease where the elimination of damaged or superfluous cells helps to ensure organismal health [1].
There are two apoptotic pathways—the extrinsic pathway (activated by ligand engagement of cell surface death receptors) and the intrinsic (mitochondrial) pathway. This review focuses on the BCL-2 family of proteins that regulate activation of the intrinsic apoptotic pathway in response to cellular stresses such as DNA damage, γ-irradiation, oncogene activation and growth factor withdrawal.
Recent pharmaceutical advances have allowed the specific targeting of protein–protein interactions in the BCL-2 (B-cell lymphoma 2) family [2–4]. Early clinical results in haematological cancers show considerable promise. This review will summarize apoptotic pathway regulation by the BCL-2 family, their perturbation in cancer and utility as therapeutic targets.
2. The BCL-2 family
The founder member, BCL-2, was first identified through chromosomal mapping in follicular lymphoma where constitutive BCL-2 expression is driven from the immunoglobulin locus by the t[12;18] translocation [5–7]. Unlike the cell growth and proliferative functions of other known oncoproteins at that time, BCL-2 was found to facilitate oncogenesis through cell death resistance [8,9]. In the following years over 15 proteins have been added to this family, each containing one or more BCL-2 homology (BH) domain and functional studies have allowed grouping into three classes (figure 1).
Figure 1.
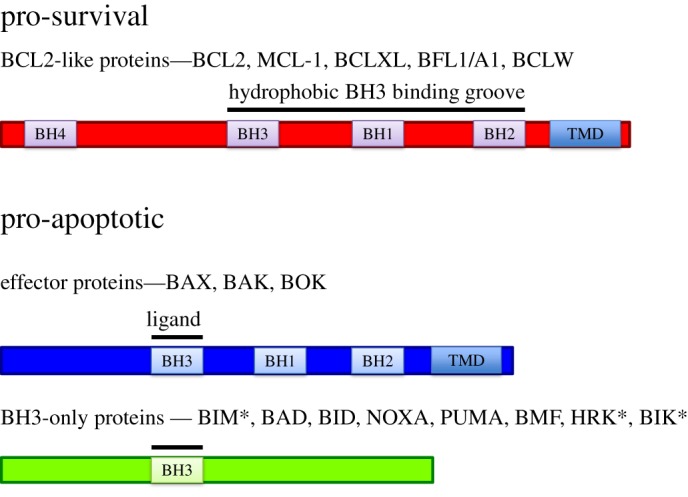
The BCL-2 family is composed of pro-survival and pro-apoptotic proteins. BCL-2 family members show sequence homology to BCL-2 in one or more BH (BCL-2 homology) domain. These proteins can be divided into pro-survival and pro-apoptotic proteins. Within the pro-apoptotic members there is a further subdivision between the multi-BH domain containing effector proteins and those proteins whose only region of homology to BCL-2 is BH3 (known as BH3-only proteins). Membrane insertion is mediated by transmembrane domains (TMD) present in pro-survival, effector and some BH3-only proteins (*BIM, BIK and HRK).
A clear division in the family exists between members that function to prevent apoptosis (pro-survival or anti-apoptotic) and those that induce apoptosis (pro-apoptotic). The pro-apoptotic BCL-2 family members can be further divided into the multi-BH-domain effector proteins (containing BH1, BH2 and BH3 domains) and BH3-only proteins (only region of homology to BCL-2 is BH3) (figure 1).
Physical interaction between pro-survival and pro-apoptotic family members can buffer the cell against the onset of mitochondrial-mediated apoptosis. Structural studies have revealed that BH1, BH2 and BH3 regions together form a hydrophobic pocket that can be filled by the amphipathic α-helical BH3 domain of pro-apoptotic BCL-2 proteins [10,11]. The balance of this interaction ensures appropriate apoptotic regulation in response to development cues and cellular stresses. In a simple model, when pro-survival proteins predominate, apoptosis is held in check: when pro-apoptotic proteins predominate, apoptosis is triggered. However, localization and conformation of BCL-2 proteins is also important in regulation of activity.
3. Pro-survival BCL-2 proteins
Pro-survival proteins such as BCL-XL, MCL-1, BFL1 (A1 in mouse) and BCL-W contain multiple regions of homology to BCL-2 (figure 2). Each of these proteins is found in many cell types/tissues and co-expression of multiple pro-survival proteins often occurs. The relative expression levels can vary in a cell type and developmental manner and are perhaps best characterized in the haematopoietic system. For example, dynamic patterns of pro-survival BCL-2 gene expression occurs during B lymphocyte development with Bclx being expressed early in B-cell development, Mcl1 and Bcl2 expression generally increasing with B-cell maturity and A1 levels peaking in the intermediate stages [12]. In this way, different pro-survivals play key roles at distinct stages of development.
Figure 2.
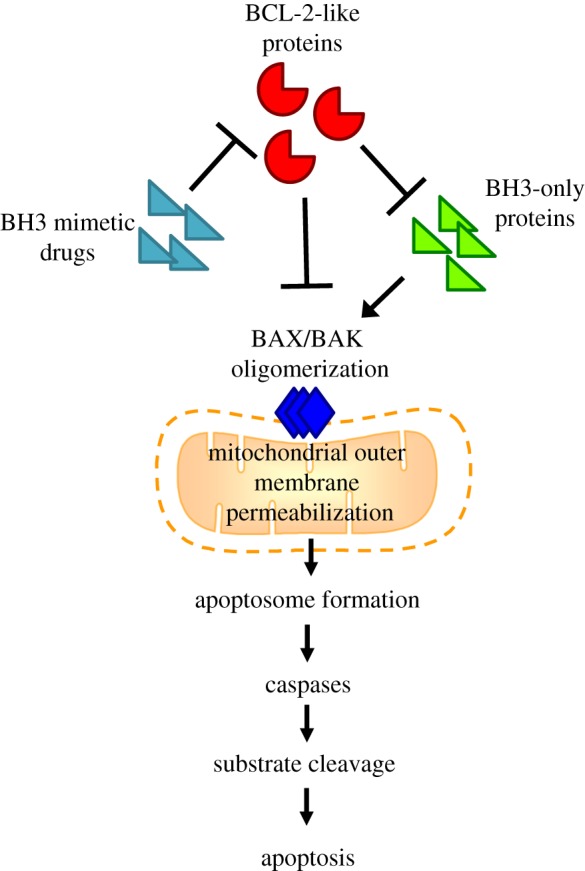
BCL-2 family interactions regulate mitochondrial outer membrane permeabilization (MOMP). Interaction between pro-survival and pro-apoptotic BCL-2 proteins sets a threshold for activation of apoptosis. BCL-2-like pro-survival proteins inhibit BAX/BAK activation whereas BH3-only proteins promote BAX/BAK oligomerization. Drugs mimicking the action of BH3-only proteins indirectly lead to BAX/BAK activation. This allows MOMP, apoptosome formation and subsequent caspase activation and apoptosis.
Levels of pro-survival BCL-2 proteins can also be regulated by protein turnover. BCL-2 and BCL-XL are relatively stable proteins (e.g. the half-life of BCL-2 approx. 20 h) [13]. By contrast, MCL-1 and A1 protein turnover is constitutive through polyubiquitination and proteosomal degradation (reflected in their short half-lives approx. 30 and approx. 15 min, respectively) [14–17]. In this way, levels of MCL-1 and A1 help facilitate dynamic responses to cell death stimuli.
Gene deletion studies have revealed essential and non-redundant roles of pro-survival BCL-2 proteins in mice. While embryogenesis proceeds normally in the absence of Bcl2, deficient mice show postnatal growth retardation, premature greying, apoptotic involution of spleen/thymus and succumb to early mortality through polycystic kidney disease with altered renal cell differentiation and elevated apoptosis [18]. Young Bcl2−/− mice have normal haematopoietic populations, but this is not sustained, with notable loss in peripheral B and T lymphocyte populations [18,19]. These phenotypes can be reversed by loss of one or two Bim alleles (encoding a BH3-only pro-apoptotic member of the BCL-2 family), indicating that sequestration of BIM is the major function of BCL-2 [20].
Deletion of Bclx is lethal around embryonic day 13 with extensive neuronal and haematopoietic apoptosis [21], loss of Bim can rescue the haematopoetic but not neuronal phenotype in bclx null embryos [22]. In adult mice, acute deletion of Bclx is tolerated (animals were followed for 1 month) but still resulted in severe anaemia, consistent with Bclx being required for reticulocyte survival [23]. In contrast to embryogenesis, loss of bim could not restore erythropoiesis in adults [23].
Deletion studies of A1 had been incomplete until recently when knockout of all three functional isoforms of A1 in mice was achieved. Surprisingly, A1 function seems largely redundant, with only minor impact on subsets of cells in the haematopoietic system [24]. The function of BCL-W also appears mostly dispensable for normal development and health, but Bclw-deficient males are infertile due to a defect in spermatogenesis [25,26].
Across the BCL-2 family the phenotype of the Mcl1 knockout mouse is most severe. Mcl1 deficiency results in early lethality at pre-implantation stage, but these blastocysts showed no evidence of increased apoptosis [27], providing a suggestion of a non-apoptotic role for MCL-1. Conditional deletion studies in the adult mouse have also revealed an essential role for MCL-1 in numerous cell types, including T and B lymphocytes [28], haematopoietic stem cells [29], cardiomyocytes [30,31], hepatocytes [32], neuronal progenitors [33] and neutrophils, but not macrophages [34,35], mammary epithelium or megakaryocytes [36,37].
Interestingly, while MCL-1 deficiency alone in megakaryocytes had no impact, when combined with loss of Bclx this caused embryonic or pre-weaning lethality, which is also far more dramatic than the impaired platelet shedding phenotype that is found with loss of just Bclx [36,37]. This could be rescued by co-deletion of Bax/Bak [37], but clearly illustrates the co-dependence of certain cell types on multiple pro-survival proteins. This co-dependence has been shown in elegant detail in a recent study of immune populations where multiple pro-survival proteins were targeted by genetic and pharmacologic methods [38]. Therefore, while gene knockout studies have given much insight into cell types in which individual pro-survival proteins have a dominant role, there has probably been an underestimation of the extent of their contribution to cell survival in many other cell types. Indeed, the sum effect of all pro-survival proteins present may be more important for survival than expression levels of an individual protein. This is an important consideration for therapeutic targeting of pro-survival BCL-2 proteins and minimization of damage to normal tissues.
4. Pro-apoptotic BCL-2 members
Pro-apoptotic BCL-2 proteins fall into two sub-classes (figure 2). BH3-only proteins such as BIM, BAD, BID, NOXA, PUMA, BMF, HRK and BIK only show homology to the BH3 domain of BCL-2. The effector proteins BAX, BAK and BOK contain multiple BH domains and structural studies of BAX revealed that effector protein three-dimensional conformation is similar to that of pro-survival BCL-2 proteins [39]. Like the pro-survival BCL-2 proteins, multiple pro-apoptotic proteins are found expressed in cells at the same time.
Upregulation of BH3-only proteins can occur at transcriptional/post-translational levels in response to stress to trigger cell death. For example, Puma and Noxa are transcriptional targets of the p53 tumour suppressor and their expression is increased in response to cytotoxic stimuli that activate p53, although PUMA is also important in response to p53-independent apoptotic stimuli [40]. BH3-only proteins can also be regulated by post-translational modification. Phosphorylation of BAD leads to sequestration by 14-3-3 proteins in the cytosol where it cannot exert pro-apoptotic functions [41]. Other mechanisms of activation exist such as altered cellular localization. Full-length BID is located in the cytosol but upon cleavage by caspase 8 (downstream of death-receptor signalling in the extrinsic apoptotic pathway) a truncated product is formed (tBID) which is capable of locating to the mitochondria and activating apoptosis [42].
BAX and BAK contain membrane anchoring C-terminal tails and while BAK is constitutively bound to the outer mitochondrial membrane, in healthy cells BAX appears cytosolic [43]. However, BAX and (to a lesser degree) BAK are actually in a dynamic equilibrium between cytosol and membranes, and are constitutively retrotranslocated to the cytosol by pro-survival BCL-2 proteins [44–46]. In the absence of any other BCL-2 proteins BAX becomes membrane localized, like BAK [45].
The mechanism of activation of BAX/BAK by BH3-only proteins has been the subject of intense debate. Evidence exists to suggest that some BH3-only proteins are ‘activators’: in this model BIM, tBID and PUMA, can directly interact with BAX/BAK to trigger their conformational change [47,48]. The function of the other BH3-only proteins in this model is as ‘sensitizers’, whose binding to BCL2-like pro-survival proteins frees up ‘activator’ BH3-only proteins [47,48]. An alternative, ‘indirect’ model has also been proposed whereby pro-survival BCL-2 proteins exert their function through direct interaction with BAX/BAK and BH3-only proteins act to sequester pro-survival proteins away from BAX/BAK. Such interactions do not always depend on BH3 domains as transmembrane domain (TMD) dimerization also occurs in the outer mitochondrial membrane. This has been observed in non-apoptotic cells and could indicate competition between the TMDs of pro-survival proteins such as BCL-2 and BCL-XL to prevent BAX/BAK homo-oligomerization [49].
Recently, genome editing was used to disrupt all known BH3-only proteins (eight in total) in HCT116 cells rendering these cells resistant to stress-induced apoptosis [50]. Interestingly, treatments that downregulate/target MCL-1- and BCL-XL-induced apoptosis with equivalent kinetics to those seen in cells proficient for BH3-only proteins revealing that known BH3-only proteins are not required for BAX/BAK activation [50]. Instead, association with the mitochondrial outer membrane is sufficient to drive homo-oligomerization of BAX/BAK in the absence of pro-survival BCL-2 proteins [50]. Regardless of which model is active in a particular cell type/situation, the outcome of a relative increase in the levels of pro- versus anti-apoptotic BCL-2 proteins is the same and the C-termini of BAX/BAK undergo conformational change that allows dimerization [51,52]. Reciprocal interaction between the BH3 domain of one molecule and the hydrophobic groove on another results in symmetrical homodimers (although heterodimers of BAX/BAK can also form in this manner) and linkage of these leads to higher-order oligomerization [53,54]. These oligomers delineate arcs, lines or ring-like structures in the outer mitochondrial membrane [55,56]. This mitochondrial outer membrane permeabilization (MOMP) allows release of soluble proteins from the mitochondrial inner membrane space such as cytochrome c which binds to APAF-1 (apoptotic protease activating factor 1), promoting its oligomerization and binding to pro-caspase 9—forming a complex termed the apoptosome. At the apoptosome, pro-caspase-9 is activated via dimerization, which in turn cleaves and activates the executioner caspases-3 and -7 to drive mass proteolysis that leads to DNA fragmentation, chromatin condensation and the dismantling of the cell. It is important to note that the mitochondrial pathway also contributes to death-receptor-mediated apoptosis through caspase 8 cleavage of BID, indeed BAX/BAK are required in many circumstances for receptor-mediated apoptosis [57,58].
In vivo analysis suggests the function of BAX and BAK is largely redundant in physiological settings; only a minority of double knockout mice survive to adulthood [59], but one copy of either is enough to allow normal development. In the absence of BAX/BAK apoptosis is impaired in response to almost all stimuli showing their requirement for the initiation of MOMP [60]. Less well studied, BOK is an additional multi-domain pro-apoptotic BCL-2 protein that was identified through its interaction with MCL-1 [61], although subsequent studies have suggested that BOK does not interact with pro-survival BCL-2 proteins [62,63]. BOK transcripts are present in many tissues in mice [64] but the protein is short-lived due to turnover by ubiquitylation and endoplasmic reticulum-associated degradation mediated by the proteasome [63].
BOK shows strong homology to BAX and BAK, and its role in apoptotic regulation has recently been extensively investigated. BOK deficient mice appear normal [64], but enforced BOK expression can drive apoptosis in a range of cell types and there is debate over whether this requires BAX/BAK [62–67]. Endogenous BOK is found predominantly in the membranes of the Golgi apparatus and endoplasmic reticulum (ER) and a function in ER stress response has been suggested [62,65]. More recently, the ability of proteasome inhibitors to induce apoptosis in BAX/BAK-deficient MEF or HCT116 cells was shown to require BOK expression but this was independent of pro-survival BCL-2 family proteins [63]. For now, the role of BOK certainly seems unique and requires further investigation.
5. Favoured interactions
As discussed above, a level of specificity is granted by differential expression pattern, cellular localization, post-translational activation and turnover of BCL-2 family proteins. A further level of complexity was revealed by biochemical and cell biology studies that showed differential binding affinities of particular BH3-only proteins for pro-survivals [68,69]. While BH3-only proteins such as BIM, PUMA and tBID bind with high affinity to all pro-survival proteins, others are more selective in their interaction. For example, BAD preferentially interacts with BCL2, BCL-XL and BCL-W while NOXA showing a reciprocal interaction preference for only MCL-1 and A1 (figure 3). Further complexity is added by preference of BH3-only proteins for activation of BAX or BAK. While BIM and BID bind the same repertoire of pro-survival proteins (figure 3), preference of BID to mediate apoptosis through BAK has been shown [70,71], whereas BIM preference for BAX [70], or no preference [71], has been observed. Domain swap experiments have revealed that these effects are determined by the BH3 sequence of the BH3-only proteins and are dependent on cell type [71].
Figure 3.
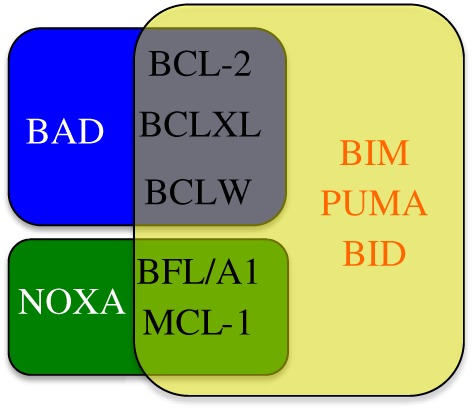
Specific interactions of BH3-only with pro-survival proteins. Some BH3-only proteins (BIM, PUMA and BID) are promiscuous and can bind all pro-survival BCL-2 proteins, whereas others (BAD and NOXA) show a more restricted binding pattern.
An additional layer of complexity is added with specificity in pro-survival proteins for BAK versus BAX activation. MCL-1 and BCL-XL constrain BAK, but BCL-2 does not [72], and it is the BH3 domain of BAK that determines these associations [71].
5.1. Increased pro-survival BCL-2 proteins in cancer
Evasion of apoptosis can aid oncogenic transformation at multiple stages through facilitating sustained tumour growth, survival during metastatic process and resistance to therapy. Therefore, it is not surprising that increased expression of pro-survival BCL-2 proteins is found in many cancer types. This upregulation can occur through a variety of mechanisms including chromosomal translocation, gene amplification, increased gene expression/translation or protein stability with various mechanisms and alternative pro-survival BCL-2 protein increases seeming more prominent in particular cancers.
Besides the Bcl2 t(12;18) translocation, first found in follicular lymphoma (and subsequently in diffuse large-cell lymphomas [73]), translocation of BCL-2 family genes is not common across different cancer types nor does it seem to occur to other pro-survival members. Interestingly, t(14;18) translocation of Bcl2 has also been found in peripheral blood lymphocytes from healthy individuals [74] and modelling of this translocation in B cells of mice is only weakly tumorigenic [75]. Together, these data suggest that this translocation is not overtly oncogenic.
Gene copy number increases in pro-survival BCL-2 members in cancer are more widespread than translocation. Transgenic modelling of increases in pro-survival proteins has mostly been limited to the haematopoietic systems where elevation of Bcl2, Mcl1 or Bclx predisposes to lymphoma development, albeit with long latency and incomplete penetrance. Amplification of MCL1 and BCL2L1 (encodes BCL-XL) were found to be among the most frequent chromosomal gains in a study of over 3000 samples representing 26 tumour types [76]. It is interesting that these amplifications were prominent outside of haematopoietic cancers, which have been the traditional niche for studies on tumorigenic roles of the BCL-2 members. Indeed, analysis of The Cancer Genome Atlas (TCGA) data through cBioportal [77] confirms the prevalence of MCL1, and to a lesser extent BCL2L1, amplification in many solid cancers (figure 4). Again, there seems specificity between family members as BCL2, BCL2A1 (BFL) and BCL2L2 (BCL-W) amplification are much rarer events. Consistent with a pro-tumour role, mutation or deletion of pro-survival BCL2 proteins was also infrequent (figure 4). While pro-survival BCL-2 expression on its own is mildly oncogenic, acquisition of additional genetic hits is clearly required for tumour formation. Co-amplification of MYC with MCL-1 or BCLX is common in cancer [76] and in cell culture and mouse models increased BCL-2-like proteins, and MYC is a potent oncogenic combination [83–87].
Figure 4.
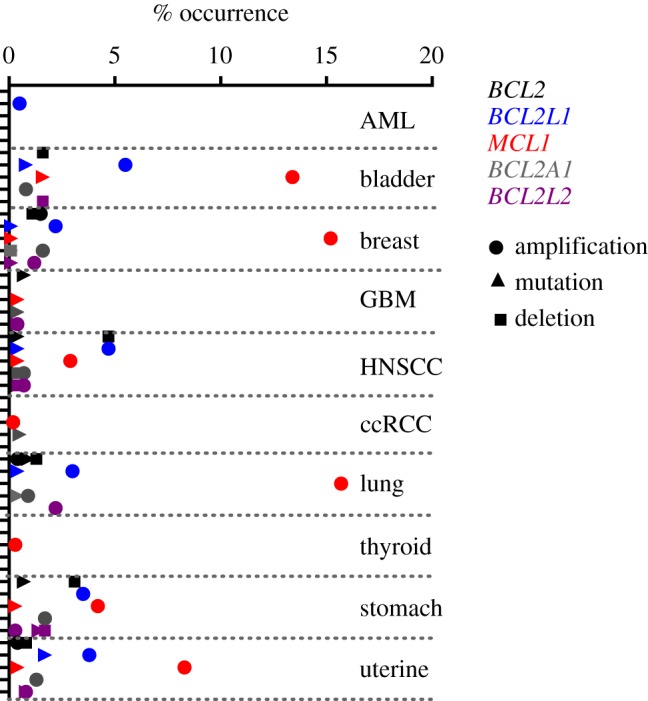
Frequency of genomic alteration of pro-survival BCL-2 proteins in cancer. Frequency of amplification (circle), mutation (triangle) or deletion (square) of pro-survival BCL-2 members in a range of cancers. Data mined from TCGA studies through cBioportal [77]. BCL-2 (black), BCL-XL (BCL2L1 blue), MCL-1 (red), BFL (BCL2A1 grey), BCL-W (BCL2L2 purple). AML, acute myeloid leukaemia [78], 173 cases. Bladder, urothelial carcinoma nature (TCGA provisional), 408 cases. Breast, invasive carcinoma (TCGA provisional), 1100 cases. GBM, glioblastoma [79], 166 cases. HNSCC, head and neck squamous cell carcinoma (TCGA provisional), 522 cases. ccRCC, kidney renal clear cell carcinoma [80], 534 cases. Lung adenocarcinoma (TCGA provisional), 517 cases. Thyroid, papillary thyroid carcinoma [81], 509 cases. Stomach adenocarcinoma (TCGA provisional), 415 cases. Uterine, corpus endometrial carcinoma [82], 177 cases.
Increased transcription of pro-survival proteins can also elevate their expression in cancer and increased transcription/translation seems important for regulation of MCL-1 levels. For example, in chronic lymphocytic leukaemia (CLL) c-ABL has been shown to drive high MCL-1 mRNA and protein expression through STAT3/NF-κB [88]. Similarly, in a mouse model of B-cell acute lymphoblastic leukaemia (B-ALL) the BCR-ABL oncoprotein was shown to drive high levels of MCL-1 expression that was essential for leukemogenesis [89].
Increased protein translation impacts on MCL-1 and BCL-XL levels and MCL-1 has been identified as a downstream mediator of the oncogenic effect of the translation initiation factor eIF4e [90]. Disruption of pathways regulating MCL-1 protein stability also occurs in cancer (e.g. loss or mutation of FBW7 inhibits MCL-1 degradation and is associated with tumorigenesis and resistance to chemotherapy [91,92]). Therefore, in addition to genetic alterations in BCL-2 family members, activation of oncogenic signalling pathways can also increase pro-survival protein levels.
It is important to consider that the observation of high levels of pro-survival BCL-2 proteins in cancer need not necessarily indicate strong apoptotic resistance. Elevation of BCL-2 actually sensitizes to apoptosis induced by the BCL-2/BCL-XL/BCL-W targeting drug ABT-737 through release of high levels of BIM that have been harboured in BIM/BCL-2 complexes [93]. In this way, a cell can be thought of as primed for death, close to the threshold required for apoptosis induction and measurement of the level of mitochondrial priming in cells can be used to predict response to chemotherapy [94,95].
Alterations in pro-survival BCL2 proteins might have importance beyond cancer genesis as high levels of MCL-1 expression at diagnosis correlate with poor prognosis in breast cancer [96], and MCL-1 amplification is prominent in treatment-resistant breast cancers [97], suggesting that this may be a source of innate and acquired resistance to cancer therapy. Such associations do not hold true for pro-survival BCL-2-like proteins in general and high levels of BCL-2 are actually associated with good prognosis in breast cancer [98–101].
5.2. Decreased pro-apoptotic BCL-2 proteins in cancer
Decreased expression of pro-apoptotic BCL-2 proteins has the same functional outcome as increased pro-survival expression in cancer. Gene deletion studies in mice do not reveal a strong oncogenic impact of decreased BH3-only protein expression. With the exception of BAD knockout mice, which succumb to late onset lymphoma [102], deficiency in individual BH3-only proteins does not predispose mice to tumour development [103]. Functional redundancy between BH3-only proteins could account for this, however, compound deletion of multiple BH3-only proteins is still only weakly tumour promoting with autoimmunity contributing to morbidity [104].
Similar to over-expression of pro-survival BCL2-like proteins, loss of BH3-only proteins is not overtly oncogenic but can dramatically accelerate lymphoma development in the context of elevated MYC. BIM seems most potent in this context, with deletion of even a single allele of BIM having dramatic effects [105].
Cancer therapeutics engaging the p53 response would be predicted to upregulate PUMA and NOXA and resistance to apoptosis could be mediated by their downregulation. Indeed, deletion of the gene encoding PUMA (Bbc3) occurs in a range of cancer types [76] and other mechanisms can decrease PUMA expression such as promoter methylation [106].
5.3. Altered expression of effector proteins
Elimination or downregulation of apoptotic effector proteins such as BAX/BAK is a potent way to disable mitochondrial-mediated apoptosis in cell culture systems. As the function of BAX and BAK are largely redundant, with one copy of either allowing normal development, abrogation of their activity would require loss of all four alleles. There is limited evidence that this occurs in cancer and BAX/BAK levels naturally decline with age in mice and humans [107]. It is important to note that localization and conformation may be even more important than absolute levels of BAX/BAK. For example, in acute myeloid leukaemia (AML) mitochondrial localized BAX is associated with both increased apoptotic sensitivity and improved patient prognosis [108]. Gene deletion studies in mice do not suggest a tumour suppressive role for BAX or BAK when knocked out individually (presumably due to their redundant roles) and double knockout results in perinatal lethality [59,109]. Using chimeric mice Bax/Bak deletion in the haematopoietic compartment drives fatal autoimmune disease [110]. The prominence of autoimmune disease in chimeric mice with Bax/Bak [111] deficient haematopoietic systems may mask tumour development. Targeted deletion mouse studies can avoid these autoimmune complications and in this setting loss of BAX/BAK in the brain and testicles results in tumours [112].
It seems pertinent loss of Bok is a relatively frequent event in a range of cancers [76] and gene silencing may result in loss of BOK protein in additional cases [111]. BOK is downregulated in non-small cell lung cancer (NSCLC) and high BOK is associated with good prognosis in lymph-node positive patients [111]. Interestingly, the tumour suppressive effect of BOK in NSCLC does not seem to be through apoptotic regulation and is instead through antagonism of TGF-β2 mediated epithelial to mesenchymal transition (EMT) and cell migration [111]. Unlike so many other BCL-2 proteins, Bok loss failed to reveal a tumour suppressive role the Eu-Myc mouse model [64], a system that is a sensitive read-out for altered apoptosis, although factors such as cooperating second hits, timing of Bok loss and cell type specificity could all be involved in a BOK effect. Compound knockout of Bax/Bak/Bok in the haematopoietic system also results in autoimmune disease rather than tumourigenesis [113]. More recently, in a chemical (DEN)-induced model of liver carcinogenesis, loss of BOK has been shown to protect against cancer. In this model, cancer can be promoted by death of hepatocytes, supporting compensatory proliferation with associated mutagenesis, in surrounding tissue that ultimately leads to live cancer [114]. Interestingly, deletion of BOK inhibited the ER-stress response and induction of pro-apoptotic BH3-only proteins BIM and PUMA, placing BOK's tumour promoting role upstream of MOMP [114]. Secondly, BOK was also shown to have an additional growth-promoting effect, though the mechanisms underlying this remain unclear it would also be expected to be pro-tumourigenic [114].
5.4. Pharmaceutical intervention to reset the balance
As the balance of BCL-2 proteins can act as a trigger for apoptosis, mechanisms to alter their expression or interaction would be expected to have clinical use. Indeed conventional anti-cancer therapies can act through disruption of the BCL-2 family. For example, DNA damaging agents such as etoposide or daunorubicin activate the p53 tumour suppressor transcription factor whose targets include PUMA and NOXA. However, the presence of elevated pro-survival BCL-2 proteins can act as a barrier to apoptosis even upon upregulation of BH3-only proteins. Neutralization of pro-survival proteins would re-sensitize to BH3-only upregulation and could even have the potential to trigger apoptosis alone.
Interest in the development of inhibitors of pro-survival BCL-2 proteins has come to fruition over the past decade following the identification of small molecule inhibitors capable of occupying the hydrophobic pocket on pro-survival proteins. The first of these BH3 mimetic drugs, ABT-737, was developed through NMR-based fragment screening. ABT-737 mimics the BH3-domain of pro-apoptotic BAD and interacts with BCL-2, BCL-XL and BCL-W (figure 5), displacing BH3-only proteins to trigger apoptosis in cell lines and restrict tumour growth in xenograft models [2]. Derivation of an orally available analogue (ABT 263/Navitoclax) allowed clinical testing [115]. On-target toxicity of Navitoclax was observed with dose-limiting thrombocytopenia occurring due to platelet dependence on BCL-XL [116,117]. This can be managed in the clinic and preclinical models have supported the use of Navitoclax in clinical trials as a combination therapy in a range of (predominantly solid) tumour types [118,119] (clinicaltrials.gov).
Figure 5.
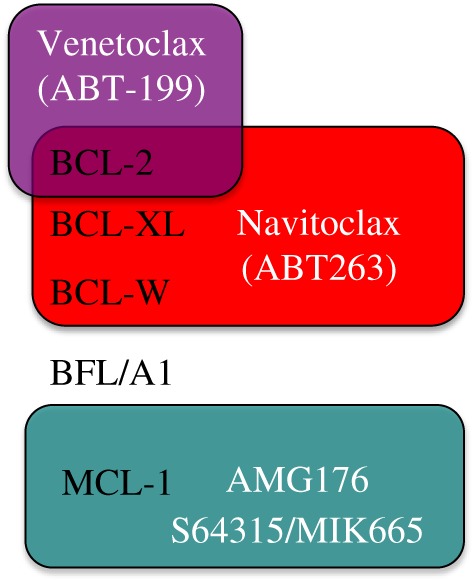
Selectivity of BH3-mimetic drugs under clinical investigation. Drugs specifically targeting BCL-2 (Venetoclax/ABT-199), BCL-2, BCL-XL and BCL-W (Navitoclax/ABT263) or MCL-1 (AMG176, S64315/MIK665) are now in clinical trial/use.
Further development has led to additional BH3 mimetics with increased specificity for individual BCL-2 proteins. The success of this approach has been shown with the BCL-2 specific BH3 mimetic ABT-199 (Venetoclax), which obtained breakthrough FDA status for use in relapsed/refractory CLL. In this disease single-agent efficacy was seen with partial response in 79% and complete response in 20% of patients [120]. Even more impressive results were seen when Venetoclax was used in combination with rituximab-complete response occurred in 51% of patients with disease-free status occurring for up to 2 years after completion of therapy [121]. Encouraging (but more modest) effects are seen in non-Hodgkin lymphomas, acute myeloid leukaemia and multiple myeloma as a single agent [122–124] or combination therapy [125]. While much is understood of the role of the BCL-2 family in cancers of the blood, the case for targeting these proteins in solid tumours, most probably in conjunction with conventional therapies, is compelling. Translocation or amplification of BCL2 itself is rarely seen in solid tumours but dependence on BCL2 has been shown in small cell lung cancer (SCLC) [126]. Efficacy of Venetoclax when used as a combination therapy has been shown in preclinical models of breast cancer [127] and Venetoclax is now in clinical trials in combination with tamoxifen in breast cancer (ISRCTN98335443).
Resistance to BCL-2 and BCL-2/BCL-XL targeting BH3 mimetics has been observed, and in vitro studies indicate that ABT-737 treatment can increase MCL-1 levels and MCL-1 expression promotes resistance to ABT737 in vitro and in vivo [128–130]. MCL-1 has been associated with resistance to cancer therapy for some time [131,132], and interest has intensified in the development of drugs that specifically target MCL-1. A number of MCL-1 inhibitors have been mooted but until very recently compounds with clear specificity and on-target effect were lacking [133]. The tide has turned and a number of robust BH3 mimetic drugs targeting MCL-1 are now available; Abbvie's A1210477 potently inhibits MCL-1 in vitro to restrict growth of diverse cancer cell lines [134,135], UMI-77 inhibits pancreatic and breast cancer cell line growth in vitro and in vivo [96,136], and the Servier compound S63845 seems particularly potent, with on-target single agent killing of leukaemia and lymphoma models in vitro and in vivo [4] and in combination with conventional cancer therapy in xenograft models of breast cancer [137]. The potential for MCL-1 specific BH3-mimetics in the clinic is now being tested with the Novartis/Servier drug S64315/MIK665 and Amgen AMG176 in phase I clinical trials for haematopoietic cancers/myelodysplastic syndrome (NCT02992483, NCT02979366, NCT02675452). Taking advantage of the short half-life of MCL-1 protein, a number of other approaches can be taken to decrease MCL-1 levels. This includes inhibition of transcription through CDK inhibition [138] or targeting translation through mTOR inhibition [139]. Beyond targeting anti-apoptotic BCL-2 proteins, increasing interest has centred on developing drugs that directly activate BAX and BAK in order to kill tumour cells [140,141]. Along these lines, a recent study has shown that a BAX-activating molecule, BTSA1, shows potent anti-tumour effects on human acute myeloid leukaemia (AML) xenografts in the absence of toxicity [142].
6. Inhibiting BCL-2 proteins in cancer prophylaxis
It is conceivable that targeting the pro-survival BCL-2 proteins could help eliminate pre-cancerous lesions or early-stage tumours. Indeed, ABT737 can act as an anti-cancer prophylactic in the Eμ-MYC mouse model of B-cell lymphoma, which has been shown to be dependent on BCL-XL [143]. Evidence for applicability beyond MYC-driven lymphoma is limited and in p53 null mice (which predominantly succumb to thymic lymphoma) prophylactic treatment with ABT737 had no impact on tumorigenesis [144]. When low-dose γ-irradiation was added to this experimental protocol to mimic environmental factors that could induce additional mutations prophylactic ABT737 treatment was shown to delay lymphoma onset in p53 null mice, but this reduction in thymic lymphoma was accompanied by increased incidence of sarcoma, and while significant, the difference in survival outcome afforded by prophylactic ABT 737 treatment was minimal [144].
Confounding factors include the altered activity of cells that have failed to be eliminated by targeting pro-survival BCL2 proteins. For example, the spontaneous apoptosis induced by targeted deletion of mcl1 in hepatocytes results in severe liver damage and increased proliferation that actually results in hepatocellular carcinoma (HCC) [145]. Such effects have also been shown in models of γ-irradiation induced thymic lymphoma where deletion of puma or over-expression of Bcl2 (normally considered as pro-cancer events) actually protects against tumour development [146]. In this scenario, γ-irradiation no longer causes depletion of bone marrow leucocytes meaning that there is no niche for the proliferation of stem/progenitor cells carrying damaged DNA that normally give rise to the thymic lymphomas in this model [146,147]. There are further indications that inhibiting the pro-survivals may not always have anti-cancer impact, in settings where ABT737 fails to induce apoptosis detrimental side effects can occur through activation of CAD and genome instability [148]. It remains to be seen whether these concerns hold true in the clinic.
In the decades following the discovery of BCL-2 (and related family members) a vast quantity of research has unravelled their role in regulating apoptosis. The functional division of this family into pro- and anti-apoptotic members and the elucidation of their structures and mechanism of interaction has allowed the pharmaceutical development of molecules to specifically inhibit these protein–protein interactions and reinstate apoptosis. Available data from clinical trials suggests good efficacy of BH3-mimetics targeting BCL-2 in some types of blood cancer. Deregulation of BCL-2 proteins is now recognized as a frequent event in many types of cancer and it seems likely that targeting pro-survival BCL-2 proteins will form a valuable adjunct to current cancer therapies. Restoration of apoptosis offers the potential to eliminate cancer cells at all stages of pathology and as BH3-mimetic drugs make their way into the clinic they could make dramatic improvements in survival outcome in cancer.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
Research in our laboratory is supported by Cancer Research UK, Breast Cancer Now and Tenovus Scotland.
References
- 1.Meier P, Finch A, Evan G. 2000. Apoptosis in development. Nature 407, 796–801. (doi:10.1038/35037734) [DOI] [PubMed] [Google Scholar]
- 2.Oltersdorf T, et al. 2005. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. (doi:10.1038/nature03579) [DOI] [PubMed] [Google Scholar]
- 3.Souers AJ, et al. 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. (doi:10.1038/nm.3048) [DOI] [PubMed] [Google Scholar]
- 4.Kotschy A, et al. 2016. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. (doi:10.1038/nature19830) [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. 1984. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099. (doi:10.1126/science.6093263) [DOI] [PubMed] [Google Scholar]
- 6.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. 1985. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 41, 899–906. (doi:10.1016/S0092-8674(85)80070-2) [DOI] [PubMed] [Google Scholar]
- 7.Cleary ML, Sklar J. 1985. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc. Natl Acad. Sci. USA 82, 7439–7443. (doi:10.1073/pnas.82.21.7439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442. (doi:10.1038/335440a0) [DOI] [PubMed] [Google Scholar]
- 9.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. 1990. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348, 334–336. (doi:10.1038/348334a0) [DOI] [PubMed] [Google Scholar]
- 10.Sattler M, et al. 1997. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986. (doi:10.1126/science.275.5302.983) [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. 2003. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity 19, 341–352. (doi:10.1016/S1074-7613(03)00234-6) [DOI] [PubMed] [Google Scholar]
- 12.Vikstrom IB, et al. 2016. MCL-1 is required throughout B-cell development and its loss sensitizes specific B-cell subsets to inhibition of BCL-2 or BCL-XL. Cell Death Dis. 7, e2345 (doi:10.1038/cddis.2016.237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blagosklonny MV, Alvarez M, Fojo A, Neckers LM. 1996. bcl-2 protein downregulation is not required for differentiation of multidrug resistant HL60 leukemia cells. Leuk. Res. 20, 101–107. (doi:10.1016/0145-2126(95)00103-4) [DOI] [PubMed] [Google Scholar]
- 14.Adams KW, Cooper GM. 2007. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J. Biol. Chem. 282, 6192–6200. (doi:10.1074/jbc.M610643200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. 2006. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760. (doi:10.1016/j.molcel.2006.02.009) [DOI] [PubMed] [Google Scholar]
- 16.Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G, Berberich I. 2006. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J. Biol. Chem. 281, 13 663–13 671. (doi:10.1074/jbc.M600266200) [DOI] [PubMed] [Google Scholar]
- 17.Zhong Q, Gao W, Du F, Wang X. 2005. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095. (doi:10.1016/j.cell.2005.06.009) [DOI] [PubMed] [Google Scholar]
- 18.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. 1993. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75, 229–240. (doi:10.1016/0092-8674(93)80065-M) [DOI] [PubMed] [Google Scholar]
- 19.Nakayama K, et al. 1993. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science 261, 1584–1588. (doi:10.1126/science.8372353) [DOI] [PubMed] [Google Scholar]
- 20.Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. 2001. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev. Cell 1, 645–653. (doi:10.1016/S1534-5807(01)00083-1) [DOI] [PubMed] [Google Scholar]
- 21.Motoyama N, et al. 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267, 1506–1510. (doi:10.1126/science.7878471) [DOI] [PubMed] [Google Scholar]
- 22.Akhtar RS, Klocke BJ, Strasser A, Roth KA. 2008. Loss of BH3-only protein Bim inhibits apoptosis of hemopoietic cells in the fetal liver and male germ cells but not neuronal cells in bcl-x-deficient mice. J. Histochem. Cytochem. 56, 921–927. (doi:10.1369/jhc.2008.951749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delbridge AR, et al. 2017. The BH3-only proteins BIM and PUMA are not critical for the reticulocyte apoptosis caused by loss of the pro-survival protein BCL-XL. Cell Death Dis. 8, e2914 (doi:10.1038/cddis.2017.304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenk RL, et al. 2017. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. 24, 534–545. (doi:10.1038/cdd.2016.156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Print CG, et al. 1998. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl Acad. Sci. USA 95, 12 424–12 431. (doi:10.1073/pnas.95.21.12424) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD, MacGregor GR. 1998. Testicular degeneration in Bclw-deficient mice. Nat. Genet. 18, 251–256. (doi:10.1038/ng0398-251) [DOI] [PubMed] [Google Scholar]
- 27.Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. 2000. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 14, 23–27. [PMC free article] [PubMed] [Google Scholar]
- 28.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. 2003. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426, 671–676. (doi:10.1038/nature02067) [DOI] [PubMed] [Google Scholar]
- 29.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307, 1101–1104. (doi:10.1126/science.1106114) [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. 2013. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 27, 1351–1364. (doi:10.1101/gad.215855.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas RL, et al. 2013. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 27, 1365–1377. (doi:10.1101/gad.215871.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vick B, et al. 2009. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology 49, 627–636. (doi:10.1002/hep.22664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arbour N, et al. 2008. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 28, 6068–6078. (doi:10.1523/JNEUROSCI.4940-07.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dzhagalov I, St John A, He YW. 2007. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109, 1620–1626. (doi:10.1182/blood-2006-03-013771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. 2009. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 113, 2805–2815. (doi:10.1182/blood-2008-05-159145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Debrincat MA, et al. 2012. Mcl-1 and Bcl-x(L) coordinately regulate megakaryocyte survival. Blood 119, 5850–5858. (doi:10.1182/blood-2011-12-398834) [DOI] [PubMed] [Google Scholar]
- 37.Kodama T, et al. 2012. Mcl-1 and Bcl-xL regulate Bak/Bax-dependent apoptosis of the megakaryocytic lineage at multistages. Cell Death Differ. 19, 1856–1869. (doi:10.1038/cdd.2012.88) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrington EM, et al. 2017. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 24, 878–888. (doi:10.1038/cdd.2017.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki M, Youle RJ, Tjandra N. 2000. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654. (doi:10.1016/S0092-8674(00)00167-7) [DOI] [PubMed] [Google Scholar]
- 40.Villunger A, et al. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302, 1036–1038. (doi:10.1126/science.1090072) [DOI] [PubMed] [Google Scholar]
- 41.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. 1996. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619–628. (doi:10.1016/S0092-8674(00)81382-3) [DOI] [PubMed] [Google Scholar]
- 42.Li H, Zhu H, Xu CJ, Yuan J. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501. (doi:10.1016/S0092-8674(00)81590-1) [DOI] [PubMed] [Google Scholar]
- 43.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. 1997. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139, 1281–1292. (doi:10.1083/jcb.139.5.1281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ. 2011. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116. (doi:10.1016/j.cell.2011.02.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schellenberg B, et al. 2013. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 49, 959–971. (doi:10.1016/j.molcel.2012.12.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todt F, et al. 2015. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 34, 67–80. (doi:10.15252/embj.201488806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. 2010. The BCL-2 family reunion. Mol. Cell 37, 299–310. (doi:10.1016/j.molcel.2010.01.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. (doi:10.1016/S1535-6108(02)00127-7) [DOI] [PubMed] [Google Scholar]
- 49.Andreu-Fernandez V, et al. 2017. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc. Natl Acad. Sci. USA 114, 310–315. (doi:10.1073/pnas.1612322114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Neill KL, Huang K, Zhang J, Chen Y, Luo X. 2016. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 30, 973–988. (doi:10.1101/gad.276725.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nechushtan A, Smith CL, Hsu YT, Youle RJ. 1999. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18, 2330–2341. (doi:10.1093/emboj/18.9.2330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144, 903–914. (doi:10.1083/jcb.144.5.903) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. 2008. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 30, 369–380. (doi:10.1016/j.molcel.2008.04.005) [DOI] [PubMed] [Google Scholar]
- 54.Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM. 2012. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19, 661–670. (doi:10.1038/cdd.2011.138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, Garcia-Saez AJ. 2016. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 35, 389–401. (doi:10.15252/embj.201593384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S. 2016. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 35, 402–413. (doi:10.15252/embj.201592789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng Y, Lin Y, Wu X. 2002. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 16, 33–45. (doi:10.1101/gad.949602) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kandasamy K, Srinivasula SM, Alnemri ES, Thompson CB, Korsmeyer SJ, Bryant JL Srivastava RK.. 2003. Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res. 63, 1712–1721. [PubMed] [Google Scholar]
- 59.Lindsten T, et al. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399. (doi:10.1016/S1097-2765(00)00136-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei MC, et al. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. (doi:10.1126/science.1059108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ. 1997. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl Acad. Sci. USA 94, 12 401–12 406. (doi:10.1073/pnas.94.23.12401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, Kaufmann T. 2013. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 20, 785–799. (doi:10.1038/cdd.2013.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Llambi F, et al. 2016. BOK Is a Non-canonical BCL-2 family effector of apoptosis regulated by er-associated degradation. Cell 165, 421–433. (doi:10.1016/j.cell.2016.02.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, Kaufmann T. 2012. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 19, 915–925. (doi:10.1038/cdd.2011.210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG. 2015. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc. Natl Acad. Sci. USA 112, 7201–7206. (doi:10.1073/pnas.1421063112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez-Marrero Y, Ke F, Echeverry N, Bouillet P, Bachmann D, Strasser A, Kaufmann T. 2016. Is BOK required for apoptosis induced by endoplasmic reticulum stress? Proc. Natl Acad. Sci. USA 113, E492–E493. (doi:10.1073/pnas.1516347113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Einsele-Scholz S, et al. 2016. Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J. Cell Sci. 129, 2213–2223. (doi:10.1242/jcs.181727) [DOI] [PubMed] [Google Scholar]
- 68.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535. (doi:10.1016/j.molcel.2005.02.003) [DOI] [PubMed] [Google Scholar]
- 69.Chen L, et al. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403. (doi:10.1016/j.molcel.2004.12.030) [DOI] [PubMed] [Google Scholar]
- 70.Sarosiek KA, et al. 2013. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 51, 751–765. (doi:10.1016/j.molcel.2013.08.048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopez J, et al. 2016. Mito-priming as a method to engineer Bcl-2 addiction. Nat. Commun. 7, 10538 (doi:10.1038/ncomms10538) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DCS. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305. (doi:10.1101/gad.1304105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weiss LM, Warnke RA, Sklar J, Cleary ML. 1987. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl. J. Med. 317, 1185–1189. (doi:10.1056/NEJM198711053171904) [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Hernandez AM, Shibata D, Cortopassi GA. 1994. BCL2 translocation frequency rises with age in humans. Proc. Natl Acad. Sci. USA 91, 8910–8914. (doi:10.1073/pnas.91.19.8910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strasser A, Harris AW, Cory S. 1993. E mu-bcl-2 transgene facilitates spontaneous transformation of early pre-B and immunoglobulin-secreting cells but not T cells. Oncogene 8, 1–9. [PubMed] [Google Scholar]
- 76.Beroukhim R, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. (doi:10.1038/nature08822) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cerami E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. (doi:10.1158/2159-8290.CD-12-0095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ley TJ, et al. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl. J. Med. 368, 2059–2074. (doi:10.1056/NEJMoa1301689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brennan CW, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155, 462–477. (doi:10.1016/j.cell.2013.09.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.The Cancer Genome Atlas Research Network. 2013. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. (doi:10.1038/nature12222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Agrawal N, et al. 2014. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690. (doi:10.1016/j.cell.2014.09.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kandoth C, et al. 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. (doi:10.1038/nature12113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strasser A, Harris AW, Bath ML, Cory S. 1990. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 331–333. (doi:10.1038/348331a0) [DOI] [PubMed] [Google Scholar]
- 84.Fanidi A, Harrington EA, Evan GI. 1992. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature 359, 554–556. (doi:10.1038/359554a0) [DOI] [PubMed] [Google Scholar]
- 85.Bissonnette RP, Echeverri F, Mahboubi A, Green DR. 1992. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature 359, 552–554. (doi:10.1038/359552a0) [DOI] [PubMed] [Google Scholar]
- 86.Swanson PJ, et al. 2004. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J. Immunol. 172, 6684–6691. (doi:10.4049/jimmunol.172.11.6684) [DOI] [PubMed] [Google Scholar]
- 87.Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL, Cory S. 2010. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood 116, 3197–3207. (doi:10.1182/blood-2010-04-281071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Allen JC, Talab F, Zuzel M, Lin K, Slupsky JR. 2011. c-Abl regulates Mcl-1 gene expression in chronic lymphocytic leukemia cells. Blood 117, 2414–2422. (doi:10.1182/blood-2010-08-301176) [DOI] [PubMed] [Google Scholar]
- 89.Koss B, Morrison J, Perciavalle RM, Singh H, Rehg JE, Williams RT, Opferman JT. 2013. Requirement for antiapoptotic MCL-1 in the survival of BCR-ABL B-lineage acute lymphoblastic leukemia. Blood 122, 1587–1598. (doi:10.1182/blood-2012-06-440230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wendel HG, et al. 2007. Dissecting eIF4E action in tumorigenesis. Genes Dev. 21, 3232–3237. (doi:10.1101/gad.1604407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wertz IE, et al. 2011. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114. (doi:10.1038/nature09779) [DOI] [PubMed] [Google Scholar]
- 92.Gasca J, Flores ML, Giraldez S, Ruiz-Borrego M, Tortolero M, Romero F, Japón MA, Sáez C. 2016. Loss of FBXW7 and accumulation of MCL1 and PLK1 promote paclitaxel resistance in breast cancer. Oncotarget 7, 52 751–52 765. (doi:10.18632/oncotarget.10481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Merino D, et al. 2012. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 119, 5807–5816. (doi:10.1182/blood-2011-12-400929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. (doi:10.1016/j.ccr.2006.03.027) [DOI] [PubMed] [Google Scholar]
- 95.Chonghaile TN, et al. 2011. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334, 1129–1133. (doi:10.1126/science.1206727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Campbell KJ, et al. 2018. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 9, 19 (doi:10.1038/s41419-017-0035-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Balko JM, et al. 2014. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 4, 232–245. (doi:10.1158/2159-8290.CD-13-0286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lipponen P, Pietilainen T, Kosma VM, Aaltomaa S, Eskelinen M, Syrjanen K. 1995. Apoptosis suppressing protein bcl-2 is expressed in well-differentiated breast carcinomas with favourable prognosis. J. Pathol. 177, 49–55. (doi:10.1002/path.1711770109) [DOI] [PubMed] [Google Scholar]
- 99.Silvestrini R, Veneroni S, Daidone MG, Benini E, Boracchi P, Mezzetti M, Di Fronzo G, Rilke F, Veronesi U. 1994. The Bcl-2 protein: a prognostic indicator strongly related to p53 protein in lymph node-negative breast cancer patients. J. Natl. Cancer Inst. 86, 499–504. (doi:10.1093/jnci/86.7.499) [DOI] [PubMed] [Google Scholar]
- 100.Callagy GM, Pharoah PD, Pinder SE, Hsu FD, Nielsen TO, Ragaz J, Ellis IO, Huntsman D, Caldas C. 2006. Bcl-2 is a prognostic marker in breast cancer independently of the Nottingham Prognostic Index. Clin. Cancer Res. 12, 2468–2475. (doi:10.1158/1078-0432.CCR-05-2719) [DOI] [PubMed] [Google Scholar]
- 101.Dawson SJ, et al. 2010. BCL2 in breast cancer: a favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br. J. Cancer 103, 668–675. (doi:10.1038/sj.bjc.6605736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ranger AM, et al. 2003. Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl Acad. Sci. USA 100, 9324–9329. (doi:10.1073/pnas.1533446100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Labi V, Erlacher M, Kiessling S, Villunger A. 2006. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 13, 1325–1338. (doi:10.1038/sj.cdd.4401940) [DOI] [PubMed] [Google Scholar]
- 104.Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, Tzankov A, Villunger A. 2014. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood 123, 2652–2662. (doi:10.1182/blood-2013-11-537217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Egle A, Harris AW, Bouillet P, Cory S. 2004. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl Acad. Sci. USA 101, 6164–6169. (doi:10.1073/pnas.0401471101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garrison SP, et al. 2008. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol. Cell. Biol. 28, 5391–5402. (doi:10.1128/MCB.00907-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarosiek KA, et al. 2017. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 31, 142–156. (doi:10.1016/j.ccell.2016.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reichenbach F, et al. 2017. Mitochondrial BAX Determines the Predisposition to Apoptosis in Human AML. Clin. Cancer Res. 23, 4805–4816. (doi:10.1158/1078-0432.CCR-16-1941) [DOI] [PubMed] [Google Scholar]
- 109.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. 1995. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270, 96–99. (doi:10.1126/science.270.5233.96) [DOI] [PubMed] [Google Scholar]
- 110.Mason KD, et al. 2013. Proapoptotic Bak and Bax guard against fatal systemic and organ-specific autoimmune disease. Proc. Natl Acad. Sci. USA 110, 2599–2604. (doi:10.1073/pnas.1215097110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moravcikova E, Krepela E, Donnenberg VS, Donnenberg AD, Benkova K, Rabachini T, Fernandez-Marrero Y, Bachmann D, Kaufmann T. 2017. BOK displays cell death-independent tumor suppressor activity in non-small-cell lung carcinoma. Int. J. Cancer 141, 2050–2061. (doi:10.1002/ijc.30906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Katz SG, Fisher JK, Correll M, Bronson RT, Ligon KL, Walensky LD. 2013. Brain and testicular tumors in mice with progenitor cells lacking BAX and BAK. Oncogene 32, 4078–4085. (doi:10.1038/onc.2012.421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ke F, Grabow S, Kelly GL, Lin A, O'Reilly LA, Strasser A. 2015. Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis. 6, e1938 (doi:10.1038/cddis.2015.304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rabachini T, et al. 2017. BOK promotes chemical-induced hepatocarcinogenesis in mice. Cell Death Differ. 25, 706–718. (doi:10.1038/s41418-017-0008-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tse C, et al. 2008. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428. (doi:10.1158/0008-5472.CAN-07-5836) [DOI] [PubMed] [Google Scholar]
- 116.Mason KD, et al. 2007. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186. (doi:10.1016/j.cell.2007.01.037) [DOI] [PubMed] [Google Scholar]
- 117.Wilson WH, et al. 2010. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159. (doi:10.1016/S1470-2045(10)70261-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oakes SR, et al. 2012. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl Acad. Sci. USA 109, 2766–2771. (doi:10.1073/pnas.1104778108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen J, et al. 2011. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 10, 2340–2349. (doi:10.1158/1535-7163.MCT-11-0415) [DOI] [PubMed] [Google Scholar]
- 120.Roberts AW, et al. 2016. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl. J. Med. 374, 311–322. (doi:10.1056/NEJMoa1513257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seymour JF, et al. 2017. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 18, 230–240. (doi:10.1016/S1470-2045(17)30012-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Davids MS, et al. 2017. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J. Clin. Oncol. 35, 826–833. (doi:10.1200/JCO.2016.70.4320) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Konopleva M, et al. 2016. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 6, 1106–1117. (doi:10.1158/2159-8290.CD-16-0313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kumar S, et al. 2017. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 130, 2401–2409. (doi:10.1182/blood-2017-06-788786) [DOI] [PubMed] [Google Scholar]
- 125.Moreau P, et al. 2017. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 130, 2392–2400. (doi:10.1182/blood-2017-06-788323) [DOI] [PubMed] [Google Scholar]
- 126.Lochmann TL, et al. 2017. Venetoclax is effective in small cell lung cancers with high BCL-2 expression. Clin. Cancer Res. 24, 360–369. (doi:10.1158/1078-0432.CCR-17-1606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vaillant F, et al. 2013. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 24, 120–129. (doi:10.1016/j.ccr.2013.06.002) [DOI] [PubMed] [Google Scholar]
- 128.Konopleva M, et al. 2006. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388. (doi:10.1016/j.ccr.2006.10.006) [DOI] [PubMed] [Google Scholar]
- 129.van Delft MF, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399. (doi:10.1016/j.ccr.2006.08.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Williams MM, et al. 2017. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol. Cancer Res. 15, 259–268. (doi:10.1158/1541-7786.MCR-16-0280-T) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, Reed JC. 1998. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 91, 991–1000. [PubMed] [Google Scholar]
- 132.Kitada S, et al. 1998. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood 91, 3379–3389. [PubMed] [Google Scholar]
- 133.Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. 2013. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 20, 1475–1484. (doi:10.1038/cdd.2013.79) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leverson JD, et al. 2015. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 7, 279ra40 (doi:10.1126/scitranslmed.aaa4642) [DOI] [PubMed] [Google Scholar]
- 135.Leverson JD, et al. 2015. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 6, e1590 (doi:10.1038/cddis.2014.561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abulwerdi F, et al. 2014. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 13, 565–575. (doi:10.1158/1535-7163.MCT-12-0767) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Merino D, et al. 2017. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 9, eaam7049 (doi:10.1126/scitranslmed.aam7049) [DOI] [PubMed] [Google Scholar]
- 138.MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. 2005. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 65, 5399–5407. (doi:10.1158/0008-5472.CAN-05-0233) [DOI] [PubMed] [Google Scholar]
- 139.Preuss E, Hugle M, Reimann R, Schlecht M, Fulda S. 2013. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J. Biol. Chem. 288, 35 287–35 296. (doi:10.1074/jbc.M113.495986) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Leshchiner ES, Braun CR, Bird GH, Walensky LD. 2013. Direct activation of full-length proapoptotic BAK. Proc. Natl Acad. Sci. USA 110, E986–E995. (doi:10.1073/pnas.1214313110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pritz JR, et al. 2017. Allosteric sensitization of proapoptotic BAX. Nat. Chem. Biol. 13, 961–967. (doi:10.1038/nchembio.2433) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Reyna DE, et al. 2017. Direct Activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 32, 490–505e10. (doi:10.1016/j.ccell.2017.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kelly PN, Grabow S, Delbridge AR, Adams JM, Strasser A. 2013. Prophylactic treatment with the BH3 mimetic ABT-737 impedes Myc-driven lymphomagenesis in mice. Cell Death Differ. 20, 57–63. (doi:10.1038/cdd.2012.92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grabow S, Waring P, Happo L, Cook M, Mason KD, Kelly PN, Strasser A. 2012. Pharmacological blockade of Bcl-2, Bcl-x(L) and Bcl-w by the BH3 mimetic ABT-737 has only minor impact on tumour development in p53-deficient mice. Cell Death Differ. 19, 623–632. (doi:10.1038/cdd.2011.133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weber A, et al. 2010. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 51, 1226–1236. (doi:10.1002/hep.23479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, Strasser A. 2010. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 24, 1608–1613. (doi:10.1101/gad.1940110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. 2010. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 24, 1602–1607. (doi:10.1101/gad.1940210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ichim G, et al. 2015. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872. (doi:10.1016/j.molcel.2015.01.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.