

Abstract
The briarane diterpenoids are a large family of marine natural products that have been primarily isolated from gorgonian octocorals around the world. Structurally, the family is characterized by a trans-fused bicyclo[8.4.0]tetradecane ring system containing a central, stereogenic, all-carbon quaternary carbon (C1) flanked by three additional stereocentres (C2, C10, C14). Many family members have demonstrated biological activity in numerous areas, including: cytotoxicity, anti-inflammatory, antiviral, antifungal, immunomodulatory and insect control. Despite their interesting structural properties and bioactivity, the briaranes have been largely overlooked by the synthetic community. However, in recent years, several research groups have reported progress toward developing a synthetic route to these natural products. Most of these efforts have focused on the stereoselective construction of the central C1–C2–C10–C14 stereotetrad. This review will discuss the various synthetic efforts aimed at the briarane diterpenoids along with the challenges that remain.
Keywords: natural products, diterpenoids, total synthesis, stereoselectivity, conformational analysis
1. Introduction and overview of the briarane diterpenoids
Marine gorgonians represent a diverse set of octocoral organisms that dominate coral reefs throughout the world. The gorgonians can be further divided into three orders: Scleraxonia, Holaxonia and Calaxonia. Owing to the corals' natural abundance and wide geographical distribution, the gorgonians have been a fruitful area of study for natural products chemists [1], and the chemical makeup of a particular gorgonian sample has often served as a taxonomic marker for gorgonian classification [2]. In this regard, diterpenoids represent one of the more important classes of natural products that have been isolated from gorgonian sources. In 2009, Berrue & Kerr discussed the isolation of 40 different diterpenoid skeletal structures from gorgonians [1]. Figure 1 shows the nine most common skeletal structures.
Figure 1.

The nine most common diterpenoid skeletons isolated from the gorgonian family.
Briarane-type diterpenoids represent one of the more common natural product families to be isolated from gorgonian octocorals. The isolation of the first family member (briarein A) was reported in 1977 [3]. Since that time, over 700 unique briarane diterpenoids have been characterized [4–9]. Briarane-type diterpenoids have been isolated from both the Scleraxonia and Calaxonia suborders, but not the Holaxonia suborder. The family Briareidae (suborder Scleraxonia) is by far the most plentiful source of briarane diterpenoid compounds. In addition to gorgonian sources, several examples have also been reported from non-gorgonian sources such as the sea pansy Renilla reniformis [10], the sea pen octocoral Scytalium tentaculatum [11] and the Mediterranean nudibranch mollusk Armina maculata [12]. It has been hypothesized [4–9] that the briaranes are biosynthetically derived from the cembrane skeleton, another commonly found skeletal structure in marine gorgonian species, through cyclization between C3 and C8 followed by further oxidative processing (figure 2). However, no biosynthetic studies have been published.
Figure 2.

Proposed biosynthesis of the briarane skeleton.
In their final form (1), briarane diterpenoids are characterized by a trans-fused bicyclo[8.4.0]tetradecane ring system. Most members also possess a γ-lactone spanning C7 and C8. Another common feature is a congested set of four, contiguous stereogenic centres comprising C1–C2–C10–C14. Oxidative processing by the organism can install oxygenation at nearly every carbon in the system, as evidenced by the representative family members shown in figure 3 [4–9].
Figure 3.

Representative members of the briarane family.
Given the size and structural diversity associated with the briarane family, it is not surprising that many family members have demonstrated biological activity in a number of areas including: cytotoxicity, anti-inflammatory, antiviral, antifungal, immunomodulatory, insect control, antifouling and ichthyotoxicity [4–9,13]. Despite the wide structural diversity and interesting biological activity associated with the briaranes, relatively little effort has been expended in devising synthetic routes to access these natural products. Only a few groups have reported synthetic progress towards any member of the briaranes. To date, no completed total synthesis has been reported; a fact that illustrates the significant challenge posed by these targets. These efforts will be the subject of this mini-review. Briarane numbering of carbon atoms will be used throughout this review.
2. Early synthetic approaches
This section will discuss early reports that were focused on constructing various small or highly simplified fragments, including the 10-membered ring and different stereogenic elements.
2.1. Procter's synthesis of briarane 10-membered ring of solenolide F
In 1995, Procter and co-workers reported the first synthetic work related to the briaranes (figure 4) [14]. Their efforts focused specifically on devising an efficient route to the bicyclo[8.4.0]tetradecane ring system. To accomplish this, they borrowed chemistry from Leyendecker and Comte, who found that vinyl sulfoxides could serve as an effective Michael acceptor for enolates [15]. Procter's work began with the Cu-catalysed conjugate addition of Grignard reagent 2 into enone 3. The resulting enolate was trapped with Me3SiCl to afford silyl enol ether 4. Addition of methyl lithium to compound 4 generated the corresponding lithium enolate, which engaged sulfoxide 5 as planned. After elimination, ketone 6 could be obtained in good overall yield, but as a 1 : 1 mixture of diastereomers at C1 and C10. Reduction of the ketone and ester groups, and hydrolysis of the ketal protecting group, provided diol 7. Protection of the primary alcohol as the tert-butyldimethylsilyl (TBS) ether allowed the two diastereomers (8a and 8b) to be separated chromatographically. The relative configuration of 8a and 8b was assigned using coupling-constant analysis and a series of nuclear Overhauser effect (NOE) experiments. The authors hypothesized that 8a would more readily undergo the required cyclization due to the trans-diaxial conformation of 8b, which would position the two sidechains far from each other (figure 4, box). Consequently, only diastereomer 8a was advanced through the synthetic sequence. This proved to be an important early insight into the conformational constraints surrounding the macrocyclization (see §4 for a similar analysis).
Figure 4.

Procter's approach to the bicyclo[8.4.0]tetradecane ring system.
After protection of the secondary alcohol in 8a as an ether, a Takai olefination was used to convert the ketone into the vinyl iodide needed for ring closure. Unfortunately, iodide 9 formed as an inseparable 1 : 1 mixture of E and Z isomers. Subsequent removal of the TBS ether and oxidation to the aldehyde gave cyclization precursor 10. After optimization, the authors found that cyclized product 11 could be obtained in low yield (1 : 1 dr at C7) by using a massive excess (200 equivalents) of CrCl2 in dimethylsulfoxide. The desired product was accompanied by ‘substantial amounts’ of a compound (12) in which the iodide had been replaced by a proton. It was hypothesized that the E-vinyl iodide gives rise to this material, as it is unable to cyclize. Although Procter and co-workers were able to successfully form the 10-membered ring, the poor diastereoselectivity at C1, C6 and C10, as well as the use of extremely large amounts of toxic, expensive CrCl2, made this route unsuitable for further optimization. No further studies by the Procter laboratory have been reported.
2.2. Nantz's synthesis of solenolide F fragments
In 1997, Nantz and co-workers reported synthetic work inspired by solenolide F [16]. As outlined in figure 5, they planned to initially construct the 10-membered ring and use compound 13 as an intermediate onto which the six-membered ring would be appended. They envisioned compound 13 arising from a McMurray coupling of dialdehyde 14. Recognizing that the two fully substituted carbon atoms in 13 (C1 and C8) both have adjacent oxygenated stereogenic carbon atoms (C2 and C7), they hypothesized that the skeleton could be broken into three fragments. Two of these fragments (15 and 16) contain all of the required stereochemical information. The third was an unidentified, four carbon fragment that could function as a nucleophile on both ends (referred to by the authors as a ‘propyne equivalent’). Interestingly, the authors proposed that fragments 15 and 16 could both be accessed through regioselective ring opening of an epoxide with a methyl anion equivalent. Furthermore, they believed that the epoxides needed to form 15 and 16 could both be derived from a compound similar to diol 17.
Figure 5.

Nantz's retrosynthetic analysis of solenolide F (DTS = dimethylthexylsilyl).
The authors began by synthesizing the diol precursor to both 15 and 16 (figure 6). Commercially available 1,3-dihydroxyacetone (18) was acetylated, and the resulting bis-acetate converted to triester 19 by means of a Horner–Emmons olefination. Cleavage of the acetates under acidic conditions led to spontaneous lactonization. The remaining primary alcohol was then benzylated to give 20 in good yield. Reduction of the lactone provided a diol that was subjected to mono-silylation with dimethylthexylsilyl chloride (DTS-Cl). After a survey of protecting groups, the authors found that the DTS group led to mono-silylated products 21 and 22 in approximately equal amounts. This would prove ideal, as alcohols 21 and 22 are useful for the preparation of different fragments [16].
Figure 6.

Nantz's synthetic route to briarane building blocks.
Alcohol 21 was subjected to a Sharpless asymmetric epoxidation to give epoxide 23 in good yield and enantiomeric purity. Swern oxidation of the primary alcohol and Wittig olefination gave vinyl epoxide 24, after removal of the DTS group. Based on results obtained in a series of model reactions, ring-opening of epoxide 24 was accomplished with trimethylaluminium. The authors were pleased to discover that desired diol 25 was the major product. They proposed that the benzyl protecting group in 24 minimized SN2′ addition, while the adjacent π-system served to minimize migration of the neighbouring groups. Finally, activation of the primary alcohol in 25, followed by treatment with KOH, formed fragment 15 [16].
A Sharpless asymmetric epoxidation was also performed on alkene 22, to give epoxide 26. A methyl group was added to the less hindered carbon of the epoxide using Me2CuLi. The resulting diol was then protected as acetonide 27. Finally, a Birch reduction removed the benzyl protecting group and a Swern oxidation produced aldehyde fragment 16 in good yield [16]. Ultimately, the Nantz group was able to construct both of the planned building blocks. Unfortunately, neither the union of fragments 15 and 16 nor the subsequent closure of the 10-membered ring have been reported.
3. Approaches to the central stereotetrad
In more recent years, synthetic efforts have been focused on constructing what is arguably the most challenging portion of the briaranes—the C1–C2–C10–C14 stereotetrad. Not only does this fragment include a stereogenic, all-carbon quaternary carbon; but it is flanked by three additional stereocentres. Correctly setting the relative configuration of these four contiguous stereocentres represents a major synthetic challenge, and attempting to ‘correct’ the configuration of any one of these will require additional synthetic steps to what will likely be an already lengthy synthesis. Nevertheless, as this structural motif is present in a majority of briarane family members, the rapid construction of the stereotetrad represents an important strategic opportunity. One could imagine that a well-designed fragment containing the complete stereotetrad might be able to serve as a building block for several different family members. This idea still has yet to be realized, though several groups have reported significant progress in this area.
3.1. Ito and Iguchi's synthesis of the briarane stereotetrad
In 2006, the Ito/Iguchi laboratory reported the first synthetic approach to the briarane stereotetrad [17]. This effort was driven by a desire to synthesize pachuclavulide B, an anticancer briarane family member isolated by the same researchers [18]. More specifically, they hypothesized that compound 38 (figure 7) could serve as a key intermediate, upon which the 10-membered ring could be appended. In examining this potential substructure, the authors identified latent symmetry within the molecule, which they hoped to exploit. To that end, they postulated that 38 could be derived from epoxide 31, which would come from 2,6-methylbenzoic acid (28).
Figure 7.

Ito and Iguchi's synthetic route to a potential briarane building block.
The authors began their synthesis by performing a Birch reduction of 2,6-dimethylbenzoic acid, followed by conversion to methyl ester 29. After some optimization, they found that the desymmetrization of diene 29 was best carried out using a stoichiometric amount of d-sorbose-derived catalyst [19] 30. Under these conditions, epoxide 31 could be obtained in 44% yield with 81% de and 87% ee. Ester 31 was then reduced with DIBAL-H and the resulting alcohol protected to give silyl ether 32. Recrystallization at this stage improved the enantiopurity to 99% ee. Epoxide 32 was then converted to iodo acetal 33 over two steps. A radical cyclization was then used to construct the all-carbon quaternary carbon at C1. The formation of the C1 stereocentre was, presumably, dictated by the configuration at C14. Unfortunately, the desired reaction pathway (5-exo-trig) was accompanied by significant amounts of iodine-reduction and 6-endo-trig cyclization. Nevertheless, the authors elected to carry the material forward [17].
Having formed three of the four stereocentres, the authors turned their attention to generating the aldehyde needed to form the C2 stereocentre. In order to accomplish this transformation, one carbon would need to be excised. To this end, a Jones oxidation was used to convert the radical cyclization product into lactone 34. The direct, α-hydroxylation of lactone 34 proved to be quite difficult and the authors speculated that the bulky tert-butyldiphenylsilyl (TBDPS) group was the culprit. Based on this hypothesis, the silyl ether was removed. Subsequent hydroxylation of the lactone enolate with Davis' oxaziridine (35) proceeded smoothly and provided lactone 36 after reinstallation of the TBDPS ether. Reduction of lactone 36 with LiAlH4 and oxidative cleavage of the diol afforded aldehyde 37. Finally, addition of vinylmagnesium chloride led to the isolation of diol 38 in quantitative yield as a single diastereomer. The authors confirmed the stereochemistry through NOE correlations of the corresponding acetonide (not shown). The authors proposed a chelation model (39) in order to rationalize the high level of stereocontrol. Coordination with the magnesium alkoxide of the C14 hydroxyl group would lock the aldehyde in a conformation wherein one face is effectively blocked by the TBDBS group. This leads to selective addition of the Grignard reagent from the Si face [17]. Through this route, the authors were able to access the stereotetrad core of the briarane diterpenoids, in enantioenriched form, in 18 steps from 2,6-methylbenzoic acid. Also notable is their incorporation of a double bond between C11 and C12. Many briarane family members contain a double bond at this position, or have functionality (hydroxyl groups, epoxides) that could arise from an alkene in this position. No further synthetic efforts from this group have been reported.
3.2. Bates’ synthesis of the briarane stereotetrad
As in the previous report by Ito and Iguchi, Bates and co-workers identified that a subunit containing an intact stereotetrad could serve as a common building block for numerous family members. Their approach hinged on setting the stereocentre at C1 through a sigmatropic rearrangement, and the use of a Diels–Alder reaction to form the six-membered ring [20]. The authors began by synthesizing a diene that would not only react with the planned crotonate dienophile, but also provide for facile installation of the allylic alcohol needed for the planned sigmatropic rearrangement. To this end, sodium p-toluenesulfinate was alkylated with crotyl chloride (40) under phase-transfer conditions (figure 8). The resulting sulfone was then converted into β-hydroxysulfone 41. Elimination of the alcohol was effected using MsCl and NEt3. The resulting diene (42) was prone to spontaneous dimerization when kept in its pure form, but could be stored at –78°C as a solution in CH2Cl2. A Lewis acid-mediated Diels–Alder reaction between diene 42 and methyl crotonate was then used to access cyclohexene 43 [20]. This reaction is notable for being an uncommon example of a [4 + 2] cycloaddition involving reaction partners that are both electron-deficient [21]. An additional complicating factor is the reluctance of crotonates to participate as dienophiles. Consequently, extended reaction times (7 days) at elevated temperatures were necessary to achieve high conversions. Nevertheless, the reaction proceeded smoothly and Diels–Alder adduct 43 was obtained as a single diastereomer.
Figure 8.

Construction of the six-membered ring using a Diels–Alder reaction.
The authors found removal of the sulfone to be quite challenging. Numerous oxidative methods were evaluated with little success. Finally, it was discovered that the double bond in alcohol 44 (prepared by reduction of ester 43) could be isomerized by heating under basic conditions (figure 9). After TBS protection of the alcohol, the sulfone was then oxidatively removed with lithium diisopropylamide and MoOPD (MoO5•Py•DMPU) [22] to give enone 45 along with a small amount of over-oxidized products. Luche reduction of the enone provided equatorial alcohol 46. The allylic alcohol proved to be quite recalcitrant to the planned sigmatropic rearrangement. Eventually, the authors found an Eschenmoser–Claisen rearrangement to be effective. Thus, treating allylic alcohol 46 with dimethylacetamide dimethylacetal in xylene under microwave irradiation gave desired product 47 in moderate, but acceptable yield [20]. The difficulties experienced during the various attempts at using a sigmatropic rearrangement to generate the C1 stereocentre can be attributed to unfavourable conformational restraints in ketene aminal 48, the reactive species in the rearrangement. Two half-chair conformers are especially germane to our discussion. Conformer 48a is likely the most favoured as all groups can be placed in equatorial positions. However, in this conformation, the ketene aminal moiety is not appropriately positioned to engage the alkene for the rearrangement. By contrast, conformer 48b places the ketene aminal in an appropriate position (axial) to react with the alkene, but all of the other groups will also be in an axial position, making conformer 48b higher in energy than 48a.
Figure 9.

Bates’ synthesis of a potential briarane building block.
With the C1 and C10 stereocentres established with the correct relative configuration, the authors' attention turned to installing the C2 and C14 stereocentres. To this end, an iodolactonization was performed on amide 47, establishing the C14 stereocentre. Elimination of the resulting iodide with 1,8-diazabicyclo[5.4.0]undec-7-ene provided alkene 49, the assignment of which was confirmed by X-ray crystallography. α-Oxygenation of lactone 49 with lithium bis(trimethylsilyl)amide (LiHMDS) and MoOPD then gave alcohol 50. Unfortunately, the C2 stereocentre in compound 50 was formed with the configuration opposite to what is required by the natural products. Consequently, a two-step sequence was needed to invert the configuration. This was accomplished through oxidation of alcohol 50 with 2-iodoxybenzoic acid and reduction of the ketone with NaBH4. With the C2 stereocentre in place, lactone 51 was reduced with LiAlH4 to provide triol 52. The primary alcohol in 52 could be selectively converted to an iodide (53) by treating with I2 and PPh3. The remainder of the side-chain was installed through a Keck allylation to give briarane building block 54 in 16 steps [20]. No further progress has been reported by the Bates laboratory. Compared to compound 38 synthesized by Ito and Iguchi, the building block constructed by Bates contains a C2–C5 fragment that is well positioned for eventual ring closure; presumably through a ring-closing metathesis (RCM) reaction. However, the placement of the alkene in the six-membered ring of 54 is, arguably, not as ideal as that seen with compound 38.
3.3. Harned's synthesis of the briarane stereotetrad
In 2015, Moon & Harned reported a very rapid entry into the briarane stereotetrad [23]. Their approach was predicated on the densely functionalized nature of, and the predictable reactivity patterns afforded by, 2,5-cyclohexadienones. Furthermore, in contrast to the work of Ito/Iguchi and Bates, the reaction Harned and Moon used to generate the C1 stereocentre was not directed by an existing C14 stereocentre.
Oxidation of methyl salicylate 55 with the indicated μ-oxobridged diiodide provided cyclohexadienone 56 in good yield (figure 10). Subsequent conjugate addition of an aluminium acetylide into the more electron-deficient alkene generated β-ketoester 57. In order to avoid potential rearomatization reactions, intermediate 57 was immediately alkylated with methyl iodide. The moderate overall yield of the conjugate addition/alkylation sequence was offset by the significant increase in molecular complexity the sequence provided. Compound 58 was isolated as a single diastereomer containing the correct relative configuration at C1 and C10 [23]. Molecular modelling calculations were performed to better understand the factors influencing the stereochemical outcome of the alkylation reaction [24]. To a first approximation, the stereoselectivity is consistent with Houk's torsional steering model [25]. In this case, TS1 is responsible for forming the observed stereoisomer and is favoured as it leads to a product with a staggered conformation. In contrast, diastereomeric transition state TS2 leads to an eclipsed conformation, and was found to be higher in energy.
Figure 10.

Harned's formation of the C1 and C10 stereocentres.
Having formed the C1 and C10 stereocentres, the authors' attention turned to the stereoselective reduction of the C14 ketone (figure 11). First, enone 58 was treated with metallic zinc in order to effect the reductive cleavage of the methoxy group at C11. Many conditions were screened for their ability to convert ketone 59 into desired alcohol 60a. Simple reducing agents, such as LiAlH(Ot-Bu)3 and NaBH4, revealed a strong preference for forming the undesired diastereomer (60b). Fortunately, including stoichiometric Y(OTf)3 as a Lewis acidic additive with NaBH4 provided both diastereomeric alcohols in roughly equal amounts [23]. At the time, the strong preference for forming 60b was seen as a nuisance. However, subsequent work by Crimmins (§4) has revealed that 60b may eventually prove useful.
Figure 11.

Harned's completion of the briarane stereotetrad.
With alcohol 60a in hand, the authors then set out to install the final stereocentre at C2. Based on the work of Ito/Iguchi (§3.1), it was anticipated that addition of a suitable organometallic reagent into β-hydroxyaldehyde 61 would lead to the selective formation of desired diastereomer 62a (after acetonide formation). In practice, the addition of allylmagnesium bromide was poorly selective and provided a diastereoselectivity of about 3 : 1 under optimal conditions. The authors identified the strong axial preference of the alkyne as one contributing factor for the poor selectivity. Such a conformational preference might disrupt the preferential chelation that was the basis of the model proposed by Ito/Iguchi [23]. At the same time, the high reactivity displayed by allyl Grignard reagents [26] may also contribute to the diminished stereoselectivity. Although further work is required to address the different stereochemical problems associated with this route, it illustrates that the densely functionalized nature of cyclohexadienones can be leveraged into a very rapid construction of the requisite stereotetrad.
4. Construction of the trans-fused ring system
In 2017, Crimmins et al. reported the most significant progress to date in the quest to develop a synthetic route to the briarane diterpenoids [27]. In this work, they were able to successfully form the bicyclo[8.4.0]tetradecane ring system; however, unlike Procter's work [14], Crimmins was able to access the 6,10 ring system with a complete stereotetrad. Although, the Crimmins work would ultimately fall short of realizing a completed total synthesis, it serves as an important foundation for future work by uncovering important reactivity patterns that will prove useful in future synthetic efforts. By disconnecting at the C1–C10 bond, Crimmins and co-workers took a very different approach to forming the all-carbon quaternary stereocentre.
The authors began with thiazolidenthione 63, which was employed in an asymmetric aldol reaction with aldehyde 64 (figure 12). Aldol product 65 was then converted into ester 66 upon treatment with the indicated alcohol under basic conditions. Based on methodology developed by the Kurth [28] and Crimmins [29] groups, double deprotonation of ester 66 with LiHMDS initiated a dianionic Claisen rearrangement to deliver carboxylic acid 68, which was immediately converted into ester 69 using diazomethane. The rearrangement proceeded with excellent stereocontrol and ester 69 was obtained as a single stereoisomer [27]. The high degree of stereocontrol was rationalized by considering transition states 67a and 67b. Of these two, chair-like transition state 67a was expected to be favoured, as boat-like transition state 67b places the alkenyl methyl group and the R2 group in unfavourable steric environments. Similarly, the R1 group of the chelated enolate forces the rearrangement to occur on only one face of the enolate [28,29].
Figure 12.

Crimmins’ use of a dianionic Ireland–Claisen rearrangement to set the C1 and C10 stereocentres.
With the C1 stereocentre set, Crimmins and co-workers turned their attention to constructing the six-membered ring (figure 13). The secondary alcohol in compound 69 was protected as a TBS ether and the ester was reduced to primary alcohol 70. Swern oxidation and addition of allylmagnesium bromide provided alcohol 71 as a 1 : 1 mixture [26] of diastereomers. Fortunately, alcohols 71a (corresponding to the natural configuration at C14) and 71b could be separated by column chromatography and could be recycled in either direction though an oxidation/reduction sequence. The two alcohols were separately carried though a five-step sequence in order to access cyclohexene 74. First, a RCM was performed by exposing alcohol 71 to the second-generation Grubbs catalyst. Cleavage of the silyl ethers and selective protection of the resulting 1,3-diol gave acetonide 73. Swern oxidation of the primary alcohol and addition of allylmagnesium bromide then provided alcohols 74a (from 71a) and 74b (from 71b) as a mixture of stereoisomers at C8 [27].
Figure 13.

Crimmins’ use of ring-closing metathesis to form the 6,10 ring system of the briaranes.
With acetonides 74a and 74b in hand, the authors were poised to close the 10-membered ring. For this, they planned to carry out another RCM reaction. Considering the limited number of examples of RCM being used to form 10-membered carbocyclic rings [30–32], this would be a difficult task. Curiously, the C8 acetate derived from acetonide 74a failed to undergo ring closure, even when other protecting groups were used instead of the acetonide. However, the C8 acetate derived from acetonide 74b smoothly reacted to give compound 75 in 91% yield. Much like the analysis used by Procter (§2.1), Crimmins and co-workers rationalized the reactivity, or lack thereof, of acetonides 74a and 74b by considering their conformational preferences (figure 14). They thought that substrate 74a-OAc (14S) might adopt one of two conformers, both of which place the ends of the two alkene chains far away from each other. In contrast, substrate 74b-OAc (14R) is able to adopt a conformation in which the two alkene chains are proximal to one another, thereby facilitating ring closure [27].
Figure 14.

Crimmins’ use of ring-closing metathesis to form the 6,10 ring system of the briaranes.
Unfortunately, the configuration at C14 in compound 75 does not correspond to the natural stereocentre. Fortunately, this could be remedied by first replacing the acetonide between C2 and C14 with an acetal between C2 and C3. Oxidation of the C14 hydroxyl group delivered ketone 76. Stereoselective reduction of the ketone restored the C14 stereocentre, now with the correct configuration. Reprotection of the C14 hydroxyl group was followed by cleavage of the C8 acetate and oxidation to ketone 77. Seemingly, the C8 ketone present in compound 77 is well positioned for installation of the lactone present in many briarane family members. However, none of the attempts at generating the butenolide from the corresponding α-hydroxyketone were fruitful (details not provided) [27]. Although this was a setback to Crimmins and co-workers, this observation is of critical importance for future synthetic endeavours as it suggests that the C7–C8 γ-lactone may need to be generated before closing the 10-membered ring.
5. Conclusion
In conclusion, the briarane diterpenoids are an intriguing class of bioactive synthetic targets. Several exploratory routes have been reported with varying degrees of success. Although this work has yet to produce a completed total synthesis, several key observations have been made that will undoubtedly inspire future efforts in this area. For example, Crimmins' successful use of an RCM to close the ten-membered ring, and the associated stereochemical constraints, is a major development that will likely be an important disconnection moving forward. At the same time, the problems experienced by Crimmins and co-workers when trying to install the C7–C8 lactone at a late stage is a major impediment that will need to be addressed in the future.
As mentioned earlier, the C1–C2–C10–C14 stereotetrad arguably represents the biggest synthetic challenge associated with the briarane family. Accessing this motif quickly and with minimal corrections to the relative stereochemistry will be crucial in order to realize an efficient and tractable synthesis. As summarized in figure 15, three different C–C bond disconnections have been used to construct the stereogenic quaternary carbon at C1. Other disconnections likely will be reported in the future, and it will be interesting to see what advantages they offer. It will be interesting to see how future work makes constructive use of the conformational preferences of ring-closure explored by Procter and Crimmins.
Figure 15.
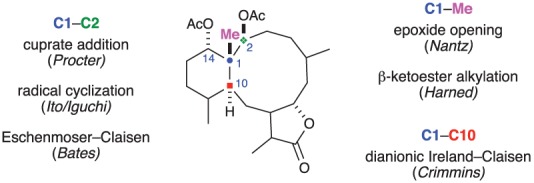
Summary of strategies used to construct the C1 stereocentre.
Data accessibility
This article has no additional data.
Authors' contributions
N.G.M. and A.M.H. collected references and wrote the manuscript. All authors gave final approval for publication.
Competing interests
We declare we have no competing financial interests.
Funding
We thank Texas Tech University and the National Science Foundation (CHE-1541685) for supporting our efforts in this area.
References
- 1.Berrue F, Kerr RG. 2009. Diterpenes from gorgonian corals. Nat. Prod. Rep. 26, 681–710. (doi:10.1039/B821918B) [DOI] [PubMed] [Google Scholar]
- 2.Dorta E, Díaz-Marrero AR, Brito I, Cueto M, D'Croz L, Darias J. 2007. The oxidation profile at C-18 of furanocembranolides may provide a taxonomical marker for several genera of octocorals. Tetrahedron 63, 9057–9062. (doi:10.1016/j.tet.2007.06.080) [Google Scholar]
- 3.Burks JE, Van der Helm D, Chang CY, Ciereszko LS. 1977. The crystal and molecular structure of briarein A, a diterpenoid from the Gorgonian Briareum asbestinum. Acta Cryst. B33, 704–709. (doi:10.1107/S0567740877004518) [Google Scholar]
- 4.Sung PJ, Sheu JH, Xu JP. 2002. Survey of briarane-type diterpenoids of marine origin. Heterocycles 57, 535–579. (doi:10.3987/REV-01-546) [Google Scholar]
- 5.Sung PJ, Chang PC, Fang LS, Sheu JH, Chen WC, Chen YP, Lin MR. 2005. Survey of briarane-related diterpenoids—Part II. Heterocycles 65, 195–204. (doi:10.3987/REV-04-589) [Google Scholar]
- 6.Sung PJ, et al. 2008. Survey of briarane-type diterpenoids—Part III. Heterocycles 75, 2627–2648. (doi:10.3987/REV-08-633) [Google Scholar]
- 7.Sung PJ, et al. 2011. Survey of briarane-type diterpenoids—Part IV. Heterocycles 83, 1241–1258. (doi:10.3987/REV-11-695) [Google Scholar]
- 8.Sheu JH, et al. 2014. Briarane diterpenoids isolated from gorgonian corals between 2011 and 2013. Mar. Drugs 12, 2164–2181. (doi:10.3390/md12042164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su YD, Su JH, Hwang TL, Wen ZH, Sheu JH, Wu YC, Sung PJ. 2017. Briarane diterpenoids isolated from octocorals between 2014 and 2016. Mar. Drugs 15, 44 (doi:10.3390/md15020044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keifer PA, Rinehart KL, Hooper IR. 1986. Renillafoulins, antifouling diterpenes from the sea pansy Renilla reniformis (Octocorallia). J. Org. Chem. 51, 4450–4454. (doi:10.1021/jo00373a020) [Google Scholar]
- 11.Ravi BN, Marwood JF, Wells RJ. 1980. Three new diterpenes from the sea pen Scytalium tentaculatum . Aust. J. Chem. 33, 2307–2316. (doi:10.1071/CH9802307) [Google Scholar]
- 12.Guerriero A, D'Ambrosio M, Pietra F. 1987. Verecynarmin A, a novel briarane diterpenoid isolated from both the mediterranean nudibranch molluse Armina maculata and its prey, the pennatulacean octocoral veretillum cynomorium. Helv. Chim. Acta 70, 984–991. (doi:10.1002/hlca.19870700408) [Google Scholar]
- 13.Liu Y, Lin X, Yang B, Liu J, Zhou X, Peng Y. 2013. Cytotoxic briarane-type diterpenoids. In Marine pharmacognosy (ed. SK Kim.), pp. 53–63. Boca Raton, FL: CRC Press. [Google Scholar]
- 14.Roe MB, Whittaker M, Procter G. 1995. Studies on the synthesis of solenolide F.; a Cr(II)-mediated cyclization to form the ten-membered ring. Tetrahedron Lett. 36, 8103–8106. (doi:10.1016/0040-4039(95)01687-D) [Google Scholar]
- 15.Leyendecker F, Comte MT. 1986. Chimie des fragrances. Partie I: nouvelle strategies de synthese de α-(2-oxocyclohexyl)-acrylates de methyl. Tetrahedron 42, 1413–1421. (doi:10.1016/S0040-4020(01)87361-0) [Google Scholar]
- 16.Balasubramaniam RP, Moss DK, Wyatt JK, Spence JD, Gee A, Nantz MH. 1997. Methylation-ring opening of 3,3-disubstituted 2,3-epoxy alcohols. Synthesis of chiral quaternary fragments for assembly of briaran diterpenes. Tetrahedron 53, 7429–7444. (doi:10.1016/S0040-4020(97)00453-5) [Google Scholar]
- 17.Iwasaki J, Ito H, Nakamura M, Iguchi K. 2006. A synthetic study of briarane-type marine diterpenoid, pachyclavulide B. Tetrahedron Lett. 47, 1483–1486. (doi:10.1016/j.tetlet.2005.12.099) [Google Scholar]
- 18.Iwasaki J, Ito H, Aoyagi M, Sato Y, Iguchi K. 2006. Briarane-type diterpenoids from the okinawan soft coral pachyclavularia violacea. J. Nat. Prod. 69, 2–6. (doi:10.1021/np0580661) [DOI] [PubMed] [Google Scholar]
- 19.Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. 1997. An efficient catalytic asymmetric epoxidation method. J. Am. Chem. Soc. 119, 11 224–11 235. (doi:10.1021/ja972272g) [Google Scholar]
- 20.Bates RW, Pinsa A, Kan X. 2010. Synthesis of the northern hemisphere of the briaranes. Tetrahedron 66, 6340–6348. (doi:10.1016/j.tet.2010.04.003) [Google Scholar]
- 21.Bäckvall JE, Juntunen SK. 1987. 2-(Phenylsulfonyl)-1,3-dienes as versatile synthons in organic transformation. Multicoupling regents and Diels–Alder dienes with a dual electron demand. J. Am. Chem. Soc. 109, 6396–6403. (doi:10.1021/ja00255a027) [Google Scholar]
- 22.Anderson JC, Smith SC. 1990. Oxodiperoxymolybdenum(pyridine)-1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidone (MoO5•Py•DMPU): a safer alternative to MoOPH for the α-hydroxylation of carbonyl compounds. Synlett 1990, 107–108. (doi:10.1055/s-1990-21003) [Google Scholar]
- 23.Moon NG, Harned AM. 2015. A concise synthetic route to the stereotetrad core of the briarane diterpenoids. Org. Lett. 17, 2218–2221. (doi:10.1021/acs.orglett.5b00816) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon NG, Harned AM. 2017. Torsional steering as friend and foe: development of a synthetic route to the briarane diterpenoid stereotetrad. Org. Biomol. Chem. 15, 1876–1888. (doi:10.1039/c7ob00124j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Houk KN. 2014. Torsional control of stereoselectivities in electrophilic additions and cycloadditions to alkenes. Chem. Sci. 5, 462–470. (doi:10.1039/C3SC52538D) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Read JA, Woerpel KA. 2017. Allylmagnesium halides do not react chemoselectively because reaction rates approach the diffusion limit. J. Org. Chem. 82, 2300–2305. (doi:10.1021/acs.joc.7b00053), and references therein. [DOI] [PubMed] [Google Scholar]
- 27.Crimmins MT, Zhang Y, Williams PS. 2017. Approach to the synthesis of briarane diterpenes through a dianionic Claisen rearrangement and ring-closing metathesis. Org. Lett. 19, 3907–3910. (doi:10.1021/acs.orglett.7b01806) [DOI] [PubMed] [Google Scholar]
- 28.Kurth MJ, Yu CM. 1985. Acyclic stereocontrol through the dianionic Claisen rearrangement of β-hydroxy esters: synthesis of (±)-botryodiplodin. J. Org. Chem. 50, 1840–1845. (doi:10.1021/jo00211a010) [Google Scholar]
- 29.Crimmins MT, Knight JD, Williams PS, Zhang Y. 2014. Stereoselective synthesis of quaternary carbons via the dianionic Ireland–Claisen rearrangement. Org. Lett. 16, 2458–2461. (doi:10.1021/ol5008422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larrosa I, et al. 2006. Highly convergent three component benzyne coupling: the total synthesis of ent-clavilactone B. J. Am. Chem. Soc. 128, 14 042–14 043. (doi:10.1021/ja0662671) [DOI] [PubMed] [Google Scholar]
- 31.Nakamura T, Oshida M, Nomura T, Nakazaki A, Kobayashi S. 2007. Synthetic study of diversifolin: the construction of 11-oxabicyclo[6.2.1]undec-3-ene core using ring-closing metathesis. Org. Lett. 9, 5533–5536. (doi:10.1021/ol7025119) [DOI] [PubMed] [Google Scholar]
- 32.Nakamura T, Tsuboi K, Oshida M, Nomura T, Nakazaki A, Kobayashi S. 2009. Total synthesis of (–)-diversifolin. Tetrahedron Lett. 50, 2835–2839. (doi:10.1016/j.tetlet.2009.03.192) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.