

Abstract
A totally synthetic microperoxidase-11 (MP-11) is reported. Accordingly, the undecapeptide (VQKCAQCHTVE) was synthesized by solid-phase peptide synthesis followed by the thiol-ene click reaction with haemin for reconstitution. High-speed atomic force microscopy measurement conducted in water confirmed the protein reconstitution by visualizing the morphological differences as animated molecular images. The synthetic MP-11 showed a considerable magnitude of catalytic activity (27%) against the natural MP-11 in the oxidation of 3,3′,5,5′-tetramethylbenzidine by hydrogen peroxide, whereas it showed very low (2.7%) activity of a synthetic variant with a point mutation (VQKCAQCMTVE, H8M). Slab waveguide spectroscopic measurements revealed that the ferrous/ferric redox reaction occurred by the direct electron transfer with specific spectral changes. Indeed, if hydrogen peroxide existed in the solution phase, the peroxidase-modified electrode showed catalytic current–voltage behaviour regardless of whether it was prepared using natural MP-11 or the synthetic MP-11. If a substrate recycling reaction was assumed, computer simulation well reproduced the experimental curves to give a global set of electrocatalytic reaction parameters. In any of the experiments, the synthetic MP-11 and natural MP-11 gave almost identical results. Our approach will be a convenient means of preparing MP-11, as well as its mutants, that does not rely on nature.
Keywords: microperoxidase, total synthesis, high-speed atomic force microscopy, enzymatic assay, spectroelectrochemistry, electrocatalytic reaction
1. Background
Microperoxidases (MPs) have been establishing themselves as an attractive class of alternatives to haemperoxidases [1]. For example, a naturally occurring undecamer (VQKCAQCHTVE, MP-11) obtained proteolytically from cytochrome c contains a haem group that covalently bonds to the polypeptide chain via two thioester bonds of cysteine residues. Besides, the imidazole side chain of histidine binds to the haem iron to give the corresponding five-coordinate, high-spin complex. Even though the structure is minimal, MP-11 and its homologues show a substantial degree of peroxidase activity. Therefore, they have been widely used as models for haem active sites and for investigations of the oxidoreductase reaction mechanism [2–6]. MPs have further stirred intensive research for their use as oxidation catalysts for various chemical/biochemical purposes including biosensing [7–9], energy conversion [10,11] and biofuel cells [12].
Functional modification of MP-11 is also forming an interesting research field; for the oxidoreduction function not found in the original, demetalation of the Fe(III)-haem followed by complexation with Mn(III) [13] or Co(III) [14] has been examined. MPs with different amino acid sequences could be obtained from Shewanella oneidensis [15] or Marinobacter hydrocarbonoclasticus [16]. In vivo expression technology has produced various MP variants with peptide sequences that do not occur naturally [17]. Recently, Bren et al. extended to a biosynthesis method that does not rely on cytochromes c expression [18]. Working constantly on those naturally originating polypeptides, we can also expect any MP with an unusual function to be available.
While cytochromes occupy an important position as the raw material of MPs, solid-phase peptide synthesis (SPPS) is the primary tool for creating diverse polypeptides. As a result, synthetic methods are also yielding significant results; various peptide–porphyrin conjugates were studied and some of them achieved peroxidase-like activity, i.e. miniaturized metalloenzymes [19,20]. In addition to SPPS, highly substituted porphyrin derivatives contribute greatly by acting as covalently bound, catalytic sites. Currently though, installing of haem c (haemin) into host peptides has not been established.
Here, we report a totally synthetic approach that yields MP-11; SPPS was used to synthesize the undecapeptide as the N-acetylated form, followed by the thiol-ene click reaction with haemin for reconstitution (NAcMP, figure 1). On the one hand, we expanded early research [21] by synthesizing an example of mutants, VQKCAQCMTVE (H8M); mitochondrial cytochromes c contain a c-type haeme with axial His18 and Met80 ligands and therefore, after reconstitution, H8M represents a homologous MP-11 having the opposite axial ligand other than His. On the other hand, we characterized the reconstitution reaction through single-molecule visualization using atomic force microscopy (AFM). Moreover, we collected information on the ferrous-to-ferric oxidoreduction reaction in detail, which becomes important in bioanalysis applications. In any of the experiments, results of the synthetic and the natural MP-11 were almost identical. Therefore, we concluded that our approach is a convenient means of MP-11 synthesis that does not rely on nature.
Figure 1.

(a) The synthetic scheme for the initial SPPS of N-acetylated undecapeptides, followed by the thiol-ene click reaction for reconstitution. (b) A three-dimensional model of a theoretically optimized structure (Gaussian 09/LanL2DZ).
2. Experimental
2.1. Synthesis of MPs-11
Two-types of undecapeptide, VQKCAQCHTVE and VQKCAQCMTVE, were synthesized by Fmoc SPPS in a microwave synthesis system (Discover® SP, CEM Co., Matthews, NC, USA). The N-terminal of the final product was acetylated to improve the solubility and to prevent aggregation after being reconstituted to the holocompound [22]. The undecapeptides were further subjected to the thiol-ene click reaction [23] with haemin. The detailed synthetic procedures, including analytical data, and UV–visible spectra (electronic supplementary material, figures S1–S4) are provided as electronic supplementary material.
2.2. Atomic force microscopy imaging
AFM imaging was carried out using a high-speed atomic force microscope (Research Institute of Biomolecule Metrology (RIBM) Co., Ltd, Tsukuba, Japan). The cantilevers were typically silicon with a tip radius of 10 nm: length 10 µm, spring constant 0.1 N m–1, resonance frequency 1500 kHz. The sample peptides were dissolved in 2,2,2-trifluoroethanol, diluted with aqueous MgCl2 solution (1 mol dm–3) to give 0.5 mg ml–1 solution. Atop freshly cleaved mica was placed a 2 µl portion of the sample solution, left for 10 min at room temperature, and then the surface was rinsed with deionized water. A 100 µl portion of deionized water was placed on the sample surface and the surface topological images were continuously acquired at 10 frames per second. All AFM data thus obtained were processed and analysed by using a copy of Gwyddion [24]. All experiments were made at room temperature, typically 22 ± 2°C.
2.3. UV–visible study of the peroxidase reaction
An aliquot of NAcMP was dissolved in a phosphate buffer solution (0.01 M, pH 7.0). The UV–visible absorption at 406 nm (A406) of the solution was linearly dependent on NAcMP concentration in a concentration range investigated (0–36 µM, r2 = 0.999). Peptide concentrations were spectrophotometrically determined (59 µM, ϵ406 = 60 300 l mol−1 cm−1). A 3 ml portion of 0.143 mM 3,3′,5,5′-tetramethylbenzidine (TMBZ) was placed into a 1 cm quartz cuvette, then a required amount of haempeptide solution was added. The reaction was initiated by adding 1.5 µl of 0.1% H2O2 and the time course of TMBZ oxidation was measured by recording the absorbance at 655 nm (ϵmax 5400 l mol−1 cm−1) [25]. Similarly, NAcMPm (58 µM) was also tested. The solution was gently agitated with a magnetic stirrer during measurement while the temperature was kept at 37°C.
2.4. Electrochemical and spectroelectrochemical measurements
Gold disc electrodes (ø1.6 mm) were used for cyclic voltammetry (CV) measurements in combination with a Pt-wire counter electrode. All electrode potentials were referenced to an Ag/AgCl electrode (3 M NaCl). For spectroelectrochemical measurements, a home-built slab optical waveguide (SOWG) spectroscopy system was used in combination with indium tin oxide (ITO) electrodes. A xenon lamp and a CCD equipped with a monochrometer were used as the light source and detector, respectively. For protein immobilization, each cleaned ITO electrode was treated with a 1.7 ml portion of NAcMP solution (103 µM in 0.01 M phosphate buffer, pH 7.0) for 30 min. Detailed experimental conditions were set according to a previous report [26]. For the direct electron transfer (DET) measurements, each cleaned Au electrode was immersed into a 10 mM ethanoic 6-mercapto-1-hexanethiol (HXT) solution for 12 h, and subsequently exposed to a 100 µl portion either of NAcMP or natural MP-11 solution for 30 min. A model ALS 750 potentiostat (BAS Inc., Tokyo, Japan) collected the electrochemical responses and its built-in software served to reproduce them. All electrochemical measurements were conducted at room temperature, typically 22 ± 2°C.
3. Results and discussion
3.1. Atomic force microscopy imaging in water
A previous report included the solution CD spectral data, which indicated that the undecapeptide became a somewhat developed secondary structure upon reconstitution: the apparent helical content improved from 13% to 14% [21]. However, the bulky prosthetic molecule haemin must have some influence on the higher-order structure of the host undecapeptide. High-speed AFM observation clearly depicted the structural changes by visualizing/animating the characteristic shape of each molecule.
As seen in figure 2, the surface of undecapeptide-treated mica sheet is covered with dozens of nanometre-sized substances. The apparent density became higher if larger amount of undecapeptide was used for sample preparations (data are not shown). Therefore, these surface bodies were expected to be undecapeptide molecules. Most of them have unique twisted coil shapes; it was about 6 nm in length and was 13 nm when it became extended. After reconstitution of haemin, most of the undecapeptide represented spherical morphology, typically 6 nm in diameter. The structural change was further evidenced by the density distribution with height. After reconstitution, the broad peak at 0.90 nm (FWMH 0.28 nm) for the undecapeptide shifts to larger direction with smaller FWMH, 1.04 nm and 0.17 nm, respectively, which evidences more compact spatial structure.
Figure 2.

Representative AFM images (500 × 500 nm) for the undecapeptide (a) and NAcMP (b) obtained in water. (c) Plots of density distribution with height for the undecapeptide (black) and NAcMP (red).
Protein structures fluctuate in conformational basins and, at the minima, are generally close to what X-ray analysis predicts. The fluctuation involves various molecular motions that exhibit a specific time scale ranging from sub-nanosecond to millisecond: the movement of the atomic groups, such as amides and methyl groups, is very fast, whereas large structural interconversions generally occur slowly in the microsecond to millisecond time scale. The animated AFM image (electronic supplementary material, figure S5) for the undecapeptide successfully visualized the temporal change of morphology. NAcMP is also active for the structural change. However, with the animated data one can notice that covalent binding of the prosthetic group markedly suppresses the conformational change of apopeptide chain.
3.2. Peroxidase activity towards oxidation of TMBZ
Next, the peroxidase activity of NAcMP was examined by taking TMBZ oxidation in the presence of H2O2 as an example. Figure 3 compares the time course of the oxidation product; results clearly showed that NAcMP catalysed the reaction. A mixture including only TMBZ and H2O2 did not yield any product. Unsurprisingly, apo-NAcMP or haemin used alone was inactive. The specific activity of NAcMP towards TMBZ oxidation was 0.43 mol min−1 mg−1, which remained at 27% of natural MP-11 (1.6 mol min−1 mg−1). Catalytic reaction of haemperoxidase is explained by the ‘push–pull’ mechanism [27], which involves a protonated-amino- or carboxylic-acid-residue as the proton donor. N-acetylation that effectively suppresses aggregation could adversely affect the reactivity of the catalyst molecule. Additionally, we obtained somewhat higher activity (43% against natural MP-11) in the previous report [21]. From the three-dimensional model (figure 1), one can find that the haem plane without an axial ligand is open towards the outside and the guest molecule can easily approach the active site to be involved in the catalytic reaction. Accordingly, we can expect that the catalytic activity depends on the primary structure in the vicinity of haem. As the thiol-ene click reaction does not have any preference in reacting with the vinyl group, either 2- or 4-position of haem, the final compound may consist of several structural isomers. Previously, Gray et al. examined a low-spin ferric cyanide derivative of MP-8 by using 1H and 13C NMR and reasonably explained the structure including the thioether bonds [28]. The primary structure of NAcMP must be examined similarly in the future for the detailed discussion of specific activity.
Figure 3.
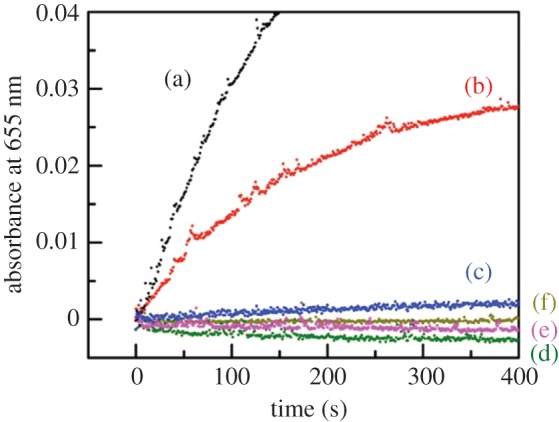
The time course of absorbance at 655 nm of 0.14 mM TMBZ solution (0.1 M phosphate buffer) in the presence of 15 µM H2O2 and each of MP-11 from cytochrome c (a), NAcMP (b), NAcMPm (c), apo-NAcMP (d), haemin (e), and without the catalytic component (f) at 37°C. The concentration of catalysis component was 0.68 µM (natural MP-11, NAcMP and NAcMPm) or 0.81 µM (apo-NAcMP).
As seen in figure 3, the peroxidase activity of NAcMPm was 2.7% of natural MP-11. UV–visible and circular dichroic spectral measurements revealed that NAcMPm preserved the higher-order structure characteristics of natural MP-11 (electronic supplementary material, figures S6 and S7). On the one hand, class I cytochromes c (His/Met coordination) exert peroxidase activity when the haem-Fe loses Met80 coordination [29]. In the ‘push–pull’ mechanism, the histidine-ligated structure has been considered to play the key role in peroxidase reactions [27]. The activity of NAcMPm was found to be considerably low comparing with that of NAcMP, which could endorse the proposed theory. On the other hand, class I cytochromes c show more positive redox potential than class III cytochromes c (His/His coordination) [30]. The initial oxidation by H2O2 proceeds more promptly as the redox potential of the haemperoxidase becomes more negative. Therefore, by examining the redox potential, one may find the cause of low peroxidase activity of NAcMPm.
The peroxidase reaction is described by a three-step mechanism involving the formation of compound (cpd) I and II; they are assigned to the iron(IV)-oxo porphyrin π-cation radical and the iron(IV)-oxo species. The overall reaction, including the ferric resting state of the enzyme (E) and the reducing agent, i.e. TMBZ (AH2), is given below:
| 3.1 |
| 3.2 |
| 3.3 |
The rate of reaction (v) is given by the steady-state kinetics described by Dunford as follows [31]:
| 3.4 |
AH2 exists at a higher concentration that leads to the assumption and therefore equation (3.4) is further simplified to a simple second-order expression:
| 3.5 |
When treating data in this way (electronic supplementary material, figure S8), simple regression analysis determined the rate constants k1 and k3 to be 2.4 × 103 M−1 s−1 and 1.1 × 103 M−1 s−1, respectively (r = 0.984). The specific rate constant of cpd I formation of NAcMP remained low compared with that of Coprinus cinereus peroxidase, (6.7 ± 0.2) × 106 M−1 s−1 [32]. Importantly though, it was of the same order as that reported for natural MP-8, 4778 ± 87 M−1 s−1 [33].
3.3. Spectroelectrochemical study for redox reaction of NAcMP
In the following sections, the electrochemical property of NAcMP is compared with that of natural MP-11. Here, by using a SOWG device, NAcMP was subjected to spectroelectrochemical measurements to probe the chemical species involved.
First, CV measurements confirmed that NAcMP undergoes DET reaction at ITO surfaces as is known for natural MP-11 (figure 4b). Figure 4c summarizes the spectral changes for the NAcMP-attached ITO at different potentials. At the initial potential (+500 mV), the NAcMP-attached ITO shows an intense UV–visible absorption with a maximum wavelength (λmax) of 385 nm. The peak moves to a longer wavelength, whereas it monotonically decreases the absorbance as the electrode potential becomes reducible. At −500 mV, λmax reached its longest wavelength of 405 nm. NAcMP dissolved in phosphate buffer (pH 7.0) showed a Soret band at 408 nm, which moved to a longer wavelength (416 nm) upon reduction by dithionite [21]. The spectroelectrochemical behaviour can be consistent with those obtained in homogeneous solution, considering the short-wavelength shift of λmax that is often seen in an adsorption system. The wavelength shift occurred reversibly and reproducibly between 385 and 405 nm even when the potential sweep was repeated dozens of times (figure 4d).
Figure 4.

Schematic illustration of spectroelectrochemical measurements using a SOWG device (a), and a representative CV for the NAcMP–ITO in 1.0 M phosphate buffer (pH 4.2) at 100 mV s−1 and 22 ± 2°C (b). The UV–visible spectra of NAcMP–ITO (c) were obtained at various potentials at +500 mV (a), +250 mV (b), 0 mV (c), –250 mV (d) and –500 mV (e), and the time course of λmax with repeated potential scanning (d).
As the basis of the electrocatalytic reaction to be studied next, the DET reaction was further examined by conventional CV measurements. We modified Au-disc electrode surfaces with HXT self-assembled monolayers to conveniently accommodate NAcMP through hydrophobic interaction (electronic supplementary material, figure S9). Analysis of the CV data determined the electrochemical parameters as summarized in table 1; they were almost consistent with those previously reported [35,36]. Comparing with the SOWG data, one may notice that the formal potential E°′, taken as (Epc + Epa)/2, shifts by 340 mV to the negative direction, which could be primarily explained by the pH dependence of E°′ (59 mV/pH) [37]. It is also shown that the reduction potential of haem c confined in cytochromes c can be modified by roughly 500 mV through variations in the degree of haem exposure to solvent [38]. At the HXT-monolayer surface, MP-11 and NAcMP showed a considerably negative formal potential, which is presumably solvent-exposed to a great extent. Contrastingly, we can expect that the ITO-adsorbed state effectively hinders the solvent access to the haem as cytochromes c attain.
Table 1.
Electrochemical parameters for the haemprotein–HXT–Au electrode in 0.1 M phosphate buffer solution (pH 7.0) at 25°C.
| entry | NAcMP | MP-11 (Cyt c) |
|---|---|---|
| Γ/pmol cm–2 a | 20 | 23 |
| E°′/mV (Ag/AgCl) | –335 | –327 |
| ΔEp/mV | 69 | 123 |
| kDET/s–1 b | 1.7 | 0.71 |
aThe value was determined by integration of the charge under the cathodic peak of CV.
bThe rate constant was calculated by the literature method [34].
3.4. Electrocatalytic reaction involving H2O2
As shown in figure 5, the NAcMP–Au electrode accepts H2O2 as an oxidizing agent to give characteristic CV responses. The initially observed, peak-shaped cathodic current (equation (3.6)) increased to a considerable extent and changed to a sigmoidal curve, which is typical of a steady-state reaction. Iron(II) is oxidized to a ferryl group by H2O2 in several reactions [39]. Therefore, as the primary component, the one-electron-reduced product of haemin can account for the behaviour (equation (3.7)):
| 3.6 |
| 3.7 |
| 3.8 |
Because the haem ferryl group has a far more positive E° value than the ferric haem, it is immediately reduced at the electrode potential (equation (3.8)). The regenerated substrates NAcMP-Fe(III) are again involved in equation (3.6) to achieve a steady state.
Figure 5.
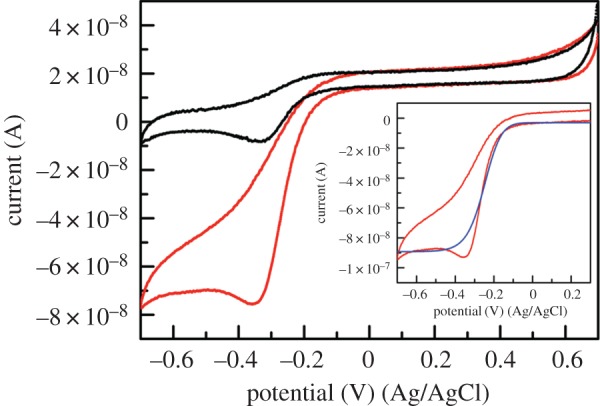
Representative CVs for a NAcMP–HXT–Au electrode in 0.1 M phosphate buffer (pH 7) (black lines) or in the presence of 0.33 µM H2O2 (red lines). Scan rate 10 mV s−1, temperature 22 ± 2°C. The inset compares the background-subtracted experimental CV (red lines) and the theoretical current–voltage curve during the forward scan (blue line). Owing to a quasi-reversible direct electron transfer reaction, replication of the reversed-scan wave was unsatisfactory.
We simply used digital simulations for characterizing the substrate-recycling DET reaction adopting an assumption. The redox-active species exist only at about 10–12 mol cm–2 level on the electrode surface, which is extremely small when compared with H2O2 dissolved in the solution phase. Given this, the second-order interfacial reaction was simplified to a pseudo-first-order reaction with the rate constant kex (equation (3.7′)):
| 3.7′ |
The parameter sets that were determined from the best-fit data are summarized in table 2. It is apparent that E°′ of NAcMP shifted to the positive direction by 135 mV, indicating that catalytic action was occurring. It is reported that coordination of the haem with certain types of the sixth-ligands including N-acetylmethionine (AcMet) caused a positive shift of the E°′ of the haem. An E°′ is the expression of the relative stability of the two redox partners under the particular conditions including the medium used for measurements. Currently, we are not aware whether the H2O2 coordination occurs preferentially to the haem-Fe(II) or the haem-Fe(III) in NAcMP. Yet, we expect that, similarly with AcMet, the affinity of H2O2 coordination with haem-Fe(II) is considerably higher than that of the oxidized peptide, which increased the haem reduction potential. The DET rate constant was classified as moderate reaction similar to the noncatalytic system. The rate constant for substrate regeneration, 1.8 s−1, represents a rather slow turnover frequency. In table 2 are given the electrocatalytic parameters obtained for natural MP-11 (electronic supplementary material, figure S10). In previous reports, Willner et al. examined the maximum current at H2O2 saturation that corresponds to vmax in the Michaelis–Menten model; the MP-11-modified electrode was determined to be −1.0 µA [40]. Later, a bis-histidine-ligated unfolded cytochrome c mutant was examined in a similar way by Scheller et al.; it was found to be 5.18 µA cm−2 [41]. As can be seen from the unit of current density, if the Faraday and the electroactive surface concentration divide the data, it becomes the pseudo-first-order rate constant. Comparison of the data concluded that the rate constant for both NAcMP and natural MP-11 was one order larger than that of the previous report, and was of the same order as the unfolded cytochrome c mutant.
Table 2.
Global parameters estimated by theoretical simulation of the background-subtracted experimental current–voltage profiles.
| entry | NAcMP | MP-11 (Cyt c) |
|---|---|---|
| Γ/pmol cm–2 a | 20 | 23 |
| E°′app/mV (Ag/AgCl) | –200 | –230 |
| kDET/s–1 | 0.70 | 0.60 |
| α b | 0.5 | 0.5 |
| Cdl/µF a | 0.3 | 0.5 |
| kex/s–1 | 1.8 | 1.6 |
aThese values were determined from CVs obtained in 0.1 M phosphate buffer solution without H2O2.
bThe parameter was fixed at 0.5 by assuming a symmetrical energy barrier of the electrode reaction.
4. Conclusion
Here, we reported the first totally synthetic MP-11. By taking advantage of the thiol-ene click reaction, we have successfully reconstituted haem c into the host peptide, which one could not achieve so far. We have also succeeded to visualize, with animated AFM images, that the spatial structure of protein remarkably changes before and after the treatment. Oxidation of TMBZ by hydrogen peroxide confirmed that the synthetic haemperoxidase displayed a similar degree of enzymatic activity to that of natural MP-11. Additionally, the synthetic material achieved an electrochemical catalytic system that will be useful for H2O2-sensing platform. Computer simulation by trial and error well reproduced the catalytic current–voltage curve and simultaneously, estimated the catalytic rate constant that seemed appropriate. In all experiments, the results of the synthetic MP-11 were almost identical to those of its naturally occurring counterpart. Therefore, we concluded that our approach is a convenient means of MP-11 synthesis that has no reliance on nature. From the viewpoint of improved efficiency of enzymatic reaction, a synthetic mutant, NAcMPm, was disappointing. However, NAcMPm seems an example of proof that our approach allows us various peptides in hand. We expect that our totally synthetic strategy will be a useful means in preparing MPs regardless of natural or nonnatural sequence.
Supplementary Material
Supplementary Material
Acknowledgements
We thank the Kyushu University Program for Leading Graduate Schools: Advanced Graduate Course on Molecular Systems for Devices for the access to the AFM instrument.
Data accessibility
The electronic supplementary material, including the synthetic procedures, analysis data, UV–visible spectra, CD spectra and cyclic voltammograms, is available to support the paper. Additionally, the animated versions of AFM images for both the undecapeptide and the synthetic MP-11 are accessible as a PowerPoint slideshow.
Authors' contributions
K.N. conceived the project, designed methods, analysed the results and wrote the manuscript. J.T. performed the synthetic work and acquired the UV–visible, CD, electrochemical and spectroelectrochemical data. R.H. synthesized the mutant haempeptide and made the spectral measurements. T.H. performed the AFM imaging. H.O. and N.M. contributed by obtaining and analysing SOWG data. R.I. and T.I. assisted with the validation of the data. All authors have given approval for publication.
Competing interests
The authors declare no competing interests.
Funding
This work was financially supported, in part, by a grant from JSPS KAKENHI (grant nos. 25620115 and 15H01713 (K.N.)).
References
- 1.Adams PA. 1999. Microperoxidases and iron porphyrins. In Peroxidases in chemistry and biology, vol. II (eds Everse J, Everse KE, Grisham MB), pp. 171–200. Boca Raton, FL: CRC Press. [Google Scholar]
- 2.Primus JL, Grunenwald S, Hagedoorn PL, Albrecht-Gary AM, Mandon D, Veeger C. 2002. The nature of the intermediates in the reactions of Fe(III)- and Mn(III)-micro-peroxidase-8 with H2O2: a rapid kinetics study. J. Am. Chem. Soc. 124, 1214–1221. (doi:10.1021/ja016907u) [DOI] [PubMed] [Google Scholar]
- 3.Dallacosta C, Monzani E, Casella L. 2003. Reactivity study on microperoxidase-8. J. Biol. Inorg. Chem. 8, 770–776. (doi:10.1007/s00775-003-0478-z) [DOI] [PubMed] [Google Scholar]
- 4.Cowley AB, Lukat-Rodgers GS, Rodgers KR, Benson DR. 2004. A possible role for the covalent heme-protein linkage in cytochrome c revealed via comparison of N-acetylmicroperoxidase-8 and a synthetic, monohistidine-coordinated heme peptide. Biochemistry 43, 1656–1666. (doi:10.1021/bi035531p) [DOI] [PubMed] [Google Scholar]
- 5.Zhong FF, Lisi GP, Collins DP, Dawson JH, Pletneva EV. 2014. Redox-dependent stability, protonation, and reactivity of cysteine-bound heme proteins. Proc. Natl Acad. Sci. USA 111, E306–E315. (doi:10.1073/pnas.1317173111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ascenzi P, Sbardella D, Fiocchetti M, Santucci R, Coletta M. 2015. NO2−-mediated nitrosylation of ferrous microperoxidase-11. J. Inorg. Biochem. 153, 121–127. (doi:10.1016/j.jinorgbio.2015.06.022) [DOI] [PubMed] [Google Scholar]
- 7.Katz E, Baron R, Willner I. 2005. Magnetoswitchable electrochemistry gated by alkyl-chain-functionalized magnetic nanoparticles: control of diffusional and surface-confined electrochemical processes. J. Am. Chem. Soc. 127, 4060–4070. (doi:10.1021/ja042910c) [DOI] [PubMed] [Google Scholar]
- 8.Yarman A, Badalyan A, Gajoviv-Eichelmann N, Wollenberger U, Scheller FW. 2011. Enzyme electrode for aromatic compounds exploiting the catalytic activities of microperoxidase-11. Biosens. Bioelectron. 30, 320–323. (doi:10.1016/j.bios.2011.09.004) [DOI] [PubMed] [Google Scholar]
- 9.Zhang B, Zhou J, Li S, Zhang X, Huang D, He Y, Wang M, Yang G, Shen Y. 2015. Hydrogen peroxide sensor based on microperoxidase-11 immobilized on flexible MWCNTs-BC nanocomposite film. Talanta 131, 243–248. (doi:10.1016/j.talanta.2014.07.027) [DOI] [PubMed] [Google Scholar]
- 10.Renault C, Andrieux CP, Tucker RT, Brett MJ, Balland V, Limoges B. 2012. Unraveling the mechanism of catalytic reduction of O2 by microperoxidase-11 adsorbed within a transparent 3D-nanoporous ITO film. J. Am. Chem. Soc. 134, 6834–6845. (doi:10.1021/ja301193s) [DOI] [PubMed] [Google Scholar]
- 11.Xuan Y, Huang X, Su B. 2015. Biomimetic oxygen reduction reaction catalyzed by microperoxidase-11 at liquid/liquid interfaces. J. Phys. Chem. C 119, 11 685–11 693. (doi:10.1021/acs.jpcc.5b02131) [Google Scholar]
- 12.Ramanavicius A, Kausaite A, Ramanaviciene A. 2005. Biofuel cell based on direct bioelectrocatalysis. Biosens. Bioelectron. 20, 1962–1967. (doi:10.1016/j.bios.2004.08.032) [DOI] [PubMed] [Google Scholar]
- 13.Low DW, Abedin S, Yang G, Winkler JR, Gray B. 1998. Manganese microperoxidase-8. Inorg. Chem. 37, 1841–1843. (doi:10.1021/ic971166c) [Google Scholar]
- 14.Kleingardner JG, Kandemir B, Bren KL. 2014. Hydrogen evolution from neutral water under aerobic conditions catalyzed by cobalt microperoxidase-11. J. Am. Chem. Soc. 136, 4–7. (doi:10.1021/ja406818h) [DOI] [PubMed] [Google Scholar]
- 15.Yang F, et al. 2005. Characterization of purified c- type heme-attachment sites in Shewanella oneidensis cytochromes using mass spectrometry. J. Proteome Res. 4, 846–854. (doi:10.1021/pr0497475) [DOI] [PubMed] [Google Scholar]
- 16.Caputi L, Di Tullio A, Di Leandro L, De Angelis F, Malatesta F. 2005. A new microperoxidase from Marinobacter hydrocarbonoclasticus. Biochim. Biophys. Acta Gen. Subj. 1725, 71–80. (doi:10.1016/j.bbagen.2005.05.023) [DOI] [PubMed] [Google Scholar]
- 17.Braun M, Thöny-Meyer L. 2004. Biosynthesis of artificial microperoxidases by exploiting the secretion and cytochrome c maturation apparatuses of Escherichia coli. Proc. Natl Acad. Sci. USA 101, 12 830–12 835. (doi:10.1073/pnas.0402435101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleingardner EC, Asher WB, Bren KL. 2017. Efficient and flexible preparation of biosynthetic microperoxidases. Biochemistry 56, 143–148. (doi:10.1021/acs.biochem.6b00915) [DOI] [PubMed] [Google Scholar]
- 19.Lombardi A, Nastri F, Pavone V. 2001. Peptide-based heme-protein models. Chem. Rev. 101, 3165–3189. (doi:10.1021/cr000055j) [DOI] [PubMed] [Google Scholar]
- 20.Nastri F, et al. 2011. A heme–peptide metalloenzyme mimetic with natural peroxidase-like activity. Chem. Eur. J. 17, 4444–4453. (doi:10.1002/chem.201003485) [DOI] [PubMed] [Google Scholar]
- 21.Nakano K, Tanabe J, Ishimatsu R, Imato T. 2017. Monolithic peptide-nucleic acid hybrid functioning as an artificial microperoxidase. Bioconjugate Chem. 28, 2031–2034. (doi:10.1021/acs.bioconjchem.7b00216) [DOI] [PubMed] [Google Scholar]
- 22.Marques HM, Perry CB. 1999. Hemepeptide models for hemoproteins: the behavior of N-acetylmicroperoxidase-11 in aqueous solution. J. Inorg. Biochem. 75, 281–291. (doi:10.1016/S0162-0134(99)00100-2) [DOI] [PubMed] [Google Scholar]
- 23.Hoppmann C, Schmieder P, Heinrich N, Beyermann M. 2011. Photoswitchable click amino acids: light control of conformation and bioactivity. ChemBioChem 12, 2555–2559. (doi:10.1002/cbic.201100578) [DOI] [PubMed] [Google Scholar]
- 24.Nečas D, Klapetek P. 2012. Gwyddion: an open-source software for SPM data analysis. Cent. Eur. J. Phys. 10, 181–188. (doi:10.2478/s11534-011-0096-2) [Google Scholar]
- 25.Holland VR, Saunders BC, Rose FL, Walpole AL. 1974. A safer substitute for benzidine in the detection of blood. Tetrahedron 30, 3299–3302. (doi:10.1016/S0040-4020(01)97504-0) [Google Scholar]
- 26.Matsuda N, Okabe H, Omura A, Nakano M, Miyake K. 2017. In situ observation of direct electron transfer reaction of cytochrome c immobilized on ITO electrode modified with 11-{2-[2-(2-methoxyethoxy)-ethoxy] ethoxy}undecylphosphonic acid self-assembled monolayer film by electrochemical slab optical waveguide spectroscopy. Anal. Sci. 33, 469–472. (doi:10.2116/analsci.33.469) [DOI] [PubMed] [Google Scholar]
- 27.Dawson JH. 1988. Probing structure-function relationships in heme-containing oxygenases and peroxidases. Science 240, 433–439. (doi:10.1126/science.3358128) [DOI] [PubMed] [Google Scholar]
- 28.Low DM, Gray HB, Duus JØ. 1997. Paramagnetic NMR spectroscopy of microperoxidase-8. J. Am. Chem. Soc. 119, 1–5. (doi:10.1021/ja962948s) [Google Scholar]
- 29.Diederix REM, Ubbink M, Canters GW. 2002. Effect of the protein matrix of cytochrome c in suppressing the inherent peroxidase activity of its heme prosthetic group. ChemBioChem 3, 110–112. (doi:10.1002/1439-7633(20020104)3:1<110::AID-CBIC110>3.0.CO;2-2) [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y. 2014. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469. (doi:10.1021/cr400479b) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunford HB. 1999. Heme peroxidases, pp. 9–17. New York, NY: Wiley. [Google Scholar]
- 32.Abelskov AK, Smith AT, Rasmussen CB, Dunford HB, Welinder KG. 1997. pH dependence and structural interpretation of the reactions of Coprinus cinereus peroxidase with hydrogen peroxide, ferulic acid, and 2,2'-azinobis(3-ethylbenzthiazoline-6-sulfonic acid). Biochemistry 36, 9453–9463. (doi:10.1021/bi970387r) [DOI] [PubMed] [Google Scholar]
- 33.Adams PA. 1990. The peroxidase activity of the heam octapeptide microperoxidase-8 (MP-8): the kinetic mechanism of the catalytic reduction of H2O2 by MP-8 using 2,2'-azinobis-(3-ethylbenzothiazothiazoline-6-sulphonate) (ATBS) as reducing substrate. J. Chem. Soc. Perkin Trans. 2, 1407–1414. (doi:10.1039/P29900001407) [Google Scholar]
- 34.Laviron E. 1979. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 101, 19–28. (doi:10.1016/S0022-0728(79)80075-3) [Google Scholar]
- 35.Santucci R, Reinhard H, Brunori M. 1988. Direct electrochemistry of the undecapeptide from cytochrome c (microperoxidase) at a glassy carbon electrode. J. Am. Chem. Soc. 110, 8536–8537. (doi:10.1021/ja00233a035) [Google Scholar]
- 36.Ruzgas T, Gaigalas A, Gorton L. 1999. Diffusionless electron transfer of microperoxidase-11 on gold electrodes. J. Electroanal. Chem. 469, 123–131. (doi:10.1016/S0022-0728(99)00194-1) [Google Scholar]
- 37.Das DK, Medhi OK. 1998. Effect of surfactant and pH on the redox potential of microperoxidase 11 in aqueous micellar solutions. J. Chem. Soc. Dalton Trans. 1693–1698. (doi:10.1039/A708732B) [Google Scholar]
- 38.Tezcan FA, Winkler JR, Gray HB. 1998. Effects of ligation and folding on reduction potentials of heme proteins. J. Am. Chem. Soc. 120, 13 383–13 388. (doi:10.1021/ja982536e) [Google Scholar]
- 39.Kremer ML. 1999. Mechanism of the Fenton reaction. Evidence for a new intermediate. Phys. Chem. Chem. Phys. 1, 3595–3605. (doi:10.1039/A903915E) [Google Scholar]
- 40.Moore ANJ, Katz E, Willner I. 1996. Electrocatalytic reduction of organic peroxides in organic solvents by microperoxidase-11 immobilized as a monolayer on a gold electrode. J. Electroanal. Chem. 417, 189–192. (doi:10.1016/S0022-0728(96)04736-5) [Google Scholar]
- 41.Ranieri A, Battistuzzi G, Borsari M, Bortolotti CA, Rocco GD, Monari S, Sola M. 2012. A bis-histidine-ligand unfolded cytochrome c immobilized on anionic SAM shows pseudo-peroxidase activity. Electrochem. Commun. 14, 29–31. (doi:10.1016/j.elecom.2011.10.021) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The electronic supplementary material, including the synthetic procedures, analysis data, UV–visible spectra, CD spectra and cyclic voltammograms, is available to support the paper. Additionally, the animated versions of AFM images for both the undecapeptide and the synthetic MP-11 are accessible as a PowerPoint slideshow.