

Abstract

Model coliphages (e.g., ΦX174, MS2, and PRD1) have been widely used as surrogates to study the fate and transport of pathogenic viruses in the environment and during wastewater treatment. Two groups of coliphages (F-specific and somatic) are being explored as indicators of viral fecal pollution in ambient water. However, the detection and quantification of coliphages still largely rely on time-consuming culture-based plaque assays. In this study, we developed an in-gel loop-mediated isothermal amplification (gLAMP) system enabling coliphage MS2 quantification within 30 min using standard laboratory devices. Viral particles (MS2) were immobilized with LAMP reagents in polyethylene glycol hydrogel, and then viral RNAs were amplified through a LAMP reaction. Due to the restriction effect of the hydrogel matrix, one viral particle would only produce one amplicon dot. Therefore, the sample virus concentrations can be determined based on the number of fluorescent amplicon dots using a smartphone for imaging. The method was validated by using artificially spiked and naturally contaminated water samples. gLAMP results were shown to correlate well with plaque assay counts (R2 = 0.984, p < 0.05) and achieved similar sensitivity to quantitative reverse-transcription polymerase chain reaction (RT-qPCR; 1 plaque-forming unit per reaction). Moreover, gLAMP demonstrated a high level of tolerance against inhibitors naturally present in wastewater, in which RT-qPCR was completely inhibited. Besides MS2, gLAMP can also be used for the quantification of other microbial targets (e.g., Escherichia coli and Salmonella). Considering its simplicity, sensitivity, rapidity, and versatility, gLAMP holds great potential for microbial water-quality analysis, especially in resource-limited settings.
Introduction
Human pathogenic enteric viruses (e.g., adenovirus, enterovirus, and norovirus) found in domestic wastewater have been identified as important causative agents responsible for a wide range of infections in humans.1 Previous studies suggest that traditional fecal indicator bacteria (e.g., Escherichia coli and Enterococcus) do not adequately predict the fate of human viral pathogens because they respond differently to wastewater-treatment processes and environmental degradation processes from viruses.2 However, the direct detection and quantification of specific viral pathogens in environmental water samples is challenging due to methodological limitations.3 Therefore, coliphages (viruses that infect E. coli cells) are being explored as indicators of actual viral pathogens.4 Coliphages are not pathogenic to humans but are similar to pathogenic enteric viruses in terms of size, morphology, surface properties, and genetic structures. Model coliphages (e.g., ΦX174, MS2, and PRD1) are also widely employed as process indicators to evaluate the viral removal efficiency of various water treatment processes, such as sand filtration,5 reverse osmosis,6 UV,7 and electrochemical disinfection.8 In 2015, the U.S. Environmental Protection Agency (U.S. EPA) initiated a criteria-development process considering the use of F-specific and somatic coliphages as possible viral indicators of fecal contamination in ambient water.3
A variety of methods are available for bacteriophage detection. These include traditional culture-based plaque assays and molecular-based methods. Two culture-based methods were approved by the U.S. EPA for coliphage monitoring in groundwater (U.S. EPA methods 1601 and 1602). Depending on the incubation time, these methods require 18 to 72 h to obtain the final results. A genetic modified E. coli strain has recently been developed to detect somatic coliphages based on the color changes of the growth media triggered by the phage-mediated release of intracellular enzyme β-glucuronidase. The method reduces the culture time to between 3.5 and 5.5 h, which is by far the fastest reported culture-based detection method.9 In contrast, molecular-based methods, represented by quantitative polymerase chain reaction (qPCR), provide better sensitivity, specificity, and a much-shorter sample-to-result time (1 to 4 h).10 Despite its wide acceptance, qPCR is limited by the reliance on standard reference materials (standard curve) for quantification. Unreliable and inconsistent commercial standard reference materials were reported to affect the accuracy of qPCR quantification.11,12 Also, qPCR is prone to inhibition caused by substances naturally present in environmental samples (e.g., heavy metals and organic matter), thereby leading to inaccurate target quantification or false-negative results. Compared to qPCR, the cutting-edge digital PCR technique has shown to be a more-robust solution for virus detection in environmental samples.11,13 A recent study by Cao et al. highlighted that digital PCR was unaffected by humic acid (HA) at concentrations up to 17.5 ng/μL, while the HA tolerance level of qPCR was only 0.5 ng/μL.11 However, the implementation of digital PCR methods to point-of-use applications is challenging because it requires costly high-end instruments, a well-equipped laboratory environment, and highly trained personnel to conduct the assay. These factors severely restrict the method’s accessibility and adoption in resource-limited settings.
Alternatives to PCR-based nucleic acid amplification and detection techniques, isothermal amplification methods such as loop-mediated isothermal amplification (LAMP),14 helicase-dependent amplification (HDA),15 multiple-displacement amplification (MDA),16 and rolling circle amplification (RCA),17 offer the opportunity to deliver the benefits of molecular assays beyond centralized laboratories. With no need for thermal cycling, isothermal reactions are more suitable for coupling with miniaturized, portable, and battery-powered “lab-on-a-chip” platforms.18 Initially described in 2000,19 LAMP has become the most-popular isothermal amplification technique, covering most microbial pathogens relevant to sanitation.20−22 LAMP is capable of amplifying a target DNA template 109 times in less than 60 min at a temperature around 65 °C.19 Similar to PCR, LAMP products can be detected by fluorescence using intercalating dyes (e.g., EvaGreen, Sybr Green, and SYTO9) or with unaided eyes through turbidity changes caused by magnesium pyrophosphate precipitation as a byproduct of amplification.14 Many portable devices have been developed to facilitate the application of LAMP in point-of-care disease diagnostics.18,23 In contrast, the application of LAMP in environmental studies is lagging behind, with recent work by Martzy et al., who developed a LAMP assay for the detection of Enterococcus spp. in water, being a notable exception.21 This is likely because most LAMP assays are qualitative but microbial water-quality analysis generally requires quantitative data. Although a few quantitative LAMP assays have been reported in real-time or digital formats, they all require complex instruments (i.e., real-time fluorescence detection devices)24 or customized microfluidic chips (e.g., Slipchip25 and DropChip),26 making them hard to be adopted by a broader user community.
Our vision is to take advantage of LAMP to develop a quantitative, low-cost, and rapid coliphage detection tool that can be easily adopted in resource-limited settings. Inspired by earlier work on in situ PCR,27 immobilization of microbes in hydrogels,28 PCR amplification in polyacrylamide gels,29 and MDA amplification in polyethylene glycol (PEG) hydrogels,16 we have developed a smartphone-based in-gel LAMP (gLAMP) system capable of quantifying coliphage MS2 in environmental water samples within 30 min. gLAMP requires no specialized equipment, no microfluidic chips, and limited personnel training. It is worth to mention that the gLAMP system is not restricted to MS2 detection. It is a nucleic acid amplification testing platform, like qPCR, that can also be used for the quantification of many other microbial targets (e.g., E. coli and Salmonella).
Materials and Methods
Model Coliphage MS2 Preparation
Coliphage MS2 (ATCC 15597-B1) was chosen as the model virus for the method development. For phage propagation, 0.1 mL [107 plaque-forming units (PFU)/mL] of MS2 was inoculated into 20 mL of actively growing E. coli-3000 (ATCC 15597) host suspension in Luria–Bertani medium. The infected bacteria were continuously aerated at 37 °C for 36 h. The host-associated MS2 suspension was then centrifuged at 3000g for 10 min to pellet the bacterial cells and debris. The supernatant, containing the MS2 virions, was further purified by 0.2 μm syringe filter (GE Whatman, Pittsburgh, PA). The filtrate was diluted 1000× in 1× PBS (pH of 7.5) (Corning, New York, NY) and used as MS2 stock for seeding studies. The concentration of MS2 stock was titrated by the double-agar-layer method.30 An AllPrep PowerViral DNA/RNA Kit (Qiagen, Germantown, MD) was used for MS2 RNA extraction per the manufacturer’s protocol.
gLAMP Assay Design
Two types of hydrogels were initially tested as the matrix for gLAMP. The polyacrylamide (PA) gel was formed through the cross-linking between acrylamide and bis-acrylamide (acrylamide/Bis 19:1) (Bio-Rad, Hercules, CA) using 0.05% (w/v) ammonium persulfate (Bio-Rad, Hercules, CA) as initiator and catalyzed by 0.05% (w/v) tetramethylethylenediamine (TEMED) (Bio-Rad). The PEG gel was formed through Michael addition between the four-arm PEG acrylate [molecular weight (MW) of 10 000] and thiol-PEG-thiol (MW of 3400; Laysan Bio, Arab, AL) at a mole ratio of 1:2. The MS2 LAMP primers and probes originally developed by Ball et al.31 were used and optimized in the current study (Table S1). For each gLAMP assay (25 μL), the optimized hydrogel reaction mix had the following composition: 10% (w/v) hydrogel, 12.5 μL of 2×WarmStart LAMP Mastermix (a blend of Bst 2.0 WarmStart DNA polymerase and WarmStart RTx reverse transcriptase; New England Biolabs, Ipswich, MA), 1.25 μL of 20× virus primer mix (the final concentrations of F3/B3, FIP/BIP and LF/LB were 0.2, 1.6, and 0.4 μM, respectively), and 2 μL of MS2 RNA templates or 2 μL of water sample. For reactions using the complementary fluorescent probe and quencher primers, quencher primer (qFIP-3′IBFQ) was added (final concentration 3.2 μM) when fluorophore-labeled primer (5′FAM-FIP) was used to substitute the regular FIP primer. The above-described 25 μL of hydrogel reaction mix was loaded into an in situ PCR frame seal chamber (9 × 9 mm; Bio-Rad) on a glass slide and then covered with a transparent qPCR film (Sorenson, Salt Lake City, UT). The hydrogel was polymerized at room temperature (21 °C) for 5–15 min and then incubated on a PCR machine (MJ Research PTC-100, Watertown, MA) or a mini dry bath (Benchmark, Edison, NJ) at 65 °C for 25 min. After amplification, the gel was stained with 0.5× LAMP dye (included in the WarmStart LAMP kit) in the dark for 15 min and then washed twice with 2× TE buffer (pH of 7.8; Corning, New York, NY). For reactions using the complementary fluorescent probe and quencher primers, no post-reaction staining was needed. The slides were illuminated with an E-Gel Safe Imager (Invitrogen, Carlsbad, CA), and the amplicon dots were documented with an iPhone 6s Plus. To verify the sensitivity of the smartphone detection system, the slides were also imaged using a fluorescence microscope (Leica DMi8; Leica Co., Germany).
gLAMP Assay Optimization
Initial gLAMP development was carried out using extracted MS2 viral RNA. Assays were conducted to find the optimal staining strategy (post-reaction staining with LAMP dye or using fluorescent probe), incubation time (20, 25, and 30 min), and assay dynamic range (low, 1–20 copies per reaction; medium, 20–200 copies per reaction; and high, 200–2000 copies per reaction). Subsequently, with the intention of simplifying the RNA extraction step, we also explored simple heating (95 °C, 5 min) as a pretreatment procedure or direct detection of MS2 viral particles without RNA extraction. The assay sensitivity and dynamic range were compared to RT-qPCR using Eppendorf RealPlex2 (Hamburg, Germany). The primers and probe and reaction conditions of the one-step RT-qPCR are provided in Table S2.
Tolerance of gLAMP to Inhibitors Present in Environmental Water Samples
A total of three environmental water samples were tested in the present study to evaluate the tolerance of gLAMP to inhibitors naturally present in environmental wasters. Lake water (LW) was collected from Echo Park Lake (Los Angeles, CA), which functions primarily as a detention basin in the city’s storm-drain system while providing recreational benefits and wildlife habitat. Pond water (PW) was collected from the Turtle Pond at the California Institute of Technology (Caltech). Wastewater (WW) was collected from the sedimentation and storage tank of a pilot-scale solar-powered mobile toilet system also located on the Caltech campus. WW is composed of urine, feces, and hand-washing and toilet-flushing water. More details about the design and operational conditions of the toilet system were reported in previous studies.8,32 Basic water-quality parameters of these samples are summarized in Table S3. The dissolved organic matter (DOM) in the environmental samples were characterized by excitation–emission matrix (EEM) using a fluorescence spectroscopy (Shimadzu RF-6000, Kyoto, Japan). Corrected EEMs were generated from raw scans (excitation wavelengths: 250–550 nm, 5 nm interval; emission wavelengths: 300–600 nm, 2 nm interval) and used to estimate the various DOM components (see Figure S5 for a graphical illustration of these components). The concentrations of indigenous MS2 (without preconcentration) in all these samples were below the detection limit of plaque assays (1 PFU/mL) and RT-qPCR (1 plaque-forming unit per reaction). Therefore, pure cultured MS2 was spiked to these samples and a PBS buffer solution at the final concentration of 2 × 103-2 × 104 PFU/mL (equaling 10–100 plaque-forming unit per reaction). Spiked water samples were allowed to equilibrate for 1 h before being directly analyzed with gLAMP, in-tube real-time LAMP (see Table S1 for more details), and RT-qPCR without RNA extraction. The MS2-spiked PBS served as a control because no inhibition was expected in this buffer solution. Inhibition effect was evaluated by comparing results from environmental samples with those obtained from PBS: in gLAMP, inhibition was reflected as fewer fluorescent dot counts in environmental samples than those in PBS, while for in-tube LAMP and qPCR, it was shown as increased time-to-detection and larger quantitation cycle (Cq) values, respectively.
Detection of MS2 in Primary Effluent Samples
To demonstrate the detection of MS2 in nonspiked natural water, primary effluent wastewater sample was collected from a local wastewater treatment plant serving 150 000 people. A 20 mL water sample was filtered with 0.22 μm syringe filters (GE Whatman, Pittsburgh, PA) to remove bacteria and debris before further analysis. For double-layer plaque assays,30 F-specific coliphages were enumerated using E. coli Famp (ATCC 700891) as the bacterial host, while E. coli C3000 (ATCC 15597) was used for total (somatic and F-specific) coliphage enumeration. A total of 15 mL of the filtrate was further concentrated to 150 μL using an Amicon Ultra-15 Centrifugal Filter (30 KDa nominal molecular weight limit) (Millipore, Burlington, MA). Virus RNA was extracted from 100 μL of concentrate using the AllPrep PowerViral DNA/RNA Kit (Qiagen, Germantown, MD) and then analyzed by gLAMP and RT-qPCR.
Results and Discussion
Gel Selection
Clear polyacrylamide gels were formed within 10–15 min, while the formation of PEG gel was faster, taking 3–5 min at alkaline pH (the pH of the LAMP reaction mix is 8.8). Both gels showed no fluorescent background, and gLAMP was successfully carried out in either case (Figure 1). We found that when the amplicon dot sizes were smaller than 20 μm (diameter), the detection would require a fluorescence microscope. To facilitate the results reading with a smartphone camera while still maintaining a practical assay dynamic range, dot sizes between 50 to 200 μm were preferred in the current study. The size of amplicon dots was mainly decided by the restriction effect of the gel matrix. Ideally, the gel matrix should allow the free diffusion of small molecules (molecular weight (MW) of <100 kDa) such as water, ions, primers (<50 bp, MW < 15 kDa), and enzymes (Bst: 67 kDa) but restrict the movement of DNA and RNA templates and the amplicons (>150 bp, MW > 100 kDa). This can be achieved by tuning the gel cross-linking degree and the length of cross-linkers to control the gel mesh size and, thus, the macroscopic gel properties (i.e., diffusion). Mitra et al. found that 514, 234, and 120 bp templates produced uniform PCR amplicon dots of 100, 400, and 800 μm in polyacrylamide hydrogel, respectively.29 It should be noted that, unlike single length PCR amplicons, products of LAMP are a mixture of concatemers of the target region with various sizes. Based on the agarose gel electrophoresis profile (Figure S1), the shortest MS2 LAMP amplicons were about 90 bp, while the longest amplicons were up to several thousand base pairs. During gLAMP, longer amplicons were retarded by the hydrogel matrix, but shorter amplicons diffused away from the initial templates (the center of the dot) and served as templates for further amplification until they reached the diffusion limit or the LAMP reagents (e.g., enzyme, primer, and dNTP) in the vicinity were depleted. Due to the stochastic nature of LAMP, dots of different sizes were produced in the hydrogels. According to previous studies, the mesh size of the 2 hydrogels were similar and in the range of 20–25 nm.33,34 However, the amplicon dots in the PEG gel (Figure 1B) were significantly smaller and more-uniform than those formed in polyacrylamide gel (Figure 1A). The results showed that the PEG gel had a better restriction effect on the smaller amplicons. Therefore, besides size exclusion, other interactions (i.e., charge interaction) between the polymers and the DNA templates may also affect the diffusion coefficient. Given the better template-restriction effect, PEG hydrogel was chosen for further method development.
Figure 1.

gLAMP hydrogel selection. Assays using (A) polyacrylamide and (B, C) polyethylene glycol hydrogels. LAMP amplicon dots were stained with 0.5× LAMP dye (A, B) after incubation or (C) using the QUASR primers without post-reaction staining. The images were taken by an iPhone 6s Plus. Extracted MS2 RNAs were used as templates, and the reaction time was 25 min.
gLAMP Amplicon Staining Strategies
Clear MS2 amplicon dot profiles were obtained through post-reaction gel staining with intercalating LAMP dye (Figure 1B). Similar profiles were obtained in gLAMP assays for E. coli and Salmonella (see Table S4 and Figure S2 for more details). The results highlight the feasibility of adapting established qualitative LAMP assays into quantitative assays via the gLAMP system. However, opening the frame seal chamber and staining the gel after amplification added extra complexity to the assay and may result in amplicon contamination to the surrounding working environment. We also found that adding intercalating fluorescent dyes into the reaction mix before heat incubation was not an option because it resulted in a high level of fluorescent background.
To develop a simpler gLAMP without the need for post-reaction staining, a primer-dye and primer-quencher duplex, previously reported as quenching of unincorporated amplification signal reporters (QUASR) by Ball et al.,31 was adopted and optimized in this study. In QUASR, the forward internal primer (FIP) is labeled with a fluorophore at the 5′ prime end (5′FAM-FIP). The probe is quenched by a complementary primer with a quencher (Iowa Black FQ) at the 3′ prime end (qFIP-3′IBFQ). Because the melting temperature (Tm) of the complex is 5–10 °C lower than the reaction temperature (65 °C), the 5′FAM-FIPs are released and behave like regular FIPs during the LAMP reaction. 5′FAM-FIPs are incorporated into the LAMP amplicons when there are target templates present in the sample. After the reaction, extra unincorporated 5′FAM-FIPs are quenched again by the complementary quencher primer qFIP-3′IBFQs. In contrast, 5′FAM-FIPs incorporated into LAMP amplicons would not be quenched because they already form a stable double-strand DNA structure during the LAMP reaction. Compared with nonspecific DNA intercalating dyes (i.e., the LAMP dye), QUASR significantly reduces the issue of false positive results associated with LAMP assays.31 However, QUASR cannot turn into a quantitative assay in a real-time LAMP scheme because the fluorescent intensity of the reaction mix is constantly at the highest level (all 5′FAM-FIP released) instead of progressively increasing during heat incubation. Therefore, it can only be used as a qualitative assay for end point determination. In preliminary experiments, we found that the QUASR primers did not reduce gLAMP amplification efficiency, although a higher concentration of quencher primer (2× of the complementary probe primer) was needed to maintain a clean gel background at the end of the gLAMP reaction. These results suggest that the PEG gel allowed the free movement of the dye-labeled short oligonucleotides, even though the diffusion coefficient would be smaller in the gel matrix than that in a solution. As 5′FAM-FIPs were incorporated into the amplicons and accumulated around the initial templates, bright and defined amplicon dots could be directly visualized with a smartphone camera upon blue light exposure (Figure 1C). Therefore, QUASR primers were used for further gLAMP optimization.
gLAMP optimization
Amplicon dots were visible as early as 20 min under a fluorescence microscope (Figure 2A). The dots developed to about 156 ± 33 μm (diameter) after 25 min, and the fluorescence intensity was strong enough to be detected with a smartphone camera (Figure 3). Although the amplicon dots kept increasing in size and reached 212 ± 50 μm after 30 min, the number of dots stayed similar to those at 25 min. Hence, 25 min was chosen as optimal reaction time for MS2 gLAMP. The amplicon dot sizes showed no significant difference at low (1–20 copies per reaction) and medium template (20–200 copies per reaction) concentrations (Figure 2B). Under these conditions, amplicon dots were far from each other and had very limited interactions. The dot sizes represented the largest size that the amplicons could develop within the given reaction time, while the size variability in a single gel may result from variable initial template conformation, the degree of template denaturation or from local inhomogeneities in the hydrogel structure (due to the free dangling ends, self-looping, or entanglements of macromers). In contrast, the size of amplicon dots at high concentration (200–2000 copies per reaction) were significantly smaller than those formed at the low and medium concentrations (Figure 2B). Similar template concentration-dependent amplicon size variations were reported for an in-gel MDA assay.35 Xu et al. concluded that the smaller amplicon sizes at higher template concentration was due to a global autoinhibition, especially due to a drop in pH.35 In gLAMP, however, we think that local competition for enzymes, primers, and dNTP existed, as clear separations were developed among the amplicons close to each other (Figure 3G,H). The smaller amplicon sizes plus the clear boundaries developed at higher template concentration benefited the assay’s dynamic range by improving fluorescent dot identifiability. For smartphone camera reading, the optimal assay dynamic range was 1–1000 dots per reaction. When a fluorescence microscope was used for reading results, each gel can accommodate as many as 5000 dots without compromising the precision. Automatic amplicon analysis for microscope and smartphone images was realized by CellProfiler 2.2.0. The results of each key step are shown in Figure S3. With appropriate threshold settings, the difference between automatic and manual counting was less than 5%.
Figure 2.

gLAMP optimization. Effect of (A) reaction time and (B) template concentration on the size of QUASR gLAMP amplicon dots. Box plots with the original data of the amplicon dot diameters on the left side. Different letters indicate significant differences at the p < 0.05 level according to one-way ANOVA followed by a Tukey’s post-hoc test. Template concentration definition: low, 1–20 copies per reaction; medium, 20–200 copies per reaction; and high, 200–2000 copies per reaction. A medium template concentration was used in panel A, while the reaction time shown in panel B was 25 min. Extracted MS2 RNA was used as templates.
Figure 3.

Impact of template concentration on the size of gLAMP amplicon dots. (A, B) No template control; (C, D) low template concentration of 1–20 copies per reaction; (E, F) medium template concentration of 20–200 copies per reaction; and (G, H) high template concentration of 200–2000 copies per reaction. Top panel images were taken by an iPhone 6s Plus, while the bottom panel images were taken by fluorescent microscope for the same gel (scale bar of 1 mm). Extract MS2 RNA was used as templates, and the reaction time was 25 min.
For nucleic-acid-based detection methods, a simple DNA and RNA extraction procedure is preferred in point-of-use applications. In gLAMP analysis of MS2-spiked PBS solution, crude samples, samples after a simple heating (95 °C, 5 min) pretreatment, and samples extracted with commercial RNA extraction kit showed no significant differences in terms of amplicon dot counts (Figure S4, ANOVA, p > 0.05), dot sizes, and the amplicon fluorescence intensity. Simple heating pretreatment was previously reported to improve the detection of bacteria in LAMP assays because the compromised cell membranes were more-permeable to LAMP reagents and the denatured DNA can facilitate the strand displacement activity of the Bst enzyme.36 However, the current results indicate that the LAMP primers and enzymes (RTx reverse transcriptase and Bst 2.0 DNA polymerase) were able to penetrate the viral capsid at the reaction temperature (65 °C), and denaturing may not be necessary because the viral genome is much smaller compared to that of bacteria.
To evaluate the sensitivity of direct gLAMP, we compared it with traditional plaque assays and RT-qPCR (Figure 4). gLAMP amplicon counts showed a good correlation to plaque assay counts (R2 = 0.984, p < 0.05). The regression line (slope of 1.036 and intercept of −0.290) indicates that 1 gel amplicon dot was closely equal to 1 PFU. gLAMP achieved a similar lower limit of detection (0.7 plaque-forming units per reaction) compared to that of RT-qPCR (0.4 plaque-forming units per reaction), while RT-qPCR still showed the advantage of a larger upper detection limit. As discussed before, the dynamic range of gLAMP (1–1000 plaque-forming units per reaction) could be increased by reducing the amplicon dot sizes. Accommodating more amplicon dots in a single gel would be desirable for applications such as mutation detection and in-gel sequencing.37 However, the ability to distinguish amplicon dots from other contaminating fluorescent signals (i.e., autofluorescent substances) may suffer at small dot sizes. Consequently, the precision at low concentration (<20 plaque-forming units per reaction) would be compromised.16 Because high-concentration samples can be easily diluted, we think maintaining the precision at low concentration is more valuable than expanding the upper detection limit of gLAMP.
Figure 4.
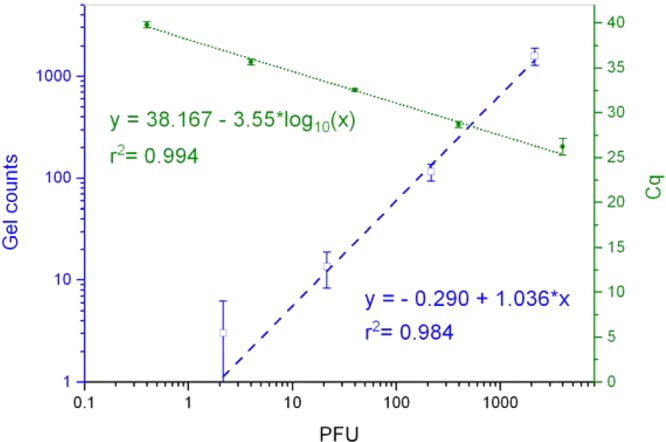
Direct detection of MS2 in PBS solution without RNA extraction. Correlation analysis indicates significant linear relationship between direct gLAMP counts with traditional plaque assay counts (r2 = 0.984, p < 0.05). A similar relationship was also found between log10-transformed plaque assay counts and the Cq values of RT-qPCR (r2 = 0.994, p < 0.05). Error bars represent the standard deviation of triplicate independent experiments.
Tolerance to Inhibitors
Enzyme-driven nucleic-acid amplification processes are susceptible to various inhibitory substances (e.g., organic matter and heavy metals) commonly found in environment samples.11 WW was yellow-brownish and had a chemical oxygen demand (COD) level of 821 mg/L, representing highly contaminated water. LW and PW were clear and contained fewer organic contaminants, with COD levels of 63 and 75 mg/L, respectively. gLAMP assays were successfully carried out in all MS2-spiked environmental water samples (spiking levels of 2 × 103 to 2 × 104 PFU/mL, equaling 10–100 plaque-forming units per reaction) without RNA extraction. No inhibition was observed because there were no significant differences between the environmental samples and the PBS control in terms of amplicon dot counts (p > 0.05) (Figure 5A) as well as dot morphologies. For in-tube real-time LAMP assay, no significant inhibition was found in LW and PW (p > 0.05). However, 4 out of 6 in-tube real-time LAMP assays were completely inhibited in WW, as no amplification was observed at the end of the reaction (60 min) (Figure 5B). For RT-qPCR, the assay was completely inhibited in WW because the Cq was beyond the lower limit of detection (Cqmax = 40) (Figure 5C). In general, LAMP assays have a more-robust chemistry than PCR in terms of handling complex crude samples because: (1) it employs six primers to initiate the amplification compared with two primers in PCR, (2) the smaller 67 kDa Bst polymerase may enter target cells and viral particles more easily than the 94 kDa Taq DNA polymerase used in PCR,38 and (3) the yields of LAMP (10–20 micrograms per reaction) are about 50–100 times higher than those of PCR (0.2 micrograms per reaction).22 Several studies have reported LAMP assays with crude samples.39,40 It should be noted that, similar to the in-tube real-time LAMP assay demonstrated in this study, many of these assays are qualitative or semiquantitative. The use of crude samples may not compromise the lower limit of detection (still detectable), but it usually resulted in an increase in the time-to-detection39 or a decrease in the signal-to-noise ratio40 at the end of the reaction. In RT-qPCR, similar delayed amplification would result in the increase of Cq values and, therefore, underestimate the target concentration.
Figure 5.

Direct detection of MS2 in spiked PBS, lake water (LW), pond water (PW), and wastewater (WW) without RNA extraction. (A) gLAMP counts, (B) time to detection in in-tube real-time LAMP, and (C) Cq values in RT-qPCR. Error bars represent standard errors of the means. Different letters indicate significant differences at the p < 0.05 level according to one-way ANOVA followed by a Tukey’s post-hoc test. MS2 was spiked at the concentration of 2 × 103 to 2 × 104 PFU/mL, equaling 10–100 plaque-forming units per reaction.
Figure S5 shows the EEM profiles of the LW, PW, and WW samples. The primary fluorescent DOM peaks for PW and WW were the C-peak and the M-peak, which were associated with humic-like components. The concentration of humic-like DOM in WW was 10–15 times higher than that in LW and PW, which is in agreement with the COD and DOC data. WW also contained low levels of proteinaceous material, as represented by the B-peak and the T-peak. Considering the source of WW, the inhibitors were likely to be organic in origin, similar to those found in urine and feces samples. Urea present in urine samples is known to prevent the noncovalent binding of polymerase enzymes and interferes with primer annealing.41 The inhibition concentration of urea in PCR was as low as 50 mM,42 while the tolerance of LAMP to urea was reported to be up to 1.8 M.43 However, the better performance of the gLAMP in WW cannot be simply attributed to a more-robust LAMP chemistry. In fact, we speculate that the gel matrix played a more-important role in the enhanced tolerance against inhibitors in WW. First, similar to digital PCR, gLAMP is an end-point amplification-detection assay, counting the final amplification products. Therefore, its quantification is less-affected by amplification efficiency. Second, because the DNA and RNA templates were spatially isolated, substrate competition during amplification should be minimized. Moreover, depending on their molecular weight, the movement of large-molecular-weight organic inhibitors would be restricted by the gel matrix, and thus, the local inhibitor concentrations close to the templates are reduced.
MS2 in Primary Effluent
MS2 was successfully detected in the RNA extracted from the primary effluent sample by gLAMP (7.8 ± 7.7 PFU/mL). A similar result was obtained in RT-qPCR (1.13 ± 0.98 PFU/mL), which confirms the sensitivity and specificity of the gLAMP assay. However, gLAMP detection of MS2 in the concentrated primary effluent sample without RNA extraction was failed due to the interference of co-concentrated autofluorescent substances (a highly fluorescent background). This issue could be alleviated by using other virus concentration methods (i.e., the adsorption–elution method);44,45 however, the optimization of the concentration step to facilitate the direct detection of MS2 in this specific sample is beyond the scope of this work. Culture-based plaque assays generated much higher counts using E. coli C3000 [(6.9 ± 0.4) × 103 PFU/mL] and E. coli Famp [(2.6 ± 0.7) × 103 PFU/mL] as host cells. The discrepancy was because the bacterial hosts used in culture-based plaque assays were susceptible to a wide range of coliphages contained in the sample, while the gLAMP and RT-qPCR assays were specific to MS2.
Perspectives of gLAMP in Environmental Monitoring
In a recent meta-analysis, Amarasiri et al. concluded that MS2 is the best validation and operational monitoring indicator for membrane bioreactors (MBR) because the log removal values (LRVs) of MS2 in MBR were shown to be lower than those of human enteric viruses, while other bacteriophages (T4, somatic, and F-specific) provided higher LRVs.46 MS2 may also be employed as a microbial tracer in field studies to understand the environmental fate of enteric viruses.47,48 The MS2 gLAMP assay, demonstrated in this study, can be readily used for these type of applications. In terms of using coliphages as indicators for fecal contamination, gLAMP assays targeting certain groups of coliphages would be more useful than an assay specific to MS2. It was suggested that F-specific RNA coliphage genogroups II (GII) and (GIII) are more frequently found in human excreta, while the other two genogroups (GI and GIV) are specific to animal excreta.49 Similar to RT-qPCR assays detecting individual F-specific RNA genogroup,49 the design of new gLAMP assays targeting similar genes is feasible in the future.
Currently, only one molecular-based method (U.S. EPA method 1611, qPCR for Enterococcus) has been certified for ambient water-quality analysis. The high capital investment and the complexity of data interpretation are likely the main challenges thwarting the application of molecular-based detection methods for routine microbial water quality analysis. Table S5 compares the gLAMP system with traditional culture-based plaque assays and the cutting-edge digital PCR system. As shown in the graphic abstract, gLAMP can be carried out with standard laboratory devices. A portable hand-held heating and fluorescence detection device is under development. Lyophilized LAMP reagents are also being tested to facilitate the field-scale applications. Moreover, gLAMP is noticeably faster than other available methods, taking less than 30 min compared with 4 h for RT-qPCR and 24 h for plaque assays. The amplified gel slides can be stored at room temperature for more than 1 month without affecting the florescent-dot visualization (Figure S6). This indicates that the gel matrix provides a good protection for the amplicons, which would allow for sample shipment in case further analysis is required. Considering its outstanding simplicity, sensitivity, rapidity, and versatility, the gLAMP system presented in this study holds great potential for microbial water-quality analysis, especially in resource-limited settings.
Acknowledgments
The authors acknowledge the financial support provided by the Bill and Melinda Gates Foundation (grant no. OPP1111252).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.8b00241.
Tables showing primer and probe sequences, water-quality parameters, and a comparison of microbial water-quality monitoring tools. Figures showing agarose gel electrophoresis, gLAMP detection of E. coli and Salmonella, automatic dot counting, the effect of sample pretreatment procedures on gLAMP counting, excitation emission fluorescent spectra, and the protection of gel matrix to the amplicon dots. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Montazeri N.; Goettert D.; Achberger E. C.; Johnson C. N.; Prinyawiwatkul W.; Janes M. E. Pathogenic enteric viruses and microbial indicators during secondary treatment of municipal wastewater. Appl. Environ. Microbiol. 2015, 81 (18), 6436–6445. 10.1128/AEM.01218-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc H.; Edberg S.; Pierzo V.; Delattre J. Bacteriophages as indicators of enteric viruses and public health risk in groundwaters. J. Appl. Microbiol. 2000, 88 (1), 5–21. 10.1046/j.1365-2672.2000.00949.x. [DOI] [PubMed] [Google Scholar]
- Bell K. Y.; da Silva T. A.. Status of EPA Development of New Ambient Water Quality Criteria for Bacteriophage; IUVA News: Bethesda, MD, 18( (2), ), 2016, . [Google Scholar]
- Worley-Morse T. O.; Mann M. A.; Gonzalez R.. Potential USEPA Coliphages Criteria: Key Knowledge Gaps and Coliphage Fate in Conventional and Advanced WRRFs. In Proceedings of the Water Environment Federation; WEFTEC: Alexandria, VA, 1133–1139, 2017. (Session 220 through Session 227). [Google Scholar]
- Attinti R.; Wei J.; Kniel K.; Sims J. T.; Jin Y. Virus’(MS2, φX174, and Aichi) attachment on sand measured by atomic force microscopy and their transport through sand columns. Environ. Sci. Technol. 2010, 44 (7), 2426–2432. 10.1021/es903221p. [DOI] [PubMed] [Google Scholar]
- Pype M.-L.; Lawrence M. G.; Keller J.; Gernjak W. Reverse osmosis integrity monitoring in water reuse: The challenge to verify virus removal–A review. Water Res. 2016, 98, 384–395. 10.1016/j.watres.2016.04.040. [DOI] [PubMed] [Google Scholar]
- Sun P.; Tyree C.; Huang C.-H. Inactivation of Escherichia coli, bacteriophage MS2, and Bacillus spores under UV/H2O2 and UV/peroxydisulfate advanced disinfection conditions. Environ. Sci. Technol. 2016, 50 (8), 4448–4458. 10.1021/acs.est.5b06097. [DOI] [PubMed] [Google Scholar]
- Huang X.; Qu Y.; Cid C. A.; Finke C.; Hoffmann M. R.; Lim K.; Jiang S. C. Electrochemical disinfection of toilet wastewater using wastewater electrolysis cell. Water Res. 2016, 92, 164–172. 10.1016/j.watres.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniesa M.; Ballesté E.; Imamovic L.; Pascual-Benito M.; Toribio-Avedillo D.; Lucena F.; Blanch A.; Jofre J. Bluephage: A rapid method for the detection of somatic coliphages used as indicators of fecal pollution in water. Water Res. 2018, 128, 10–19. 10.1016/j.watres.2017.10.030. [DOI] [PubMed] [Google Scholar]
- Gunda N. S. K.; Mitra S. K. Rapid Water Quality Monitoring for Microbial Contamination. Electrochem. Soc. Interface 2016, 25 (4), 73–78. 10.1149/2.F06164if. [DOI] [Google Scholar]
- Cao Y.; Raith M. R.; Griffith J. F. Droplet digital PCR for simultaneous quantification of general and human-associated fecal indicators for water quality assessment. Water Res. 2015, 70, 337–349. 10.1016/j.watres.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Sivaganesan M.; Siefring S.; Varma M.; Haugland R. A. MPN estimation of qPCR target sequence recoveries from whole cell calibrator samples. J. Microbiol. Methods 2011, 87 (3), 343–349. 10.1016/j.mimet.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Yang R.; Paparini A.; Monis P.; Ryan U. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples. Int. J. Parasitol. 2014, 44 (14), 1105–1113. 10.1016/j.ijpara.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Lowe S. B.; Gooding J. J. Brief review of monitoring methods for loop-mediated isothermal amplification (LAMP). Biosens. Bioelectron. 2014, 61, 491–499. 10.1016/j.bios.2014.05.039. [DOI] [PubMed] [Google Scholar]
- Kolm C.; Martzy R.; Brunner K.; Mach R. L.; Krska R.; Heinze G.; Sommer R.; Reischer G. H.; Farnleitner A. H. A Complementary Isothermal Amplification Method to the US EPA Quantitative Polymerase Chain Reaction Approach for the Detection of Enterococci in Environmental Waters. Environ. Sci. Technol. 2017, 51 (12), 7028–7035. 10.1021/acs.est.7b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Brito I. L.; Alm E. J.; Blainey P. C. Virtual microfluidics for digital quantification and single-cell sequencing. Nat. Methods 2016, 13 (9), 759–762. 10.1038/nmeth.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsen M. G.; Kool E. T. The Discovery of Rolling Circle Amplification and Rolling Circle Transcription. Acc. Chem. Res. 2016, 49 (11), 2540–2550. 10.1021/acs.accounts.6b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craw P.; Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip 2012, 12 (14), 2469–2486. 10.1039/c2lc40100b. [DOI] [PubMed] [Google Scholar]
- Notomi T.; Okayama H.; Masubuchi H.; Yonekawa T.; Watanabe K.; Amino N.; Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28 (12), e63. 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priye A.; Bird S. W.; Light Y. K.; Ball C. S.; Negrete O. A.; Meagher R. J. A smartphone-based diagnostic platform for rapid detection of Zika, chikungunya, and dengue viruses. Sci. Rep. 2017, 7, 44778. 10.1038/srep44778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martzy R.; Kolm C.; Brunner K.; Mach R. L.; Krska R.; Šinkovec H.; Sommer R.; Farnleitner A. H.; Reischer G. H. A loop-mediated isothermal amplification (LAMP) assay for the rapid detection of Enterococcus spp. in water. Water Res. 2017, 122, 62–69. 10.1016/j.watres.2017.05.023. [DOI] [PubMed] [Google Scholar]
- Parida M.; Sannarangaiah S.; Dash P. K.; Rao P.; Morita K. Loop mediated isothermal amplification (LAMP): a new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol. 2008, 18 (6), 407–421. 10.1002/rmv.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njiru Z. K. Loop-mediated isothermal amplification technology: towards point of care diagnostics. PLoS Neglected Trop. Dis. 2012, 6 (6), e1572. 10.1371/journal.pntd.0001572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedtfeld R. D.; Stedtfeld T. M.; Kronlein M.; Seyrig G.; Steffan R. J.; Cupples A. M.; Hashsham S. A. DNA extraction-free quantification of Dehalococcoides spp. in groundwater using a hand-held device. Environ. Sci. Technol. 2014, 48 (23), 13855–13863. 10.1021/es503472h. [DOI] [PubMed] [Google Scholar]
- Schoepp N. G.; Schlappi T. S.; Curtis M. S.; Butkovich S. S.; Miller S.; Humphries R. M.; Ismagilov R. F. Rapid pathogen-specific phenotypic antibiotic susceptibility testing using digital LAMP quantification in clinical samples. Sci. Transl. Med. 2017, 9 (410), eaal3693. 10.1126/scitranslmed.aal3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler F.; Siber C.; Hin S.; Wadle S.; Paust N.; Zengerle R.; von Stetten F. Digital droplet LAMP as a microfluidic app on standard laboratory devices. Anal. Methods 2016, 8 (13), 2750–2755. 10.1039/C6AY00600K. [DOI] [Google Scholar]
- Nuovo G. J.PCR in situ hybridization: protocols and applications; Raven Press: New York, 1994. [Google Scholar]
- Heo J.; Thomas K. J.; Seong G. H.; Crooks R. M. A Microfluidic Bioreactor Based on Hydrogel-Entrapped E. c oli: Cell Viability, Lysis, and Intracellular Enzyme Reactions. Anal. Chem. 2003, 75 (1), 22–26. 10.1021/ac0259717. [DOI] [PubMed] [Google Scholar]
- Mitra R. D.; Church G. M. In situ localized amplification and contact replication of many individual DNA molecules. Nucleic Acids Res. 1999, 27 (24), e34–e39. 10.1093/nar/27.24.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropinski A. M.; Mazzocco A.; Waddell T. E.; Lingohr E.; Johnson R. P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol. 2009, 501, 69–76. 10.1007/978-1-60327-164-6_7. [DOI] [PubMed] [Google Scholar]
- Kropinski A. M., Mazzocco A., Waddell T. E., Lingohr E., Johnson R. P.. Enumeration of Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages. Methods in Molecular Biology; Clokie M. R., Kropinski A. M., Eds.; Humana Press: New York, NY, 2009; vol 501. [DOI] [PubMed] [Google Scholar]
- Cho K.; Qu Y.; Kwon D.; Zhang H.; Cid C. A.; Aryanfar A.; Hoffmann M. R. Effects of Anodic Potential and Chloride Ion on Overall Reactivity in Electrochemical Reactors Designed for Solar-Powered Wastewater Treatment. Environ. Sci. Technol. 2014, 48 (4), 2377–2384. 10.1021/es404137u. [DOI] [PubMed] [Google Scholar]
- Raeber G.; Lutolf M.; Hubbell J. Molecularly engineered PEG hydrogels: a novel model system for proteolytically mediated cell migration. Biophys. J. 2005, 89 (2), 1374–1388. 10.1529/biophysj.104.050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen N. C. Apparent pore size of polyacrylamide gels: Comparison of gels cast and run in Tris-acetate-EDTA and Tris-borate-EDTA buffers. Electrophoresis 1998, 19 (10), 1542–1547. 10.1002/elps.1150191004. [DOI] [PubMed] [Google Scholar]
- Xu P.; Janex M.-L.; Savoye P.; Cockx A.; Lazarova V. Wastewater disinfection by ozone: main parameters for process design. Water Res. 2002, 36 (4), 1043–1055. 10.1016/S0043-1354(01)00298-6. [DOI] [PubMed] [Google Scholar]
- Tomita N.; Mori Y.; Kanda H.; Notomi T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 2008, 3 (5), 877–882. 10.1038/nprot.2008.57. [DOI] [PubMed] [Google Scholar]
- Shendure J.; Porreca G. J.; Reppas N. B.; Lin X.; McCutcheon J. P.; Rosenbaum A. M.; Wang M. D.; Zhang K.; Mitra R. D.; Church G. M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309 (5741), 1728–1732. [DOI] [PubMed] [Google Scholar]
- Maruyama F.; Kenzaka T.; Yamaguchi N.; Tani K.; Nasu M. Detection of bacteria carrying the stx2 gene by in situ loop-mediated isothermal amplification. Appl. Environ. Microbiol. 2003, 69 (8), 5023–5028. 10.1128/AEM.69.8.5023-5028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R.; Fukuta S.; Matsumoto Y.; Hasegawa T.; Kojima H.; Hotta M.; Miyake N. Development of reverse transcription loop-mediated isothermal amplification assay as a simple detection method of Chrysanthemum stem necrosis virus in chrysanthemum and tomato. J. Virol. Methods 2016, 236, 29–34. 10.1016/j.jviromet.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Patterson A. S.; Heithoff D. M.; Ferguson B. S.; Soh H. T.; Mahan M. J.; Plaxco K. W. Microfluidic chip-based detection and intraspecies strain discrimination of Salmonella serovars derived from whole blood of septic mice. Appl. Environ. Microbiol. 2013, 79 (7), 2302–2311. 10.1128/AEM.03882-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedman J., Rådström P.. Overcoming Inhibition in Real-Time Diagnostic PCR. In PCR Detection of Microbial Pathogens. Methods in Molecular Biology (Methods and Protocols); Wilks M., Ed.; Humana Press: Totowa, NJ, 2013; vol 943. [DOI] [PubMed] [Google Scholar]
- Khan G.; Kangro H.; Coates P.; Heath R. Inhibitory effects of urine on the polymerase chain reaction for cytomegalovirus DNA. J. Clin. Pathol. 1991, 44 (5), 360–365. 10.1136/jcp.44.5.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T.; Burke P. A.; Smalley H. B.; Gillies L.; Hobbs G. Loop-mediated isothermal amplification test for detection of Neisseria gonorrhoeae in urine samples and tolerance of the assay to the presence of urea. Journal of clinical microbiology 2014, 52 (6), 2163–2165. 10.1128/JCM.00314-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J.; Audicana A.; Isern A.; Llaneza J.; Moreno B.; Tarancón M. a. L.; Jofre J.; Lucena F. Standardised evaluation of the performance of a simple membrane filtration-elution method to concentrate bacteriophages from drinking water. J. Virol. Methods 2004, 117 (1), 19–25. 10.1016/j.jviromet.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Li D.; Shi H.-c.; Jiang S. C. Concentration of viruses from environmental waters using nanoalumina fiber filters. J. Microbiol. Methods 2010, 81 (1), 33–38. 10.1016/j.mimet.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Amarasiri M.; Kitajima M.; Nguyen T. H.; Okabe S.; Sano D. Bacteriophage removal efficiency as a validation and operational monitoring tool for virus reduction in wastewater reclamation. Water Res. 2017, 121, 258–269. 10.1016/j.watres.2017.05.035. [DOI] [PubMed] [Google Scholar]
- Pang L.; Close M.; Goltz M.; Noonan M.; Sinton L. Filtration and transport of Bacillus subtilis spores and the F-RNA phage MS2 in a coarse alluvial gravel aquifer: Implications in the estimation of setback distances. J. Contam. Hydrol. 2005, 77 (3), 165–194. 10.1016/j.jconhyd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Wall K.; Pang L.; Sinton L.; Close M. Transport and attenuation of microbial tracers and effluent microorganisms in saturated pumice sand aquifer material. Water, Air, Soil Pollut. 2008, 188 (1–4), 213–224. 10.1007/s11270-007-9537-3. [DOI] [Google Scholar]
- Ogorzaly L.; Gantzer C. Development of real-time RT-PCR methods for specific detection of F-specific RNA bacteriophage genogroups: application to urban raw wastewater. J. Virol. Methods 2006, 138 (1–2), 131–139. 10.1016/j.jviromet.2006.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.