

ABSTRACT
The antigenic makeup of tumour cells can have a profound effect on the progression of cancer and success of immunotherapies. Therefore, one strategy to improve the efficacy of cancer treatments is to augment the antigens displayed by tumours. The present study explores how the recognition of tumour cells may be altered by non-cytotoxic concentrations of gemcitabine (GEM). Testing a panel of chemotherapeutics in human cancer cell lines in vitro, it was found that GEM increased surface expression of HLA-A,B,C and that underlying this were specific increases in β-2-microglobulin and immunoproteasome subunit proteins. Furthermore, the peptide antigen repertoire displayed on HLA class I was altered, revealing a number of novel antigens, many of which that were derived from proteins involved in the DNA-damage response. Changes in the nature of the peptide antigens eluted from HLA-A,B,C after GEM treatment consisted of amino acid anchor-residue modifications and changes in peptide length which rendered peptides likely to favour alternative HLA-alleles and increased their predicted immunogenicity. Signalling through the MAPK/ERK and NFκB/RelB pathways was associated with these changes. These data may explain observations made in previous in vivo studies, advise as to which antigens should be used in future vaccination protocols and reinforce the idea that chemotherapy and immunotherapy could be used in combination.
Introduction
The recognition of antigenic molecules on the surface of tumour cells plays an important role in CD8+ T-cell-mediated clearance of cancer. Part of this recognition is dependent upon the MHC class I-antigen complex which indicates the health of the cell by displaying signs of aberrant protein expression to the immune system. Loss of MHC class I can hide tumour cells from the adaptive immune response, preventing detection by removing antigenic evidence of cancer-related proteins from the plasma membrane. HLA class I downregulation has been observed in numerous human tumour types, 1-4 and can be mediated through defects in α-heavy chain or β-2-microglobulin (β2 m). 5 , 6 Downregulation of MHC class I is strongly associated with poor prognosis in cancer, 7-9 and so reversing this is a promising strategy to enhance or reengage an anti-cancer immune response, especially in cancers characterised by low MHC class I, such as colorectal cancer. 10 MHC class I expression has been shown to be increased on tumour cells in response to stress stimuli, including chemotherapeutic treatments such as 5-fluoracil (5-FU) and gemcitabine (GEM). 11 , 12
In addition to the absolute level of MHC class I, the peptide antigens expressed in conjunction with MHC class I are vital in the detection of cancer by immune cells and as such, antigen-specific tumour immunotherapy will be enhanced by the identification of putative tumour-associated immunogenic HLA-ligands. Many factors influence the make-up of these peptide ligands but an important part of this process is the cleavage of peptide bonds which can be catalysed by constitutively expressed proteasomal subunits or the interferon (IFN)-γ-inducible immunoproteasomal subunits LMP2 (β1i), MECL-1 (β2i) and LMP7 (β5i). 13 , 14 Compared with their constitutively expressed counterparts, immunoproteasomal subunits confer increased trypsin and chymotrypsin-like activity and generate peptides with distinctive C-termini. 15-17
GEM is a nucleoside analogue that has a broad spectrum of anti-tumour activity against solid tumours, it exerts its antiproliferative effects via “masked-termination” of DNA replication and targeting of ribonucleotide reductase, an enzyme required for DNA replication and repair. 18 GEM has been successfully combined with a number of different immunotherapies in cancer. It is reported that GEM improves dendritic cell (DC) vaccination in clinical and pre-clinical settings, possibly by encouraging a cytotoxic T-cell response against subdominant immune epitopes. 19-23 GEM selectively removes myeloid-derived suppressor cells (MDSC) in mice; 24 , 25 and this may link to the potentiation of immunotherapy that is observed in combination with GEM. However, this has not been extensively studied in humans where there are conflicting reports on the ability of treatments involving GEM to reduce the percentage of Lin−DR−CD11b+ MDSC in patients with advanced adenocarcinoma. 26 GEM is not associated with suppression of lymphatic activity in cancer patients, 27-29 and is shown to expand the T-lymphocyte subset and increase tumour infiltration in mice by enhancing cross-priming of tumour-specific CD8+ T-cells. 30 Additionally, GEM increases the absolute numbers and percentage of peripheral CD14+ monocytes and DCs in pancreatic cancer patients, 31 and in mice broadens the range of tumour antigens seen by CD8+ T-cells by shifting the CD8+ T-cell response towards subdominant epitopes. 32
Considering the capacity of GEM to upregulate MHC class I on the surface of tumour cells in in vitro and in vivo settings; 12 and the coordinated regulation of MHC class I and the antigen processing machinery (APM), we suggested that in addition to influencing MHC class I expression, other changes in antigen presentation may be caused GEM. In the present study we confirm GEM-mediated upregulation of cell surface HLA-A,B,C and demonstrate that this is influenced by altered expression of β2 m. Moreover, consistent with our hypothesis, GEM also induced upregulation of immunoproteasomal subunits and altered the peptide antigens displayed by tumour cells in in vitro cell cultures.
Results
GEM altered expression of HLA-A,B,C at the surface of tumour cells
Surface expression of HLA-A,B,C was measured on a panel of tumour cell lines after culturing with equi-active concentrations of chemotherapeutic drugs for 24 hours. Representative plots are shown in Fig. 1a. GEM significantly increased expression of HLA-A,B,C in all three cell lines (Fig. 1b). The mean change in HLA-A,B,C expression in response to culture with GEM, as measured by a change in MFI from untreated controls, was 81.3% for HCT116 cells, 67.7% for A549 cells and 41.5% for MCF-7 cells. For comparison, 1000 IU/ml IFNγ increased HLA-A,B,C by 62.2% in HCT116 cells, 142.9% in A549 cells and 367% in MCF-7 cells (data not shown). Cyclophosphamide (CPM) did not influence HLA-A,B,C expression while oxaliplatin (OXP) had no effect on expression of HLA-A,B,C in A549 and HCT116 cells but reduced expression in MCF-7 cells by 24.8%. Increased surface expression of HLA-A,B,C was associated with increased levels of intracellular β2 m but not α-heavy chain proteins. Western blots showed that the amount of β2 m protein detected increased in response to GEM in all three cell lines (Fig. 1c). In contrast expression of HLA class I α-heavy chains A, B and C were not altered by GEM treatment. Real-time PCR and microarray data, performed only on HCT116 cells (Fig. 1d and e), suggested that the increase in β2 m may be partially due to increased gene transcription. These gene analyses also corroboratively indicated that α-heavy chain expression remained unchanged by culturing with GEM. Transfection of A549, HCT116 and MCF-7 cells with a β2 m-expressing plasmid was sufficient to increase surface expression of HLA-A,B,C (Fig. 1f). Generally, transfected cells displayed a smaller increase in HLA-class I compared to GEM-treated cells.
Figure 1.
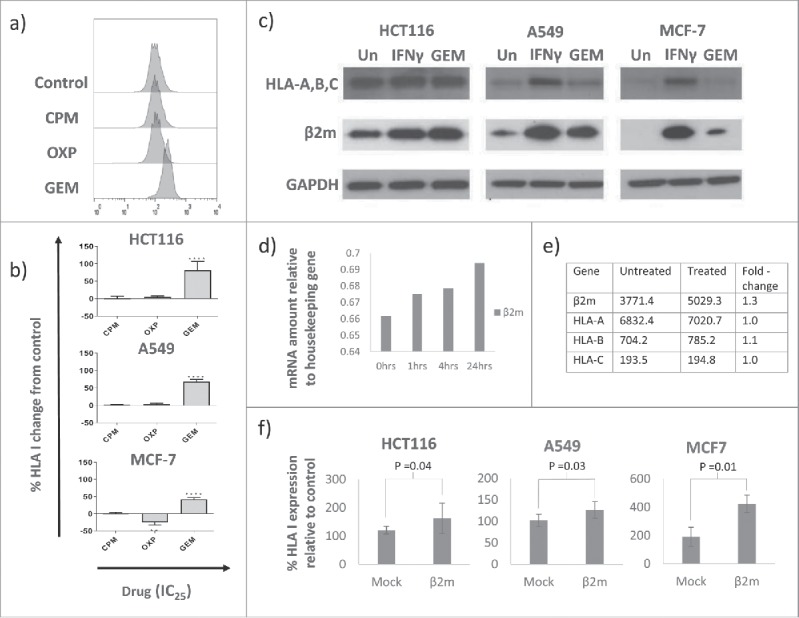
GEM increased HLA class I expression on tumour cells in a β2 m-associated manner. a) Representative histograms showing change in HLA-A,B,C MFI in response to culture with drugs at IC25. b) The effect of chemotherapy drugs on surface expression of HLA-A,B,C on tumour cells as measured by flow cytometry. Data are expressed relative to untreated controls. Mean and standard deviation is plotted and values are significantly different (**** = p<0.0001, ** = p<0.01) to controls by one-way ANOVA with Dunnett's multiple comparisons test. n = 3. c) Blots representative of three experiments showing expression of HLA α-heavy chains and β2 m proteins in untreated (Un), IFNγ-treated (1000 IU/ml) or GEM-treated (100 nM) tumour cells. d) Transcription of the β2 m gene was increased in HCT116 cells in response to GEM, as assessed by qPCR. n = 1. e) Mean fold-change in mRNA for HLA class I genes in response to GEM. n = 3. f) Tumour cells transfected with human β2 m expressing plasmid had increased surface expression of HLA class I 48 hours after transfection, as measured by flow cytometry. Means and standard deviations are plotted and mock-transfected and β2 m-transfected are significantly different by student's paired t-test. For A549 and MCF-7, n = 3, for HCT116 n = 6.
Immunoproteasome subunits are expressed upon treatment with GEM
In addition to changes in HLA-A,B,C, expression of the immunoproteasome catalytic subunits LMP2 and MECL-1 was also induced by GEM in all three cell lines (Fig. 2a). Expression of the third immunoproteasome subunit, LMP7, was not detectably increased at this concentration and time-point. Proteasome pull-outs from HCT116 cells showed that LMP2 and MECL-1 were incorporated into proteasomes in increasing amounts after GEM treatment. Fig. 2b shows expression of the constitutive proteasome subunits β1, β2 and β5, the immunoproteasome subunits LMP2, LMP7 and MECL-1, and α2, which is present in both forms of proteasome, in control and HCT116 cells treated with GEM for 24 hours. Shown are the relative amounts of each protein in crude cell lysate, unbound and proteasomal fractions. LMP2 and MECL-1 coimmunoprecipitated with proteasomes in GEM-treated cells, LMP7 was also present in this fraction. Subunit α2 was used as a loading control, indicating the total amount of proteasome present. There was little change in the expression of the constitutive proteasome subunits, β1, β2 and β5 in the crude lysate or proteasomal fractions.
Figure 2.
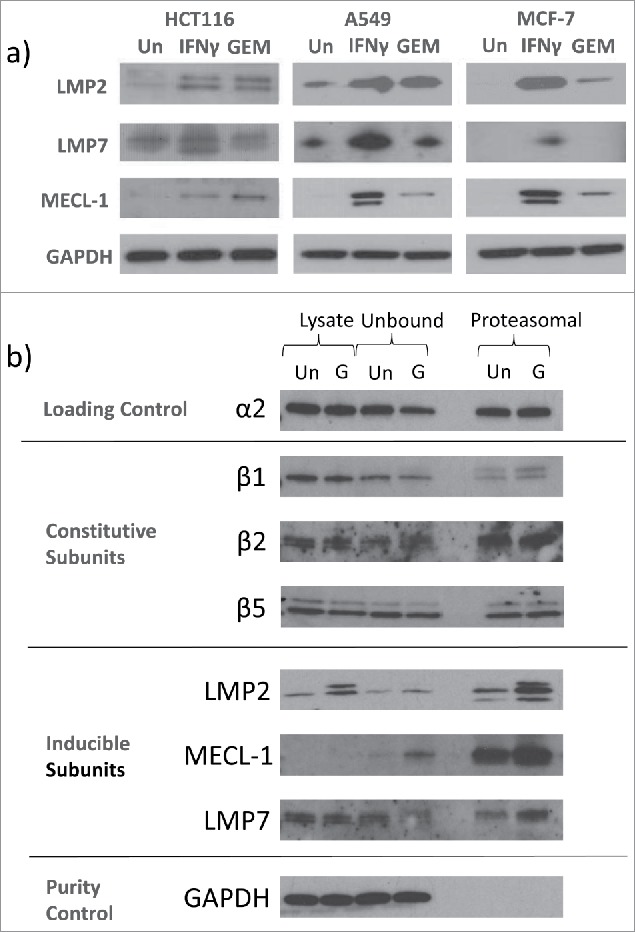
GEM induced immunoproteasome subunits in tumour cells. a) Immunoproteasome expression was assessed by Western blot after 24 hour culture with 100 nM GEM. IFNγ (1000 IU/ml) was used as a positive control. Blots are representative of three separate experiments. b) Proteasomes were isolated from HCT116 cells either untreated (Un) or treated with 100 nM GEM (G) for 24 hours. Expression of proteasome and immunoproteasome subunits in crude cell lysate, unbound and proteasome pull-out fractions was then measured. Representative blots from three separate experiments are shown.
The types of peptides displayed on HLA-A,B,C are altered by GEM
The induction of LMP2 and MECL-1 in tumour cells in response to culture with GEM suggests that there may also be changes to the HLA-expressed peptidome of these cells. To assess this, HCT116 cells were cultured alone or with GEM and peptides eluted from HLA-A,B,C molecules before being sequenced by mass spectrometry. The sequences derived were analysed by comparing the peptide antigens present only on the surface of the GEM-treated cells versus those found on only control cells.
In the presence of GEM, the peptide repertoire displayed on the surface of tumour cells was altered. There was a noticeable change in the length of peptides, with 8-mers making up 9.8% of peptides exclusively displayed on control cells but 17.1% of peptides exclusively displayed on GEM-treated cells. In addition, there were fewer peptides with a length of 10 amino acids or more after GEM treatment (26.2% versus 16.1%) (Fig. 3a). GEM also increased the proportion of leucine and phenylalanine residues at the C-terminus of the peptide sequences (Fig. 3b). Of the peptides that appeared only on control cells, 7.6% had C-terminal leucine. For peptides that appeared only after GEM treatment, this was increased to 12.2%. The proportion of phenylalanine residues at the C-terminus was increased to an even greater extent after GEM-treatment, from 2.2% to 8.0% and this change reached statistical significance. Conversely, after treatment with GEM the numbers of peptides with alanine or proline at the C-terminus was decreased, from 27.8% to 13.2% and 10.1% to 5.0%, respectively, the latter reaching statistical significance.
Figure 3.

Peptide ligands eluted from HLA-A,B,C on HCT116 cells treated with 100 nM GEM were different from those found on untreated control cells. All panels represent mean and standard deviation values of three separate peptide elution experiments and show differences in the proportion of peptides with various characteristics between those found exclusively on treated or exclusively on control cells in terms of: a) Peptide length, b) C-terminal amino acid, c) Predicted HLA-allele binding, d) Predicted immunogenicity. Values significant different from controls by student's t-test are indicated (** = p<0.01). Due to the anomalous appearance of a number of 11 and 12mer peptides in one of the three peptide elution experiments, data from this particular experiment was removed from the analysis of a) and replaced by peptide length data from a small pilot study.
HLA-binding preferences for individual peptides were analysed using HLA binding prediction servers IEDB analysis resource and SYFPEITHI. The percentage of peptides predicted to bind most strongly to HLA-B*18:01 was larger in the GEM-treated group than the untreated group (Fig. 3c). On average, 10.6% of peptides were predicted to bind to HLA-B*18:01 in control cells but this was increased to 24.9% in GEM-treated cells. Conversely, the percentage of peptides predicted to bind most strongly to HLA-B*45:01 decreased from 38.7% in control cells to 18.7% in GEM-treated cells. This preference for peptides to bind HLA-B*18:01 after GEM was underpinned by changes in the anchor residues occurring at binding position P2 and was especially true for peptides with an F or L residue at the C-terminal position which were now more likely to be associated with an acidic amino acid at the P2 position (Supplemental Figure S1). A prediction model was used to assess the immunogenicity of the eluted peptides. 33 Peptides displayed on HLA class I exclusively after culture with GEM were, on average, predicted to be more immunogenic than those peptides found on control cells (Fig. 3d).
GEM treatment causes new protein representation in the peptidome
Eluted peptides were used to identify proteins expressed in HCT116 cells and then we asked whether new epitopes were generated from within these proteins in the presence of GEM. Table 1 lists a sample of proteins from which the peptidome of HCT116 cells was derived. Only proteins with novel GEM-exclusive peptides found in at least two of three peptide elution experiments are shown.
Table 1.
How proteins were represented in the immunopeptidome of HCT116 cells was altered by GEM, which induced new peptide antigens from proteins which were displayed on HLA-A,B,C. In addition to proteins having altered peptide representation after GEM treatment, new proteins were also represented in the immunopeptidome of HCT116 cells after GEM treatment. Microarray data is also shown indicating the relative amount of mRNA present for each gene. A sample of proteins with consistently expressed GEM-novel peptides in at least two peptide elution experiments are shown.
| Protein | Peptide(s) from Control | Peptide(s) from Treated | Microarray score Control | Microarray score Treated |
|---|---|---|---|---|
| Oncoprotein Mdm2 | SEQETLVRP | SEQETLVRP | 144.1 | 207.5 |
| DEVYQVTVY | ||||
| YTMKEVLFY | ||||
| DEKQQHIVY | ||||
| Replication protein A 70 kDa DNA-binding subunit | AEAILGQNAA | AEAILGQNAA | 2977.9 | 3072.2 |
| DTEFPNFKY | DTEFPNFKY | |||
| TEFPNFKY | ||||
| Pyruvate dehydrogenase [acetyl-transferring]]-phosphatase 1 | VGDPNSFLNY | VGDPNSFLNY | 1672.9 | 1389.5 |
| SLLPHETLL | ||||
| DNA Topoisomerase 1 | SQIEADFRL | SQIEADFRL | 390.7 | 365.5 |
| YLDPRITVA | ||||
| Cullin-9 | LLLDLERVL | 189.4 | 211.8 | |
| Kanadaptin | EENPIVLEF | 255.8 | 226.8 | |
| Serine/threonine-protein kinase PLK2 | NEDRISTTF | 235.9 | 246.3 | |
| Exosome component 10 | DEYDFYRSF | 1608.8 | 1580.5 | |
| Spermatogenesis-associated serine-rich protein 2 | SSEKGGMNGY | 864.9 | 715.0 | |
| Dynamin-2 | TLIDLPGITKV | 292.8 | 286.8 | |
| ETERIVTTY | ||||
| Antigen peptide transporter 1 (TAP 1) | LLYESPERY | 726.0 | 1235.8 | |
| Sestrin-1 | SLAELVHAV | 212.0 | 639.4 | |
| QMDGPLPLHY |
The peptide SEQETLVRP from the oncoprotein Mdm2, was found before and after GEM treatment. However, after cells were cultured with GEM, additional peptides from Mdm2 were also detected in the immunopeptidome of HCT116 cells: DEVYQVTVY, YTMKEVLFY and DEKQQHIVY. Using peptide spectral matches (PSMs) as a measure of abundance, SEQETLVRP made up 0.26% of peptides eluted from control cells but 0.56% in GEM-treated cells (Fig. 4a). Intracellular expression of MDM2 remained unchanged by GEM treatment (Fig. 4c), though transcription may have increased, 144.1 versus 207.5 units, measured by microarray.
Figure 4.
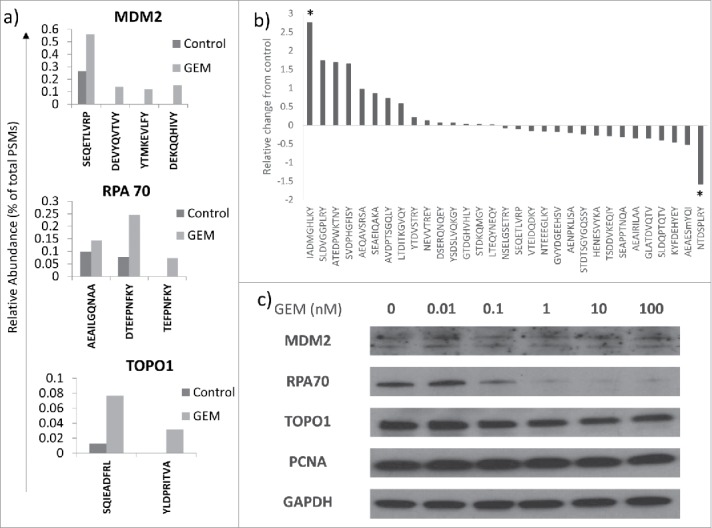
GEM alters the immunopeptidome of tumour cells. a) Relative abundance of peptides from MDM2, RPA70 and TOPO1 proteins was assessed in control and GEM-treated HCT116 cells. n = 2. b) The relative abundance of all peptides present in both control and GEM treated cells in all three experiments is shown. Values significantly different from controls by student's paired t-test are indicated (* = p<0.05). c) Western blots showing the expression of proteins that have an altered peptide representation in GEM-treated cells are shown. Blots are representative of three separate experiments.
The presentation of new antigens to the immune system was also observed with other proteins, including DNA Topoisomerase 1 (TOPO1). Expression of TOPO1 decreased slightly in the presence of GEM, as assessed by microarray (390.7 versus 365.5) and Western blot (Fig. 4c), but an additional peptide, YLDPRITVA, was generated in GEM-treated cells (Table 1). Interestingly, in control cells peptides from TOPO1 made up only 0.013% of total peptides but with GEM treatment this was increased nearly tenfold to 0.11% (Fig. 4a). These data imply that expression level alone does not drive the appearance of new epitopes within individual proteins. As well as an increasing diversity of peptides from a particular protein, peptide antigens from proteins not represented on HLA-A,B,C at the surface of control HCT116 cells also appeared after culturing cells with GEM. For example, the lung cancer oncoprotein, kanadaptin, was not represented in the immunopeptidome of HCT116 cells under basal conditions, however, upon culture with GEM the peptide EENPIVLEF was displayed on HLA class I (Table 1). No peptide exclusive to GEM treatment was found in all three peptide elution experiments.
However, there were 31 peptide sequences present on both control and GEM-treated HCT116 cells that were conserved between all three experimental repeats. The relative fold-change in the abundance of these peptides in response to GEM (as measured by changes in PSMs) is shown in Fig. 4b. Two peptides showed a fold-difference greater than two and were statistically different from no change, NTDSPLRY, from 40 S ribosomal protein SA, which was downregulated in response to GEM treatment, and IADMGHLKY, from the protein proliferating cell nuclear antigen (PCNA), which was upregulated. The intracellular expression of PCNA was unchanged by GEM treatment (Fig. 4c).
Of the proteins measured by Western blot, only RPA70 showed major changes in expression in response to GEM treatment (Fig. 4c). The loss of RPA70 protein alongside increases in the amount of RPA70 peptides at the surface of the cell upon GEM treatment may suggest increased degradation of this protein as part of the response to GEM.
Table 2 lists the peptides found exclusively after GEM treatment in more than one of the peptide elution experiments. References to these sequences were then investigated in the scientific literature. Of 86 GEM-specific peptides conserved between two experiments, 53 (61.63%) were novel (no reference could be found), whereas 33 had been reported previously.
Table 2.
References to the sequences of peptides exclusive to GEM treatment and conserved in two of three experiments were searched for in the scientific literature. “Novel“ indicates that the present study is the first time the peptide has been reported. Otherwise, peptides found previously in: A = Patent - Cytotoxic T-lymphocyte-inducing immunogens for prevention treatment and diagnosis of cancer, 56 B = Patent - Comparative ligand mapping from MHC class I positive cells, 57 C = Toward a Definition of Self: Proteomic Evaluation of the Class I Peptide Repertoire, 58 D = Characterization of spontaneous tumor antigen-reactive T cell responses in melanoma patients and treatment of human melanoma with optimized T cell receptor transgenic T cells in a xenotransplantation model, 59 E = Patent - MHC molecule-binding tumor-associated peptides, 60 F = Features of TAP-independent MHC class I ligands revealed by quantitative mass spectrometry. 61 IEDB = Found in the immune epitope database.
| Peptide | Protein | Description | |
|---|---|---|---|
| 1 | LSLENLEKI | Phosphatidylinositide phosphatase SAC2 | A (2008) |
| 2 | NEDRISTTF | Serine/threonine-protein kinase PLK2 | Novel |
| 3 | DEYDFYRSF | Exosome component 10. | B and C |
| 4 | SLAELVHAV | p53 regulated PA26 nuclear protein | Known immune epitope IEDB |
| 5 | DEFEFLEKA | E3 ubiquitin/ISG15 ligase TRIM25 | B and C |
| 6 | DEVYQVTVY | E3 ubiquitin-protein ligase Mdm2 | B and known CLL ligand |
| 7 | YTMKEVLFY | E3 ubiquitin-protein ligase Mdm2 | Novel |
| 8 | DEGLIIHVF | Protein kinase C, zeta | Novel |
| 9 | SSEKGGMNGY | Spermatogenesis-associated serine-rich protein 2 | Novel |
| 10 | VVEQLKDWLY | MAD2 mitotic arrest deficient-like 1 | Novel |
| 11 | MEVEVDGQKF | Interleukin enhancer-binding factor 3 | Known immune epitope IEDB |
| 12 | SEIELFRVF | U5 small nuclear ribonucleoprotein 200 kDa helicase | known immune epitope IEDB and A |
| 13 | TLWVDPYEV | B-cell Translocation Gene 1 | Known immune epitope IEDB |
| 14 | NEAIMHQY | Protein FAM111B | Novel |
| 15 | MEQVIFKY | ARP3 actin-related protein 3 homolog; | B |
| 16 | ETERIVTTY | Dynamin 2 | Novel |
| 17 | NQVIFPVSY | Mak3 homolog | Known immune epitope IEDB |
| 18 | EENPIVLEF | Kanadaptin | Known immune epitope IEDB |
| 19 | LTEIKGSVY | Zinc finger with UFM1-specific peptidase domain protein | Novel |
| 20 | ADKVHLMY | E3 ubiquitin/ISG15 ligase, Tripartite motif-containing 25 | Novel |
| 21 | TVDDPYATFV | Cofilin-1 | Novel |
| 22 | DVDPETLSY | Exonuclease I | Novel |
| 23 | TEFPNFKY | Replication protein A 70 kDa DNA-binding subunit | Novel |
| 24 | TVDPASLWEY | Fascin homolog 1 | Novel |
| 25 | VEIITKEF | PMPCA protein | Novel |
| 26 | YTELLAQVY | Solute carrier family 25 member 35 | Novel |
| 27 | ASDGTVRL | Heterogeneous nuclear ribonucleoprotein H3 | Novel |
| 28 | ASEIAVGHQY | Predicted Putative solute carrier family 25 member 35 | Novel |
| 29 | DENFILKH | Peptidyl-prolyl cis-trans isomerase A | B |
| 30 | LLYESPERY | Antigen peptide transporter 1 | Known immune epitope IEDB |
| 31 | NEYLNPEL | Histone chaperone ASF1B | Novel |
| 32 | DEAGGRFVAF | Ubiquitin fusion degradation 1 like | Novel |
| 33 | DEWKAIQN | SERPINE1 mRNA binding protein 1 | Novel |
| 34 | EEFETIERF | Chromodomain-helicase-DNA-binding protein 1 | B and C |
| 35 | DEKQQHIVY | E3 ubiquitin-protein ligase Mdm2 | D and known CLL ligand |
| 36 | AEQKKLEAA | NAD(P)H dehydrogenase [quinone] 1 | Novel |
| 37 | VTEAIQAVL | WD repeat-containing protein 72 | Novel |
| 38 | NLAEKLIGV | Ral GTPase-activating protein subunit beta | A (2011) |
| 39 | DEKSIITY | Plectin | E |
| 40 | KLLEVQILE | GRIP & coiled-coil domain-containing protein 2 | A (2011) |
| 41 | FGGLGGGSVR | Keratin, type I cytoskeletal 19 | Novel |
| 42 | GLGGGSVRFG P | Keratin, type I cytoskeletal 19 | Novel |
| 43 | YTSGPGSRIS | Keratin, type II cytoskeletal 8 | Novel |
| 44 | YTSGPGSRISS | Keratin, type II cytoskeletal 8 | Novel |
| 45 | VKLAKAGKN | Nucleolin | Found on HEK293 cells |
| 46 | ILIDWLVQV | G2/mitotic-specific cyclin-B1 | Known immune epitope |
| 41 | KMDASLGNLFA | Protein FAM3 C | F |
| 42 | DEKPLVLEm | N-acetyltransferase 14 | Novel |
| 43 | AEISAMLKA | Pop1 | Novel |
| 44 | RTLAEIAKV | Non-POU domain-containing octamer-binding protein | Known immune epitope IEDB |
| 45 | KIFEMGPVFTL | Cytochrome c oxidase subunit II | Novel |
| 46 | SEIYIHGL | Ribonuclease P protein subunit p20 | Novel |
| 47 | QAEFQILKA | MORC family CW-type zinc finger protein 4 | Novel |
| 48 | KEMPVKVEA | Importin-8 isoform 2 | Novel |
| 49 | TELLIRKL | Histone H3.3 C | Known immune epitope IEDB |
| 50 | SEYQWITSP | Centrosomal protein of 78 kDa | Novel |
| 51 | GSDDGTVKL | 38kDa splicing factor | Novel |
| 52 | TLTEEGVIKV | GTP binding protein 4 | Known immune epitope IEDB |
| 53 | DEVVWVRA | Aspartyl-tRNA synthetase | Novel |
| 54 | DEMNVKVL | MYL6 protein | Found in thymus |
| 55 | SEAEIFYNA | Plakophilin 3 | Novel |
| 56 | LTDDDLLRY | 1,4-alpha-glucan-branching enzyme | Known immune epitope IEDB |
| 57 | NETDILSQY | Metastasis-associated protein MTA2 | Novel |
| 58 | LEAHRDAPGA | Serine/threonine-protein phosphatase 6 regulatory subunit 2 | Novel |
| 59 | QEYSEFVKA | PTPL1-associated RhoGAP 1 variant | Novel |
| 60 | AEILSEMRA | 2 '-5 ' oligoadenylate synthetase 3 | Novel |
| 61 | ATEYKNEEY | YTH domain-containing protein 1 isoform2 | Known immune epitope IEDB |
| 62 | VEHKVETF | 40 S ribosomal protein S7 | Known immune epitope IEDB |
| 63 | NEVPVKEL | Leucine-rich repeat-containing protein 59-like | Novel |
| 64 | DELEVIHL | Ro ribonucleoprotein | Known immune epitope IEDB |
| 65 | FLLGPRLVLA | transmembrane emp24 domain-containing protein 10 precursor | Novel |
| 66 | EEEFFYEKA | BRCA2 and CDKN1 A interacting protein | Novel |
| 67 | LLLDLERVL | Cullin-9 | Novel |
| 68 | VMAPRTLVL | HLA class I histocompatibility antigen, A-2 alpha chain precursor | Known immune epitope IEDB – HLA-E binding HLA-2 leader sequence |
| 69 | PDPIRGFGS | Putative poly(ADP-ribosyl) transferase | Novel |
| 70 | KTDKTLVLL | Profilin-1 | A (2011) |
| 71 | DEHEGPALY | Proteasome subunit beta type-2 | Known immune epitope IEDB and B |
| 72 | QMDGPLPLHY | Sestrin-1 | Novel |
| 73 | SSDRHLTQY | Chromodomain helicase DNA binding protein 1 | Novel |
| 74 | GEKRFADAA | Nuclear pore complex protein Nup85 | Known immune epitope IEDB |
| 75 | SLLPHETLL | Pyruvate dehyrogenase phosphatase catalytic subunit 1 | Novel |
| 76 | HSDPSILGY | GIGYF1 protein | Novel |
| 77 | YLDPRITVA | DNA topoisomerase | Novel |
| 78 | AVLELKNEL | Protein-tyrosine kinase 2-beta | Novel |
| 79 | EPAQVSLLY | Ubiquitin carboxyl-terminal hydrolase 40 | Novel |
| 80 | ATLVRSPGP | Junction-mediating and -regulatory protein | Novel |
MEK and NFκB are involved in GEM mediated HLA-A,B,C upregulation
In accordance with previous reports, culturing tumour cells with GEM was found to activate the MEK signalling pathway, increasing phosphorylation of ERK1/2 (Fig. 5a). To test whether this was involved in upregulation of HLA-A,B,C and the changes in the peptide repertoire, phosphorylation of ERK1/2 was inhibited by U0126 in GEM-treated HCT116 cells and the effect on HLA-A,B,C surface expression and intracellular LMP2 measured. Inhibition of ERK1/2 phosphorylation was demonstrable by Western blot (Fig. 5d) and reduced GEM-induced HLA-A,B,C in a dose-dependent manner (Fig. 5b). GEM alone increased expression of HLA-A,B,C by a mean of 78% compared to matched untreated control but this was reduced to 57% when 1 μM U0126 was added to the culture medium and 23% at 10 μM of U0126. It should be noted that treatment with U0126 alone did increase basal expression of HLA-A,B,C.
Figure 5.
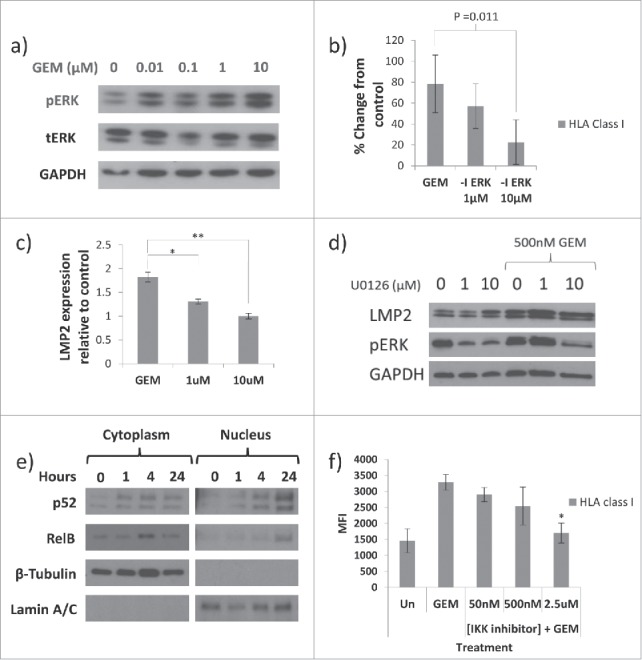
MEK and NFκB are associated with GEM-mediated HLA-A,B,C upregulation. For all panels n = 3 and involve HCT116 cells unless otherwise stated. a) Phosphorylation of ERK1/2 was increased in response to GEM in HCT116, A549 and MCF-7 cells. Representative blots from MCF-7 cells are shown. b) GEM-mediated HLA-A,B,C upregulation was reduced by inhibiting ERK signalling. HCT116 cells were untreated (Un) or cultured with 100 nM GEM, +/− ERK inhibitor U0126 (-I ERK) at 1 μM or 10 μM and HLA-A,B,C measured by flow cytometry. n = 4. c) As b) but measuring intracellular levels of LMP2 expression by Western blot. d) Representative blot showing the effect of U0126 on expression of LMP2 and pERK. e) Expression of p52 and RelB were measured in the cytoplasmic and nuclear fractions of HCT116 cells treated with GEM for various durations. Representative blots are shown. Anti-β-tubulin and lamin A/C antibodies used as loading and purity controls for cytoplasmic and nuclear fractions, respectively. f) HCT116 tumour cells were treated with 100 nM GEM +/−IKK-specific NFκB inhibitor. The effect on upregulation of HLA-A,B,C at the surface of the tumour cells was then assessed. b), c) and f) Values significantly different from GEM-treated by one-way ANOVA with Dunnett's test for multiple comparisons are shown. * = p<0.05, ** = p<0.01.
Western blots showed that GEM-mediated LMP2 upregulation was also reduced when inhibiting phosphorylation of ERK1/2 (Fig. 5c and d). GEM alone increased relative expression of LMP2 1.8-fold above untreated controls, however, this was reduced to 1.3-fold and to basal levels by 1 μM and 10 μM U0126, respectively. As with HLA-A,B,C, use of the MEK inhibitor alone was associated with increased LMP2 (Fig. 5d).
In addition to the MEK pathway, NFκB was also involved in GEM-mediated HLA-A,B,C upregulation. Culture with GEM caused nuclear translocation of the NFκB subunits RelB and p52 in HCT116 cells. No effect on the p65 subunit of NFκB was observed (data not shown). The involvement of NFκB in GEM-mediated HLA-A,B,C upregulation was confirmed by inhibiting NFκB with IKK-16. Expression of HLA-A,B,C was reduced from a mean MFI of 3288 units with GEM alone to 1698 units with 2.5 μM IKK-16 also present. These data suggest that GEM activates non-canonical NFκB signalling and that this is important in the regulation of HLA-A,B,C at the surface of tumour cells.
Discussion
In this study it was found that the chemotherapeutic drug, GEM, increased expression of HLA-A,B,C on the surface of a number of different tumour cell lines. This upregulation occurred using low concentrations of GEM and was associated with increased levels of β2 m. Moreover, this study demonstrated that GEM induced immunoproteasome subunit expression and that this corresponded to changes in the immunopeptidome of HCT116 cells, which included the display of novel potential T-cell epitopes not present on control cells.
MHC class I is often down-modulated in tumour cells, rendering them less susceptible to CD8+ T-cell-mediated killing, and it is presently unclear whether the changes in HLA-A,B,C observed in response to GEM represent an increase above basal levels or some correction of previous downregulation. However, culturing cell lines with GEM upregulated β2 m but not α-heavy chain expression and reduction of MHC class I expression in cancer frequently occurs through defects in β2 m which are often reversible. 34 , 35 It is interesting to note that transfection with β2 m protein alone was able to increase HLA-A,B,C at the surface of the tumour cells tested here. Because of the role of β2 m in the MHC class I folding, mutation of the β2 m gene leads to the gradual loss of HLA class I and immune escape during tumour progression. 36 β2 m deficiency, through mutation or other means, is reported in a number of cancer types 37-39 and is proposed as a reason for the failure of check-point blockade therapy. 40 Our data demonstrate that through modulation of β2 m, GEM increases MHC class I expression, suggesting the potential to correct non-mutational MHC class I deficiency in tumour cells by using GEM.
MHC class I expression increases in response to various chemotherapy drugs and radiation. 11 , 12 , 41 , 42 This may form part of a normal stress response and represents an increase in the quantity of the MHC class I-antigen complex on the surface of tumours. Increasing the number of antigens on a tumour cell is, on balance, a good strategy to elicit an immune response against tumour. However, what may be equally or even more important than the quantity of the antigens presented, is the quality of the antigens and a major finding of the present study is that culturing with GEM altered the nature of peptide antigens displayed on HLA class I by tumour cells.
Exposure to 100 nM GEM increased expression of the immunoproteasomal subunits LMP2 and MECL-1 in tumour cells and these proteins were detected in proteasomes using α4 pull-outs. No increases were found in the total amount of LMP7 present in tumour cells in response to GEM, though LMP7 was found in proteasomes. Proteasomes can exist as constitutive, mixed, or immunoproteasomes, dependent on which catalytic subunits are present and so the precise composition of proteasomes in GEM-treated HCT116 cells is not currently known. However, the induction of immunoproteasomal subunits by GEM suggests an alteration in the way in which proteins are degraded and may help explain the new pattern of peptides within the tumour cell immunopeptidome. 43 It is suggested that immunoproteasomally cleaved peptides may be distinguished by their tendency to have particular hydrophobic amino acids, such as phenylalanine or leucine, at the C-terminus. 44-46 Data presented in the present study also indicate an increase in the proportion of peptides with phenylalanine or leucine at the C-terminus eluted from HLA-A,B,C after culture with GEM. An interesting further study would be to compare the peptide antigens eluted from GEM-treated cells to those eluted from IFNγ-treated cells which should have a full immunoproteasome.
Peptides eluted from GEM-treated HCT116 cells had shifted in character so that a higher proportion were predicted to bind HLA-B*18. This may be a consequence of an increase in shorter peptides found after GEM-treatment, as HLA-B*18 molecules have a preference for binding shorter peptides in their binding clefts. 47 Shorter peptides may allow a broader response, as is the case for an HLA-B*18 bound 8-mer from EBV which has been reported to initiate stronger T-cell responses to a wider range of peptide epitopes compared to the 12-mer peptide displayed on HLA-B*44. 48 Additionally, CTLs have been shown to lyse cells expressing HLA-B*18 bound 8-mer peptides more strongly than other peptides bound to other HLA-allotypes. 49 HLA-B*18 may be of particular importance in immunity. It is associated with autoimmune diseases such as multiple sclerosis, coeliac disease and diabetes mellitus; 50-52 and multiple cancers present with a loss or downregulation of HLA-B*18 expression. 53-55 Increasing the proportion of peptides that bind to “immune-relevant” alleles may enhance immunity. However, GEM-induced changes to antigen binding could also be detrimental to an immune response if the resultant peptides are designated for HLA alleles lost in loss of heterozygosity mutations. Here, peptides would be generated but unable to be displayed efficiently, potentially undermining an antigen-specific immune response. These types of mutations are common in cancer and may limit the use of GEM as an immune potentiator in this disease. 62 , 63
Altered proteasomal cleavage specificity may explain the differences observed in peptides eluted from control or GEM-treated cells; however, TAP-independent mechanisms may play a role, as could changes in proteasome entry requirements or changes to peptide trimming, transport and loading. 64-66 Whatever the true mechanism, GEM-induced changes to the conformation of HLA-ligands have the potential to increase the ability of the adaptive immune system to propagate an effective immune response towards them.
It is reported that a discrepancy exists between the epitopes cross-presented by DCs and those displayed on tumour cells. 67 GEM may overcome this by inducing immunoproteasomes in cancer cells and shifting the peptides displayed on HLA class I molecules to be more similar to those generated and cross-presented by APCs. Interestingly, in the above study involving NY-ESO-1, the efficiently cross-presented, immunoproteasomally generated NY-ESO-188-96 peptide was HLA-B*18 restricted, while the non-immunogenic, NY-ESO-1157-165, was HLA-A*02 restricted.
Numerous peptides were exclusively found after GEM treatment and not on control cells, raising the possibility of using some of these novel, GEM-inducible antigens in future immunotherapy-GEM combinations. Some GEM-inducible peptides were derived from tumour-associated proteins, suggesting that GEM-treatment may reveal tumour antigens to immune cells. There were also a number of novel peptides from proteins intimately associated with the DNA-damage response (DDR) and these may represent a signal to the immune system further to the well-known innate signals induced by DNA-damage, such as increased expression of CD95 or NKG2D ligands. Many of the peptides found only on GEM-treated cells have previously been observed on other cell types, including: patented immunogens and tumour-associated peptides. The GEM-inducible peptide, ILIDWLVQV, has been identified by reverse immunology and shown to elicit spontaneous T-cell reactivity in blood from cancer patients and healthy donors. Additionally, there were GEM-inducible peptides previously described as “unique” to HIV MN-1-infected human T-cells; an interesting finding in the context of the work presented here, as GEM-treatment and viral-infection both induce immunoproteasomes and provide danger signals to immune cells. Further to those previously reported immunogens, the 53 novel peptides found in response to GEM represent new HLA-ligands that may be chemotherapy-specific and novel epitopes for cytotoxic T-cells.
A primary function of the adaptive immune response is to protect the integrity of the genome and as such the antigenic targets of T-cells are often evidence of some defect in the DNA of a cell, the most pertinent example of this in cancer being the “targetability” of neoantigens. Neoantigens, tumour-associated antigens, and antigens corresponding to other forms of DNA damage have consistently been shown to elicit robust and effective CD8+ T-cell responses. In the present study, many of the GEM-inducible novel peptides appearing on HLA-A,B,C were linked to the DDR, which may suggest that self-antigens from DDR proteins are immunogenic. A theory conceptually advantageous as it would allow rapid responses by a less clonally diverse pool of T-cells against cells harbouring DNA damage, and plausible, given that these antigens will often be perceived by the immune system under inflammatory conditions and the existence of T-cells specific to DDR-related protein antigens such as MDM2 and TOPO1 in the periphery. 68 , 69 If GEM is providing a mechanism for these danger signals to be displayed on tumour cells then this helps explain the reported synergy between the drug and immunotherapy. Renovating the peptidome in this way may be a prime strategy to reengage immune responses against tumours and could also play a role in autoimmune disease and toxicities associated with GEM.
GEM-mediated cellular damage, genomic or otherwise, may generate peptides normally only seen on diseased or damaged cells making these cells targetable by the immune system. This concept increases understanding about the improved efficacy of chemotherapies in immunocompetent individuals and helps to identify mechanisms by which GEM has been shown to alter the antigen-specificity of primed CD8+ T-cells towards subdominant epitopes. GEM may not be unique in this action but understanding the relationship between drug treatment and altered immunopeptidome may be important in boosting T-cell responses to cancer and poses questions as to which antigens should be chosen to combine with chemotherapy. This work suggests a rationale for combining GEM-treatment with immunotherapies, such as anti-PD-1, as increasing the antigenicity of the tumour will render it more susceptible to antigen-specific CD8+ T-cells that may be unconstrained by checkpoint blockade therapy.
Methods
Tumour cell lines
The human cancer cell lines: A549 (lung), HCT116 (colon) and MCF7 (breast) (all Public Health England, Porton down, UK), were grown in DMEM (Sigma-Aldrich, Dorset, UK) medium supplemented with 10% foetal bovine serum (Invitrogen, Paisley, UK) and 1% penicillin/streptomycin (Sigma). All cell lines were incubated in a humidified atmosphere with 5% CO2 in air at 37 °C. For in vitro experiments, cells were seeded at 1 × 105 cells/ml and allowed to attach overnight before addition of experimental reagents. All experiments represent 24 hour treatment of cells with 100 nM GEM unless otherwise stated.
Drugs, cytokines and inhibitors
GEM, OXP, CPM (all Sigma) and IFNγ (R & D biosystems, Abingdon, UK) were reconstituted in phosphate buffered saline (PBS) (Sigma). IC25 values used for GEM, OXP and CPM were: 100 nM, 500 nM and 1μM, respectively. Cell signalling was inhibited by U0126 (New England Biolabs (NEB), Hitchin, UK) and NFκB by IKK-16 (Selleck Chemicals).
Flow cytometry
Cells were stained with HLA-A,B,C-FITC (Becton Dickinson (BD), Oxford, UK) using manufacturer's instructions prior to analysing with LSRII flow cytometer (BD Biosciences).
Immunoblotting
Cells were solubilised in lysis buffer (NEB) and resolved by tris-glycine electrophoresis. Following transfer of proteins onto nitrocellulose membranes (ThermoFisher Scientific, Dartford, UK), staining was performed with primary and then horseradish peroxidase (HRP)-conjugated anti-species secondary antibodies. Primary antibodies used were specific for: proteasome subunits α2, β1 and β2, β5 (all Enzo Life Sciences, Exeter, UK), β2 m (Abcam, Cambridge, UK), HLA-A, B and C (all Santa-Cruz Biotechnologies, California, USA), LMP2 (β1i), LMP7 (β5i) and MECL-1 (β2i) (all Abcam), pERK, tERK, RelB, p50, p52, p65 (all NEB). Bands were visualised on Hyperfilm™ ECL (GE Healthcare, Buckinghamshire, UK) using SuperSignal™ West Pico chemiluminescent substrate (Thermo). The house-keeping protein, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (NEB) was used as a loading control.
Separation of Nuclear and Cytoplasmic Fractions
Cell lysates were separated into nuclear and cytoplasmic fractions using NE-PER® Nuclear and Cytoplasmic Extraction reagents (Thermo) and following manufacturer's instructions. Briefly, Cytoplasmic Reagent (CER) I was added to cells before vortexing vigorously and addition of CER II. Cells were then centrifuged at 16000 g for 5 minutes and the supernatant (cytoplasmic extract) isolated and stored. The cell pellets were resuspended in Nuclear Extraction Reagent and incubated on ice for 40 minutes with vigorous vortexing at regular intervals. The resulting lysate was then centrifuged at 16000 g for 10 minutes and the supernatant (nuclear extract) stored at -80oC. After separation, cytoplasmic and nuclear fractions were treated as total cellular lysates in the previous section. Lamin A/C (NEB) and β-tubulin (NEB) were used to determine the quality of the separation and as loading controls for the nuclear and cytoplasmic protein fractions, respectively.
Real-time reverse transcriptase polymerase chain reaction
High purity RNA was isolated from HCT116 cells with the Qiagen RNA extraction mini-prep kit (Qiagen, Manchester, UK) using manufacturer's instructions. This was converted to cDNA by reverse transcription using precision RT all-in-one mix (Primer Design Ltd, Southampton, UK). Gene expression of β2 m was then measured by qPCR, using QuantiTect® Sybr® Green-based qPCR primers (Qiagen, Manchester, UK). Approximately 40 ng cDNA (4 μl), 1 μl QuantiTect® primer and 10 μl SsoFast™ EvaGreen® Supermix (Bio-rad, Hertfordshire, UK) were added to the qPCR reaction mix. 18 S ribosomal RNA was used as the reference gene.
Illumina microarray
RNA was isolated from HCT116 cells using the Qiagen mini-kit protocol as before. Microarrays were performed by Dr Jayne Dennis at the SGUL Biomics Centre as described previously. 70
Transfections
Tumour cells were transfected with a human β2 m (NM_004048) expressing, pCMV6-XL5 plasmid (OriGene, Herford, Germany) using Lipofectamine® LTX (HCT116 cells) or Lipofectamine® 3000 (A549 and MCF-7 cells) (Invitogen) and following manufacturer's instructions. Briefly, DNA-lipid complexes were added to cells before incubating for 48 hours at 37oC and 5% CO2 in a humidified atmosphere. Expression of HLA-A,B,C at the cell surface was then determined by flow cytometry. Cells “mock transfected” with a non-encoding plasmid were used as a negative control
HLA ligand elution and sequencing
HCT116 cells were lysed in buffer containing PBS, 0.6% CHAPS, and complete protease inhibitor (Roche). Following centrifugation to remove debris supernatant was applied on affinity columns overnight. Columns were prepared by coupling W6/32 antibodies to CNBr-activated Sepharose (GE Healthcare) (1 mg antibody/40 mg Sepharose). On the second day the columns were eluted in 8 steps using 0.2% TFA. Filtration of the eluate through a 10 kDa filter (Merck Millipore, Darmstadt, Germany) yielded the HLA ligands in solution. The filtrate was desalted with C18 ZipTips (Merck) and subsequently concentrated using a vacuum centrifuge (Bachofer, München, Germany). Sample volume was adjusted for measurement by adding 1% ACN/0.05% TFA (v/v). With an injection volume of 5 µl HLA ligands were loaded (100 µm x 2 cm, C18, 5 µm, 100 Å) and separated (50 µm x 25 cm, C18, 3 µm, 100 Å) on Acclaim Pepmap100 columns (Dionex, Sunnyvale, CA) using an Ultimate 3000 RLSCnano uHPLC system (Dionex). A gradient ranging from 2.4-32% of ACN/H2O with 0.1% formic acid was used to elute the peptides from the columns over 90 min at a flow rate of 300 nl/min. Online electrospray ionization (ESI) was followed by tandem MS analysis in a LTQ Orbitrap XL instrument (Thermo Fisher Scientific, Bremen, Germany). Survey scans were acquired in the Orbitrap mass analyser with a resolution of 60.000 and a mass range of 400 to 650 m/z. Peptides with a charge state other than 2+ or 3+ were rejected from fragmentation. Fragment mass spectra of the 5 most intense ions of each scan cycle were recorded in the linear ion trap (top5 CID). Normalized collision energy of 35%, activation time of 30 ms and isolation width of 2 m/z was utilized for fragment mass analysis. Dynamic exclusion was set to 1s. The RAW files were processed against the human proteome as comprised in the Swiss-Prot database (www.uniprot.org, status: Dec12th, 2012; 20.225 reviewed sequences contained) using MASCOT server version 2.3.04 (Matrix Science, Boston, MA) and Proteome Discoverer 1.4 (Thermo). Oxidation of methionine was allowed as dynamic peptide modification. A mass tolerance of 5 ppm or 0.5 Da was allowed for parent and fragment masses respectively. Filtering parameters were set to a Mascot Score<20, search engine rank = 1, peptide length of 8–12 AA, achieving a false discovery rate (FDR) of 5% as determined by an inverse decoy database search.
Peptide sequence analysis
Peptides was stratified into two groups: those sequences that were unique to control HCT116 cells and those that were unique to GEM-treated HCT116 cells. These peptide groups were then subjected to various tests, including analysis of: length, C-terminal amino acid, predicted HLA-allele binding and predicted immunogenicity. The length and C-terminal amino acid of each peptide was determined manually. MHC class I binding predictions were made with IEDB Analysis Resource (http://tools.immuneepitope.org/mhci/) and SYFPEITHI Epitope Prediction (http://www.syfpeithi.de/bin/MHCServer.dll/EpitopePrediction.htm) using the HLA-alleles HLA-A*01:01, HLA-A*02:01, HLA-B*18:01 and HLA-B*45:01. Predicted Immunogenicity of the peptides was determined using a model, IEDB Analysis Resource (http://tools.iedb.org/immunogenicity/), where the peptides were analysed for the presence of particular immunogenicity-associated amino acids at specific positions in the peptide sequence. 33 N.B. Due to a technical issue with the peptide length data in one of the three peptide elution experiments, this data was removed from the analysis of Fig. 3a and replaced by peptide length data from a small pilot study.
Proteasome isolation
Proteasomes were purified using the Enzo Protein Purification Kit (Enzo Life Sciences, Exeter, UK) and following manufacturer's instructions. Briefly, cells were lysed for 15 minutes on ice before centrifuging at high speed to clear the supernatant. A sample of this crude lysate was put aside for analysis and proteasome purification matrix (agarose immobilised anti-proteasome subunit-α4 antibodies) added to the remaining supernatant (25 μl matrix/100 μg protein). After incubation overnight, the matrix was isolated by centrifugation for 30 seconds at 5000 g and the supernatant (unbound fraction) set aside for analysis. Proteasomes were eluted from the matrix by the addition of LDS and boiling for 10 minutes.
Supplementary Material
Funding Statement
Institute for Cancer Vaccines and Immunotherapy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was kindly supported by the Institute for Cancer Vaccines and Immunotherapy (ICVI). Peptide elution work was completed at the University of Tübingen and supported by the Deutsche Forschungsgemeinschaft (DFG, SFB 685).
References
- 1. Zhao X, Sun Q, Tian H, Cong B, Jiang X, Peng C. Loss of heterozygosity at 6p21 and HLA class I expression in esophageal squamous cell carcinomas in China. Asian Pac J Cancer Prev. 2011;12:2741–2745. [PubMed] [Google Scholar]
- 2. Kaneko K, Ishigami S, Kijima Y, Funasako Y, Hirata M, Okumura H, Shinchi H, Koriyama C, Ueno S, Yoshinaka H, et al.. Clinical implication of HLA class I expression in breast cancer. BMC Cancer. 2011;11:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Romero JM, Jiménez P, Cabrera T, Cózar JM, Pedrinaci S, Tallada M, Garrido F, Ruiz-Cabello F. Coordinated downregulation of the antigen presentation machinery and HLA class I/beta2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int J Cancer 2005; 113:605–610. [DOI] [PubMed] [Google Scholar]
- 4. Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195:346–355. [DOI] [PubMed] [Google Scholar]
- 5. Nie Y, Yang G, Song Y, Zhao X, So C, Liao J, Wang LD, Yang CS. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22:1615–1623. [DOI] [PubMed] [Google Scholar]
- 6. Imanishi T, Kamigaki T, Nakamura T, Hayashi S, Yasuda T, Kawasaki K, Takase S, Ajiki T, Kuroda Y. Correlation between expression of major histocompatibility complex class I and that of antigen presenting machineries in carcinoma cell lines of the pancreas, biliary tract and colon. Kobe J Med Sci. 2006;52:85–95. [PubMed] [Google Scholar]
- 7. Cabrera T, Lara E, Romero JM, , Maleno I, Real LM, Ruiz-Cabello F, Valero P, Camacho FM, Garrido F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol Immunother. 2007;56:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seliger B, Stoehr R, Handke D, Mueller A, Ferrone S, Wullich B, Tannapfel A, Hofstaedter F, Hartmann A. Association of HLA class I antigen abnormalities with disease progression and early recurrence in prostate cancer. Cancer Immunol Immunother. 2010;59:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeestraten EC, Reimers MS, Saadatmand S, Goossens-Beumer IJ, Dekker JW, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br J Cancer. 2014;110:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohtsukasa S, Okabe S, Yamashita H, Iwai T, Sugihara K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J Cancer Res Clin Oncol. 2003;129:719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rammensee HG, Falk K, Rötzschke O. Peptides naturally presented by MHC class I molecules. Annu Rev Immunol. 1993;11:213–244. [DOI] [PubMed] [Google Scholar]
- 14. Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL. 26 S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J. 2001;20:2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997;272:25200–25209. [DOI] [PubMed] [Google Scholar]
- 16. Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci. 2011;68:1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999;17:739–779. [DOI] [PubMed] [Google Scholar]
- 18. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 19. Bauer C, Bauernfeind F, Sterzik A, Orban M, Schnurr M, Lehr HA, Endres S, Eigler A, Dauer M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut. 2007;56:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghansah T, Vohra N, Kinney K, Weber A, Kodumudi K, Springett G, Sarnaik AA, Pilon-Thomas S. Dendritic cell immunotherapy combined with gemcitabine chemotherapy enhances survival in a murine model of pancreatic carcinoma. Cancer Immunol Immunother. 2013;62:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kimura Y, Tsukada J, Tomoda T, Takahashi H, Imai K, Shimamura K, Sunamura M, Yonemitsu Y, Shimodaira S, Koido S, et al.. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas. 2012;41:195–205. [DOI] [PubMed] [Google Scholar]
- 22. Hirooka Y, Itoh A, Kawashima H, Hara K, Nonogaki K, Kasugai T, Ohno E, Ishikawa T, Matsubara H, Ishigami M, et al.. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas. 2009;38:e69–74. [DOI] [PubMed] [Google Scholar]
- 23. Mayanagi S, Kitago M, Sakurai T, Matsuda T, Fujita T, Higuchi H, Taguchi J, Takeuchi H, Itano O, Aiura K, et al.. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015;106:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. [DOI] [PubMed] [Google Scholar]
- 25. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–909. [DOI] [PubMed] [Google Scholar]
- 26. Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D, et al.. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother. 2014;63:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daikeler T, Maas K, Hartmann JT, Kanz L, Bokemeyer C. Weekly short infusions of gemcitabine are not associated with suppression of lymphatic activity in patients with solid tumors. Anticancer Drugs. 1997;8:643–644. [DOI] [PubMed] [Google Scholar]
- 28. Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res. 2002;62:2353–2358. [PubMed] [Google Scholar]
- 29. Plate JM, Plate AE, Shott S, Bograd S, Harris JE. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother. 2005;54:915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, Frelinger JA, Robinson BW. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170:4905–4913. [DOI] [PubMed] [Google Scholar]
- 31. Soeda A, Morita-Hoshi Y, Makiyama H, et al.. Regular dose of gemcitabine induces an increase in CD14+ monocytes and CD11 c+ dendritic cells in patients with advanced pancreatic cancer. Jpn J Clin Oncol. 2009;39:797–806. [DOI] [PubMed] [Google Scholar]
- 32. Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol Immunother. 2012;61:2343–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD Sette A, Keşmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9:e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garrido F, Cabrera T, Aptsiauri N “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer. 2010;127:249–256. [DOI] [PubMed] [Google Scholar]
- 35. Bicknell DC, Rowan A, Bodmer WF. Beta 2-microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proc Natl Acad Sci U S A. 1994;91:4751–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. del Campo AB, Kyte JA, Carretero J, Zinchencko S, Méndez R, González-Aseguinolaza G, Ruiz-Cabello F, Aamdal S, Gaudernack G, Garrido F, et al.. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer. 2014;134:102–113. [DOI] [PubMed] [Google Scholar]
- 37. Chang CC, Ogino T, Mullins DW, Oliver JL, Yamshchikov GV, Bandoh N, Slingluff CL Jr, Ferrone S. Defective human leukocyte antigen class I-associated antigen presentation caused by a novel beta2-microglobulin loss-of-function in melanoma cells. J Biol Chem. 2006;281:18763–18773. [DOI] [PubMed] [Google Scholar]
- 38. Browning M, Petronzelli F, Bicknell D et al.. Mechanisms of loss of HLA class I expression on colorectal tumor cells. Tissue Antigens. 1996;47:364–371. [DOI] [PubMed] [Google Scholar]
- 39. Bernal M, Ruiz-Cabello F, Concha A, Paschen A, Garrido F. Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol Immunother. 2012;61:1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al.. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tseng CW, Hung CF, Alvarez RD, Trimble C, Huh WK, Kim D, Chuang CM, Lin CT, Tsai YC, He L, et al.. Pretreatment with cisplatin enhances E7-specific CD8+ T-Cell-mediated antitumor immunity induced by DNA vaccination. Clin Cancer Res. 2008;14:3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One. 2012;7:e32542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vigneron N, Van den Eynde BJ. Proteasome subtypes and the processing of tumor antigens: increasing antigenic diversity. Curr Opin Immunol. 2012;24:84–91. [DOI] [PubMed] [Google Scholar]
- 44. Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, Laplace C, Zwinderman A, Dick TP, Müller J, et al.. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med 2001; 194:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kesmir C, van Noort V, de Boer RJ, Hogeweg P. Bioinformatic analysis of functional differences between the immunoproteasome and the constitutive proteasome. Immunogenetics. 2003;55:437–449. [DOI] [PubMed] [Google Scholar]
- 46. Tenzer S, Stoltze L, Schönfisch B, et al.. Quantitative analysis of prion-protein degradation by constitutive and immuno-20 S proteasomes indicates differences correlated with disease susceptibility. J Immunol. 2004;172:1083–1091. [DOI] [PubMed] [Google Scholar]
- 47. Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, Sullivan LC, Burrows JM, Miles JJ, Brennan RM, et al.. HLA peptide length preferences control CD8+ T cell responses. J Immunol. 2013;191:561–571. [DOI] [PubMed] [Google Scholar]
- 48. Burrows SR, Rossjohn J, McCluskey J. Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 2006;27:11–16. [DOI] [PubMed] [Google Scholar]
- 49. Bourgault Villada I, Bénéton N, Bony C, Connan F, Monsonego J, Bianchi A, Saiag P, Lévy JP, Guillet JG, Choppin J. Identification in humans of HPV-16 E6 and E7 protein epitopes recognized by cytolytic T lymphocytes in association with HLA-B18 and determination of the HLA-B18-specific binding motif. Eur J Immunol. 2000;30:2281–2289. [DOI] [PubMed] [Google Scholar]
- 50. Trouillas P, Betuel H. Hypocomplementaemic and normocomplementaemic multiple sclerosis. Genetic determinism and association with specific HLA determinants (B18 and B7). J Neurol Sci. 1977;32:425–435. [DOI] [PubMed] [Google Scholar]
- 51. Congia M, Frau F, Lampis R, Frau R, Mele R, Cucca F, Muntoni F, Porcu S, Boi F, Contu L, et al.. A high frequency of the A30, B18, DR3, DRw52, DQw2 extended haplotype in Sardinian celiac disease patients: further evidence that disease susceptibility is conferred by DQ A1*0501, B1*0201. Tissue Antigens. 1992;39:78–83. [DOI] [PubMed] [Google Scholar]
- 52. Larsen CE, Alper CA. The genetics of HLA-associated disease. Curr Opin Immunol. 2004;16:660–667. [DOI] [PubMed] [Google Scholar]
- 53. Torres MJ, Ruiz-Cabello F, Skoudy A, Berrozpe G, Jimenez P, Serrano A, Real FX, Garrido F. Loss of an HLA haplotype in pancreas cancer tissue and its corresponding tumor derived cell line. Tissue Antigens. 1996;47:372–381. [DOI] [PubMed] [Google Scholar]
- 54. Cabrera T, Salinero J, Fernandez MA, Garrido A, Esquivias J, Garrido F. High frequency of altered HLA class I phenotypes in laryngeal carcinomas. Hum Immunol. 2000;61:499–506. [DOI] [PubMed] [Google Scholar]
- 55. Brady CS, Bartholomew JS, Burt DJ, Duggan-Keen MF, Glenville S, Telford N, Little AM, Davidson JA, Jimenez P, Ruiz-Cabello F, et al.. Multiple mechanisms underlie HLA dysregulation in cervical cancer. Tissue Antigens. 2000;55:401–411. [DOI] [PubMed] [Google Scholar]
- 56.Ramakrishna V, Ross MM, Philip R, Keller LH. Cytotoxic T-lympho-cyte-inducing immunogens for prevention, treatment, and diagnosis of cancer. 2011. [Google Scholar]
- 57.Hildebrand WH, Hawkins O. Comparative ligand mapping from MHC class I positive cells. 2011. [DOI] [PubMed] [Google Scholar]
- 58.Hickman HD, Luis AD, Buchli R, Few SR, Sathiamurthy M, Van-Gundy RS, Giberson CF, Hildebrand WH. Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J Immunol. 2004;172:2944–2952. [DOI] [PubMed] [Google Scholar]
- 59.Pfirschke C. Characterization of spontaneous tumor antigen-reactive T cell responses in melanoma patients and treatment of human mela- 885 noma with optimized T cell receptor transgenic T cells in a xenotransplantation model. Combined Faculties for the Natural Sciences and for Mathematics: University of Heidelberg. 2011;241. [Google Scholar]
- 60.Weinschenk T, Lemmel C, Rammensee H-G, Stevanovic S. Mhc mole-cule-binding tumor-associated peptides. 2005. [Google Scholar]
- 61.Weinzierl AO, Rudolf D, Hillen N, Tenzer S, van Endert P, Schild H, Rammensee HG, Stevanović S. Features of TAP-independent MHC class I ligands revealed by quantitative mass spectrometry. Eur J Immunol. 2008;38:1503–1510. [DOI] [PubMed] [Google Scholar]
- 62.Perea F, Bernal M, Sánchez-Palencia A, Carretero J, Torres C, Bayarri C, Gómez-Morales M, Garrido F, Ruiz-Cabello F. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int J Cancer 2017; 140:888–899. [DOI] [PubMed] [Google Scholar]
- 63.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–1271.e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.James E, Bailey I, Sugiyarto G, Elliott T. Induction of protective antitu- mor immunity through attenuation of ERAAP function. J Immunol. 2013;190:5839–5846. [DOI] [PubMed] [Google Scholar]
- 66.Schölz C, Tampé R. The peptide-loading complex–antigen translocation and MHC class I loading. Biol Chem. 2009;390:783–794. [DOI] [PubMed] [Google Scholar]
- 67.Zhao RY, Mifsud NA, Xiao K, Chan KF, Oveissi S, Jackson HM, Dimopoulos N, Guillaume P, Knights AJ, Lowen T, et al. A novel HLA-B18 restricted CD8C T cell epitope is efficiently cross-presented by dendritic cells from soluble tumor antigen. PLoS One. 2012;7:e44707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bendle GM, Xue SA, HollerA, Stauss HJ. A study of T cell tolerance to the tumor-associated antigen MDM2: cytokines can restore antigen responsiveness, but not high avidity T cell function. PLoS One. 2007;2:e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boin F, Wigley FM, Schneck JP, Oelke M, Rosen A. Evaluation of topoisomerase-1-specific CD8C T-cell response in systemic sclerosis. Ann N Y Acad Sci. 2005;1062:137–145. [DOI] [PubMed] [Google Scholar]
- 70.Liu WM, Dennis JL, Gravett AM, Chanthirakumar C, Kaminska E, Coulton G, Fowler DW, Bodman-Smith M, Dalgleish AG. Supernatants derived from chemotherapy-treated cancer cell lines can modify angiogenesis. Br J Cancer. 2012;106:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.